引用本文:
李刚, 陈烨, 夏纪宝. 过渡金属催化的基于季铵盐C-N键断裂的偶联反应研究进展[J]. 有机化学,
2018, 38(8): 1949-1962.
doi:
10.6023/cjoc201803013 Citation:
Li Gang, Chen Ye, Xia Jibao. Progress on Transition-Metal-Catalyzed Cross-Coupling Reactions of Ammonium Salts via C-N Bond Cleavage[J]. Chinese Journal of Organic Chemistry,
2018, 38(8): 1949-1962.
doi:
10.6023/cjoc201803013
School Chemistry of Chemical Engineering, Guizhou University, Guiyang 550025
b.
State Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of LICP, Lanzhou Institute of Chemical Physics(LICP), Chinese Academy of Sciences, Lanzhou 730000
Received Date:
12 March 2018 Revised Date:
08 April 2018 Available Online:
01 August 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21702212, 21772208, 21602230) and the Natural Science Foundation of Jiangsu Province (No. BK20161260)
Abstract:
Amines containing carbon-nitrogen (C-N) bonds are widely distributed in natural products, drug molecules and functional materials.C-N bonds are one of the most abundant and inert bonds in organic molecules.Selective cleavage of C-N bonds to construct carbon-carbon (C-C) and carbon-heteroatom (C-X) bonds represents a new synthetic method in organic synthesis.It is difficult for the direct cleavage of C-N bond.Ammonium salts are a series of stable compounds easily obtained from amines.The recent progress of transition-metal-catalyzed cross-coupling reactions via C-N bond cleavage using ammonium salts as starting materials is summerized.
(a) Paul, F. ; Patt, J. ; Hartwig, J. F. J. Am. Chem. Soc. 1994, 116, 5969. (b) Guram, A. S. ; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 7901. (c) Muci, A. R. ; Buchwald, S. L. Top. Curr. Chem. 2002, 219, 131. (d) Hartwig, J. F. Acc. Chem. Res. 2008, 41, 1534.
[2]
(a) Blanksby, S. J. ; Ellison, G. B. Acc. Chem. Res. 2003, 36, 255. (b) Yao, X. -Q. ; Hou, X. -J. ; Jiao, H. ; Xiang, H. -W. ; Li, Y. -W. J. Phys. Chem. A2003, 107, 9991.
[3]
(a) Bonanno, J. B. ; Henry, T. P. ; Neithamer, D. R. ; Wolczanski, P. T. ; Lobkovsky, E. B. J. Am. Chem. Soc. 1996, 118, 5132. (b) Ueno, S. ; Chatani, N. ; Kakiuchi, F. J. Am. Chem. Soc. 2007, 129, 6098. (c) Koreeda, T. ; Kochi, T. ; Kakiuchi, F. J. Am. Chem. Soc. 2009, 131, 7238. (d) Tobisu, M. ; Nakamura, K. ; Chatani, N. J. Am. Chem. Soc. 2014, 136, 5587. (e) Zhao, Y. ; Snieckus, V. Org. Lett. 2014, 16, 3200. (f) Cong, X. ; Fan, F. ; Ma, P. ; Luo, M. ; Chen, H. ; Zeng, X. J. Am. Chem. Soc. 2017, 139, 15182.
[4]
(a) Geng, W. ; Zhang, W. -X. ; Hao, W. ; Xi, Z. J. Am. Chem. Soc. 2012, 134, 20230. (b) Xie, Y. ; Hu, J. ; Wang, Y. ; Xia, C. ; Huang, H. J. Am. Chem. Soc. 2012, 134, 20613. (c) Hao, W. ; Geng, W. ; Zhang, W. X. ; Xi, Z. Chem. -Eur. J. 2014, 20, 2605.
(a) Ouyang, K. ; Hao, W. ; Zhang, W. -X. ; Xi, Z. Chem. Rev. 2015, 115, 12045. (b) Wang, Q. ; Su, Y. ; Li, L. ; Huang, H. Chem. Soc. Rev. 2016, 45, 1257. (c) Dander, J. E. ; Garg, N. K. ACS Catal. 2017, 7, 1413. (d) Takise, R. ; Muto, K. ; Yamaguchi, J. Chem. Soc. Rev. 2017, 46, 5864. (e) Gao, Y. ; Ji, C. -L. ; Hong, X. Sci. China: Chem. 2017, 60, 1413. (f) Liu, J. ; Yuan, S. ; Song, X. ; Qiu, G. Chin. J. Org. Chem. 2016, 1790. (g) Tang, H. ; Huo, X. ; Meng, Q. ; Zhang, W. Acta Chim. Sinica2016, 74, 219(in Chinese). (汤淏溟, 霍小红, 孟庆华, 张万斌, 化学学报, 2016, 74, 219. ) (h) Zeng, M. ; Song, C. ; Cui, D. Chin. J. Org. Chem. 2017, 37, 1352(in Chinese) (曾明, 宋婵, 崔冬梅, 有机化学, 2017, 37, 1352.
[8]
(a) Eds. : de Meijere, A. ; Diederich, F. ; Metal-Catalyzed CrossCoupling Reactions, Wiley-VCH, Weinheim, 2004, p. 437. (b) Eds. : de Meijere, A. ; Braese, S. ; Oestreich, M. Metal-Catalyzed Cross-Coupling Reactions And More, Wiley-VCH, Weinheim, 2014.
(a) Wenkert, E. ; Michelotti, E. L. ; Swindell, C. S. J. Am. Chem. Soc. 1979, 101, 2246. (b) Yu, D. -G. ; Li, B. -J. ; Shi, Z. -J. Acc. Chem. Res. 2010, 43, 1486. (c) Rosen, B. M. ; Quasdorf, K. W. ; Wilson, D. A. ; Zhang, N. ; Resmerita, A. -M. ; Garg, N. K. ; Percec, V. Chem. Rev. 2011, 111, 1346. (d) Su, B. ; Cao, Z. -C. ; Shi, Z. -J. Acc. Chem. Res. 2015, 48, 886. (e) Tobisu, M. ; Chatani, N. Acc. Chem. Res. 2015, 48, 1717.
[14]
(a) Clot, E. ; Eisenstein, O. ; Jasim, N. ; MacGregor, S. A. ; McGrady, J. E. ; Perutz, R. N. Acc. Chem. Res. 2011, 44, 333. (b) Keyes, L. ; Love, J. A. RSC Catal. Ser. 2013, 11, 159. (c) Yang, S. -D. In Homogeneous Transition-Metal-Catalyzed C-F Sctivation, John Wiley & Sons, New York, 2015, pp. 203~268.
(a) Katritzky, A. R. ; Gruntz, U. ; Kenny, D. H. ; Rezende, M. C. ; Sheikh, H. J. Chem. Soc., Perkin Trans. 11979, 430. (b) Katritzky, A. R. ; Marson, C. M. Angew. Chem., Int. Ed. 1984, 23, 420.
[33]
(a) Shao, Z. ; Li, X. ; Chang, H. ; Wei, W. Chemistry2013, 76, 704. (b) Dilman, A. D. ; Levin, V. V. Tetrahedron Lett. 2016, 57, 3986.
Yamamura, M. ; Moritani, I. ; Murahashi, S. -I. J. Organomet. Chem. 1975, 91, C39. (b) Murahashi, S. ; Yamamura, M. ; Yanagisawa, K. ; Mita, N. ; Kondo, K. J. Org. Chem. 1979, 44, 2408. (c) Giannerini, M. ; Fañanás-Mastral, M. ; Feringa, B. L. Nat. Chem. 2013, 5, 667.
(a) Gambarotta, S. ; Alper, H. J. Organomet. Chem. 1980, 194, C19. (b) Lei, Y. ; Zhang, R. ; Wu, Q. ; Mei, H. ; Xiao, B. ; Li, G. J. Mol. Catal. A 2014, 381, 120. (c) Lei, Y. ; Zhang, R. ; Wu, L. ; Wu, Q. ; Mei, H. ; Li, G. Appl. Organomet. Chem. 2014, 28, 310.
(a) Said, S. A. ; Fiksdahl, A. Tetrahedron: Asymmetry2001, 12, 1947. (b) Gui, Y. ; Tian, S. K. Org. Lett. 2017, 19, 1554. (c) Erika, B. ; Istvan, G. ; Gyorgy, K. Lett. Org. Chem. 2011, 8, 22.


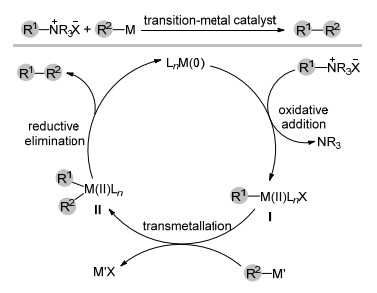
 下载:
下载:










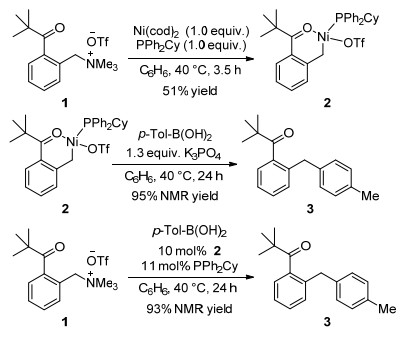








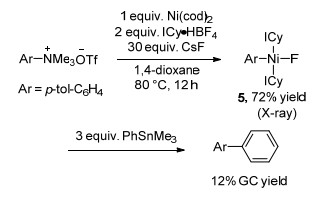













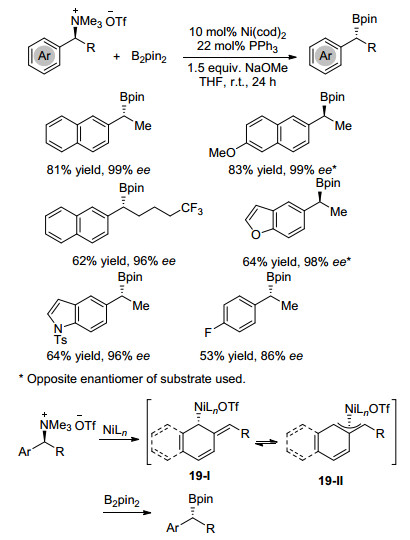
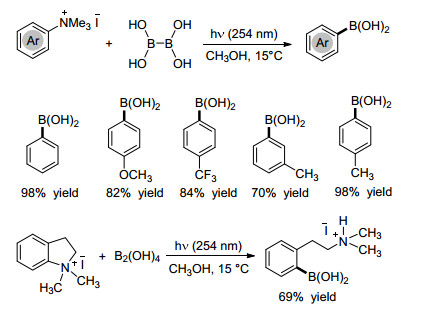


 下载:
下载:






 下载:
下载:
