图式 1.
铑催化喹啉氮原子导向C—C键断裂反应
Scheme 1.
Rhodium-catalyzed C—C activation reaction with quinoline directing group

惰性化学键选择性切断或活化近年来受到有机化学家的广泛关注, 是非常重要且极具挑战性的科学研究领域之一[1].在新反应及过程的设计中应用新型的、廉价的环境友好原料, 提高反应的原子经济性, 减少污染物的生成与排放, 成为当代化学工作者的主要任务之一.近年来, 金属促进或催化的有机反应为实现环境友好的高效、高选择性合成化学提供更好的机会, 在实现化学键的选择性切断与形成方面具有不可比拟的优势.其中碳氢键以及碳碳键骨架的活化及其在合成化学中的应用研究已经受到了广泛重视, 被称为“化学中的圣杯”[2].经过有机化学家们多年的努力, 过渡金属(如钌、铑、钯催化剂)催化碳-氢键活化反应取得了突破性的进展.碳-氢键被切断后可以加成到多种底物, 例如碳-碳双键或碳-碳叁键, 是非常经济有效的构筑新的碳-碳键骨架的方法.与碳-氢键的选择性切断反应相比, 碳-碳单键的切断、活化反应报道较少.这是由于在多数情况下碳-氢键的切断反应与碳-碳键的切断反应是竞争反应, 碳-氢键的切断要比碳-碳键的切断更为有利.碳-碳键活化通常有两条重要的活化模式:一个是金属插入到碳-碳键中(氧化加成); 另一个是通过金属有机化合物的β-碳脱除反应.碳-碳键由于具有较高的热力学稳定性和动力学惰性, 以及碳-碳σ键轨道的扭曲趋向性等原因, 在没有官能团活化的情况下通常很难发生化学反应[3].所以, 首要解决的难题是活性问题, 其次是反应的选择性问题, 大部分情况下有机分子中含有不同化学性质的碳-碳键, 如何选择性地将其中一个或数个碳-碳键活化是核心问题[4].目前, 实现这一目的的方法主要是采用一些特殊的底物.例如张力分子(如环丙烷衍生物), 由于环的高张力相对容易发生碳-碳键的断裂, 而引起化学家的兴趣.相对于张力分子, 非张力分子发生碳-碳键断裂反应的一种策略是利用配位基团或螯合基团的辅助作用.即在底物中引入具有配位能力的基团, 使其与过渡金属预配位, 将金属中心拉到一个指定的碳-碳键的近距离内, 从而发生插入反应, 使碳-碳键的活化反应在动力学上更有利.本文主要从两方面综述了1999年至2018年间过渡金属(Rh, Ni, Pd和Ru)催化基于氮原子导向基团化合物的碳-碳键选择性切断反应研究进展, 并对相关反应机理进行了探讨.为便于更全面地理解这一领域的发展, 本文也包括1999年前发表的部分重要结果.第一部分主要综述了过渡金属(Rh, Ni, Pd和Ru)催化带有氮原子导向基团的化合物(如酮、酯和醇)的碳-碳键选择性切断反应研究进展; 第二部分主要综述了铑催化外加配位或螯合辅助导向基团的化合物(如酮、醇等)的碳-碳键选择性切断反应研究进展.
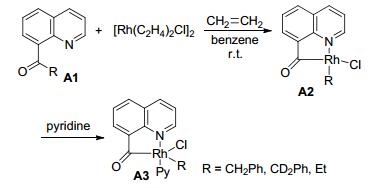
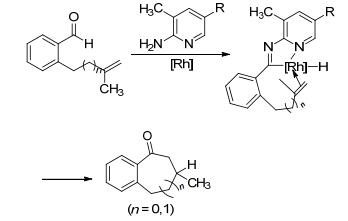
1984年, Suggs和Jun等[5]报道了化学计量的铑催化剂参与8-喹啉酮的碳-碳键断裂反应.由于喹啉上氮原子的配位作用, 使碳-碳键活化反应具有动力学上的优势.在金属化反应中, 五员环的铑金属杂环是有利的, 可以补偿碳-碳键活化过程的高能垒障碍, 使这一过程在热力学上更加可行.研究表明, 在反应过程中没有发现碳-氢键的断裂.反应可能经过铑直接进攻羰基形成四面体中间态, 而直接断裂碳-碳键.在反应前后, 羰基α-碳的立体构型保持不变, 说明羰基的存在可弱化其与邻位的碳-碳键作用, 有利于碳-碳键活化反应的进行(Scheme 1).
如果A4上的R官能团中有β-氢, 则可以与烯烃化合物发生化学计量学催化反应. Suggs和Jun等[6]报道了Rh(Ⅰ)催化8-喹啉丁基酮与乙烯气体的反应, 得到8-喹啉乙基酮和1-丁烯.该研究提出了相应的反应机理, 首先Rh(Ⅰ)对羰基α-位C—C键的插入, 生成稳定的五元铑金属环化合物A5, 然后通过β-氢消除得到A6和1-丁烯, 最后A6与乙烯反应得到产物A7 (Scheme 2).
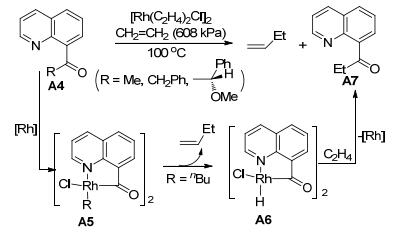
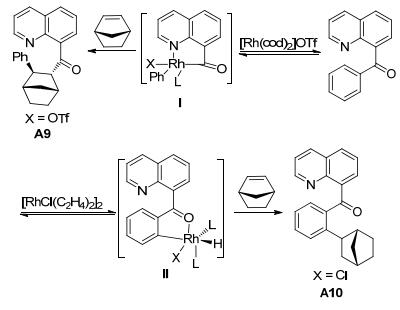
Douglas课题组对8-喹啉酮类化合物的碳-碳键断裂反应也进行了一系列研究. 2009年, 他们[7]利用8-喹啉酮化合物作为反应底物, 与烯烃类化合物A8进行反应(Eq. 1).研究发现, 在[Rh(cod)2]OTf为催化剂, 四氢呋喃(THF)为溶剂, 100 ℃加热搅拌, 只有C—C键活化的产物A9生成; 在[RhCl(C2H4)2]2为催化剂, 甲苯为溶剂, 130 ℃加热搅拌, 只有C—H键活化的产物A10生成.该反应在不同的催化条件下, 产生两种不同的反应中间体(Scheme 3).当[Rh(cod)2]OTf作为催化剂时, 铑与羰基碳、喹啉氮原子配位得到五元铑金属杂环化合物Ⅰ.当[RhCl(C2H4)2]2为催化剂时, 8-喹啉酮羰基上的氧原子与Rh配位形成络合物Ⅱ, 具有导向作用.因此, 催化剂和溶剂的选择在反应中是至关重要的.
|
|
(1) |
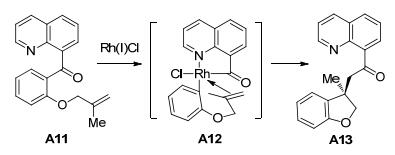
随后, Douglas课题组[8]报道了分子内烯烃插入、碳-碳键活化反应(Scheme 4).该反应具有较好的底物普适性.当Rh(OTf)(COD)2作为催化剂时, 产物的产率为25%.如果底物A11的醚基氧被氮取代, 要想发生碳-碳键活化反应需要使用威尔金逊催化剂.如果底物中的烯烃不与杂原子相连, 则以93%的产率得到五元碳环化合物.扩展底物烯烃链的长度, 以81%的产率得到六元杂环化合物.该研究提出了相应的反应机理: Rh(Ⅰ)首先插入到C—C键中, 与N原子配位, 得到五元铑杂环中间体A12.分子内烯烃与铑配位, 最后还原消除, 得到目标产物A13.
接着, Johnson课题组[9]对该反应的反应机理进行了深入研究.结合动力学和NMR研究, 了解该反应的反应动态和反应速率, 12C/13C的同位素效应及2AZDSS活化参数, 最后确定Rh(Ⅰ)插入到C—C键中是该反应的决速步骤.
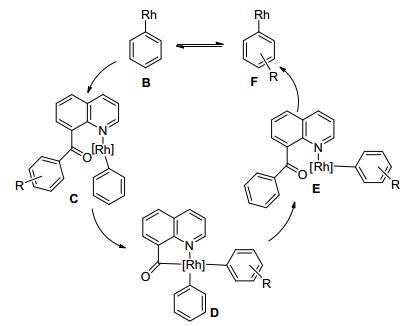
2016年, Johnson课题组[10]报道了铑催化喹啉芳基酮、烷基酮与硼酸的C—C键活化反应(Eq. 2).结果表明, 喹啉芳基酮在[Rh(C2H4)2Cl]2 作催化剂, 不加任何添加剂, 与带有吸电子基团的苯硼酸反应以较高转化率得到相应的偶联产物, 但是带有给电子基团底物的反应产率较低.喹啉烷基酮在[Rh(C2H4)2Cl]2作催化剂条件下, 不发生C—C键活化偶联反应, 需要用Rh(PPh3)3Cl作催化剂才能反应.喹啉芳基酮与甲硼酸在[Rh(C2H4)2Cl]2作催化剂, 碳酸钾作添加剂的条件下才发生C—C键活化偶联反应.该研究提出了相应的反应机理(Scheme 5):铑金属与硼酸先发生转金属生成Rh(Ⅰ)-芳基中间体B, Rh(Ⅰ)物种与路易斯碱喹啉氮原子配位形成中间体C, 然后经过氧化加成得到二芳基Rh(Ⅲ)金属五员环D, 最后还原消除, 解离得到最终产物和Rh(Ⅰ)-芳基物种F, Rh(Ⅰ)-芳基物种与另外的硼酸转金属B, 进入下一轮的催化循环.
|
|
(2) |
2011年, 施章杰课题组[11]报道了吡啶氮原子导向的仲醇C—C键断裂的反应(Scheme 6).该反应利用2-苯基吡啶的仲醇类化合物A14作为底物, 与烯烃化合物进行反应.该研究提出了可能的反应机理, Rh(Ⅲ)首先与N原子、氧原子配位形成醇铑中间体A15, 碳酸银作为碱起着去质子化、促进β-C消除释放苯甲醛的作用, 得到五元铑杂环化合物A16, 然后烯烃插入得到A17, 再经β-H消除得到烯基化产物A18和Rh(Ⅰ), Rh(Ⅰ)在碳酸银作用下氧化再次得到Rh(Ⅲ).
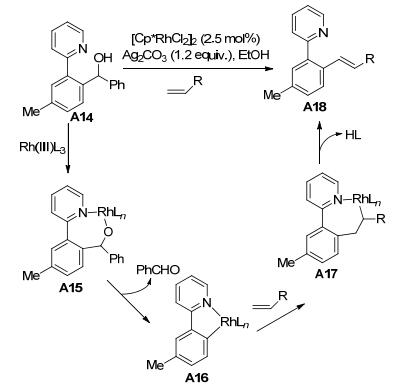
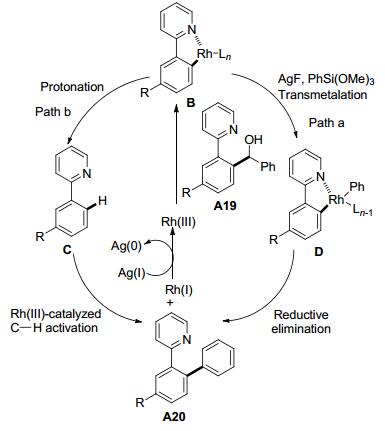
随后, 施章杰课题组[12]利用2-苯基吡啶的仲醇类化合物A19作为底物, 在[Cp*Rh(CH3CN)3][SbF6]2催化作用下, 与芳基硅化合物发生反应, 通过C—C键断裂, 得到相应的偶联产物(Eq. 3).值得注意的是, 叔醇在该催化体系下也以较高的产率得到C—C键断裂的产物, 而伯醇则以较低的产率得到C—C键断裂的产物.该课题组提出了两种可能的反应机理(Scheme 7): (1)仲醇A19先与铑催化剂生成五元铑杂环中间体B, 然后与硅试剂发生转金属化得到D, 最后发生还原消除得到偶联产物A20; (2)仲醇先与铑催化剂生成五元铑杂环中间体B, 然后质子化生成2-苯基吡啶的衍生物C, 再通过Rh(Ⅲ)催化C—H活化与硅试剂交叉偶联生成联芳基化合物A20.
|
|
(3) |
施章杰课题组[13]利用伯、仲、叔醇类化合物作为反应底物, 与亚胺类化合物的反应也进行了研究(Scheme 8).结果表明, 叔、仲醇反应产率较高, 而伯醇反应产率较低.仲醇与吸电子的醛在相同的反应条件下, 也可以发生C—C键断裂得到另外一种苄醇.该反应提出了可能的反应机理:仲醇先与铑金属生成七元铑杂环中间体, 然后β-C消除, 苯甲醛离去生成五元铑杂环中间体, 五元铑杂环中间体可以经过两种途径, 一种是与亚胺进行加成, 另一种是质子化得到2-苯基吡啶, 最后经过C—H键活化得到最终的产物.该反应条件简洁, 不需要任何添加剂, 为胺类和苄醇类化合物的合成提供了原子经济性高、绿色高效的合成方法.
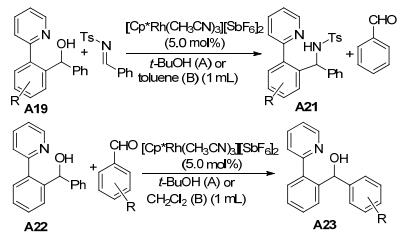
施章杰课题组[14]还发现在氢气条件下, 仲醇的C—C键断裂还原反应(Eq. 4).该反应探索了多种反应底物, 吡啶作为导向基团时, 2-苯基吡啶的苯环上带有给电子基团和吸电子基团的底物都能得到较高产率的目标产物.但是, 带有叔醇基团的底物得到目标产物的产率较低.仲醇右边苯环上带有吸电子基团能得到较高产率的目标产物, 但是带有给电子基团, 需要延长反应时间才能得到中等产率的目标产物.烷基仲醇也在该反应中适用.为了拓展该反应的应用性, 对不同的导向基团(吡唑)进行了研究, 以中等至较好的产率得到目标产物.反应过程经历七元铑杂环中间体、五元铑杂环中间体, 最后经氢气还原得到Rh(Ⅲ)氢物种. Rh(Ⅲ)氢物种可以促进C—C键断裂和醛的还原.为了解释2-苯基吡啶和醇中质子的来源, 该课题组利用不同的氘代试剂, 如D2、[D6]-EtOH、[D2]-CD2Cl2, 在相同的反应条件下, 对反应产物进行了监测与分析.
|
|
(4) |
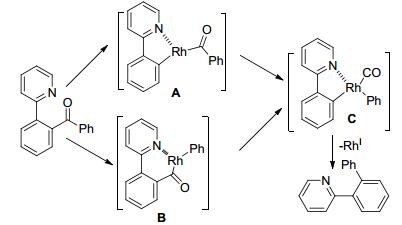
2012年, 施章杰课题组[15]利用芳基酮作为反应底物, 在[(CO)2Rh(acac)]催化作用下, 放出CO, 分子内发生C—C键断裂偶联反应(Eq. 5).结果表明, 2-苯基吡啶的苯环上无论连有给电子基团还是吸电子基团, 都能得到较高产率的脱羰基产物.苯并喹啉酮也进行了测试, 以91%的产率得到相应的目标产物.其他的氮原子导向基团, 如喹啉基、吡唑基、噁唑基, 也可以发生脱羰基反应, 得到中等产率的目标产物.邻位二酮在相同的反应条件下, 以89%的产率得到脱两个羰基的目标产物.该反应可能的反应机理是, Rh(Ⅰ)与酰基、吡啶氮原子配位生成酰基Rh(Ⅲ)物种, 然后羰基迁移插入, 最后还原消除得到最终产物(Scheme 9).
|
|
(5) |
2015年, 施章杰课题组[16]实现了2-苯基吡啶芳基酮、烷基酮与苯甲酸衍生物的脱羰基偶联反应.在铑催化条件下, 可以得到分子内脱羰基产物和分子间芳基化产物的混合物.为了提高反应的化学选择性, 需选取空间位阻大的反应底物.由于空间位阻能降低C—C键的断裂速率, 需要延长反应时间达到完全转化.从反应活性、效率和选择性综合考虑, 最终选取烷基酮A26作为反应底物, 与一系列羧酸进行反应, 主要得到分子间脱羰基偶联的产物.该反应对于官能团具有很好的兼容性.反应底物范围广, 其他的氮原子导向基团, 如喹啉、苯并喹啉、吡唑、噁唑基, 都能得到脱羰基偶联产物.作者进一步探讨了反应机理.通过密度泛函理论(DFT)计算, 对反应机理的可行性提供了一个可靠的依据.动力学结果表明, 脱羰基是反应过程的决速步骤(Eq. 6).
|
|
(6) |
2012年, 王建辉课题组[17]报道了喹啉酮与芳基硼酸的C—C键活化偶联反应(Eq. 7).在Rh(PPh3)3Cl催化剂、碘化亚铜和碳酸钾存在的条件下, 8-酰基喹啉与苯硼酸发生反应, 以较高的产率得到另一种喹啉酮化合物. 8-苯甲酰基喹啉衍生物被广泛用于设计医药和相关化合物的核心结构.例如微管蛋白抑制剂[18], 大麻素受体配体[19], 用于治疗骨代谢紊乱的药物[20], 抗溃疡剂[21].该实验方法提供了一种构建类似化合物有效的合成路径.底物拓展结果表明, 带吸电子基的苯硼酸与8-乙酰基喹啉反应的效果没有与供电子基反应的效果好. 2-噻吩硼酸与8-乙酰基喹啉反应没有得到预想的产物, 可能是由于硫原子与8-乙酰基喹啉的导向基团都可以与过渡金属配位, 有一定的竞争.邻甲基苯硼酸与8-乙酰基喹啉也不反应, 表明空间位阻在反应过程中起着重要的作用. 5-苯并喹啉-8-乙酰基酮参与反应, 虽然反应体系中有其它副产物生成, 但是仍然得到57%的目标产物. 8-乙酰基喹喔啉也能够与苯硼酸发生C—C键断裂反应, 得到目标产物的产率为89%.表明带有N-导向基团的乙酰基酮都能够使甲基官能团定向交换成芳基官能团, 在优化的反应条件下, 与一系列芳基硼酸反应得到相应的目标产物.当2-(二苯基膦)苯乙酮作为反应底物与苯硼酸进行偶联反应, 未监测到预想的目标产物, 这说明P原子作为导向基团, 在该反应中是不适用的.接着, 进一步研究了sp2-C—sp2-C键断裂反应.选用8-苯甲酰基喹啉作为反应底物, 与带有不同取代基的苯硼酸直接反应.令人遗憾的是, 相较于供电子基的苯硼酸, 带有吸电子基的苯硼酸与8-苯甲酰基喹啉都不反应, 没有预想的产物生成. 8-(4-氟苯甲酰基)喹啉与带有供电子基的苯硼酸也能够反应, 得到相应的目标产物的产率为61%~70%.相反, 与带有吸电子基的苯硼酸反应, 如4-硝基苯硼酸, 没有相应的产物被得到.但是, 当喹啉酮上带有强吸电子基团, 如8-(4-硝基苯甲酰基)喹啉, 与苯硼酸或4-甲氧基苯硼酸反应, 得到目标产物的产率明显提高, 分别为93%和91%.
|
|
(7) |
机理研究表明(Scheme 10), 催化剂Rh(PPh3)3Cl首先与8-乙酰基喹啉酮中乙酰基的C—C键和N原子螯合配位得到五元铑杂环化合物A[22]; 苯硼酸中的苯基与A中的卤素交换得到铑中间体B, 铑金属中心带有甲基和苯基官能团; 然后, 一个膦配体的插入, 使得B中的甲基和苯基官能团以甲苯的形式脱除, 得到Rh(Ⅰ)化合物C; 在氧气、碘化亚铜反应体系中[23], Rh(Ⅰ)化合物C氧化成Rh(Ⅲ)化合物D; Rh(Ⅲ)化合物D中的卤素再次与苯硼酸中的苯基交换得到络合物E; 最后, 络合物E还原消除得到产物3a, Rh(Ⅰ)化合物再次与两个膦配体配位, 进入下一轮的催化循环.

随后, 王建辉课题组[24]又报道了铑催化苯并喹啉酸乙酯与芳基硼酸的sp2-C—COOEt键的活化反应, 生成相应的10-芳基苯并喹啉化合物(Eq. 8).底物拓展结果表明, 带有供电子基的苯硼酸对于反应的进行没有明显的影响.带有吸电子基的苯硼酸, 如F, CF3, OCF3, NO2, 分别与原料苯并喹啉酸乙酯反应时, 得到产物的产率为83%~96%.当对羧酸乙酯的苯硼酸与苯并喹啉酸乙酯反应, 得到相应产物的产率较低, 仅有50%.在这个反应体系中, 苯并喹啉酸乙酯中COOEt作为离去基团, 而苯硼酸中的官能团COOEt得到保留, 这表明sp2-C—COOEt键断裂过程中, 配位N原子起着重要的作用.当苯并喹啉酸乙酯与带有卤素的苯硼酸反应, 氯原子与溴原子在反应体系中也得到保留, 可以对产物作进一步的修饰. β-萘硼酸在该催化体系中也显示了较好的反应活性, 得到目标产物的产率较高, 为87%.相反, α-萘硼酸不能发生反应, 表明空间位阻在反应体系中扮演着重要的角色.令人遗憾的是, 2-吡啶硼酸也不能与苯并喹啉酸乙酯反应, 可能是由于吡啶环上的氮原子和底物上的氮原子都能与金属配位, 具有一定的竞争力.2-甲基烯丙基苯硼酸也没有分离到相应的目标产物.
|
|
(8) |
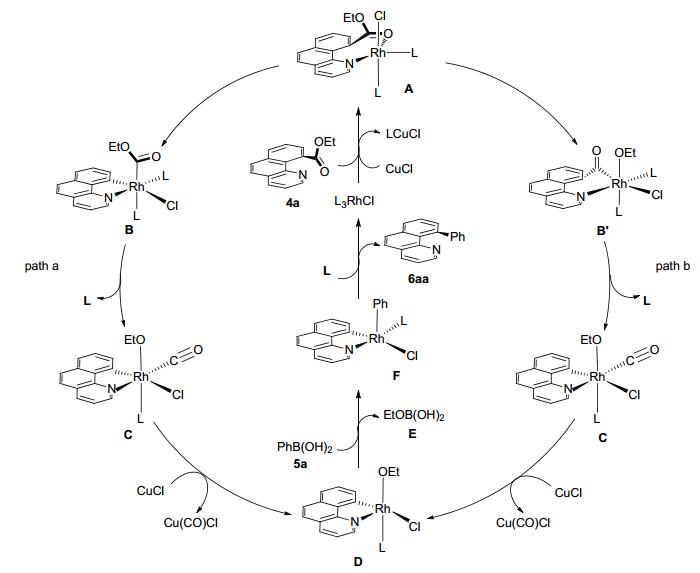
该反应提出了两种可行的机理(Scheme 11):首先, 在氯化亚铜存在条件下[25], 底物4a上的氮原子与铑催化剂配位得到络合物A; 然后, C—C键氧化加成到Rh(Ⅰ)金属中心生成Rh(Ⅲ)金属络合物B; 伴随PPh3的消除, B中酯片段中的乙氧基迁移到Rh(Ⅲ)金属中心生成Rh(Ⅲ)金属络合物C, 在C中, Rh(Ⅲ)金属与N原子、一个碳负离子、一个乙氧基官能团、一个Cl原子、一个CO和一个PPh3配位生成一种八面体结构(路径a). Rh(Ⅲ)金属络合物C经过另外一种途径生成:第一步, 在络合物A 中的Rh(Ⅰ)金属插入C(O)—OEt中得到中间体B'; 第二步, 伴随着PPh3的消除, 中间体B'酯片段中的CO迁移到Rh(Ⅲ)金属中心生成Rh(Ⅲ)金属络合物C(路径b).然后, 在CuCl作用下, C中的CO解离形成中间体D, 这个过程可能是CuCl与CO络合得到热力学稳定的络合物Cu(CO)Cl; 中间体D 中的EtO与苯硼酸中的Ph交换, 得到铑金属络合物F; 最后, 中间体F中的苯基官能团与苯并喹啉环还原消除得到目标产物6aa, 催化中间体进一步与底物4a配位得到A, 反应进入下一个循环过程.通过DFT计算, 对反应机理的可行性提供了一个可靠的依据.计算结果表明, 在路径b中的自由能明显高于路径a中的自由能.路径b中通过C—O键氧化加成的反应在动力学和热力学上来说是一种不利的反应途径.路径a则有利于反应的进行, 并在目标产物的形成过程中起着重要的作用.
2016年, 王建辉课题组[26]实现了Rh(PPh3)3Cl催化带有导向基团的伯醇衍生物发生C—C键活化反应, 并与芳基硼酸偶联制备各种联芳基化合物(Eq. 9).底物拓展结果表明, 间位Me, OMe, OCF3和NO2取代的苯硼酸, 在反应中有很好的相容性, 能以80%~92%的产率得到目标产物.邻位甲基取代的苯硼酸会受到空间位阻效应的影响, 只能以15%的产率得到目标产物.对位取代的各种苯硼酸也用于此反应中, 甲基和叔丁基取代的苯硼酸, 分别能获得93%和91%产率的产物.甲氧基和卤素取代的苯硼酸能产生70%~87%产率的产物.而乙酯基取代的苯硼酸只能获得44%的产物.官能团多取代苯硼酸也能顺利地进行此反应, 产生80%~95%产率的产物.在不同位置取代的苯并喹啉-10-甲醇, 如甲基、甲氧基、苯基、2-萘基, 都能很好的发生反应, 得到80%~90%产率的产物.说明苯并喹啉环上取代基的性质对反应没有太大影响. 2-(2-吡啶基)苯甲醇用于此反应过程, 只能得到较低产率的产物.
|
|
(9) |
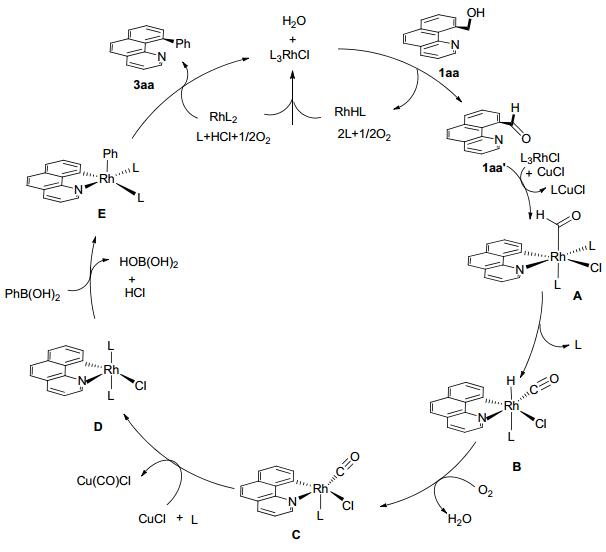
机理研究表明, 在Rh(PPh3)3Cl作用下, 原料1aa脱氢形成1aa', 随后在Rh(PPh3)3Cl和CuCl共同作用下形成中间体A, 醛内的氢迁移到Rh(Ⅲ)中心形成中间体B, 氢与金属中心共配位, 得到中间体C, 在CuCl的作用下, CO脱除形成中间体D, 加入苯硼酸形成中间体E, 最后E发生还原消除得到目标产物和Rh(0)配合物. Rh(0)配合物在氯化氢和水存在的条件下氧化为Rh(PPh3)3Cl重新进入催化反应体系(Scheme 12).
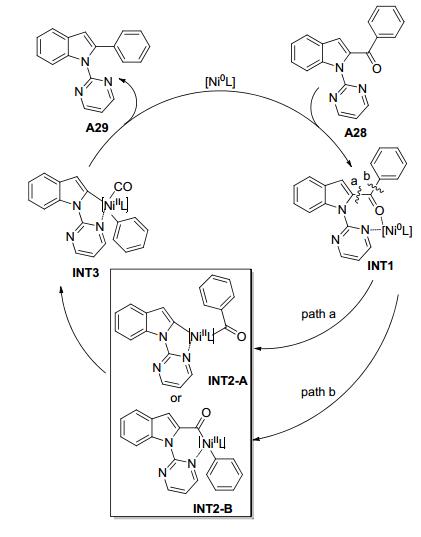
2018年, 魏颢课题组[27]实现了镍催化带有导向基团的芳基酮脱羰基C—C键断裂反应(Scheme 13).底物拓展结果表明, N-嘧啶基-2-苯甲酰基吲哚苯环对位上带有给电子基团(如甲基、乙基、异丙基、甲氧基、羟基和叔丁基)和吸电子基团(如酯基、苯基和三氟甲基)的底物都能得到较高产率的脱羰基产物.空间位阻对反应速率没有明显的影响, 在苯环的邻、间、对位连有取代基的底物都可以得到类似产率的目标产物.菲和萘环酮分别以86%和88%的产率得到脱羰基产物.富电子的杂环如呋喃和吡咯也分别以85%和82%的产率得到相应的目标产物.烷基酮没有监测到脱羰基产物.吲哚环上的取代基也进行了测试.吲哚环上连有甲氧基、甲基、酯基、氟、氰基的底物以较高产率得到相应的脱羰基产物.二酮(C7和C2上有C=O)发生反应, 只有C2上的羰基被脱掉, 表现出完全的区域选择性. 2-苯基吡啶的芳基酮在镍催化条件下也可以发生脱羰基反应.该反应提出了相应的反应机理:首先, Ni(0)与羰基氧和嘧啶氮原子配位得到七元镍(0)杂环中间体INT1;第二步有两种路径, 一个是C(吲哚)—C(=O)氧化断裂得到五元镍(Ⅱ)杂环中间体INT2-A, 另一个是C(=O)—C(芳基)氧化断裂得到五元镍(Ⅱ)杂环中间体INT2-B; 然后脱羰基产生相应的二芳基镍络合物INT3; 最后, 还原消除得到二芳基产物A29和Ni(0), Ni(0)进入下一轮催化循环.为了验证反应机理的可行性, 该课题组进行了DFT计算. DFT计算结果表明, INT1经历路径b得到INT2-B需要克服活化能垒34.0 kcal/mol, 比路径a高, 因此, 路径b可以排除.
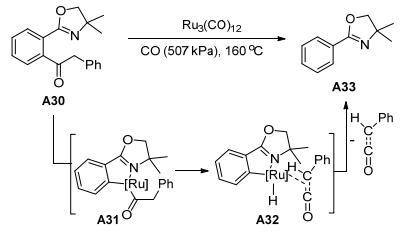
1999年, Murai课题组[28]也报道了一篇关于导向基团的C—C键活化反应(Scheme 14).底物拓展结果表明, 噁唑基氮原子作为导向基团, 甲基酮、丙酮、戊酮、苄基酮和甲硅烷基乙基酮得到高收率的脱羰基产物.十六烷基酮除了得到脱羰基产物, 并伴有77%的副产物十六碳烯.相应的环戊基酮、叔丁基酮和苯基酮未能反应.这可能是由于酮部分空间位阻大所致.使用吡啶环代替噁唑啉环, 也取得了较好的结果.值得注意的是, 导向基团的存在对于脱羰反应至关重要.二酮的反应涉及反应位点, 得到的是邻位脱羰基的产物. α, β-不饱和酮也可以在相同的反应条件下得到90%的脱羰基产物.该研究提出了相应的反应机理, 利用A30作为反应底物, 在CO (507 kPa)条件下, Ru3(CO)12为催化剂, 通过A31 β-氢消除和A32还原消除得到C—C键断裂产物A33.
2007年, Sames等[29]报道了一篇利用Ru3(CO)12作为催化剂, 一系列酯的衍生物和芳基硼酸偶联, 通过sp3-C—COOEt活化反应构建新的C—C键, 得到了相应的芳基化合物的文章(Eq. 10).研究表明, 在反应过程中并未发现有C—H键活化产物生成.
|
|
(10) |
2007年, Oshima等[30]利用带有螯合配位官能团的叔醇类衍生物作为反应底物, 在Pd(OCOCF3)2催化作用下, 与卤代苯发生C—C键断裂反应.在反应过程中, 氧化加成后, 经过配体交换, 钯与N、O原子配位得到六元钯杂环金属中间体, 经过β-碳消除, C(sp3)—C(sp3)键断裂得到胺钯中间体, 然后sp2芳基C—Pd键, 最后芳基钯物种还原消除得到2-苄基吡啶衍生物(Scheme 15).
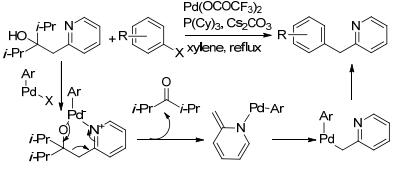
对于本身不含有导向基团的反应物, 可以通过向反应体系加入适当的配位或螯合试剂辅助反应的发生.但是, 加入的螯合试剂必须通过简单的方法易于消除.例如: 3-甲基-2-氨基吡啶则是一种很好的临时螯合辅助试剂.
Jun等[31]对这种需要加入临时螯合试剂才能发生C—C键活化反应的研究报道较多. 1999年, Jun等[32]用苄基丙酮与叔丁基乙烯反应, 在Rh(PPh3)3Cl和2-甲基- 3-氨基吡啶共催化作用下, 发生C—C键断裂反应, 得到一种烷基交换的产物分子和苯乙烯(Eq. 11).在反应过程中, 苄基丙酮首先与2-氨基-3-甲基吡啶缩合得到亚胺, 然后铑与吡啶的氮原子发生配位, 接着插入到亚胺α位C—C键形成五元环铑(Ⅲ)杂环配合物, 初步实现C—C键的选择性断裂, 继而β-氢消除释放出一分子苯乙烯, 并产生五元铑氢中间体.叔丁基乙烯插入铑氢中间体, 再还原消除形成烷基交换的新的亚胺中间体, 同时亚胺水解, 最终得到烷基交换的酮, 释放出2-氨基吡啶进入下一个催化循环.
|
|
(11) |
2001年, 在类似催化体系下, Jun等[33]将加入临时螯合辅助试剂的策略应用到醇类化合物, 使其发生预想的C—C键活化反应.该反应包含两个连续反应, 一个是仲醇氢的转移, 另一个则是螯合辅助的C—C键活化.碳酸钾在反应中起着加速反应速率的作用, 应该是一种有助于氢转移的辅助催化剂(Scheme 16).
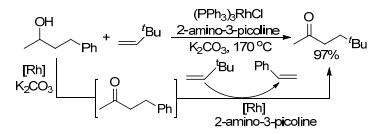
接着, Jun等[34]研究了环庚酮在[Rh(C8H14)2Cl]2和3-甲基-2-氨基吡啶作用下, 通过骨架重排得到C—C键断裂缩环产物的反应(Eq. 12).通过表征发现, 该反应生成五元环和六元环两种产物, 两种产物的比率为24:76, 六元环为主要产物.
Jun等[35]还利用C—C键活化和C—H键活化完成了等价于甲醛的烯丙基胺的官能团化反应(Scheme 17).该反应提出了相应的反应机理:烯丙基胺B1在Rh(Ⅰ)作用下异构化得到醛亚胺B2, 通过螯合作用发生C—H键活化生成五元铑杂环金属物种B3, 插入一分子叔丁基乙烯后还原消除得到酮亚胺B4, 此类型亚胺在高于80 ℃时会发生顺反异构化形成中间体B5, 同时在Rh(Ⅰ)与另一分子叔丁基乙烯作用下, 发生C—C键活化, 得到对称的二烷基亚胺B6, 继而经水解得到对称的二烷基酮B7.
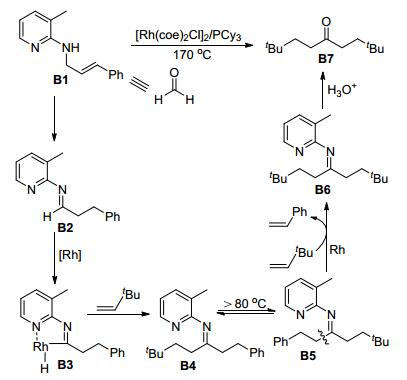
|
|
(12) |
螯合辅助C—C键断裂反应需要高温条件进行, 反应时间长, 速度慢. 2006年, Jun等[36]在无溶剂微波辐射条件下, 使C—C键断裂反应速率显著增加(Eq. 13).苄基丙酮与降冰片烯在5 mol% Rh(PPh3)3Cl、20 mol%的3-甲基-2-氨基吡啶和20 mol%环己胺共催化作用下, 微波加热200 ℃, 仅需5 min就能得到相应C—C键断裂产物和烯烃.加入催化量的环己胺的目的是进一步加速反应速率.该反应若使用常规油浴加热, 相同的反应条件和时间, 仅形成16%的产物酮.
2011年, Breit课题组[37]报道了铑金属和双功能助催化剂共同催化醛和烯烃的分子间加氢酰化反应(Scheme 18).相对于3-甲基-2-氨基吡啶, 3-甲基-6-(二苯基膦甲基)-2-氨基吡啶在加氢酰化反应中提高了助催化剂的效率.底物拓展结果表明, 苯甲醛与各种末端烯烃反应, 如酯、羧基、羟基和烷基, 以51%~83%的产率得到目标产物.苯甲醛苯环上无论带有给电子基团还是吸电子基团都能得到相应的目标产物.分子内加氢酰化利用邻乙烯基苯甲醛作为反应底物, 在苯环上带有给电子基团, 吸电子基团和中性基团, 都具有很好的官能团适应性. 2-乙烯基吡啶-3-甲醛不发生反应, 可能是由于吡啶氮原子与配体氮原子都能与铑配位, 具有一定的竞争力.
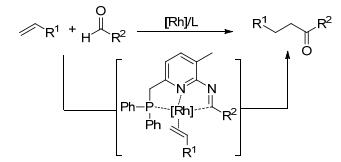
|
|
(13) |
2012年, Douglas课题组[38]报道了铑金属催化分子内加氢酰化反应合成六、七元环酮的方法(Scheme 19).在反应中, 需要加入临时螯合辅助试剂2-甲基-3-氨基吡啶类化合物, 该试剂可以有效地避免醛脱羰基分解和烯烃异构化.底物拓展结果表明, 在10 mol%共催化剂2-甲基-3-氨基吡啶条件下, 苯环上连有给电子基团(CH3、OCH3)和吸电子基团(CF3、F)的底物, 都能以76%~85%的产率得到加氢酰化产物.烯烃部分连有甲基和乙基的底物也能得到较高产率的目标产物.若烯烃部分连有苯基, 则需要25 mol%共催化剂2-甲基-3-氨基-5-吡咯烷基吡啶才能以77%的产率得到环庚酮化合物.烯烃和醛基连在杂环上, 如吡咯和吲哚, 反应速率慢, 需要25 mol% 2-甲基-3-氨基-5-吡咯烷基吡啶, 5 mol% [RhCl(coe)2]2和10 mol% PPh3才能得到相应的目标产物.该研究也对手性配体进行了探索, 虽然反应活性低, 但也以82%的产率和31% ee得到加氢酰化产物.
综上所述, 近年来化学工作者们在过渡金属(Rh, Ni, Pd和Ru)催化带有导向基团化合物的C—C键活化反应方面取得了一些成果, 为高效、高选择性地构建更多结构新颖的C—C键化合物提供了新的方法.但仍有一些问题值得深入研究.例如贵金属(如铑、钯和钌等)催化剂的使用不可避免, 这些金属虽然仅用到催化量, 但使得合成成本高昂, 难回收利用, 易造成环境污染和资源浪费.如果能使用铜、铁等金属作催化剂, 可在很大程度上节约成本.碳-碳键本身反应活性低, 目前所使用的反应条件大多比较苛刻(如高温), 主要实现的是导向基团化合物官能团邻位C—C键活化反应, 对于远离官能团的其它位置的C—C键活化非常困难, 远未达到随心所欲的水平.如果能在温和的条件下, 高效、高选择性地实现惰性碳-碳键活化, 构建结构多样的化合物具有非常重要的意义.此外, 这一领域研究的多数是消旋化反应, 对于不对称催化涉及较少.设计合成结构简单的手性辅助分子或配体, 在温和的条件下, 实现不对称碳-碳键选择性活化反应, 为构建结构新颖的手性分子提供新的合成策略.因此, 过渡金属催化C—C键活化反应仍是未来的研究热点之一, 也是合成科学面临的极具挑战性的前沿学科问题之一.
Reviews/Books on C-C bond or C-H bond activation:
(a) Yu, J. Q. ; Shi, Z. J. Topics in Current Chemistry, Springer-Verlag, New York, 2010.
(b) Dong, G. C-C Bond Activation, Springer Verlag, Berlin/Heidelberg, 2014.
(c) Chen, F. ; Wang, T. ; Jiao, N. Chem. Rev. 2014, 114, 8613.
(d) Dermenci, A. ; Coe, J. W. ; Dong, G. Org. Chem. Front. 2014, 1, 567.
(e) Liu, H. ; Feng, M. ; Jiang, X. Chem. -Asian J. 2014, 9, 3360.
(f) Souillart, L. ; Cramer, N. Chem. Rev. 2015, 115, 9410.
(g) Souillart, L. ; Cramer, N. Chem. Rev. 2015, 115, 9410.
(h) Murakami, M. ; Chatani, N. Cleavage of Carbon-Carbon Single Bonds by Transition Metals, John Wiley & Sons Ltd., Chichester, U. K., 2016.
(i) Murakami, M. ; Ishida, N. J. Am. Chem. Soc. 2016, 138, 13759.
(j) Zheng, Q. -Z. ; Jiao, N. Chem. Soc. Rev. 2016, 45, 4590.
(k) Kondo, T. Eur. J. Org. Chem. 2016, 1232.
(l) Murakami, M. ; Ishida, N. J. Am. Chem. Soc. 2016, 138, 13759.
(m) Kim, D. -S. ; Park, W. -J. ; Jun, C. -H. Chem. Rev. 2017, 117, 8977.
(n) Somerville, R. J. ; Martin, R. Angew. Chem., Int. Ed. 2017, 56, 6708.
(o) Yang, J. ; Hu, J. ; Huang, Y. ; Xu, X. ; Qing, F. Chin. J. Chem. 2017, 35, 867.
(p) Zhang, J. -R. ; Xu, L. ; Liao, Y. -Y. ; Deng, J. -C. ; Tang, R. -Y. Chin. J. Chem. 2017, 35, 271.
(q) Tang, M. -M. ; Huo, X. -H. ; Meng, Q. -H. ; Zhang, W. -B. Acta Chim. Sinica 2016, 74, 219 (in Chinese).
(汤淏溟, 霍小红, 孟庆华, 张万斌, 化学学报, 2016, 74, 219. )
(r) Zhou, Q. -Q. ; Liu, D. Xiao, W. -J. ; Lu, L. -Q. Acta Chim. Sinica 2017, 75, 110 (in Chinese).
(周泉泉, 刘丹, 肖文精, 陆良秋, 化学学报, 2017, 75, 110. )
(s) Liang, T. -T. ; Jiang, L. ; Gan, M. -M. ; Su, X. ; Li, Z. -N. Chin. J. Org. Chem. 2017, 37, 3096 (in Chinese).
(梁婷婷, 姜岚, 干苗苗, 苏鑫, 李争宁, 有机化学, 2017, 37, 3096. )
Bard, A. J.; Whitesides, G. M.; Zare, R. N.; McLaffertv, F. W. Acc. Chem. Res. 1995, 28, 91. doi: 10.1021/ar00051a001
Zhang, L. -H. Advances in Chemical Science, Chemical Industry Press, Beijing, 2005, pp. 85~102 (in Chinese).
(张礼和, 化学学科进展, 化学工业出版社, 北京, pp. 85~102. )
许泽君, 焦宁, 中国科学:化学, 2013, 43, 1121.Xu, Z.-J.; Jiao, N. Sci. Sin. Chim. 2013, 43, 1121 (in Chinese).
Suggs, J. W.; Jun, C.-H. J. Am. Chem. Soc. 1984, 106, 3054. doi: 10.1021/ja00322a063
Suggs, J. W.; Jun, C.-H. J. Chem. Soc., Chem. Commun. 1985, 92. doi: 10.1039/c39850000092
Wentzel, M. T.; Reddy, V. J.; Hyster, T. K.; Douglas, C. J. Angew. Chem., Int. Ed. 2009, 48, 6121. doi: 10.1002/anie.v48:33
Dreis, A. M.; Douglas, C. J. J. Am. Chem. Soc. 2009, 131, 412. doi: 10.1021/ja8066308
Rathbun, C. M.; Johnson, J. B. J. Am. Chem. Soc. 2011, 133, 2031. doi: 10.1021/ja109686v
Dennis, J. M.; Compager, C. T.; Dorn, S. K.; Johnson, J. B. Org. Lett. 2016, 18, 3334. doi: 10.1021/acs.orglett.6b01434
Li, H.; Zhang, X.-S.; Chen, K.; Wang, X. Shi, Z.-J. J. Am. Chem. Soc. 2011, 133, 15244. doi: 10.1021/ja205228y
Chen, K.; Li, H.; Li, Y.; Zhang, X.-S.; Lei, Z.-Q.; Shi, Z.-J. Chem. Sci. 2012, 3, 1645. doi: 10.1039/c2sc00923d
Zhang, X.-S.; Li, Y.; Li, H.; Chen, K.; Lei, Z.-Q.; Shi, Z.-J. Chem. Eur. J. 2012, 18, 16214. doi: 10.1002/chem.v18.50
Chen, K.; Li, H.; Lei, Z.-Q.; Li, Y.; Ye, W.-H.; Zhang, L.-S.; Sun, J.; Shi, Z.-J. Angew. Chem., Int. Ed. 2012, 51, 9851. doi: 10.1002/anie.201204338
Lei, Z.-Q.; Li, H.; Li, Y.; Zhang, X.-S.; Chen, K.; Wang, X.; Sun, J.; Shi, Z.-J. Angew. Chem., Int. Ed. 2012, 51, 2690. doi: 10.1002/anie.201107136
Lei, Z.-Q; Pan, F.; Li, H.; Li, Y.; Zhang, X.-S.; Chen, K.; Wang, X.; Li, Y.-X.; Sun, J.; Shi, Z.-J. J. Am. Chem. Soc. 2015, 137, 5012. doi: 10.1021/ja512003d
Wang, J.; Chen, W.; Zuo, S.; Liu, L.; Zhang, X.; Wang, J. Angew. Chem., Int. Ed. 2012, 51, 12334. doi: 10.1002/anie.v51.49
(a) Zarghi, A. ; Ghodsi, R. Bioorg. Med. Chem. 2010, 18, 5855.
(b) Nien, C. -Y. ; Chen, Y. -C. ; Kuo, C. -C. ; Hsieh, H. -P. ; Chang, C. -Y. ; Wu, J. -S. ; Wu, S. -Y. ; Liou, J. -P. ; Chang, J. -Y. J. Med. Chem. 2010, 53, 2309.
Thomas, B. C. ; James, C. A. ; Karol, D. E. ; Ulrich, S. WO 2002042248, 2002 [Chem. Abstr. 2002, 137, 6003].
Teruo, O. ; Shigeki, S. ; Takayuki, I. ; Yasuji, U. ; Tatsuya, Y. ; Noriko, Y. JP 10291988A, 1998 [Chem. Abstr. 1998, 130, 24979].
Yasuo, O. ; Juji, N. ; Haruki, T. ; Naokatsu, S. ; Hiroshi, K. ; Shunei, Y. ; Akio, I. JP 07173138A, 1995[Chem. Abstr. 1995, 123, 313777].
(a) Dowerah, D. ; Radonovich, L. J. ; Woolsey, N. F. Organometallics 1990, 9, 614.
(b) Omae, I. Chem. Rev. 1979, 79, 287.
(a) Ueura, K. ; Satoh, T. ; Miura, M. J. Org. Chem. 2007, 72, 5362.
(b) Ueura, K. ; Satoh, T. ; Miura, M. Org. Lett. 2007, 9, 1407.
(c) Kisenyi, J. M. ; Sunley, G. J. ; Cabeza, J. A. ; Smith, A. J. ; Adams, H. ; Salt, N. J. ; Maitlis, P. M. J. Chem. Soc., Dalton Trans. 1987, 2459.
Wang, J.; Liu, B.; Zhao, H.; Wang, J. Organometallics 2012, 31, 8598. doi: 10.1021/om300994j
Lipshutz, B. H.; Frieman, B.; Birkedal, H. Org. Lett. 2004, 6, 2305. doi: 10.1021/ol049681j
Yu, X. B.; Wang, J. J.; Guo, W. J.; Tian, Y.; Wang, J. Organometallics 2016, 35, 1876. doi: 10.1021/acs.organomet.6b00238
Zhao, T.-T.; Xu, W.-H.; Zheng, Z.-J.; Xu, P.-F.; Wei, H. J. Am. Chem. Soc. 2018, 140, 586. doi: 10.1021/jacs.7b11591
Chatani, N.; Ie, Y.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 1999, 121, 8645. doi: 10.1021/ja992048m
Gribkov, D. V.; Pastine, S. J.; Schnürch, M.; Sames, D. J. Am. Chem. Soc. 2007, 129, 11750. doi: 10.1021/ja072577n
Niwa, T.; Yorimitsu, H.; Oshima, K. Angew. Chem., Int. Ed. 2007, 46, 2643. doi: 10.1002/(ISSN)1521-3773
(a) Jun, C. -H. ; Lee, H. ; Hong, J. -B. J. Org. Chem. 1997, 62, 1200.
(b) Ahn, J. -A. ; Chang, D. -H. ; Park, Y. J. ; Yon, Y. R. ; Loupy, A. ; Jun, C. -H. Adv. Synth. Catal. 2006, 348, 55.
(c) Lee, D. -Y. ; Kim, I. -J. ; Jun, C. -H. Angew. Chem., Int. Ed. 2002, 41, 3031.
(d) Jun, C. -H. Chem. Soc. Rev. 2004, 33, 610.
(e) Park, Y. G. ; Park, J. -W. ; Jun, C. -H. Acc. Chem. Res. 2008, 41, 222.
Jun, C.-H.; Lee, H. J. Am. Chem. Soc. 1999, 121, 880. doi: 10.1021/ja983197s
Jun, C.-H.; Lee, D.-Y.; Kim, Y.-H.; Lee, H. Organometallics 2001, 20, 2928. doi: 10.1021/om010087c
Jun, C.-H.; Lee, H.; Lim, S.-G. J. Am. Chem. Soc. 2001, 123, 751. doi: 10.1021/ja0033537
(a) Jun, C. -H. ; Hong, J. -B. Org. Lett. 1999, 1, 887.
(b) Jun, C. -H. ; Lee, H. ; Park, J. -B. ; Lee, D. -Y. Org. Lett. 1999, 1, 2161.
(c) Wang, X. M. S. Thesis, Huaibei Normal University, Huaibei, 2014 (in Chinese).
(汪欣, 硕士论文, 淮北师范大学, 淮北, 2014. )
Ahn, J. A.; Chang, D. H.; Park, Y. J.; Yon, Y. R.; Loupy, A.; Jun, C H. Adv. Synth. Catal. 2006, 348, 55. doi: 10.1002/(ISSN)1615-4169
Vautravers, N. R.; Regent, D. D.; Breit, B. Chem. Commun. 2011, 47, 6635. doi: 10.1039/c1cc10683j
Beletskiy, E. V.; Sudheer, C.; Douglas, C. J. J. Org. Chem. 2012, 77, 5884. doi: 10.1021/jo300779q

图式 1 铑催化喹啉氮原子导向C—C键断裂反应
Scheme 1 Rhodium-catalyzed C—C activation reaction with quinoline directing group

图式 2 铑催化8-酰基喹啉C—C键活化反应
Scheme 2 Rhodium-catalyzed the C—C bond activation of 8-acylquinolines

图式 3 分子间C—C键和和C—H键活化反应机理
Scheme 3 Mechanism for intermolecular carboacylation and hydroarylation

图式 5 铑催化喹啉导向C—C键活化偶联反应机理
Scheme 5 Mechanism for rhodium-catalyzed 8-acylquinoline directed C—C bond coupling reaction

图式 6 铑催化仲醇C—C键断裂烯基化反应
Scheme 6 Rhodium-catalyzed C—C bond cleavage oefination reaction of secondary alcohols

图式 7 铑催化仲醇C—C键断裂芳基化反应机理
Scheme 7 Mechanism for rhodium-catalyzed sec-alcohols C—C bond cleavage arylation reaction

图式 8 铑催化仲醇C—C键断裂反应
Scheme 8 Rhodium-catalyzed C—C bond cleavage reaction of secondary alcohols

图式 9 铑催化芳基酮脱羰基反应机理
Scheme 9 Mechanism for rhodium-catalyzed aryl ketones decarbonylation reaction

图式 10 催化8-酰基喹啉与芳基硼酸偶联反应机理
Scheme 10 Mechanism for rhodium-catalyzed coupling reaction of 8-acylquinoline with aryl boronic acid

图式 11 铑催化苯并喹啉酸乙酯与芳基硼酸偶联反应机理
Scheme 11 Mechanism for rhodium-catalyzed coupling reaction of ethyl benzo[h]quinoline-10-carboxylate with aryl boronic acid

图式 12 铑催化伯醇衍生物与芳基硼酸偶联反应机理
Scheme 12 Mechanism for rhodium-catalyzed coupling reactions of primary alcohols and arylboronic acids

图式 13 镍催化芳基酮C—C键断裂反应
Scheme 13 Nickel-catalyzed C—C bond cleavage reaction of aryl ketones

图式 14 钌催化烷基苯基酮C—C键断裂反应
Scheme 14 Ruthenium-catalyzed C—C Bond cleavage reaction of alkyl phenyl ketones

图式 15 钯催化叔醇的C—C键活化反应
Scheme 15 Palladium-catalyzed C—C bond activation reaction of tertiary alcohols

图式 16 铑催化不含导向基团醇的C—C键活化反应
Scheme 16 Rhodium-catalyzed C—C bond activation of alcohols without directing group

图式 17 铑催化烯丙基胺的C—C键活化反应
Scheme 17 Rhodium-catalyzed C—C bond activation of allyl amines

图式 18 铑催化烯烃分子间和分子内加氢酰化反应
Scheme 18 Rhodium-catalyzed inter- and intramolecular hydroacylation of alkenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:













 下载:
下载:


 下载:
下载: