表 1
诺贝尔化学奖:绿色合成方法
Table 1.
Nobel prize in chemistry: green synthetic methods

药效基团(pharmacophore)是指药物分子产生特定药理活性所必需的原子、基团或片断[1].卤素、氮、氧、硫原子、羟基、羰基(羧基)以及芳(杂)环等是与受体结合时的特征元素, 本质上药效基团可作为(a)氢键的接受体和给予体、(b)电荷的中心(如季铵盐、羧基和四唑基)、(c)芳杂环中心及(d)疏水中心.而导向基团(directing group)则是有机分子中的定位导向的官能团, 组成上两者有着相同和不同之处.近年来直接对惰性的C—H键进行活化和官能团化, 进而避免使用预官能化的底物, 使得合成路线更简洁, 原子经济性更趋合理是当前有机化学的研究前沿[2~5], 也是热点和难点之一[6, 7].利用导向基团进行的惰性C—H活化的策略取得了很大的进展[8~12].鉴于导向基团通常是一类具有配位能力的杂原子或官能团, 因此从化学结构及组成来看, 药效基团和C—H活化导向基团具有很大的关联度, 而新药研发离不开有机合成革命性技术的突破[13].
近年来的大数据显示药物化学家把Suzuki偶联反应当作很有效的合成工具[14], 经典的交叉偶联反应主要是钯催化的Suzuki反应、Heck反应以及Negishi反应(表 1), 发明该反应的化学家也因此荣获2010年的诺贝尔化学奖.目前, 以上反应均已实现规模工业化生产, 据统计全世界25%以上的合成药品都由此三种反应中的一种制成[15], 对制药工业具有举足轻重的作用.但是, 经典的交叉偶联反应是基于有机卤化物及拟卤化物, 这些底物在自然界中并非广泛存在, 制备成本相对比较高昂.
 下载:
导出CSV
下载:
导出CSV
| 时间 | 方法 | 获奖理由和工业化应用 |
| 2001年 | 威廉·诺尔斯(美) 野依良治(日) |
手性催化还原反应 广泛应用于制药工业 实现了大规模工业化应用 |
| 巴里·夏普莱斯(美) | 手性催化氧化反应 广泛应用于制药工业 实现了大规模工业化应用 |
|
| 2005年 | 罗伯特·格拉布(美) 理查德·施罗克(美) 伊夫·肖万(法) |
烯烃复分解反应 广泛应用于制药工业 实现了大规模工业化生产 |
| 2010年 | Richard F. Heck(美) Ei-ichi Negishi(美) Akira Suzuki(日) |
钯催化交叉偶联 广泛应用于制药工业实现了规模的工业应用 |
| 20??年 | Who? | 催化的碳氢活化反应 迄今为止, 未见广泛的工业化应用 |
通过C—H键活化是最有可能实现替代有机卤化物的方案, 目前C—H键活化实现交叉偶联获得了广泛的关注, 也取得了极大的进展.但迄今为止尚没有C—H活化的直接偶联反应用于药品的工业化生产[16], 主要原因首先是C—H的键能较大, 较难活化.同时, 过渡金属催化C—H键的活化往往选择性不太好.而通过官能团导向的C—H键活化反应则很好地解决了偶联位置的异构化这一问题.
一般而言, 当底物的C—H键因不同位置的取代基而导致电性不同时, 可以控制实现具体位置的选择性官能团化.而对于某些底物, 可以通过引入Lewis碱导向基团对钯催化剂进行配位, 从而控制分子内C—H键的选择性官能团化[17].其中后一种方法更为常用.常见的导向基团为烷基甲酰基类、烷基甲酰胺基类、烷基甲酰氧基类以及羧基类, 其实质都是通过羰基氧原子与过渡金属的配位作用, 起到导向作用.另一类主要是吡啶基、亚胺/肟醚类、噁唑啉类、氨基酰基类以及氰基类, 其实质是通过氮原子与过渡金属的配位作用, 起到导向作用(Scheme 1).
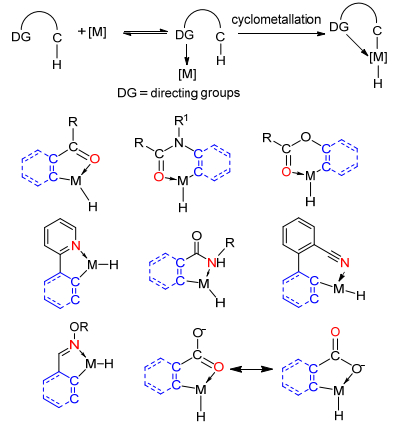
导向基团的引入虽然解决了C—H键活化的区域选择性的问题, 但其缺陷也是明显的:由于C—H活化常使用钯等金属配体催化剂, 这使得其会与杂原子(N、O、S、P)或杂环形成强配位键从而导致催化剂中毒失效, 催化剂的配体有时较为复杂不易制备, 催化剂的用量较大(通常大于20 mol%), 以及催化活性降低后影响反应的进程和收率等; 另外有些导向基团是通过官能团转化而引进分子的, 一旦完成C—H官能团化反应后, 导向基团即难以从分子中离去或不易进行进一步的结构修饰.虽然可离去和可化学修饰导向基团的研究解决了上述一般导向基团的不足, 引起了有机化学家一定的关注[18], 但无形中导向基团等同了所谓的“保护基”——既要引入又要除去, 显然有违绿色化学的原理和原子经济的规则.鉴于此, 如何解决这些挑战也是摆在化学工作者面前的一大难题.
通过引入导向基团实现C—H键的活化继而除去导向基团实现特定位置的定向官能团化, 这毫无疑问是C—H活化的一个重要研究领域, 然而此方法并不具有原子经济性.在现代药物工业中, 能否通过药物分子中已有活性基团来实现特定位置的C—H键活化并进而实现具体位置的官能团化, 从而实现官能团的双重身份, 即既是药效基团又是定向基团, 起到“一箭双雕”的作用呢?在大量文献调研中, 不难发现这样的可能性并进而提出以下理念, 即在药物化学以及药物生产工艺中充分利用药物分子中已有的药效官能团来充当C—H活化的导向基团, 进而实现具体位置的选择性官能团化来获得目标药物.本文对这一思路进行具体阐述, 通过寻找药物化学中起到重要作用的药效基团, 同时进一步查找这些药效基团是否能在C—H活化中起到导向基团的作用, 从而寻找最有希望同时充当药效基团与导向基团的官能团, 最终目的是实现这些官能团在药物化学和有机化学中进一步得到广泛应用, 并最终实现药物的规模化工业生产, 实现药物化学与有机化学的完美结合[19].
本文从两个方面对这一问题进行阐述, 首先介绍在药物化学中得到广泛应用的药效基团, 这些主要包括吡啶氮原子、氰基、羧基以及羧基的等排体, 如四氮唑以及磺酰胺等基团, 继而将阐述该类官能团在碳氢活化中的广泛应用, 最后将进一步说明利用这类药效基团与C—H活化导向基团相统一的官能团在药物化学工业中的具体应用以及在制药工业中的应用前景.限于篇幅并考虑到大多数药物分子的基本结构特征, 本文仅对以芳香(包括杂芳香)体系为骨架的小分子的C(sp2)—H活化方法学以及在药物研发方面的应用做一评述.
药物化学中进行药物设计与结构优化时, 将芳香环或芳杂环体系的一个C原子置换为N原子是常用的方法, 并且经常可以起到意想不到的效果, 因为N原子的引入会影响化合物与受体的相互作用, 进而改变化合物的生理活性, 或者因为化合物的理化性质被改变而导致其溶解性、透膜能力、蛋白结合能力以及代谢能力都不同程度地被改变, 最后的结果是药物的体内药代动力学性质的大幅改变, 进而影响药物的整体治疗效果.本文将举例介绍这类芳香环C原子被置换为N原子造成的药效改变[20].
de Esch等[21]于2012年报道了一系列5-HT3AR受体的配体筛选过程, 通过基于细胞的活性筛选获得了一系列喹啉和喹唑啉化合物(Eq. 1), 最后发现异喹啉化合物1是一个中等结合的配体(5-HT3AR, pKi=6.7), 而多一个N原子的喹唑啉化合物2与受体的结合能力则大大提高了4300倍(5-HT3AR, pKi=10).
|
|
(1) |
Müller等[22]在寻找嘌呤类G蛋白偶联受体(GPCR)配体的过程中发现1-或者3-去氮杂嘌呤显示弱的键合能力(rAde1R, Ki=11和6.8 μmol·L-1), 7-去氮杂嘌呤或者9-去氮杂嘌呤(3)则没有活性(rAde1R, Ki>100 μmol· L-1), 而将化合物3中的C原子置换为N原子, 得到的嘌呤4 (Eq. 2)对受体的活性相对于化合物3则增加了3400倍(rAde1R, Ki=0.029 μmol·L-1).
|
|
(2) |
Zhang等[23]基于纳曲酮(μ-OR, Ki=0.26 nmol·L-1)的结构进一步筛选μ-阿片样物质受体的配体, 他们发现将C-14位的羟基成苯甲酸酯得到化合物5, 该化合物仅有中等结合作用(μ-OR, Ki=120 nmol·L-1), 但是将化合物5的C-14苯甲酸酯环做一个“N-扫描”, 得到的2-氮杂类似物6 (Eq. 3)相对于化合物5的活性提高了880倍(μ-OR, Ki=0.14 nmol·L-1), 相对于3-氮杂及4-氮杂类似物活性也分别提高了11倍和40倍(μ-OR, Ki=1.6和5.6 nmol·L-1).并且化合物6相对于κ-OR及δ-OR具有更好的选择性(κ-OR Ki=26 nmol·L-1; δ-OR Ki=120 nmol·L-1), 分子对接研究表明化合物6通过N原子与μ-OR的谷氨酰胺残基发生氢键作用, 而对于κ-OR及δ-OR则不存在这种结合作用, 因此选择性大为提高.
|
|
(3) |
Verhoest等[24]在研究选择性磷酸二酯酶10 (PDE10A)的过程中发现4-苯基化合物7 (PDE10A, IC50=260 nmol·L-1)相对于4-吡啶基类似物8 (PDE10A, IC50=0.42 nmol·L-1)的活性相差610倍, 研究表明该差异的存在主要是由于化合物8 (Eq. 4)上吡啶的氮原子通过一分子水桥接与酶骨架形成氢键.有意思的是, 8的嘧啶类似物以及哒嗪类似物的活性又大为下降(PDE10A, IC50=27, 12 nmol·L-1), 这主要也是因为其不能形成氢键.化合物8相对于其他PDEs也有>1000倍的选择性.
|
|
(4) |
Vanotti等[25]首次报道了寻找第一个Cdc7激酶的工作, 通过筛选获得化合物10 (Cdc7, IC50=0.010 μmol· L-1), 其相对于苯基类似物9 (Cdc7, IC50>5.0 μmol·L-1)的活性提高约500倍以上, 分子对接研究表明化合物10 (Eq. 5)的吡啶4-位氮原子刚好与Cdc7的铰链区发生作用, 而化合物9则没有, 即使是化合物9的3-吡啶类似物也因为无法形成类似键合作用而活性降低(Cdc7, IC50>5.0 μmol·L-1).
|
|
(5) |
在寻找一类膜结合的O-乙酰基转移酶抑制剂的过程中, Ho等[26]通过高通量筛选发现吡咯化合物11 (HEK293-STF3A, IC50>50 μmol·L-1)相比咪唑类化合物12 (HEK293-STF3A, IC50=0.015 μmol·L-1)的细胞活性弱了3300倍.分子模拟研究表明化合物12 (Eq. 6)的咪唑氮原子充当了一个很好的氢键受体, 因此也大幅增加了细胞活性.
Hodgetts等[27]报道了一类TRPV1拮抗剂的研发过程, 为提高口服暴露量、降低致突变风险以及增加分子新颖性, 对于喹唑啉先导化合物13进行了一系列结构优化, 其中包括N-筛选策略, 最终成功得到一类结构新颖的吡啶并吡嗪化合物14 (Eq. 7), 其中化合物14的活性(hTRPV1, IC50=0.23 nmol·L-1)相比化合物13 (hTRPV1, IC50=310 nmol·L-1)大幅提高1300倍.
|
|
(6) |
|
|
(7) |
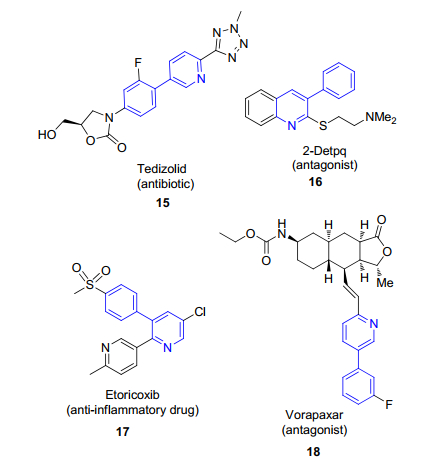
N(杂环)原子是很重要的C—H活化导向基[3], 已报道的导向官能团包括氨基、重氮、亚胺、肟、吡啶、吡唑、吡嗪等.其中, 吡啶是非常重要的含氮杂环化合物, 在天然产物、药物以及功能材料中广泛存在.例如许多临床药物都含有3-苯基吡啶这样的药效基团或官能团(图 1), 这里我们主要以吡啶这个具有广泛生物活性的分子为例来说明氮原子作为导向基团进而发生C—H活化的反应.在该类C—H活化反应中, 主要的是钯催化的C—H活化反应, 其次是铜催化, 其他过渡金属也多有报道. C—H活化可以形成各种新的C—C键、C—N键、C—O键以及其他包括C—F键、C—S键、C—P键等.吡啶氮原子作为导向基团分为两种情况:一种是与吡啶相连的芳环或者取代基被活化进而发生各种取代反应, 包括邻位活化或者远程活化; 另一种是吡啶环本身可以发生碳氢活化而形成新的化学键, 但是由于吡啶N原子邻位电子云密度很低, 导致该位置很难被活化, 一般通过将吡啶N原子氧化为N-氧化物可以实现在过渡金属下的C—H活化, 并实现各种后续反应.
Sanford等[28]于2005年首先报道了钯催化的吡啶导向的C—H活化反应(Eq. 8), 以二苯基碘盐作为芳基化底物, 采用Pd(Ⅱ)/Pd(Ⅳ)催化体系, 可以成功地将吡啶邻位芳基的2-位活化并引入芳香基团.
|
|
(8) |
Daugulis等[29]使用吡啶作为导向基团, 成功完成了钯催化的芳基碘苯作为偶联试剂的芳基化反应(Scheme 2), 如果碘苯的取代基是吸电子基团则需要长的反应时间.之后Yu等报道了酰基过氧化物以及Sun等[30, 31]报道了芳基三甲氧基硅烷作为芳基化试剂的C—H活化偶联反应.
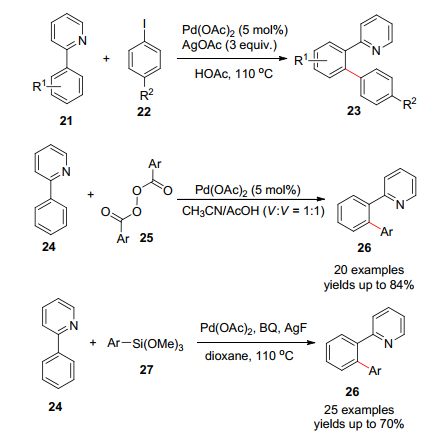
2006年, Che和Yu等[32]完成了钯催化的C—H活化的直接酰胺化反应(Eq. 9).其中, 不同的酰胺包括氨基甲酸酯、乙酰氨基或者磺酰胺基都是很好的氨基化试剂, 收率较好.
Sanford等[33]开拓了钯催化的吡啶导向的乙酰氧基化反应(Eq. 10), 底物上苯环间位被各种官能团取代, 且具有较好的耐受性.
|
|
(9) |
|
|
(10) |
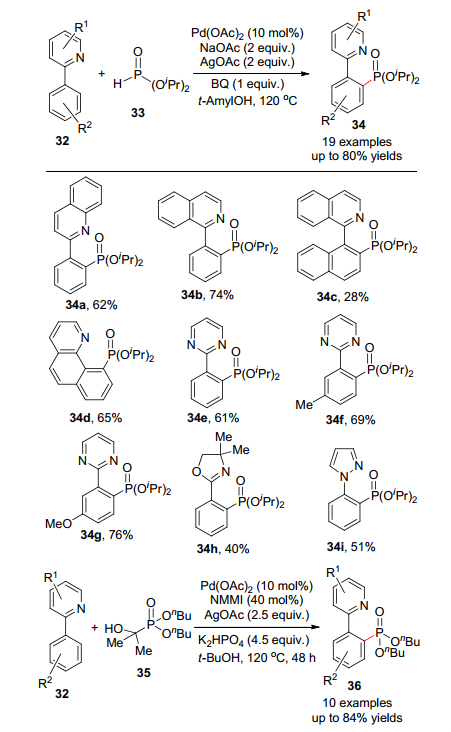
2013年, 余金权等[34]首先发展了钯催化吡啶导向C—H活化的磷酸化反应(Scheme 3).从反应的可能机理来分析, 不难发现吡啶上N原子和钯配位络合是芳基上发生C—H活化进而导致磷酸化的原因.这一假设也从各种含N芳香杂环化合物也能进行相似的磷酸化反应得到了很好的佐证.这是一种将各种含氮杂环药效基团和磷酸前药官能团联系起来的一种高效新方法.磷酸酯必须缓慢加入, 避免与催化剂的反应而导致失活. Murakami等[35]也报道了2-芳基吡啶的类似反应, 不同之处在于使用α-羟基烷基磷酸酯作为磷酸化底物.
2005年, 同样利用C—H活化反应, Fagnou等[36a]发现了钯催化的吡啶-N-氧化物的区域选择性芳基取代(Scheme 4), 可以与一系列芳基溴化物发生取代反应, 并且选择性很好.吡啶-N-氧化物上的吸电子或供电子取代基对于转化效率影响不大.值得一提的是, 芳基化也可以发生在吡啶-N-氧化物的2-甲基上, 因为这个甲基在碱的作用下更容易去质子化, 进而发生后续反应[36b].
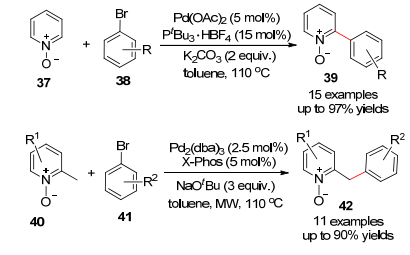
2008年Daugulis等[37]引入了CuI催化的吡啶-N-氧化物与芳基碘化物的偶联反应(Eq. 11), 其中主要是吡啶的2-位被芳基化, 如果底物酸性较弱则需要更强的锂化物作碱.碘化物也可以用芳基溴化物来替代.值得强调的是, 这样的铜催化的反应体系也适合富电子的杂环化合物, 如苯并噻唑、苯并呋喃、咖啡因、咪唑等的直接芳基化反应, 这个方法具有实验条件温和(100~125 ℃)、反应时间短(5~12 h)和产率高(高达91%)的优点.
|
|
(11) |
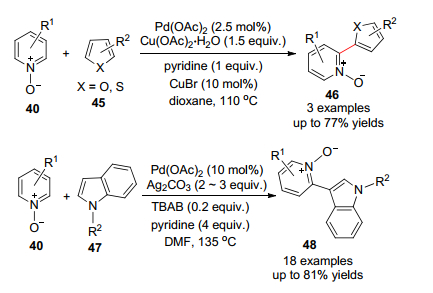
2008~2010年间, 游劲松等[38]发展了Pd催化吡啶-N-氧化物C—H活化与噻吩或者呋喃发生偶联的反应(Scheme 5), 这里需要定量的Cu(Ⅱ)盐以及催化量的CuBr来增加反应的活性.随后Li等[39]之后报道了吡啶-N-氧化物活化上引入吲哚.
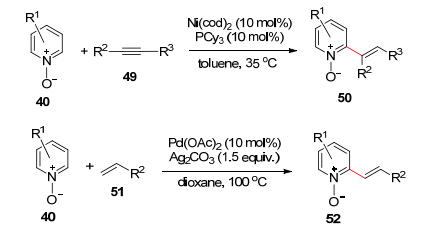
2007年, Nakao和Hiyama等[40]成功完成了Ni催化的吡啶-N-氧化物的邻位烯基化反应(Scheme 6), 接下来可以采用PCl3脱氧从而得到吡啶衍生物. 2008年, Chang等[41]报道了钯催化的吡啶-N-氧化物邻位选择性烯烃化反应, 这个反应对于底物的适应性很广, 而且没有双取代的产物, 展示出良好的化学选择性.
2014年, Hartwig等[42]通过非常巧妙的C—H活化反应, 可以不用将吡啶的N原子氧化为氮氧化物也能在吡啶邻位引入一系列官能团(Eq. 12).主要是通过两步反应来实现的:第一步是通过AgF2可以在吡啶N原子的邻位引入F原子, 第二步与亲核试剂发生反应得到目标产物.
|
|
(12) |
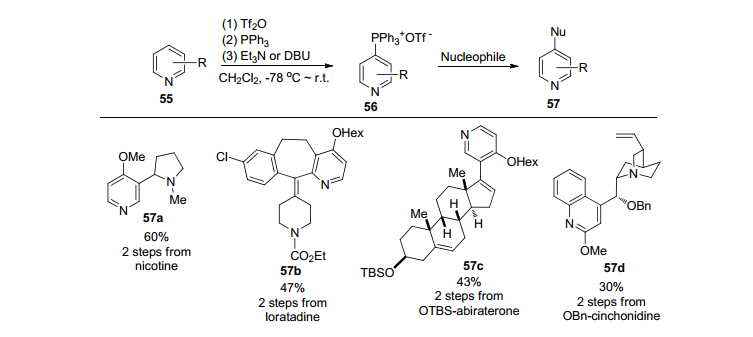
2016年, McNally和合作者[43]也报道了经过两步实现在吡啶4位上进行官能团化的新方法(Scheme 7).首先吡啶在三氟甲磺酸酐的存在下和三苯基磷形成了季鏻盐, 然后和各种亲核试剂如烷氧基负离子、硫醇盐等发生SNAr反应.一些有生物活性的分子能够方便有效地进行C-4官能团化, 有趣的是当C-4有取代基时, 反应发生在C-2的位置上.值得强调的是, 所举例子表明这一方法可以有效地对有生物活性的分子进行后续的官能团化(Late-stage functionalization).
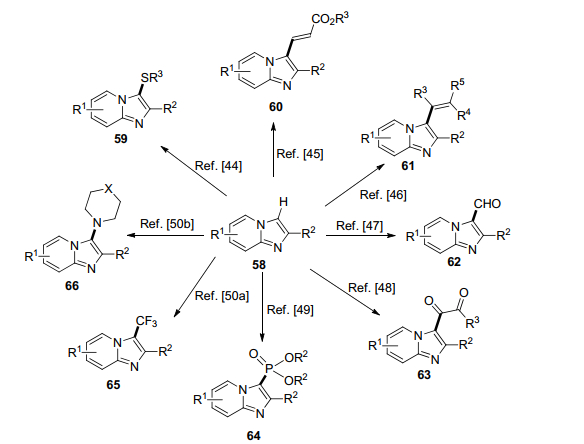
许多上市和临床药物都含有咪唑并吡啶骨架结构, 最近一系列利用C—H官能团化引入结构和性质各异的片段的新方法屡见文献报道(Scheme 8).在5 mol% CuI的催化下, 取代的咪啶并吡啶和硫醇在氧气的作用下发生氧化偶联, 有效地制备了硫醚59[44]. 2014年, Cao和合作者[45]报告了两种区域选择和立体选择的氧化烯基化的反应, 其一是采用3 mol% Ru(Ⅱ)催化剂和Cu(OAc)2, 将原料58和5 equiv.的丙烯酸酯反应, 制备了产物60, 收率在50%~75%之间; 其二是采用5 mol% Pd(OAc)2和5 mol% Ag2CO3为催化剂, 在氧气存在下反应, 产物60收率在72%~86%之间.先前的方式是采用溴代烯烃为原料, 在20 mol% Pd(OAc)2和AsPh3以及Ag2CO3的催化下, 微波加热制备化合物61[46]. Cao等[47]利用5 mol% Cu(OAc)2为催化剂采用二甲基亚砜(DMSO)为试剂和溶剂, 在氧气的存在下, 能够迅速有效地进行甲酰化反应, 以良好的收率(72%~84%)得到产物62; 值得注意的是若采用甲基酮取代DMSO, 则反应得到1, 2-二酮63, 收率在10%~93%之间[48]. 2015年, Singh等[49]成功开发了Mn(OAc)3催化, 并且利用二乙基亚磷酸酯进行咪唑吡啶的直接磷酸化反应, 得到了目标化合物64, 最高的收率可达84%.利用三氟甲基亚磺酸钠为反应试剂, 在5 mol% Cu(OAc)2的催化作用下, 能够有效地在温和条件下进行三氟甲基化反应[50a], 得到了目标化合物65, 相对Hajra等报告的反应条件, Tang等[50b]的方法更加绿色环保.
黄嘌呤和咖啡因是具有生物活性一类物质, 和苯并咪唑、苯并噻唑等杂环有一共同点就是它们都是很有用的药效基团, 其唯一的位于N杂环的C—H键是直接引入芳基的可能位点.近年来游劲松课题组[51]分别发展了高效实用的钯催化C—H/C—H交叉偶联不同的杂环和铜催化的直接芳基化(Scheme 9), 生成的产物具有良好的荧光效果, 也是潜在的非常有用的生物荧光探针的材料.
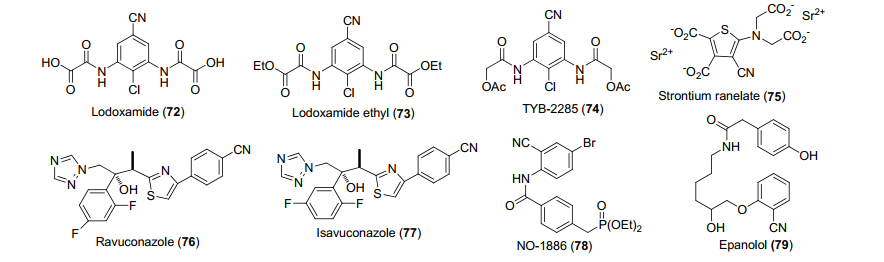
氰基是非常重要的官能团, 在体外, 氰基通常容易水解为羧酸或酰胺, 但体内的情况并不完全相同.很多药物分子中的氰基往往很稳定, 在通过人体循环时几乎保持原型不变.一般而言, 氰基可以作为羰基或者卤素的生物电子等排体与靶标发生作用, 氰基的吸电子性很强, 因此可以与氨基酸或者金属离子发生很强的偶极作用, 从而增强与靶标的作用.氰基可以作为羟基和羧酸的结合底物, 由于氰基具有强极性同时体积较小, 可以作为很强的氢键受体发生很强的溶剂化效应, 因此, 氰基可以与蛋白骨架或者氨基酸侧链以及邻近的水分子发生很强的键合力.氰基可以作为特定结构中水分子的替代物, 替代那些与底物通过水分子的桥连氢键发生作用的含氮小分子, 同时, 可以有效改变原来小分子的理化性质及吸收、分布、代谢、排泄(absorption, distribution, metabolism and excretion, ADME)性质, 从而改变化合物的代谢及毒理性质, 增强成药性.
到目前为止, 已经有30多个上市的含氰基药物以及20多个含氰基的处于临床后期的在研化合物.化合物72~79是含有氰基的上市或在研药物(图 2).如化合物72 (lodoxamide)和化合物73 (lodoxamideethyl)都是组胺抑制剂, 用于眼部抗炎[54].化合物74 (TYB-2285)用于哮喘或者过敏性皮肤炎[55].化合物75 (strontium ranelate)通过阻止骨吸收同时促进骨形成来促进骨生长[56].化合物76 (ravuconazole)是一类抗真菌剂, 分子模拟表明氰基参与受体的键合作用[57].化合物77 (isavuconazole)也是一类广谱抗真菌剂[58].化合物78 (NO-1886, ibrolipim)用于治疗高脂血症[59].化合物79 (epanolol)用于治疗心绞痛[60].以上化合物的氰基的具体作用模式未知, 但是推测氰基起到平衡芳环的电子密度、增加与受体的作用, 同时降低氧化代谢的作用.
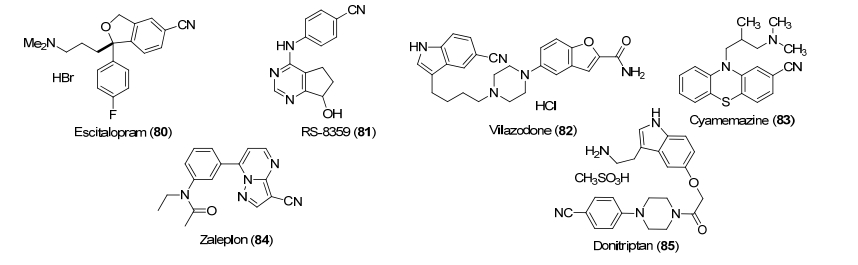
许多苯基取代氰基化合物可以用于治疗情绪紊乱症(图 3).例如化合物citalopram是一个选择性的5-羟色胺重吸收抑制剂, 最近被药效更好的单一异构体80 (escitalopram)所替代[61].分子模拟表明化合物的氰基与5-羟色胺转运体的苯丙氨酸发生作用.同样, 化合物81 (RS-8359)也含有氰基, 是可逆的单胺氧化酶抑制剂, 临床研究用于治疗抑郁[62].
化合物82 (vilazodone)是一种抗抑郁药[63], 在早期筛选过程中发现最有效的先导化合物含有吸电子基团, 其中以氰基和氟原子为最优.分子模拟表明氟原子和氰基具有相似的静电势能, 再一次证明氰基与卤素是很好的生物电子等排体.
化合物83 (cyamemazine)也是一类安定药, 用于药物上瘾的戒断[64].化合物84 (zaleplon)是一类非苯二氮䓬类抗失眠药物, 研究表明化合物选择性作用于γ-氨基丁酸α (GABAa)受体[65]. 85 (donitriptan)是一个5-羟色胺受体1b (5-HT1B)激动剂, 用于治疗偏头痛[66].
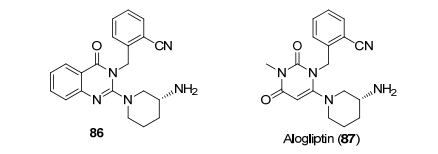
化合物87 (alogliptin, 图 4)是二肽基肽酶-4 (DPP Ⅳ)的非共价抑制剂, 用于治疗糖尿病[67], 相对二肽基肽酶-8 (DPP Ⅷ)具有10000的选择性.其先导化合物是86, 共晶结构表明化合物86的氰基与DPPⅣ的精氨酸形成氢键作用, 但是化合物86的hERG抑制太强, 代谢太快, 进一步结构优化得到化合物87.
如前所述, 氰基在药物化学中是一个很好的药效基团, 在有机化学中, 氰基又是一个使用广泛的官能团, 可以通过常用的有机化学反应轻易将氰基转化为羧基、氨基甲酰基、氨甲基、羰基以及四氮唑等杂环基团.而在C—H活化反应中, 氰基虽然与过渡金属的结合能力较弱, 但最近报道了氰基作为C—H活化定位基团的大量工作[68].氰基作为C—H活化的定位基, 可以诱导多种反应键的形成, 包括C—H键芳基化、卤化、成烯化、烷氧基化、硼酸化以及甲基化等, 催化剂主要涉及到Pd(Ⅱ), Ru(Ⅱ), Rh(Ⅲ), Ir(Ⅰ)等催化体系, 而活化位置则包括芳环的邻、间、对位, 本文主要以CN作为定位基活化芳环邻位C—H键为例来说明CN的定位作用.
2011年, Sun等[69]报道了第一例钯催化下氰基作为导向基团的苯甲腈的邻位芳基化反应(Eq. 13), 从芳基甲腈化合物和芳基碘化物出发以较好收率得到一系列双芳基-2-甲腈衍生物.相同条件下, 含吸电子基的芳基碘化物相比含供电子基的芳基碘化物的目标产物的收率要高.
|
|
(13) |
|
|
2013年, Sun等[70]报道了一种芳基腈的邻位卤化的方法(Eq. 14), 通过采用氰基导向的方法, 可以以较好的收率得到一系列碘化物、溴代物及氯代物.其中, 芳基上的取代基对反应活性没有影响, 并且, 碘代物及溴代物的反应可以放大到克级.
|
|
(14) |
|
|
2015年, Reddy等[71]报道了一种钌催化的采用氰基作为导向基团的芳环邻位烯基化反应(Eq. 15).该反应收率良好且立体选择性和区域选择性都较好.
采用C—H活化一般可以较容易地形成C—C键, 但是采用过渡金属催化的条件下一般较难形成CO键, 这主要是由于过渡金属和氧原子的电性导致两者结合能力较强从而不能继续后续反应. 2012年, Sun等[72]报道了使用氰基作为导向基团的芳基邻位的烷氧基化反应(Eq. 16).采用Pd(OAc)2作为催化剂, Na2S2O8作为氧化剂, 该反应对于多种官能团耐受, 并且得到双取代烷氧基产物的比例高于单取代烷氧基产物.
|
|
(15) |
|
|
|
|
(16) |
|
|
2005年, Smith等[73]报道了Ir催化的4-取代芳基腈的C—H活化硼酸化反应(Eq. 17).由于氰基的导向作用, 硼酸化反应主要在芳环2-位发生, 在硼酸过量的条件下, 也可以得到4-位硼酸化以及双硼酸化反应产物.这个反应所用到的苯环体系也可以扩大到杂环.
|
|
(17) |
|
|
此外, 通过精巧的设计, 氰基也可以实现芳环间位或者对位的活化, 进而得到相应的C—H活化成烯化或者芳基化产物, 但是这一类反应目前仍在持续研究与开发中, 这类氰基定位基往往不会被保留, 而仅仅做导向基团使用, 不做他用, 这样“原子经济”的理念也就无法完全实现了.
Maiti等[74]报道了钯催化的苯乙酸衍生物的间位C—H活化成烯化反应(Eq. 18), 反应收率较好, 其中的导向基团2-羟基苯氰基可以在反应最后被移除.这个作用模式是因为线性的氰基与过渡金属中心通过络合形成一个弱的相互作用中心, 这个络合物可以克服形成大环过渡态以及高能过渡态的缺陷进而促进反应进行.
|
|
(18) |
2014年, 余金权等[75]首次报道利用远程间位C—H活化引入乙酰氧基的反应(Eq. 19), 采用一个U-形的氰基配体, PhI(OAc)2作为氧化剂.底物可以是邻位、对位或者间位取代的芳基化合物, 相对而言, 当间位取代的芳基为底物时, 对应的产物收率较低.
2015年, Maiti等[76]设计了一类新的包含二苯基硅基氰的模板反应(Eq. 20), 可以用来辅助甲苯类衍生物对位C—H活化成烯反应.采用AgOAc作为氧化剂, Ac-Phe-OH作为配体, 底物无论是吸电子取代还是供电子取代都具有很好的兼容性.
羧基在生命体系中扮演非常重要的角色, 而且是一个很好的药效基团[77], 目前已经上市的含羧基的药物已超过450种[78].含羧基的药物种类繁多, 如非甾体抗炎药阿司匹林、布洛芬、萘普生、双氯芬酸那、吲哚美辛; 抗菌剂如喹诺酮类药物结构中都含有3-位的羧基, 这是它们构效关系要求的必须官能团, 如诺氟沙星、环丙沙星、司帕沙星、氧氟沙星、左氧氟沙星、洛美沙星、巴洛沙星、革替沙星, 以及传统含羧基药物如青霉素、头孢类抗生素; 血管紧张素抑制剂他丁类如氟伐他汀、阿托伐他汀、瑞舒伐他汀.
|
|
(19) |
|
|
|
|
(20) |
|
|
羧基作为定位基团具有悠久的历史, 1961年Kaeding[79]设计了一个反应可以从苯甲酸邻位选择性氧化获得苯酚, 这是报道的第一个工业化羧酸导向C—H活化的例子.羧基作为C—H活化的定位基在近年来得到了极大的关注, 该官能团相对于其他定位基团具有独到的优点, 除了该官能团在多种结构中都广泛存在, 其价格相对于其他导向基团也相对便宜, 并且能进一步衍生化, 或者羧基自身也可以通过化学转化被消除.
羧基作为C—H活化导向基团具有非常广泛的应用[80], 可以形成新的包括C—C键、C—O键、C—N键、C—卤键以及C—D键在内的各种化学键, 而在C—C键的形成过程中, 含羧基的芳烃类又可以与芳基卤代物、烷基卤代物、炔烃、烯烃以及酰胺化合物等发生反应, 本文仅举几例示之.
1998年, Miura等[81]首先实现了羧基诱导的邻位C—H键的活化:在催化量的醋酸钯和醋酸铜的共同作用下, 苯甲酸与丁基丙烯酸酯或者苯乙烯的反应, 可以获得一系列苯并内酯产物(Eq. 21).该反应仅需空气氧化催化即可, 生成的金属-烯烃络合物可以进一步与邻位的羧基发生成内酯反应, 烯烃反应则遵循反马氏规则.
|
|
(21) |
|
|
2016年Ackermann团队[82]对上述反应进行了改进和优化, 采用相对便宜和绿色的Ru催化剂, 在氧气和醇类溶剂存在下, 在温和的条件下, 以优良的收率获得了苯并内酯(Eq. 22).
|
|
(22) |
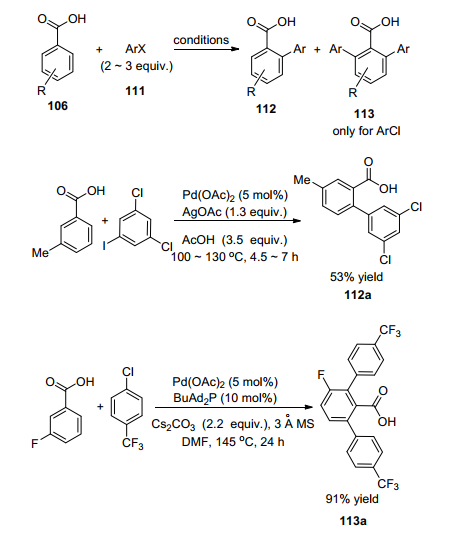
2007年, Daugulis等[83]首次报道了苯甲酸的邻位芳基化(Scheme 10), 在醋酸钯的催化下将苯甲酸与芳基卤化物偶联得到了联苯甲酸类化合物.芳基卤化物可以是氯化物或者碘化物, 前者需要配体作用.反应主要获得单芳基化合物, 但如果羧基间位是氟原子取代则主要得到双芳基化产物.
2007年和2008年, 余金权等[84a]先后报道了氧化条件下苯甲酸的邻位芳基化反应(Eq. 23), 最初采用醋酸钯催化并以苯并喹啉酮加Ag2CO3作为氧化剂, 但收率较低.后来采用苯并喹啉酮加氧气作为氧化剂, 氧气高压下可以迅速反应[84b].
2016年, Gooßen等[85]报道了第一例羧基导向的邻位苯甲酸与炔烃的反应(Eq. 24), 没有常见的内酯化产物, 同时也不需要氧化剂.
2013年, Gooßen等[86]报道了苯甲酸的邻位酰基化(Eq. 25).使用酸酐作为羰基合成子, 在[{Rh(Cod)Cl2}2]为催化剂的条件下可以中等收率获得芳基酮化合物, 但有趣的是, 条件稍微改变则可以获得苯甲酸自身酰基化的产物.这样的发展是对传统的傅-克酰基化(Friedel-Crafts acylation)反应的绿色化新探索.
|
|
(23) |
|
|
(24) |
Reinaud等[87]在1990年报道了酰胺的邻位活化羟基化反应(Scheme 11), 该反应采用N-苯氧基-2-甲基丙氨酸在化学计量的金属铜及三甲胺氧化物的作用下, 可以定量地获得邻位羟基化产物. 2005年, Que等[88]报道了通过定量Fe(Ⅱ)复合物从苯甲酸制备水杨酸.
|
|
(25) |
|
|
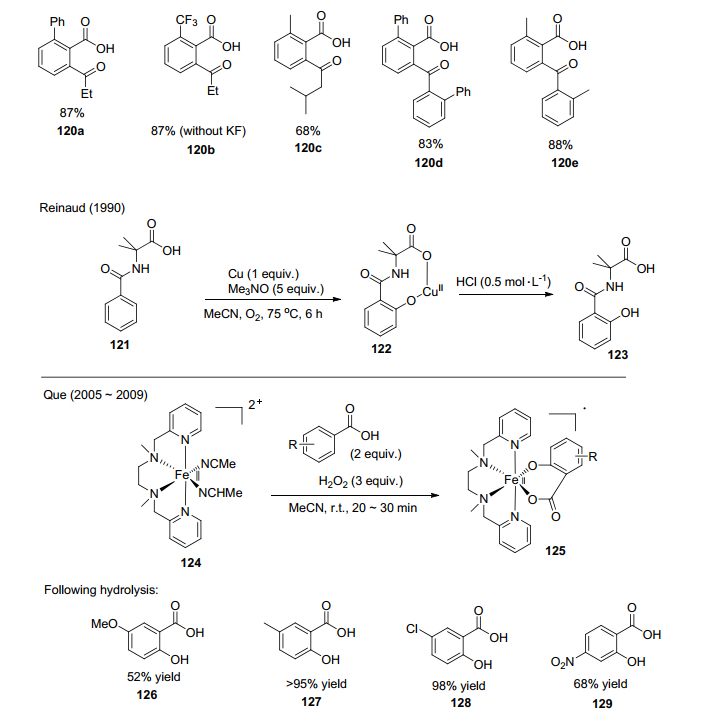
Yu等[89a]报道了Pd(OAc)2催化合成邻氨基苯甲酸的反应(Scheme 12), 这个反应的区域选择性很好, 并且官能团耐受性好.最近他们[89b]又报道了Rh催化剂用来合成邻氨基苯甲酸衍生物的方法.两者主要的不同是金属催化剂的改变以及使用不同的胺源. 2015年, Chang等[90]报道了铱催化的苯甲酸邻位引入磺酰胺基的反应(Scheme 12).

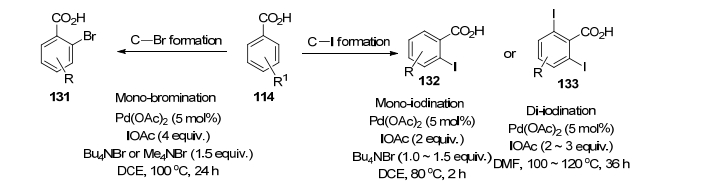
2008年, 余金权[91]报道了第一例羧酸定向的Pd催化苯甲酸邻位引入卤素的反应(Scheme 13), 可以与IOAc反应形成C—I键或者与IBr形成C—Br键.当反应在N, N-二甲基甲酰胺(DMF)中进行时, 也可以形成二碘化物, 但如果苯甲酸间位有取代, 则主要是选择性地得到单取代物.
2008年, 余金权团队[92]通过合理的逻辑分析和设计, 第一次指出N-甲氧基酰胺基如同碱金属的羧酸盐一样, 可以作为理想的C—H活化的导向基团, 该设想首先通过钯催化的带有β-C(sp3)—H键的酰胺和有机硼酸偶联反应这一里程碑式的工作获得了证实.同年, 该课题组[93]发展了分子内C(sp2)—H酰胺化反应, 采用N-甲氧基苯乙酰胺为反应底物, 有效制备了各式各样的苯并内酰胺(Scheme 14).
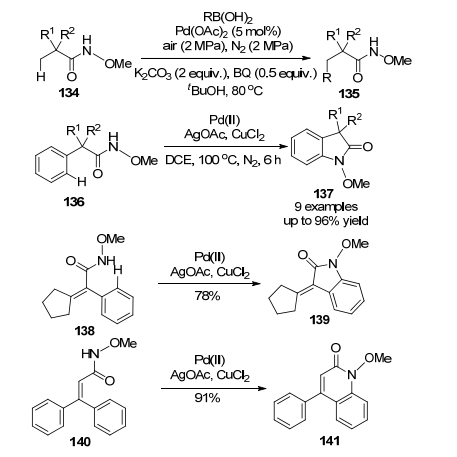
2010年, 王官武和合作者[94]报道了Pd(OAc)2催化的N-甲氧基苯甲酰胺和醇类在温和条件下(55 ℃)的直接烷氧化反应(Eq. 26), 反应的收率在20%~87 %之间.他们发现该反应是通过PdⅡ/PdⅣ的催化循环进行的, 同时确证添加强氧化剂过硫酸钾(K2S2O8)是必须的.
2011年, 王官武课题组[95]发展了钯催化的以N-甲氧基苯甲酰胺和碘苯为原料合成菲啶酮的新反应, 通过双重C—H活化, 一步形成C—C和C—N键, 得到了杂环化合物145.值得关注的是在此基础上, Cheng课题组[96]也开发了合成菲啶酮的新的有效简捷的方法(Scheme 15).这一方法尽管还是釆用了Pd(OAc)2催化剂和过硫酸钾(K2S2O8)作为氧化剂, 所不同的是化学惰性的溶剂成为了反应原料, 该过程同时破坏了三个稳定的C—H键, (90%), 反应条件也温和(25 ℃, 16 h).鉴于许多生物碱都含有菲啶酮这样的单元, 并且这些化合物都具有各样的生物活性, 同时考虑到N上甲氧基的易除特点以及这一合成新方法的高度区域选择性、非常温和的反应条件以及不需要预官能团化原料等优点, 我们相信这一新法具有非常好的应用前景.
|
|
(26) |
值得强调的是, Cheng的上述方法避免使用昂贵的Ag2O[95]更加经济实惠, 同时反应可以在室温下平稳进行.有趣的是, 2014年, Jeganmohan和Xu两个课题组[97]也几乎同时独立报道了醋酸钯/醋酸铜催化的N-甲氧基苯甲酰胺和苯炔活性中间体的成环反应, 获得了菲啶酮. Cheng课题组[98]还发展了以有机硼酸为原料、采用RhⅢ/RhI催化体系的从N-甲氧基苯甲酰胺为原料的制备菲啶酮的新方法, 在这个转化中, Ag2O作为氧化剂, 参与了两个C(sp2)—H的活化.
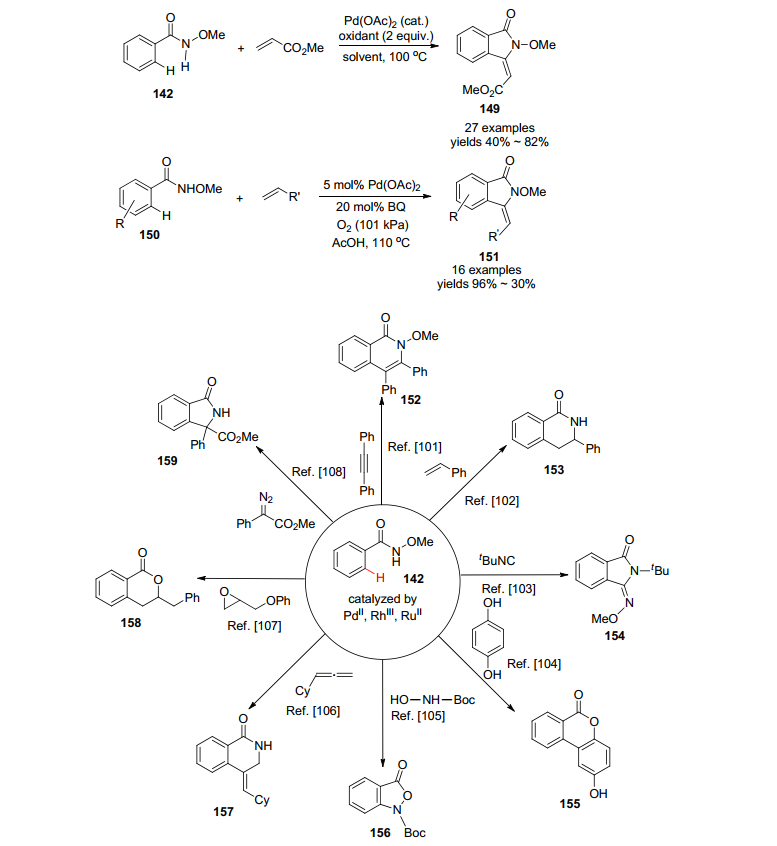
以这一简单实用的N-甲氧基苯甲酰胺出发, 近年来还发展出一系列利用C—H活化法在芳环邻位引入各种取代基和官能团, 进一步形成新的含氮杂环或内酯的新方法(Scheme 16), 例如, 2011年, 王官武等[99]再次通过钯催化活化了N-甲氧基苯甲酰胺中的C—H键, 其和丙烯酸酯/丙烯酰胺/苯乙烯的反应, 制备了异吲哚啉酮149, 几乎与此同时, Booker-Milburn等[100]也报道了相同的转化, 所不同的是反应条件和丙烯酸酯/丙烯酰胺/苯乙烯的取代基略有差异. Huang课题组[101]发展了钯催化的从N-甲氧基苯甲酰胺和双取代炔烃出发的合成异喹诺酮152的有效方法, 实验发现NaI添加剂对反应至关重要, 空气作为氧化剂, 促进了Pd(0)到Pd(Ⅱ)的再生. Glorius研究团队[102]发展了Rh(Ⅲ)催化的N-甲氧基苯甲酰胺和苯乙烯的偶联成环反应, 制备了氢化异喹诺酮化合物153, 并且通过选择不同的催化剂而实现控制区域选择性.具体全面的了解可参阅原始文献和相关综述[2b].
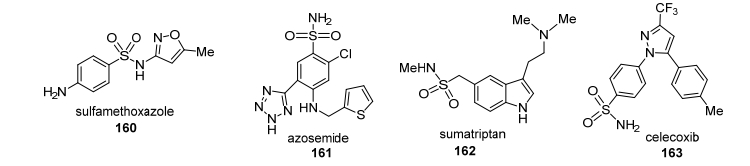
除以上几种常见官能团可用作药效基团和导向基团外, 还有更多的官能团也可以同时起到以上两方面作用.例如, 羧酸的衍生物酯基以及酰胺基在药物化学也得到了广泛的应用, 而两者同样也是C—H活化反应中极为重要的导向基团.又如磺酰胺基, 也是药物化学中一个十分重要的药效基团, 该官能团可以增强活性并且改善药物的理化性质并进而影响药物的体内吸收过程.目前市场上有约200个含有磺酰胺基的药物, 最早发现的含有磺酰胺基的药物是抗菌药磺胺甲噁唑160, 接下来有药物如利尿剂阿佐塞米(161), 抗偏头疼药物舒马曲坦(162), 非甾体抗炎药物塞来昔布(163)(图 5).
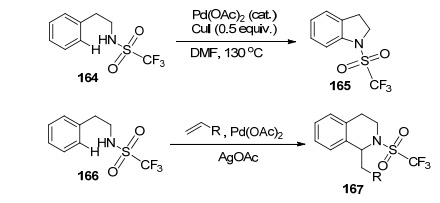
2008年, 余金权课题组[109]通过仔细的筛选和深入研究, 发现在Pd/Cu的催化下, 用三氟磺酰基保护的芳香乙胺和其他保护基所产生的有机胺有显著的区别, 这样的起始物在Pd的催化下, 通过C—H活化和一锅煮的方式, 能够有效地串联进行碘化反应和胺化反应, 利用不同的反应原料极为便利地制备了吲哚啉165、四氢喹啉和四氢异喹啉167 (Scheme 17).在这里利用三氟磺酰基增强NH组分的酸性, 进而有利于Pd—N键的形成是一个关键.
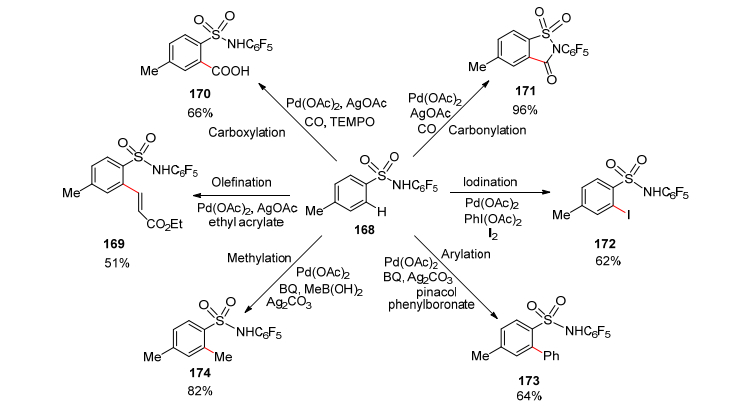
2011年, 余金权团队[110]进一步开展了磺酰胺药效基团作为导向基团用于C—H活化反应的方法学研究.通过筛选和优化, 他们发现带有SO2NHC6F5导向基团的芳香化合物, 其邻位的C—H键在二价钯/配体的共同作用和不同的反应条件下, 可以成功完成一系列反应, 包括烯基化、羧基化、羰基化、碘化、芳基化和甲基化(Scheme 18), 其中羰基化和甲基化的收率分别高达96%和82%, 这说明磺酰胺基具有很强的导向作用.此外, 产物中的N-芳基可以被三氟乙酸(TFA)移除, 进一步合成其他磺酰胺基衍生物.
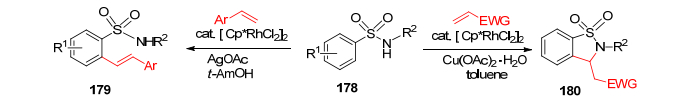
2012年, Cramer和合作者[111]通过大量的筛选和优化, 发展了以SO2NHAc为导向基团通过Rh(Ⅲ)-CuOAc催化的C—H活化过程, 和取代的炔进行加成, 一步制备了苯并磺内酰胺177 (Eq. 27, 不饱和C=C).反应具有底物选择广泛、产率高的特点.
|
|
(27) |
2014年, Wang和Li等[112]发展了铑催化的直接的C—H活化, 利用SO2NHAc为邻位导向基团, 区域选择地合成了苯并磺内酰胺180 (Scheme 19, 饱和C—C).这个氧化偶联过程具有高效、选择性好和底物选择范围广的特点.值得一提的是, 当苯乙烯为原料时, 仅仅进行成烯反应, 而没有形成闭环的产物.
随后不久, Liu和Qu等[113]报道了铑催化的以SO2-NHAc为导向基团的成烯反应(Eq. 28), 随后通过分子内的Michael协同加成, 一步合成苯并磺内酰胺183.鉴于一些在研的药物分子都含有磺内酰胺的骨架, 这一利用C—H活化制备的新方法将会更加有效地让药物合成更加简洁实用.
|
|
(28) |
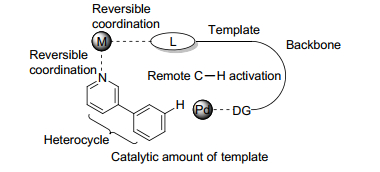
由于无需预官能团化这一优势, 从惰性C—H键活化出发构建C—C键和C—X键(X=杂原子)逐渐成为有机化学中的一个研究热点.为了解决C—H键活化中反应性和选择性的问题, 在底物结构中引入导向基团是非常重要的策略.除了常规的通过形成五元或六元金属环状中间体实现特定位点的C—H键活化外, 余金权发展了在底物结构中引入U型模板, 随后实现间位的C—H键活化.然而, 这一策略是需要先通过化学计量的化学反应将U型模板引入底物结构中, 再通过远端的导向基团实现特定位点的C—H键活化.因此, 余金权团队考虑能否通过加入催化量的特定化学结构作为模板, 实现远程的C—H键活化.最近余金权团队[114]报道了一种催化双功能模板(图 6), 一方面通过模板骨架-金属-底物的可逆配位作用结合底物, 一方面通过模板另一端的导向基团引导钯催化剂至底物远端特定位点的C—H键, 实现杂环类底物远程位点选择性的C—H键活化.
通过条件优化和筛选, 他们发现模板及配体的选择很重要, 随后在最佳条件下, 他们进行了对苯基吡啶衍生物和烯烃反应的普适性研究(Eq. 29), 发现不同底物的间位选择性和单烯基化选择性均非常好, 多种官能团如卤素和甲基醚类等都能很好地兼容.
|
|
(29) |
与此同时, 余金权的团队还对药物和天然产物分子中同样广泛存在的喹啉类结构的远程选择性C—H键烯基化也进行了考察(Eq. 30), 并且将该方法应用到了生物碱(+)-Camptothecin的后期修饰中.
|
|
(30) |
在余金权的设计下, 原有的配体催化剂的劣势成为了优势, 可逆的配位效应与远端的合成突破了现有方法学, 使得药物设计与实施出现了多样化和巨大成本优势.这个方法学设计使得药物化合物库有更多的药物结构成为备选.
现代药物的发现取决于识别先导化合物, 并迅速地合成其类似物.使用共同的药效团来指导多方位的和不同的C—H官能化来优化洐生药物分子是一个特别有吸引力的方法. 2011年余金权等[110]通过采用磺胺药效基团的药物的不同C—H功能化来证明后期官能团化的可行性, 他们开发了一组六种截然不同的磺酰胺C—H官能化反应(Scheme 20, 烯烃化、芳基化、烷基化、卤化、羧化和羰基化), 每种反应表现出不同的处理以进一步多样化的类似物.然后进行了磺胺类药物候选物的后期阶段选择性多样化.多个具有潜在反应性的C—H键直接合成新的塞来昔布类似物作为潜在的环氧合酶-Ⅱ (COX-2)抑制剂.这些磺酰胺基(CF3SO2NH, SO2NHC6F5与CONHOMe和CONHC6F5等其他近期开发的实用导向基团一起表明, 药效基团作为导向基团在新药先导化合物合成筛选中非常有用.
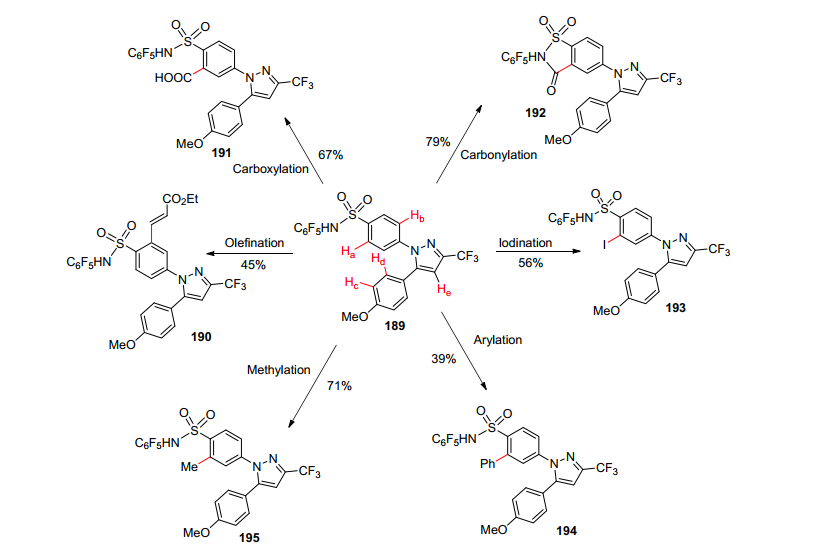
余金权等[110]利用药物塞来昔布中间体类似物(唯一的差异是塞来昔布的甲基被甲氧基取代)作为中间体, 通过磺酰胺基的导向作用, 通过C—H活化成功合成得到一系列新的塞来昔布类似物.反应中观察到在确定的反应条件下, 主要得到的都是单一位置被取代的产物, 几乎看不到其他位置被C—H活化的产物, 这说明磺酰胺基的导向作用非常好.同时, 这些新合成的中间体都是通过常规路线很难得到的, 可以进一步衍生化用来筛选更多的潜在的COX-2选择性抑制剂.
现代C—H官能化的出现使药物化学家能够更全面地考虑合成策略, 进行后续官能化, 利用药物分子的药效基团主导的C—H键作为多元化点以产生新的类似物.这种新方法提供了快速探索结构与活性构效关系(SAR)的途径, 帮助合理快速找到氧化代谢产物, 同时这些新产物也避免了代谢热点位置的氧化. C—H官能化化学是药物化学工作者新的“工具箱”[13], 这个方法必将成为影响实际的药物发现和化学生物学的有用武器.
小分子药物的合成路线中C—H直接成键一直是每个药物化学工作者梦寐以求的.直接活化引入不同的官能团、取代基, 能进一步提高SAR构效关系的研究能力, 提高药物研发的效率以及进一步缩短药物合成的步骤, 降低药物生产工业化所造成的污染环境, 同时反应无需预官能团化, 避免了卤素原子(卤化反应)和基因毒性基团如甲磺酰基(Ms)或对甲苯磺酰基(Tos)的引入和除去.鉴于C—H活化常使用钯等金属配体催化剂, 同时药物分子的药效基团如杂环中的杂原子(N、O、S)出现的频率很高, 常常和金属形成强配位作用而导致催化剂中毒失效, 催化活性大大降低而影响反应的效率.虽然目前药物化学业内还没有一个利用药效基团作为C—H活化导向基团的概念, 但实际上这种应用已经早就被药物化学家自发利用.
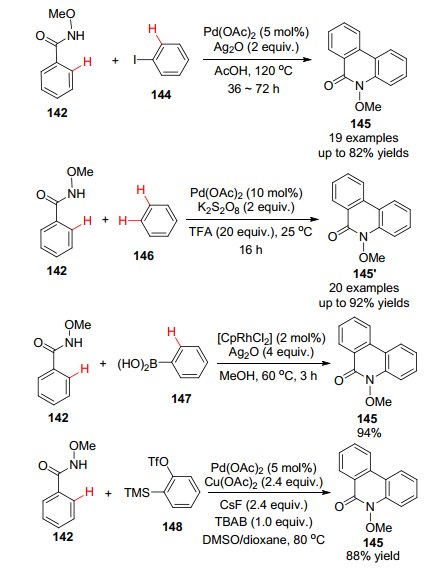
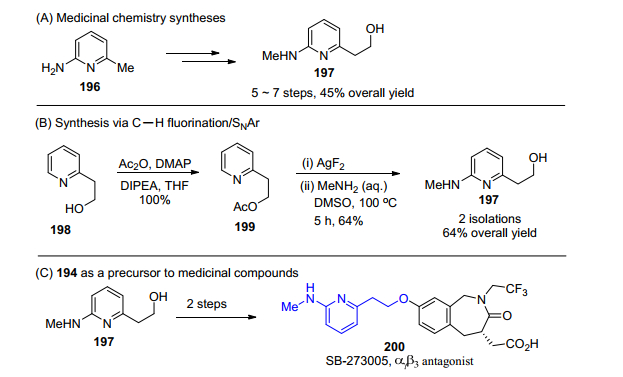
例如(6-(甲基氨基))-2-吡啶乙醇(197)是一个很重要的有机中间体, 可以作为许多药物如整合酶抑制剂SB-273005的前体.虽然结构很简单, 但之前报道的合成采用2-氨基-6-甲基吡啶(196)作原料需要7步反应, 但是如果考虑到吡啶氮原子的定位作用, 则可以采用商业购买的2-吡啶乙醇(198)作为原料, 先将乙醇基用乙酰基保护, 再利用吡啶邻位C—H键氟化并进而与MeNH2发生SNAr反应, 两步即生成目标产物(Scheme 21).总收率为64%, 相对之前以2-氨基-6-甲基吡啶(196)作为原料的反应, 合理运用C—H活化可以大大缩短反应步骤及提高反应收率[115].
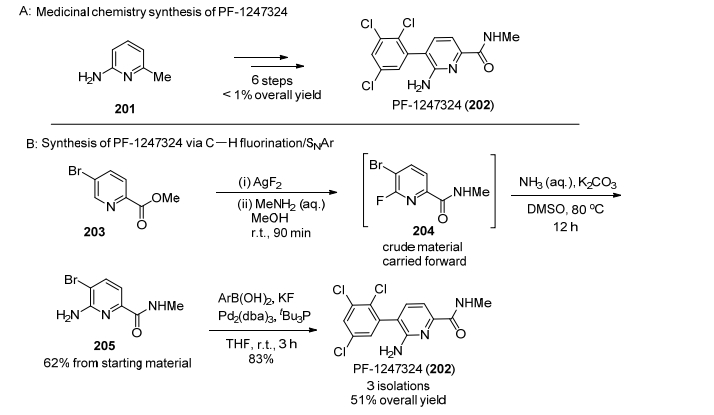
另一个实例是PF-1247324的合成, 原有的合成路线包括6步, 总收率低于1%[116].但采用C—H活化的办法, 充分利用吡啶氮原子的导向作用, 可以从5-溴-吡啶甲酸酯出发, 采用先氟化再上氨基的办法最后偶联得到202 (PF-1247324) (Scheme 22), 总收率达到51%, 反应时间也从开始的120 h缩减到18 h.
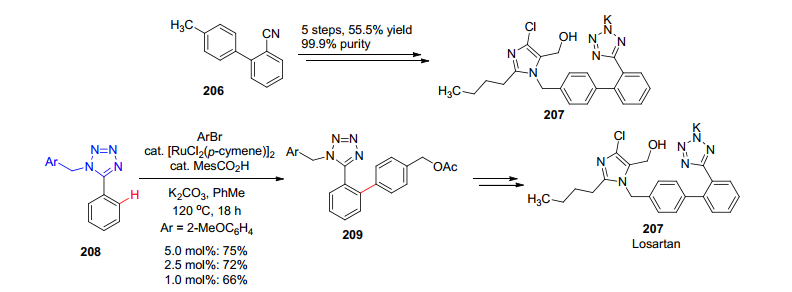
又如氯沙坦(Losartan)是一个众所周知的抗高血压药物, 一种较为简单的工业化合成方法[117]如Scheme 23所示, 采用联苯基氰为原料, 经过五步线性反应最后获得四氮唑中间体.但是如果采用四氮唑导向的C—H活化反应, 则可以在最后通过Ru催化的直接C—H芳基化反应一步将两个片段对接, 不仅遵守原子经济原则, 而且同时缩短了反应步骤, 提高了产率, 具有明显的优越性[118].
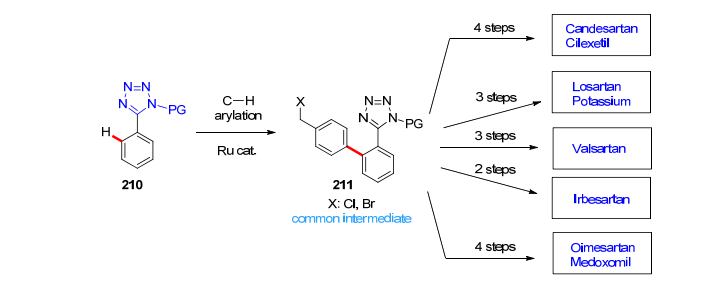
值得强调指出的是, 这样的C—H活化芳基化过程比经常使用的通过有机硼酸的Suzuki偶联反应不仅步骤少, 而且产率高, 成本低, 副产物少且无害, 同时避免了在反应的后续过程中用叠氮化钠长时间加热制备四氮唑(高危操作)的旧工艺[117].从共同的关键中间体211出发, 能够方便快捷有效地制备厄贝沙坦(Irbesartan)、氯沙坦(Losartan)、缬沙坦(Valsartan)和奥美沙坦酯(Olmesartan medoxomil)等一系列沙坦类高血压用药(Scheme 24), 其工业化生产的前景十分看好[119].
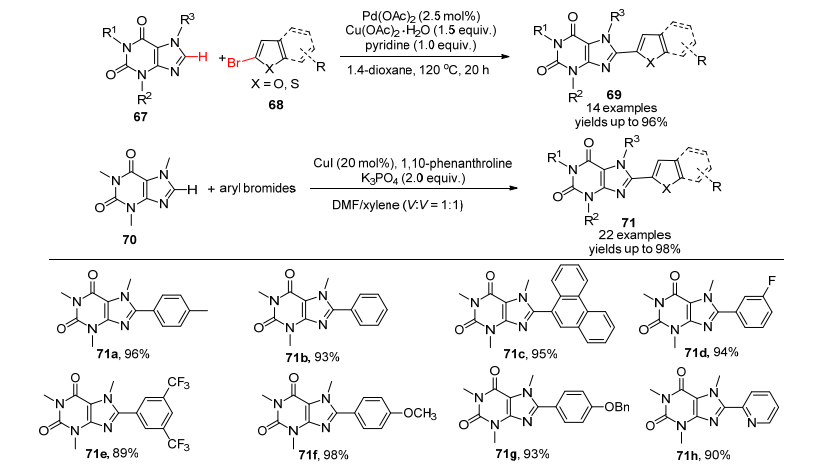
许多药物分子和临床化合物都含有苯并噻唑或苯并咪唑的药效基团或官能团, 在这些化合物的2位上引入芳基最常见的方法就是利用Suzuki偶联反应, 在药物生产过程中很少有更加原子经济的直接C—H芳基化反应的例子, 这主要是因为其化学与工艺面临着诸多的挑战, 其中包括催化剂的用量很大、反应时间很长、反应温度很高等不利因素, 在药物分子AMG 369的工艺开发中(Scheme 25), 黄金昆等[120]通过仔细的筛选, 第一次成功地发展了高度有效的PXPd/Cu(Xantphos)I共催化系统, 顺利地运用在关键中间体216制备上.值得一提的是仅用钯一种催化剂, 不添加铜催化剂的话, 反应的产率低于30%;没有钯催化剂, 仅有铜催化剂时, 几乎没有产物的生成.而PXPd/Cu(Xantphos)I共催化下, 产率达到97%.相信这一新方法在类似药物的工业化过程中会发挥重要的作用.
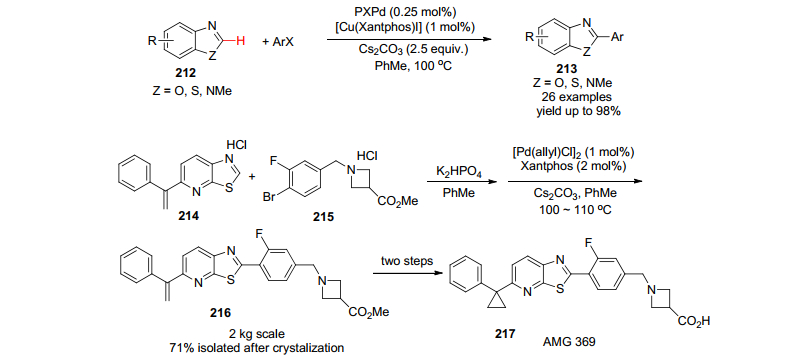
2005年, Merck制药工艺开发化学家在研究GABA激动剂时, 经过认真的分析和判断, 决定采取汇聚式合成方法[121], 这样关键反应就是钯催化的区域选择的芳基化反应(Eq. 31).值得强调的是原料稠杂环218有三个可能活化的C—H键, 另外原料219中的腈基也是邻位C—H导向基团, 经过一系列仔细的筛选卤素原子、催化剂/配体、反应溶剂和反应的温度和时间, 他们终于成功完成了高度区域选择、高效钯催化(仅仅使用了1 mol%的Pd(OAc)2和1 mol% PPh3)、高产率(86%)的芳基化反应, 一次就完成了数公斤关键化合物220的制备.
|
|
(31) |
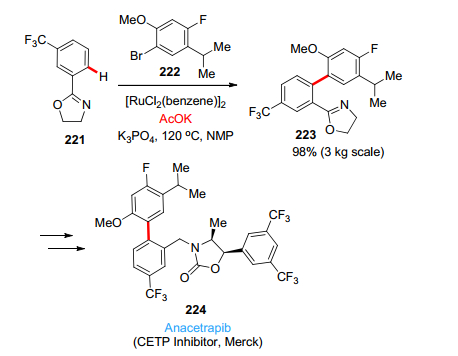
在CEPT抑制剂Anacetrapib 224的工艺化学研究中(Scheme 26), Merck的化学家们巧妙使用用了噁唑啉(通过三氟甲基苯腈和氨基乙醇制备)作为邻位导向基团, 发展了Ru催化的直接芳基化反应制备了关键中间体223, 值得关注的是, 对反应溶剂N-甲基吡咯烷酮(NMP)的杂质(含有丁内酯)研究发现添加10 mol% AcOK有预想不到的结果, 它不仅加速了反应, 而且提高了反应的转化率(大于98%)[122].这被认为是共催化剂醋酸钾承担了除去C—H键中H原子的作用, 而生成的醋酸又反过来通过氢键作用(自动催化的途径)从而加速了配体的交换.上述几个例子也反映了跨国制药的工艺开发已经有意识地利用了C—H活化这一绿色高效的合成方法.
综上所述, 在药物分子中广泛存在的药效基团, 很多其实是非常重要的C—H活化导向基团.在现代药物工业中, 通过药物分子中已有活性基团来实现特定位置的C—H键活化并进而实现具体位置的官能团化, 从而实现官能团的双重身份, 即既是药效基团又是C—H键活化导向基团, 进而实现具体位置的选择性官能团化来获得目标药物.这对于将来的药物化学以及有机合成化学包括制药化学工艺都是极为重要的, 这主要表现在以下几点: (1)开创了药物合成与工艺的新思路、新方式, 是药物合成策略上的革命性进步; (2)通过这个转化, 能够重新审视老药(包括一些由于毒性较大而临床失败的药物)的结构修饰与创新; (3)作为突破口, 在新药筛选的中后期迅速实施SAR构效关系的研究; (4)把药效基团的引入前置化, 带来合成工艺与药物生产的革命性改变; (5)快速方便地接近过去不可能制备的药物分子, 简捷地衍生化先导化合物; (6)促进新药研发包括生产工艺的绿色化发展.
C—H键活化, 包括杂环上的远程选择性的取代以及芳香化合物间位选择性的官能团化[123], C—X和C—C键的形成, 这些新的方法学需要开拓更广泛的底物适用性. C—H键活化对成型药物分子上的基团修饰也应该是研究的重点, 它的优点是可以避免为每个目标分子去设计一条反应路线, 同时药物分子中的高活性反应位点(可能产生代谢产物)的“封堵及屏蔽”恰恰是远程C—H活化触及的空间.
简而言之, 我们认为可以通过发展新的合成路线, 对已有的药物工艺进行优化, 利用药物中的药效基团如羧基、氰基、酯基、酰胺基、醛基、酮基、氨基等辅助C—H键活化, 实现某一位置的C—H官能团化, 进而大大缩短原有的合成路线, 节省成本; 对于药物备选化合物筛选过程, 可以利用某些药效基团, 通过C—H键活化方法, 引入新的官能团和取代基, 改善药物活性或者理化性质, 并进而提高药物药效或者降低药物毒性; 对于创新药物工艺路线开发, 也要有意识地观察药物分子中是否有值得利用的药效基团可以作为导向基团来使用, 这对于开发简单实用的工艺极为重要.鉴于以上几点, 我们认为今后能同时充当药效基团和C—H活化导向基团的基团, 将在药物化学和有机化学中进得到一步的应用, 并最终实现药物的规模化工业生产, 实现药物化学与有机合成化学的完美结合.
(a) Wender, P. A.; Verma, V. A.; Paxton, T. J.; Pillow, T. H. Acc. Chem. Res. 2008, 41, 40.
(b) Mason, J. S.; Morize, I.; Menard, P. R.; Cheney, D. L.; Hulme, C.; Labaudiniere, R. F. J. Med. Chem. 1999, 42, 3251.
(c) Leach, A. R.; Gillet, V. J.; Lewis, R. A.; Taylor, R. J. Med. Chem. 2010, 53, 539.
(a) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094.
(b) Zhu, R. Y.; Farmer, M. E.; Chen, Y. Q.; Yu, J.-Q. Angew. Chem., Int. Ed. 2016, 55, 2.
(a) Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624.
(b) Murakami, K.; Yamada, S.; Kaneda, T.; Itami, K. Chem. Rev. 2017, 117, 9302.
(a) Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem., Int. Ed. 2012, 51, 8960.
(b) Pulis, A. P.; Procter, D. L. Angew. Chem., Int. Ed. 2016, 55, 9842.
(a) Seregin, I. V.; Gevorgyan, V. Chem. Soc. Rev. 2007, 36, 1173.
(b) Ping, Y. Y.; Wang, L. P.; Ding, Q. P.; Peng, Y. Y. Adv. Synth. Catal. 2017, 359, 3274.
(a) Wencel-Delord, J.; Droege, T.; Liu, F.; Glorius, F. Chem. Soc. Rev. 2011, 40, 4740.
(b) Chen, Z. K.; Wang, B. J.; Zhang, J. T.; Yu, W. L.; Liu, Z. X.; Zhang, Y. H. Org. Chem. Front. 2015, 2, 1107.
(a) Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074.
(b) Aziz, J.; Piguel, S. Synlett. 2017, 49, 4562.
(a) Engle, K. M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788.
(b) Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.; Kamatani, A.; Sonoda, M.; Chatani, N. Nature 1993, 366, 529.
(c) Kakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2003, 125, 1698.
(a) Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2012, 45, 5068.
(b) Shang, M.; Sun, S. Z.; Wang, H. L.; Wang, M. M.; Dai, H. X. Synthesis 2016, 48, 4381.
(a) Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936.
(b) Biafora, A.; Gooβen, L. J. Synlett 2017, 28, 1885.
(a) Ackermann, L. Acc. Chem. Res. 2014, 47, 281.
(b) Font, M.; Quibell, J. M.; Perry, G. J. P.; Larrosa, I. Chem. Commun. 2017, 53, 5584.
(a) Ritleng, V.; Sirlin, C.; Pfeffer, M. Chem. Rev. 2012, 102, 1731.
(b) Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2012, 102, 1731.
(c) Wang, S.; Yan, F.; Wang, L. S.; Zhu, L. Chin. J. Org. Chem. 2018, 38, 291(in Chinese).
(汪珊, 严沣, 汪连生, 朱磊, 有机化学, 2018, 38, 291.
Cernak, T.; Dykstra, K. D.; Tyagarajan, S.; Vachal, P.; Krska, S. W. Chem. Soc. Rev. 2016, 45, 546. doi: 10.1039/C5CS00628G
Schneider, N.; Lowe, D. M.; Sayle, R. A.; Tarselli, M. A.; Landrum, G. A. J. Med. Chem. 2016, 59, 4385. doi: 10.1021/acs.jmedchem.6b00153
Wu, X. F.; Neumann, H.; Beller, M. Chem. Soc. Rev. 2011, 40. 4986. doi: 10.1039/c1cs15109f
(a) Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2011, 40. 5068.
(b) Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem., Int. Ed. 2012, 51, 8960.
张巍, 张家慧, 刘运奎, 有机化学, 2014, 34, 36. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract343591.shtmlZhang, W.; Zhang, J. H.; Liu, Y. K. Chin. J. Org. Chem. 2014, 34, 36(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract343591.shtml
Mousseau, J. J.; Charette, A. B. Acc. Chem. Res. 2013, 46, 412. doi: 10.1021/ar300185z
张霁, 聂飚, 张英俊, 有机化学, 2015, 35, 337. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344682.shtmlZhang, J.; Nie, B.; Zhang, Y. J. Chin. J. Org. Chem. 2015, 35, 337(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344682.shtml
Pennington, L. D.; Moustakas, D. T. J. Med. Chem. 2017, 60, 3552. doi: 10.1021/acs.jmedchem.6b01807
Verheij, M. H. P.; Thompson, A. J.; van Muijlwijk-Koezen, J. E.; Lummis, S. C. R.; Leurs, R.; de Esch, I. J. P. J. Med. Chem. 2012, 55, 8603. doi: 10.1021/jm300801u
Borrmann, T.; Abdelrahman, A.; Volpini, R.; Lambertucci, C.; Alksnis, E.; Gorzalka, S.; Knospe, M.; Schiedel, A. C.; Cristalli, G.; Müller, C. E. J. Med. Chem. 2009, 52, 5974. http://europepmc.org/abstract/MED/19731917
Yuan, Y.; Zaidi, S. A.; Elbegdorj, O.; Aschenbach, L. C. K.; Li, G.; Stevens, D. L.; Scoggins, K. L.; Dewey, W. L.; Selley, D. E.; Zhang, Y. J. Med. Chem. 2013, 56, 9156. doi: 10.1021/jm4012214
Verhoest, P. R.; Chapin, D. S.; Corman, M.; Fonseca, K.; Harms, J. F.; Hou, X.; Marr, E. S.; Menniti, F. S.; Nelson, F.; O'Connor, R.; Pandit, J.; Proulx-LaFrance, C.; Schmidt, A. W.; Schmidt, C. J.; Suiciak, J. A.; Liras, S. J. Med. Chem. 2009, 52, 5188. doi: 10.1021/jm900521k
Vanotti, E.; Amici, R.; Bargiotti, A.; Berthelsen, J.; Bosotti, R.; Ciavolella, A.; Cirla, A.; Cristiani, C.; D'Alessio, R.; Forte, B.; Isacchi, A.; Martina, K.; Menichincheri, M.; Molinari, A.; Montagnoli, A.; Orsini, P.; Pillan, A.; Roletto, F.; Scolaro, A.; Tibolla, M.; Valsasina, B.; Varasi, M.; Volpi, D.; Santocanale, C. J. Med. Chem. 2008, 51, 487. doi: 10.1021/jm700956r
Duraiswamy, A. J.; Lee, M. A.; Madan, B.; Ang, S. H.; Tan, E. S. W.; Cheong, W. W. V.; Ke, Z.; Pendharkar, V.; Ding, L. J.; Chew, Y. S.; Manoharan, V.; Sangthongpitag, K.; Alam, J.; Poulsen, A.; Ho, S. Y.; Virshup, D. M.; Keller, T. H. J. Med. Chem. 2015, 58, 5889. doi: 10.1021/acs.jmedchem.5b00507
Blum, C. A.; Caldwell, T.; Zheng, X.; Bakthavatchalam, R.; Capitosti, S.; Brielmann, H.; De Lombaert, S.; Kershaw, M. T.; Matson, D.; Krause, J. E.; Cortright, D.; Crandall, M.; Martin, W. J.; Murphy, B. A.; Boyce, S.; Jones, A. B.; Mason, G.; Rycroft, W.; Perrett, H.; Conley, R.; Burnaby-Davies, N.; Chenard, B. L.; Hodgetts, K. J. J. Med. Chem. 2010, 53, 3330. doi: 10.1021/jm100051g
Kalyani, D.; Deprez, N. R.; Desai, L.; Sanford, V. M. S. J. Am. Chem. Soc. 2005, 127, 7330. doi: 10.1021/ja051402f
Shabashov, D.; Daugulis, O. Org. Lett. 2005, 7, 3657. doi: 10.1021/ol051255q
Yu, W.-Y.; Sit, W. N.; Zhou, Z.; Chan, A. S.-C. Org. Lett. 2009, 11, 3174. doi: 10.1021/ol900756g
Li, W.; Yin, Z.; Jiang, X.; Sun, P. J. Org. Chem. 2011, 76, 8543. doi: 10.1021/jo2016168
Thu, H.-Y.; Yu, W.-Y.; Che, C.-M. J. Am. Chem. Soc. 2006, 128, 9048. doi: 10.1021/ja062856v
Kalyani, D.; Sanford, M. S. Org. Lett. 2005, 7, 4149. doi: 10.1021/ol051486x
Feng, C.-G.; Ye, M.; Xiao, K.-J.; Li, S.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 9322. doi: 10.1021/ja404526x
Li, C.; Yano, T.; Ishida, N.; Murakami, M. Angew. Chem., Int. Ed. 2013, 52, 9801. doi: 10.1002/anie.201305202
(a) Campeau, L.-C.; Rousseaux, S.; Fagnou, K. J. Am. Chem. Soc. 2005, 127, 18020.
(b) Campeau, L.-C.; Schipper, D. J.; Fagnou, K. J. Am. Chem. Soc. 2008, 130, 3266.
Do, H.-Q.; Khan, R. M. K.; Daugulis, O. J. Am. Chem. Soc. 2008, 130, 15185. doi: 10.1021/ja805688p
Xi, P.; Yang, F.; Qin, S.; Zhao, D.; Lan, J.; Gao, G.; Hu, C.; You, J. J. Am. Chem. Soc. 2010, 132, 1822. doi: 10.1021/ja909807f
Gong, X.; Song, G.; Zhang, H.; Li, X. Org. Lett. 2011, 13, 1766. doi: 10.1021/ol200306y
Kanyiva, K. S.; Nakao, Y.; Hiyama, T. Angew. Chem., Int. Ed. 2007, 46, 8872. doi: 10.1002/(ISSN)1521-3773
Cho, S. H.; Hwang, S. J.; Chang, S. J. Am. Chem. Soc. 2008, 130, 9254. doi: 10.1021/ja8026295
Patrick, F.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 10139. doi: 10.1021/ja5049303
Hilton, M. C.; Dolewski, R. D.; McNally, A. J. Am. Chem. Soc. 2016, 138, 13806 doi: 10.1021/jacs.6b08662
Cao, H.; Chen, L.; Liu, J.; Cai, H.; Deng, H.; Chen, G.; Yan, C.; Chen, Y. RSC Adv. 2015, 5, 22356. doi: 10.1039/C5RA01342A
Cao, H.; Lei, S.; Liao, J.; Huang, J.; Qiu, H.; Chen, Q.; Qiu, S.; Chen, Y. RSC Adv. 2014, 4, 50137. doi: 10.1039/C4RA09669J
Koubachi, J.; El Kazzouli, S.; Berteina-Raboin, S.; Mouaddib, A.; Guillaumet, G. Synthesis 2008, 2537.
Cao, H.; Lei, S.; Li, N.; Chen, L.; Cai, H.; Tan, J. Chem. Commun. 2015, 51, 1823. doi: 10.1039/C4CC09134E
Samanta, S.; Mondal, S.; Santra, S.; Kibriya, G.; Hajra, A. J. Org. Chem. 2016, 81, 10088. doi: 10.1021/acs.joc.6b02091
Yadav, M.; Dara, S.; Saikam, V.; Kumar, M.; Aithagani, S. K.; Paul, S.; Vishwakarma, R. A.; Singh, P. P. Eur. J. Org. Chem. 2015, 29, 6526. doi: 10.1002/ejoc.201500984/citedby
(a) Monir, K.; Bagdi, A. K.; Ghosh, M.; Hajra, A. J. Org. Chem. 2015, 80, 1332.
(b) Ji, X.-M.; Wei, L.; Chen, F.; Tang, R.-Y. RSC Adv. 2015, 5, 29766.
Zhao, D.; Wang, W.; Yang, F.; Lan, J.; Yang, L.; Gao, G.; You, J. Angew. Chem., Int. Ed. 2009, 48, 3296. doi: 10.1002/anie.v48:18
Fleming, F. F.; Yao, L.-H.; Ravikumar, P. C.; Funk, L.; Shook, B. C. J. Med. Chem. 2010, 53, 7902. doi: 10.1021/jm100762r
王江, 柳红, 有机化学, 2012, 32, 1643. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341245.shtmlWang, J.; Liu, H. Chin. J. Org. Chem. 2012, 32, 1643(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341245.shtml
Hingorani, M.; Lightman, S. Drugs 1995, 50, 208. doi: 10.2165/00003495-199550020-00002
Norman, P. Curr. Opin. Anti-Inflammatory Immunomodulatory Invest. Drugs 2000, 2, 113.
Reginster, J.-Y.; Brandi, M.-L.; Cannata-Andia, J.; Cooper, C.; Cortet, B.; Feron, J.-M.; Genant, H.; Palacios, S.; Ringe, J. D.; Rizzoli, R. Osteoporosis Int. 2015, 26, 1667. doi: 10.1007/s00198-015-3109-y
Rodriguez-Tudela, J. L.; Alcazar-Fuoli, L.; Mellado, E.; Alastruey-Izquierdo, A.; Monzon, A.; Cuenca-Estrella, M. Antimicrob. Agents Chemother. 2008, 52, 2468. doi: 10.1128/AAC.00156-08
Illnait-Zaragozi, M. T.; Martínez, G. F.; Curfs-Breuker, I.; Fernández, C. M.; Boekhout, T.; Meis, J. F. Antimicrob. Agents Chemother. 2008, 52, 1580. doi: 10.1128/AAC.01384-07
Yin, W.; Tsutsumi, K. Cardiovasc. Drug Rev. 2003, 21, 133. http://europepmc.org/abstract/MED/12847564
Hosie, J.; Scott, A. K.; Petrie, J. C.; Cockshott, I. D. Br. J. Clin. Pharmacol. 1990, 29, 333. doi: 10.1111/bcp.1990.29.issue-3
Sánchez, C.; Bøgesø, K. P.; Ebert, B.; Reines, E. H.; Braestrup, C. Psychopharmacology (Berl) 2004, 174, 163. http://europepmc.org/abstract/MED/15160261
Usui, F.; Maeda, K.; Kusai, A.; Nishimura, K.; Yamamoto, K. Int. J. Pharmol. 1997, 154, 59. doi: 10.1016/S0378-5173(97)00129-4
Hughes, Z. A.; Starr, K. R.; Langmead, C. J.; Hill, M.; Bartoszyk, G. D.; Hagan, J. J.; Middlemiss, D. N.; Dawson, L. A. Eur. J. Pharmacol. 2005, 510, 49. doi: 10.1016/j.ejphar.2005.01.018
Bourin, M.; NicDhonnchadha, B. A.; Claude, C. M.; Dib, M.; Hascoet, M. Behav. Brain Res. 2001, 124, 87. doi: 10.1016/S0166-4328(01)00238-8
Sanna, E.; Busonero, F.; Talani, G.; Carta, M.; Massa, F.; Peis, M.; Maciocco, E.; Biggio, G. Eur. J. Pharmacol. 2002, 451, 103. doi: 10.1016/S0014-2999(02)02191-X
John, G. W.; Perez, M.; Pauwels, P. J.; Le Grand, B.; Verscheure, Y.; Colpaert, F. C. CNS Drug Rev. 2000, 6, 278.
White, W. B.; Cannon, C. P.; Heller, S. R.; Nissen, S. E.; Bergenstal, R. M.; Bakris, G. L.; Perez, A. T.; Fleck, P. R.; Mehta, C. R.; Kupfer, S.; Wilson, C.; Cushman, W. C.; Zannad, F. N. Engl. J. Med. 2013, 369, 1327. doi: 10.1056/NEJMoa1305889
(a) Anbarasan, P.; Schareina, T.; Beller, M. Chem. Soc. Rev. 2011, 40, 5049.
(b) Dey, A.; Agasti, S.; Maiti, D. Org. Biomol. Chem. 2016, 14, 5440.
(c) Yang, J. Org. Biomol. Chem. 2015, 13, 1930.
(d) Qiu, G.; Wu, J. Org. Chem. Front. 2015, 2, 169.
(e) Zhang, M.; Zhang, Y.; Jie, X.; Zhao, H.; Li, G.; Su, W. Org. Chem. Front. 2014, 1, 843.
(f) Liu, J.; Chen, G.; Tan, Z. Adv. Synth. Catal. 2016, 358, 1174.
Li, W.; Xu, Z.; Sun, P.; Jiang, X.; Fang, M. Org. Lett. 2011, 13, 1286. doi: 10.1021/ol103075n
Du, B.; Jiang, X.; Sun, P. J. Org. Chem. 2013, 78, 2786 doi: 10.1021/jo302765g
Reddy, M. C.; Jeganmohan, M. Chem. Commun. 2015, 51, 10738 doi: 10.1039/C5CC03112E
Li, W.; Sun, P. J. Org. Chem. 2012, 77, 8362. doi: 10.1021/jo301384r
Chotana, G. A.; Rak, M. A.; Smith, M. R. J. Am. Chem. Soc. 2005, 127, 10539. doi: 10.1021/ja0428309
Bera, M.; Modak, A.; Patra, T.; Maji, A.; Maiti, D. Org. Lett. 2014, 16, 5760. doi: 10.1021/ol502823c
Tang, R. Y.; Li, G.; Yu, J.-Q. Nature 2014, 507, 215. doi: 10.1038/nature12963
Bag, S.; Patra, T.; Modak, A.; Deb, A.; Maity, S.; Dutta, U.; Dey, A.; Kancherla, R.; Maji, A.; Hazra, A.; Bera, M.; Maiti, D. J. Am. Chem. Soc. 2015, 137, 11888. doi: 10.1021/jacs.5b06793
Lassalas, P.; Gay, B.; Lasfargeas, C.; James, M. J.; Tran, V.; Vijayendran, K. G.; Brunden, K. R.; Kozlowski, M. C.; Thomas, C. J.; Smith Ⅲ, A. B.; Huryn, D. M.; Ballatore, C. J. Med. Chem. 2016, 59, 3183. doi: 10.1021/acs.jmedchem.5b01963
Smith, D. A. Metabolism, Pharmaco Kinetics and Toxicity of Functional Groups: Impact of the Building Blocks of Medicinal Chemistry in ADMET, Royal Society of Chemistry, Cambridge, UK, 2010, Chapter 3, pp. 99~167.
(a) Kaeding, W. J. Org. Chem. 1961, 26, 3144.
(b) Kaeding, W.; Shulgin, A. T. J. Org. Chem. 1962, 27, 3551.
(c) Kaeding, W. J. Org. Chem. 1964, 29, 2556.
(d) Kaeding, W. J. Org Chem. 1965, 30, 3750.
(e) Kaeding, W. J. Org. Chem. 1965, 30, 3754
Drapeau, M. P.; Gooβen, L. J. Chem.-Eur. J. 2016, 22, 1. doi: 10.1002/chem.201504553
Miura, M.; Tsuda, T.; Satoh, T.; Pivsa-Art, S.; Nomura, M. J. Org. Chem. 1998, 63, 5211. doi: 10.1021/jo980584b
Bechtoldt, A.; Tirler, C.; Raghuvanshi, K.; Warratz, S.; Kornhaaß, C.; Ackermann, L. Angew. Chem., Int. Ed. 2016, 55, 264. doi: 10.1002/anie.201507801
Chiong, H. A.; Pham, Q. N.; Daugulis, O. J. Am. Chem. Soc. 2007, 129, 9879. doi: 10.1021/ja071845e
(a) Giri, R.; Maugel, N.; Li, J. J.; Wang, D. H.; Breazzano, S. P.; Saunders, L. B.; Yu, J.-Q. J. Am. Chem. Soc. 2007, 129, 3510.
(b) Wang, D. H.; Mei, T.-S.; Yu, J.-Q. J. Am. Chem. Soc. 2008, 130, 17676.
Huang, L.; Biafora, A.; Zhang, G.; Bragoni, V.; Gooßen, L. J. Angew. Chem., Int. Ed. 2016, 55, 6933. doi: 10.1002/anie.201600894
Mamone, P.; Danoun, G.; Gooßen, L. Angew. Chem., Int. Ed. 2013, 52, 6704. doi: 10.1002/anie.v52.26
Reinaud, O.; Capdevielle, P.; Maumy, M. J. Chem. Soc., Chem. Commun. 1990, 566. http://www.researchgate.net/publication/250825458_Copper(II)_mediated_aromatic_hydroxylation_by_trimethylamine_N-oxide
Taktak, S.; Flook, M.; Foxman, B. M.; Que, L. Jr.; Rybak-Akimova, E. V. Chem. Commun. 2005, 42, 5301. http://www.ncbi.nlm.nih.gov/pubmed/16244735
(a) Ng, K. H.; Ng, F. N.; Yu, W. Y. Chem. Commun. 2012, 48, 11680.
(b) Ng, F. N, Zhou, Z.; Yu, W. Y. Chem.-Eur. J. 2014, 20, 4474.
Lee, D.; Chang, S. Chem.-Eur. J. 2015, 21, 5364. doi: 10.1002/chem.201500331
Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215. doi: 10.1002/anie.v47:28
Wang, D.-H.; Masa, M.; Giri, R.; Yu, J.-Q. J. Am. Chem. Soc. 2008, 130, 7190. doi: 10.1021/ja801355s
Wasa, M.; Yu, J.-Q. J. Am. Chem. Soc. 2008, 130, 14058. doi: 10.1021/ja807129e
Wang, G.-W.; Yuan, T.-T. J. Org. Chem. 2010, 75, 476. doi: 10.1021/jo902139b
Wang, G.-W.; Yuan, T.-T.; Li, D.-D. Angew. Chem., Int. Ed. 2011, 50, 1380. doi: 10.1002/anie.v50.6
Karthikeyan, J.; Cheng, C. H. Angew. Chem., Int. Ed. 2011, 50, 9880. doi: 10.1002/anie.v50.42
(a) Pimparkar, S.; Jeganmohan, M. Chem. Commun. 2014, 50, 12116.
(b) Peng X.; Wang, W.; Jiang, C.; Sun, D.; Xu, Z.; Tung, C.-H. Org. Lett. 2014, 16, 5354.
Karthikeyan, J.; Haridharan, R.; Cheng, C.-H. Angew. Chem., Int. Ed. 2012, 51, 12343. doi: 10.1002/anie.v51.49
Li, D.-D.; Yuan, T.-T.; Wang, G.-W. Chem. Commun. 2011, 47, 12789 doi: 10.1039/c1cc15897j
Wrigglesworth, J. W.; Cox, B.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. Org. Lett. 2011, 13, 5326. doi: 10.1021/ol202187h
Zhong, H.; Yang, D.; Wang, S.; Huang, J. Chem. Commun. 2012, 48, 3236 doi: 10.1039/c2cc17859a
Rakshit, S.; Grohmann, C.; Besset, T.; Glorius, F. J. Am. Chem. Soc. 2011, 133, 2350. doi: 10.1021/ja109676d
Hyster, T. K.; Ruhl, K. E.; Rovis, T. J. Am. Chem. Soc. 2013, 135, 5364. doi: 10.1021/ja402274g
Yang, W.; Wang, S.; Zhang, Q.; Liu, Q.; Xu, X. Chem. Commun. 2015, 51, 661. doi: 10.1039/C4CC08260E
Yang, W.; Sun, J.; Xu, X.; Zhang, Q.; Liu, Q. Chem. Commun. 2014, 50, 4420. doi: 10.1039/c3cc49496a
Wang, H.; Glorius, F. Angew. Chem., Int. Ed. 2012, 51, 7318. doi: 10.1002/anie.201201273
Wang, Z.; Kuninobu, Y.; Kanai, M. J. Am. Chem. Soc. 2015, 137, 6140. doi: 10.1021/jacs.5b02435
Hyster, T. K.; Ruhl, K. E.; Rovis, T. J. Am. Chem. Soc. 2013, 135, 5364. doi: 10.1021/ja402274g
Li, J.-J.; Mei, T.-S.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 6452. doi: 10.1002/anie.v47:34
Dai, H. X.; Stepan, A. F.; Plummer, M. S.; Zhang, Y. H.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 7222. doi: 10.1021/ja201708f
Pham, M. V, Ye, B.; Cramer, N. Angew. Chem., Int. Ed. 2012, 51, 10610. doi: 10.1002/anie.201206191
Xie, W.; Yang, J.; Wang, B.; Li, B. J. Org. Chem. 2014, 79, 8278. doi: 10.1021/jo5015239
Li, X.; Dong, Y.; Qu, F.; Liu, G. J. Org. Chem. 2015, 80, 790. doi: 10.1021/jo502224d
Zhang, Z.; Tanaka, K.; Yu, J.-Q. Nature 2017, 543, 538. doi: 10.1038/nature21418
Wallace, M. D.; McGuire, M. A.; Yu, M. S.; Goldfinger, L.; Liu, L.; Dai, W.; Shilcrat, S. Org. Process Res. Dev. 2004, 8, 738 doi: 10.1021/op0499021
Fray, M. J.; Gillmore, A. T.; Glossop, M. S.; McManus, D. J.; Moses, I. B.; Praquin, C. F. B.; Reeves, K. A.; Thompson, L. R. Org. Process Res. Dev. 2010, 14, 263. doi: 10.1021/op900092h
Madasu, S. B.; Vekariya, N. A.; Koteswaramma, C.; Islam, A.; Sanasi, P. D.; Korupolu, R. B. Org. Process Res. Dev. 2012, 16, 2025. doi: 10.1021/op300179u
Ackermann, L. Org. Process Res. Dev. 2015, 19, 260. doi: 10.1021/op500330g
Seki, M. Org. Process Res. Dev. 2016, 20, 867. doi: 10.1021/acs.oprd.6b00116
(a) Huang, J.; Chan, J.; Chen, Y.; Borths, C. J.; Baucom, K. D.; Larsen, R. D.; Faul, M. M. J. Am. Chem. Soc. 2010, 132, 3674.
(b) Huang, J.; Wang, X.; Chan, J. Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries, Eds.: Dunn, P. J.; Hii, K. K.; Krische, M. J.; Williams, M. T., Wiley Inc., Hoboken, New Jersey, 2013, Chapter 12.
Gauthier, D. R. Jr.; Limanto, J.; Devine, P. N.; Desmond, R. A.; Szumigala, R. H. Jr.; Foster, B. S, Volante, R. P. J. Org. Chem. 2005, 70, 5938. doi: 10.1021/jo0507035
Ouellet, S. G.; Roy, A.; Molinaro, C.; Angelaud, R.; Marcoux, J. F.; O'shea, P. D.; Davies, J. W. J. Org. Chem. 2011, 76, 1436. doi: 10.1021/jo1018574
(a) Phipps, R. J.; Gaunt, M. J. Science 2009, 323, 1593.
(b) Yang, Y.-F.; Cheng, G.-J.; Liu, P.; Leow, D.; Sun, T.-Y.; Chen, P.; Zhang, X.; Yu, J.-Q.; Wu, Y.-D.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 344.
(c) Yang, G.; Lindovska, P.; Zhu, D.; Kim, J.; Wang, P.; Tang, R.-Y.; Movassaghi, M.; Yu, J.-Q. J. Am. Chem. Soc. 2014, 136, 10807.

图式 1 导向基团与金属配位络合参与C—H活化的几种形式
Scheme 1 Models for C—H activation via directing group and metal coordination

图 1 一些含3-苯基吡啶的药物和候选药物
Figure 1 3-Phenylpyridine-containing pharmaceuticals and drug candidates

图式 2 通过钯催化的直接邻位C—H芳基化的方法
Scheme 2 Methods for palladium-catalyzed direct ortho C—H arylation

图式 3 通过C—H活化将钯催化的2-苯基吡啶直接磷酸化
Scheme 3 Palladium-catalyzed direct phosphorylation of 2-phenyl-pyridine via C—H activation

图式 4 钯催化的吡啶的区域选择的直接芳基化
Scheme 4 Palladium-catalyzed regioselective direct arylation of pyridine N-oxides

图式 5 吡啶N-氧化物与噻吩、呋喃或吲哚的钯催化的氧化交叉偶联
Scheme 5 Palladium-catalyzed oxidative cross-coupling between pyridine N-oxides and thiophenes, furans or indoles

图式 6 Ni或Pd催化的吡啶N-氧化物的烯基化
Scheme 6 Ni or Pd-catalyzed alkenylation of pyridine N-oxides

图式 7 几种生物活性分子的后期C(4)官能团化
Scheme 7 Late-stage C(4)-functionalization of several bioactive molecules

图式 8 用于咪唑并[1, 2-a]吡啶的直接C—H官能化的方法
Scheme 8 Methods for direct C—H functionalization of imidazo[1, 2-a]pyridines

图式 9 芳基溴化物直接芳基化杂环的方法
Scheme 9 Methods for direct arylation of heterocycles with aryl bromides

图式 11 Cu介导的和Fe-催化的苯甲酸的邻羟基化
Scheme 11 Cu-mediated and Fe-catalyzed ortho-hydroxylation of benzoic acids

图式 12 Pd、Rh或Ir催化C—H酰胺化合成邻氨基苯甲酸的三种新方法
Scheme 12 Three new methods for the synthesis of anthranilic acids by C—H amidation catalyzed by Pd, Rh or Ir

图式 13 Pd催化C—H官能化的苯甲酸邻卤化
Scheme 13 Pd-catalyzed ortho-halogenation of benzoic acids by C—H functionalization

图式 14 Pd催化的交叉偶联硼酸和N-甲氧基酰胺类化合物及通过C—H活化形成内酰胺
Scheme 14 Pd-catalyzed cross-coupling of boronic acids with N-methoxy amides and the formation of lactam via C—H activation

图式 15 钯催化的C—H活化N-甲氧基苯甲酰胺合成菲啶酮的新方法
Scheme 15 Palladium-catalyzed N-methoxybenzamides via C—H activation and the new synthetic method for phenanthridinones

图式 16 简单和多用途的N-甲氧基苯甲酰胺作为C—H官能团化的导向基团
Scheme 16 Simple and versatile N-methoxybenzamidesas directing group for C-H functionalizations

图 5 生物活性磺酰胺药物和活性苯并稠合五环磺酰胺
Figure 5 Biologically active sulfonamide drug and active benzofused five-ring sultam

图式 17 钯催化的通过C—H活化从三氟甲基磺酰化苯乙胺合成吲哚啉和四氢异喹啉
Scheme 17 Synthesis of indolines and tetrahydroisoquinolines from ArCH2CH2NHSO2CF3 via Pd-catalyzed C—H activation

图式 18 钯催化的通过C—H活化官能团化苯磺酰胺(ArSO2NHC6F5)
Scheme 18 Pd-catalyzed functionalization of ArSO2NHC6F5 via C—H activation

图 6 余(Yu)氏模型(U形)用于杂环类底物远程位点选择性的C—H键活化的双金属方法
Figure 6 Yu's model (U-shaped) for cooperative bimetallic approach for remote, site-selective C—H activation of heterocycles

图式 20 磺胺类药效团引导的C—H官能化:药物分子的后期多样化
Scheme 20 Divergent C—H functionalizations directed by sulfonamide pharmacophores: late-stage diversification of drug molecules

图式 21 AgF2促进氟化/胺化合成SB-273005
Scheme 21 AgF2 promoted fluorination/amination for SB-273005

图式 22 C—H氟化/胺化合成PF-1247324
Scheme 22 Synthesis of PF-1247324 via C—H fluorination/amination

图式 23 工业化合成方法和Ru催化的直接C—H芳基化反应制备氯沙坦
Scheme 23 Commercial process and Ru-catalyzed C—H arylation for Losartan

图式 24 四唑作为制备沙坦的导向基团和Ru催化的直接C—H芳基化反应
Scheme 24 Tetrazole as directing group for preparation of angiotensin Ⅱ receptor blockers

图式 25 直接C—H官能化在AMG 369的公斤级合成中的应用
Scheme 25 Application of direct C—H functionalization on kilogram scale synthesis of AMG 369

图式 26 噁唑啉作为邻位导向基团和Ru催化的直接C—H芳基化反应制备CEPT抑制剂
Scheme 26 Oxazoline as ortho-directing group and Rucatalyzed C—H arylation for CETP inhibitor
表 1 诺贝尔化学奖:绿色合成方法
Table 1. Nobel prize in chemistry: green synthetic methods
| 时间 | 方法 | 获奖理由和工业化应用 |
| 2001年 | 威廉·诺尔斯(美) 野依良治(日) |
手性催化还原反应 广泛应用于制药工业 实现了大规模工业化应用 |
| 巴里·夏普莱斯(美) | 手性催化氧化反应 广泛应用于制药工业 实现了大规模工业化应用 |
|
| 2005年 | 罗伯特·格拉布(美) 理查德·施罗克(美) 伊夫·肖万(法) |
烯烃复分解反应 广泛应用于制药工业 实现了大规模工业化生产 |
| 2010年 | Richard F. Heck(美) Ei-ichi Negishi(美) Akira Suzuki(日) |
钯催化交叉偶联 广泛应用于制药工业实现了规模的工业应用 |
| 20??年 | Who? | 催化的碳氢活化反应 迄今为止, 未见广泛的工业化应用 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们













































 下载:
下载: