Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062
b.
Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, Shanghai 200062
Received Date:
04 March 2018 Revised Date:
02 April 2018 Available Online:
01 July 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21772044), the Program of Shanghai Academic/Technology Research Leader (No. 18XD1401500), the National Young Top-Notch Talent Support Program and the Fundamental Research Funds for the Central Universities
Abstract:
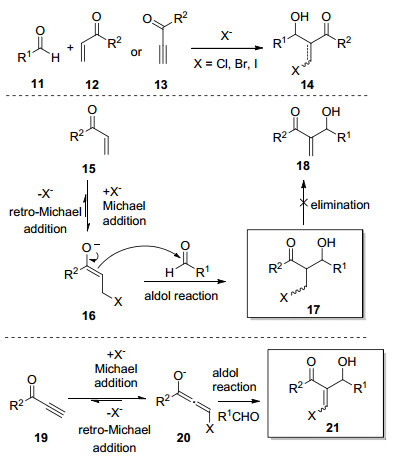
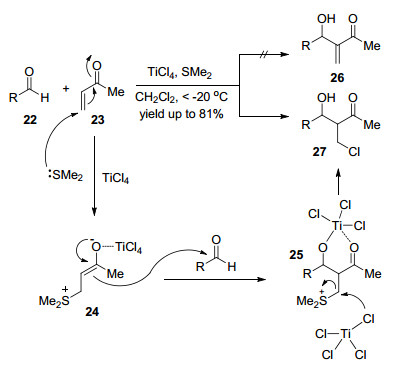
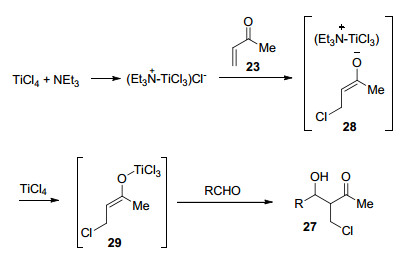
Michael addition and aldol reaction are among the most basic and commoly used reactions. Cascade halo-Michael/Aldol reaction can effectively improve the atomic economy and step economy of organic synthesis. After the Michael addition of α, β-unsaturated compound with halide ion (Cl-, Br-, I-), the reactive intermediate reacts with aldehyde through aldol reaction in the same reaction system. This process is called cascade halo-Michael/Aldol reaction. The cascade halo-Michael/Aldol reaction and its applications in the synthesis of related compounds according to the types of Lewis acid are introduced.
Medvedeva, A. S.; Demina, M. M.; Novopashin, P. S.; Sarapulova, G. I.; Afonin, A. V. Mendeleev Commun. 2002, 12, 110. doi: 10.1070/MC2002v012n03ABEH001553
[29]
Li, G.; Wei, H.-X.; Gao, J. J.; Johnson, J. Synth. Commun. 2002, 32, 1765. doi: 10.1081/SCC-120004273
(a) Wei, H. X. ; Timmons, C. ; Farag, M. A. ; Paré, P. W. ; Li, G. Org. Biomol. Chem. 2004, 2, 2893. (b) Wei, H. -X. ; Gao J. J. ; Li G. ; Paré, P. W. Tetrahedron Lett. 2002, 43, 5677.
[44]
Ciesielski, J.; Cariou, K.; Frontier, A. J. Org. Lett. 2012, 14, 4082. doi: 10.1021/ol3017116
[45]
Ciesielski, J.; Gandon, V.; Frontier, A. J. J. Org. Chem. 2013, 78, 9541. doi: 10.1021/jo4007514
[46]
Wei, H.-X.; Kim, S. H.; Li, G. Org. Lett. 2002, 4, 3691. doi: 10.1021/ol020146y
[47]
Lee, S. I.; Hwang, G. S.; Shin, S. C.; Lee, T. G.; Jo, R. H.; Ryu, D. H. Org. Lett. 2007, 9, 5087. doi: 10.1021/ol702134w
[48]
Ciesielski, J.; Leboeuf, D.; Stern, H. A.; Frontier, A. J. Adv. Synth. Catal. 2013, 355, 2077. doi: 10.1002/adsc.v355.10
Ranatunga, S.; Kim, J. S.; Pal, U.; Del Valle, J. R. J. Org. Chem. 2011, 76, 8962. doi: 10.1021/jo201727g
[65]
Ahmed, N.; Ali, H.; van Lier, J. E. J. Porphyrins Phthalocyanines 2006, 10, 1172. doi: 10.1142/S1088424606000533
[66]
Ratnayake, R.; Lacey, E.; Tennant, S.; Gill, J. H.; Capon, R. J. Chem.-Eur. J. 2007, 13, 1610. doi: 10.1002/(ISSN)1521-3765
[67]
Ratnayake, R.; Lacey, E.; Tennant, S.; Gill, J. H.; Capon, R. J. Org. Lett. 2006, 8, 5267. doi: 10.1021/ol062113e
[68]
Sloman, D. L.; Bacon, J. W.; Porco, J. A. Jr. J. Am. Chem. Soc. 2011, 133, 9952. doi: 10.1021/ja203642n
[69]
Sloman, D. L.; Mitasev, B.; Scully, S. S.; Beutler, J. A.; Porco, J. A. Jr. Angew. Chem., Int. Ed. Engl. 2011, 50, 2511. doi: 10.1002/anie.201007613
[70]
Winter, D. K.; Endoma-Arias, M. A.; Hudlicky, T.; Beutler, J. A.; Porco, J. A. Jr. J. Org. Chem. 2013, 78, 7617. doi: 10.1021/jo401169z
[71]
Butler, J. R.; Wang, C.; Bian, J.; Ready, J. M. J. Am. Chem. Soc. 2011, 133, 9956. doi: 10.1021/ja204040k
[72]
Rujirawanich, J.; Kim, S.; Ma, A. J.; Butler, J. R.; Wang, Y.; Wang, C.; Rosen, M.; Posner, B.; Nijhawan, D.; Ready, J. M. J. Am. Chem. Soc. 2016, 138, 10561. doi: 10.1021/jacs.6b05484
[73]
Sugano, M.; Sato, A.; Iijima, Y.; Furuya, K.; Haruyama, H.; Yoda, K.; Hata, T. J. Org. Chem. 1994, 59, 564. doi: 10.1021/jo00082a012
[74]
Sugano, M.; Sato, A.; Iijima, Y.; Furuya, K.; Kuwano, H.; Hata, T. J. Antibiot. 1995, 48, 1188. doi: 10.7164/antibiotics.48.1188
[75]
Sugano, M.; Sato, A.; Iijima, Y.; Oshima, T.; Furuya, K.; Kuwano, H.; Hata, T.; Hanzawa, H. J. Am. Chem. Soc. 1991, 113, 5463. doi: 10.1021/ja00014a053
[76]
Chu, M.; Patel, M. G.; Gullo, V. P.; Truumees, I.; Puar, M. S.; McPhail, A. T. J. Org. Chem. 1992, 57, 5817. doi: 10.1021/jo00048a008
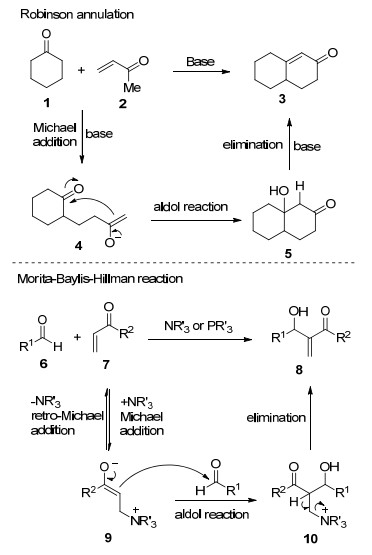
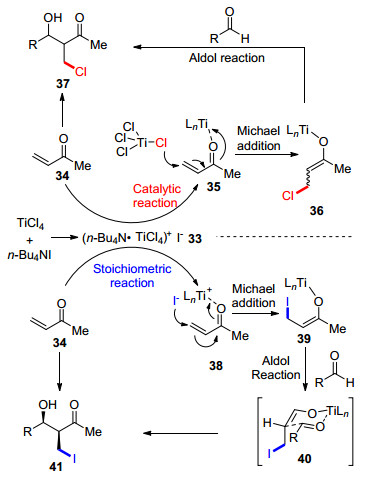
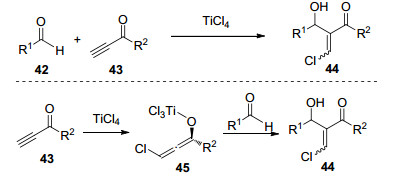
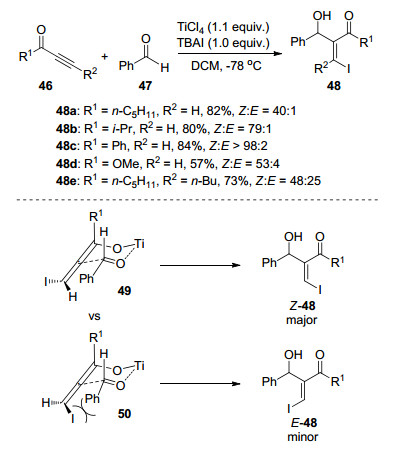
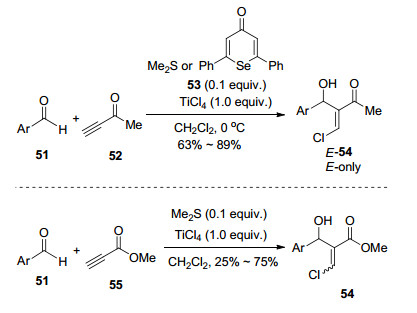
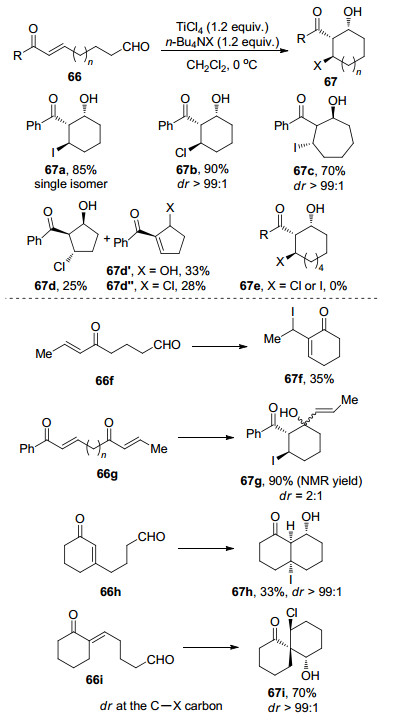
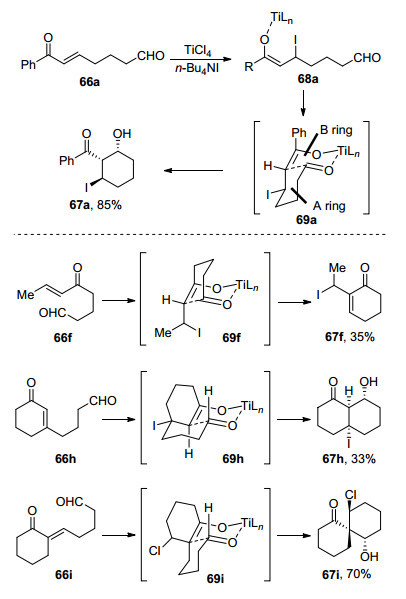
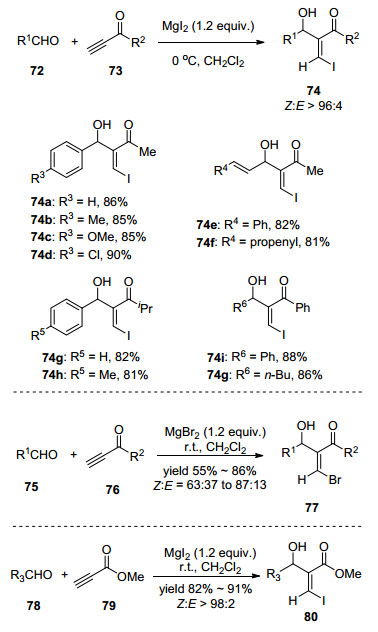
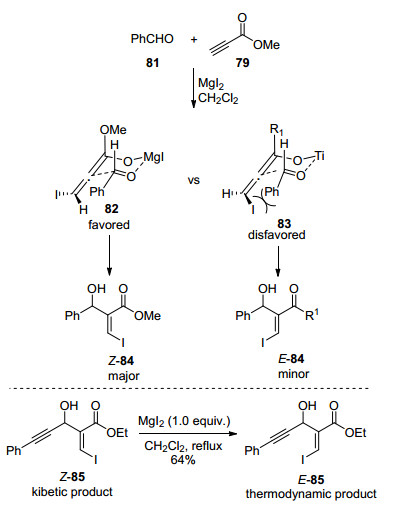
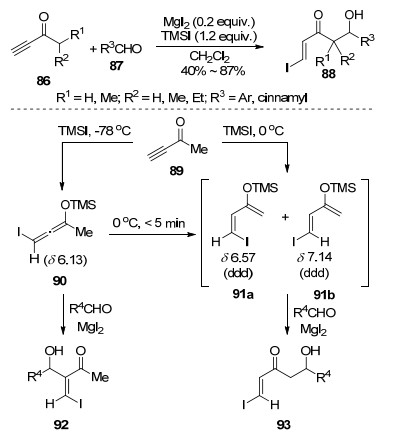
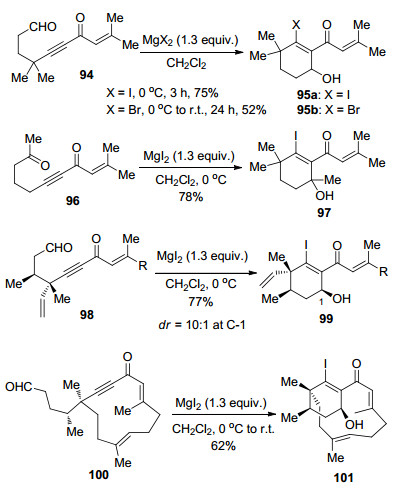
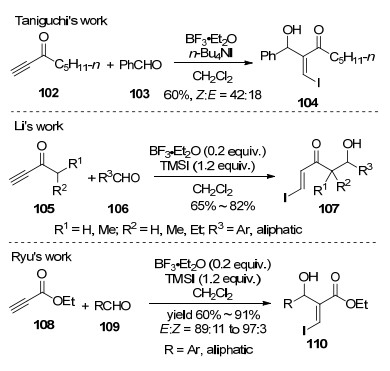
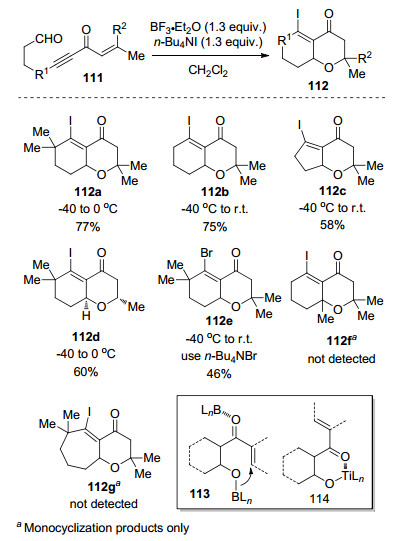
图式 1
Robinson关环反应、Morita-Baylis-Hillman反应及反应机理
Scheme 1
Robinson annulation, Morita-Baylis-Hillman reaction and their mechanism

 下载:
下载:

















 下载:
下载:






























 下载:
下载:
