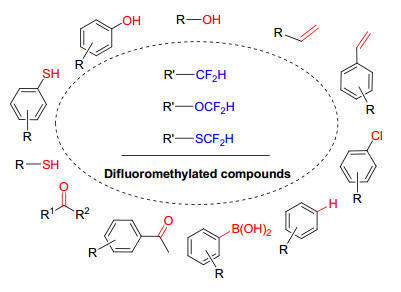
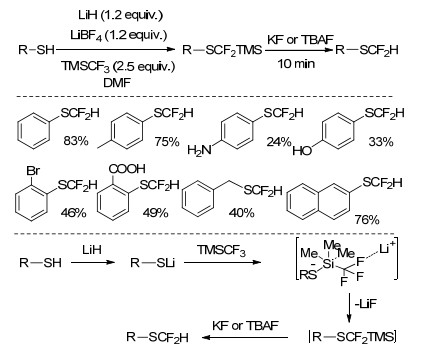
图式 1.
不同结构底物及其二氟甲基化产物
Scheme 1.
Different structural substrates and their difluoromethylated products

氟元素具有特殊的物理化学性质, 其在医药、农药等生理活性物质中占有重要地位, 据统计, 约有20%~25%的药物分子结构中含有氟原子[1].目前, 含氟医药、农药产品中大部分为含有氟原子和三氟甲基(CF3)、三氟甲硫基(SCF3)、三氟甲氧基(OCF3)的化合物, 对于该类官能团的引入手段以及相应化合物的生理活性研究已比较广泛[2~4].目前, 人们可以使用各种三氟甲基化试剂和官能化手段实现有机分子的三氟甲基化.
当前, 鉴于含氟基团在调节分子化合物固有性能方面的突出优势, 人们对于新的含氟官能团的追求是十分强烈的.近年来, 二氟甲基(CF2H)因其特殊的化学性质已经引起人们的广泛关注, CF2H和CF3均具有强的亲脂性和吸电子性, CF2H中的氢原子也可以作为氢键的给体参与氢键作用, 可以更加有效地增强有机分子的生理活性[5].因此, 对生物活性分子二氟甲基化已成为改造其生物活性的一种有效手段, 在新型药物设计和农药开发中日益受到研究人员的重视.如何向有机分子中引入二氟甲基在近些年来得到了快速的发展, 但与三氟甲基相比, 关于二氟甲基基团的引入, 相关的研究、总结较少[6].本文将以不同分子结构化合物的二氟甲基化为出发点, 对近年来二氟甲基化方法的研究进展进行归纳总结.
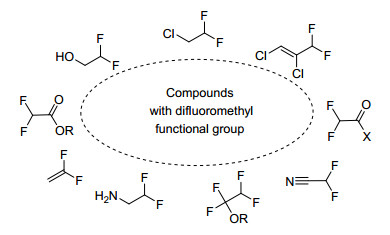
根据化合物中所含官能团的特点, 对二氟甲基化方法进行总结, 将有助于人们对不同结构有机分子的二氟甲基化过程的深入认识.按照底物结构特点, 可以二氟甲基化的化合物包括苯酚或烷基醇、硫酚或硫醇、羰基化合物、烯烃、卤代烃、芳香烃或杂环芳香烃、芳基硼酸等(Scheme 1).
在众多含氟基团中, 含氟烷氧基团(如OCF3、OCF2H、OCH2F、OCH2CF3等)普遍受到研究人员的重视[7]. OCF2H作为强吸电子基团可降低药物分子的电子云密度, 有助于抑制细胞色素P450对药物分子的氧化, 延长药物的作用时效[8].其次, 结构中的质子可作为氢键供体提高药物的靶效.基于上述独特的结构性能, 在新型医药、农药的开发领域, 将OCF2H基团引入至有机分子已成为最近十年的研究热点.
目前, 对于二氟甲基醚的制备, 大多通过酚或醇与二氟卡宾(:CF2)的反应来实现.可以制备:CF2的试剂主要有HCF2Cl[9]、BrCF2P(O)(OEt)2[10]、TMSCF2Br[11]、HFPO[12]、CF3ZnBr·2CH3CN[13]、FSO2CF2CO2H[14]等, 上述二氟甲基化试剂大多存在底物结构受限, 产率较低以及醇化合物用量较大等问题.因此, 针对醇的二氟甲基化反应, 寻求反应条件温和、对醇底物具有普适性, 且可方便制备的二氟甲基化试剂, 以及对现有反应工艺或助剂的改进仍然是人们重点关注的问题.
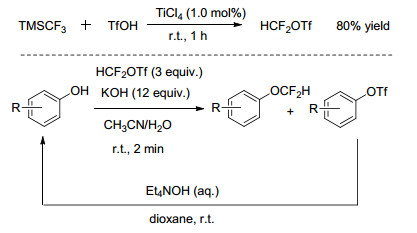
针对芳基酚的二氟甲基化, 目前使用较多的二氟甲基化试剂为HCF2Cl, 但其为消耗臭氧层物质(ODS), 开发的ODS特性的二氟甲基化试剂应用于芳基酚的二氟甲基化均需要高的反应温度和较长的反应时间, 同时适用的底物结构范围有限. 2013年, Hartwig等[15]在室温条件下, 以四氯化钛为催化剂, 通过三氟甲基三甲基硅烷(TMSCF3)和三氟甲基磺酸的反应制备了二氟甲基三氟甲基磺酸(HCF2OTf), 该试剂对空气稳定, 属非消耗臭氧层物质.以芳基酚为底物, HCF2OTf为二氟甲基化试剂, KOH为助剂, 室温条件下反应2 min即可较高收率地获得二氟甲基醚.同时, HCF2OTf反应后的产物通过在碱性条件下水解可实现原料芳基酚的回收(Scheme 2).
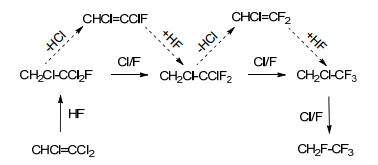
氟对氯的取代反应也是制备含氟有机化合物的常用方法.在路易斯酸或金属氧化物催化剂存在下, CCl3取代的芳香化合物经HF处理可得到CF3取代的芳香化合物.针对氯代烃或氯代烯烃和HF的气相氟化反应过程, 吕剑课题组开展了大量研究.如CHCl=CCl2(TCE)和HF反应制备CH2FCF3的反应过程, 氯代烯烃首先与HF反应生成中间体氢氟氯烃.该反应过程一般存在两种路径, 一种是通过亲核取代直接将C—Cl键转变为C—F键, 即Cl/F交换反应; 另一种是通过连续的HCl消去、HF加成反应实现上述转化.采用氘、18F/36Cl示踪、理论计算及对产物分布解析等方法对反应历程进行分析, 结果表明, TCE在氟化反应中易按亲核Cl/F交换反应路径进行(Scheme 3)[16].
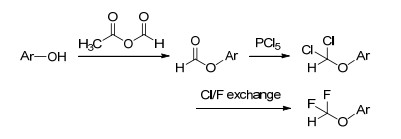
近年来, 人们也开始采用氯代化合物和HF的反应实现分子的二氟甲基化. 2012年, Brunet等[17a]采用HF对二氯甲基取代的芳香化合物处理, 得到了二氟甲基取代的芳香化合物.对于二氟甲氧基取代的芳香化合物的合成, 2014年Dolbier Jr等[17b]开发了基于氟/氯交换反应的新工艺.芳基酚首先与甲乙酐反应生成甲酸酯, 经PCl5处理生成二氯甲基醚, 再通过与氟化试剂THF-5HF、EtN-3HF或KF反应生成二氟甲基醚(Scheme 4). THF-5HF、EtN-3HF作为氟化介质, 反应可在室温下进行; KF为氟化介质, 以环丁砜为溶剂, 加热至125 ℃反应可获得较好的反应结果.两种氟/氯交换反应存在不同的反应过程, 前者主要经历碳正离子中间体, 后者为F-取代氯原子的SN2过程.该工艺可实现含不同取代基(如卤素、甲基、硝基、氰基、芳基等)的芳基酚的二氟甲基化.此外, 对于部分脂肪醇的二氟甲基化也具有适用性, 但对于氟化反应过程而言, 仅EtN-3HF具有较好的反应效果.
目前, 芳基二氟甲基醚的合成方法研究已有大量报道, 但烷基二氟甲基醚化合物的合成方法却少有报道.采用二氟卡宾合成策略制备烷基二氟甲基醚, 由于反应体系的强碱性环境通常会导致较低的产物收率. Prakash等[18]开发了醇和原位生成的亲电型二氟甲基化试剂反应生成相应的烷基二氟甲基醚的方法, 可以获得较高收率的目标产物.但是该二氟甲基化试剂稳定性较差, 同时烷基醇化合物用量需要大大过量.随后该研究小组尝试在该反应体系中使用较为稳定的亲电二氟甲基化试剂, 以此解决上述问题, 然而该类二氟甲基化试剂反应活性普遍较低.
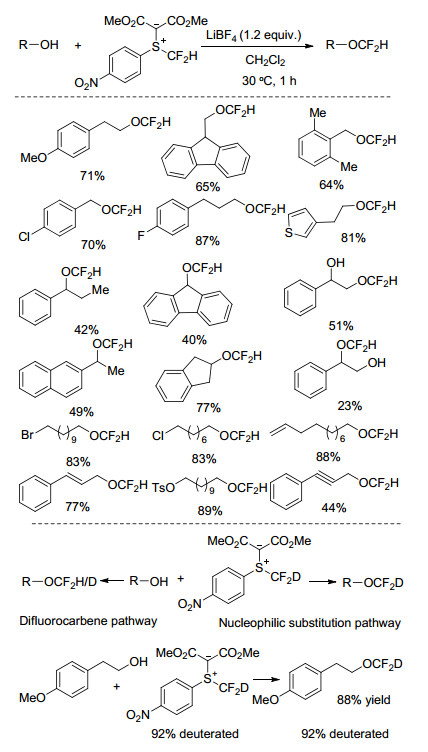
2016年, 沈其龙课题组[19]开发了一种亲电型的二氟甲基化试剂, 在路易斯酸LiBF4存在下, 可实现不同结构以及含其他官能团(卤素、烯烃、炔烃、羰基)的烷基醇化合物的二氟甲基化, 且目标产物收率较高.该二氟甲基化试剂可通过4-硝基苯基(二氟甲基硫醚)和重氮二甲酸二甲酯反应简便高效地制备.反应体系使用K2CO3, NaOH, LiOtBu或Cs2CO3等碱类助剂条件下, 目标产物收率均低于49%.然而, 以LiBF4作为反应助剂, 反应可以在非常温和的条件下进行, 二氟甲基醚收率可达到85%.反应溶剂的选择对于目标产物的收率具有重要影响, 以甲苯为反应溶剂, 产物收率为74%, 四氢呋喃为溶剂, 收率仅有13%, 乙腈、二甲基甲酰胺等极性溶剂条件下, 几乎无目标产物生成(Scheme 5).
LiBF4催化的醇的二氟甲基化反应可能经历二氟卡宾路径, 亲电二氟甲基化试剂首先脱去质子生成二氟卡宾, 随后插入到醇化合物中的O—H键生成二氟甲基醚化合物.此外, 反应也可能通过醇化合物直接亲核进攻亲电二氟甲基化试剂来进行.为进一步研究反应过程, 该研究小组合成了氘取代的亲电二氟甲基化试剂并应用于该反应体系, 如果反应经历二氟卡宾路径, 反应将会生成氢、氘取代的目标产物.研究发现, 醇和氘(D)取代的二氟甲基化试剂反应, 产物均为氘取代的二氟甲基醚, 反应条件下未发生氢和氘的交换反应.因此, 路易斯酸催化条件下, 醇的二氟甲基化过程主要为亲核取代反应路径.
TMSCF2Br作为二氟卡宾试剂在苯酚、硫醇、烯烃等化合物的二氟甲基化研究中应用较多, 胡金波课题组[20]以TMSCF2Br为二氟甲基化试剂, 对醇的二氟甲基化反应进行了大量研究, 认为:CF2中间体的生成为二氟甲基化反应过程的关键步骤. 2017年, 该课题组[21]通过在以TMSCF2Br为二氟甲基化试剂的反应体系中引入弱碱性的KOAc或弱酸性的KHF2等助剂, 实现了伯醇、仲醇、叔醇等化合物二氟甲基化过程的高效进行(Scheme 6).强碱性的NaOH为助剂, 可适用于具有富电子或较小位阻结构特点的醇类化合物的二氟甲基化.然而KOAc或KHF2适用的醇类化合物范围更广, 氰基、硝基、卤素、酯基以及被保护的氨基等基团在上述反应体系中均具有较好的耐受性, 炔基、烯基、烷基芳基醚、杂环芳香烃等基团与醇的二氟甲基化具有较好的相容性, 几乎无竞争反应.此外, 不同卤素取代的二氟甲基三甲基硅烷具有不同的反应活性, TMSCF2Br与TMSCF2Cl相比, 由于溴离子具有更好的离去性能, 使得TMSCF2Br在醇的二氟甲基化反应中表现出较好的反应性能.
研究表明, 醇的二氟甲基化过程存在浓度效应, 增加醇的浓度可大幅增加产物二氟甲基醚的收率.与苯酚不同, 醇的酸性较弱, 在强碱性条件下也难以有效生成氧负离子, 醇和氧负离子均会与:CF2反应, 醇的反应将会是主要的反应过程.醇的二氟甲基化反应存在浓度效应, 可能源于醇与苯酚负离子相比具有较低的亲核性.通过对反应体系助剂的考察, 在强碱NaOH以及弱酸KHF2等条件下, 醇的二氟甲基化反应均可高收率地得到目标产物; 而在KHF2等活化剂下, 苯酚类化合物的二氟甲基化反应以很低收率地得到目标产物.因此, 在该反应体系下, 醇可以不经脱质子化反应直接与︰CF2反应, 不同于苯酚与中间体二氟卡宾的反应路径.
与CF2H相比, SCF2H具有较高的酸性, 含二氟甲硫基(SCF2H)已成为调节有机分子生物活性的重要途径之一[22].目前, SCF2H的官能化途径主要有两种途径, 其一为硫醇及其衍生物与二氟甲基化试剂的反应; 其次为在各种有机分子结构中直接引入SCF2H[6a].两种官能化途径相比, 途径一具有相对较好的工业应用潜力.巯基具有与羟基相近的性质, 通过原位生成的二氟卡宾中间体与含巯基化合物的亲核加成反应可实现含硫化合物的二氟甲基化, 在此领域人们开展了大量研究, 文献报道的二氟卡宾前驱体主要有HCF2Cl、BrCF2P(O)(OEt)2、TMSCF2Br、HCF3[23]、FSO2CF2CO2H等.针对巯基的二氟甲基化, 近年来人们也开发了一些新的二氟甲基化试剂及其催化体系.
ClCF2COONa (SCDA)作为二氟卡宾的前体可实现对部分烯烃、卤代苯类化合物的二氟甲基化或三氟甲基化.但该试剂用于二氟甲硫基化的研究仅见于个别专利报道. 2013年, Greaney等[24]开发了以SCDA为二氟甲基试剂对硫醇化合物进行二氟甲基化的方法, 在K2CO3存在下SCDA在95 ℃下可发生脱羰基反应生成二氟卡宾中间体, 反应体系可实现芳香或杂环芳香硫醇类化合物的二氟甲基化.
2015年, Prakash等[25]首次报道了使用TMSCF3对巯基化合物进行二氟甲基化的研究.在固体碱LiH存在下, 巯基和TMSCF3反应生成五价离子中间体, 该中间体脱去LiF后生成含硅的二氟甲硫基化合物, 再经KF或四丁基氟化铵处理, 可得到相应的二氟甲硫基化合物.研究发现, 锂原子的存在有利于五价离子中间体脱除氟原子过程的进行.反应对于含NH2、OH、COOH等基团的巯基化合物的二氟甲基化具有一定的选择性, 但不适用于具有强吸电子基团取代基的巯基化合物(Scheme 7).
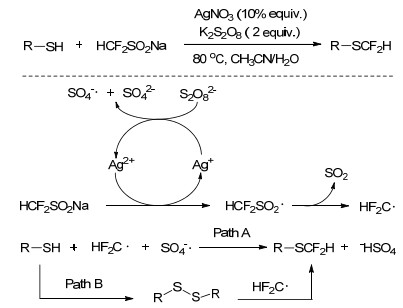
2017年, 易文斌课题组[26]报道了基于自由基反应的巯基化合物的二氟甲基化, 在Ag+/K2S2O8催化体系下, 通过巯基化合物和CF2HSO2Na的反应可实现芳香基硫醇及烷基硫醇的二氟甲基化.含有甲氧基、羟基、酰胺基、卤素等取代基的硫醇化合物, 也可以实现巯基的选择性二氟甲基化.该催化体系下, 反应过程如Scheme 8所示.首先, Ag+被过硫酸根阴离子(S2O82-)氧化产生Ag2+和硫酸根阴离子及硫酸根阴离子自由基.然后, Ag2+从CF2HSO2Na获得单个电子产生CF2HSO2自由基, 经脱除二氧化硫后生成CF2H自由基.由于在反应过程初期检测到二硫化物, 并随反应的进行浓度逐渐降低.推测反应存在两种路径, 路径A:由硫酸根阴离子自由基从硫醇化合物中夺去氢生成RS·, 该自由基通过与CF2H自由基结合生成目标产物; 路径B:反应条件下首先生成二硫化物, 该化合物再与CF2H自由基作用得到最终目标产物.
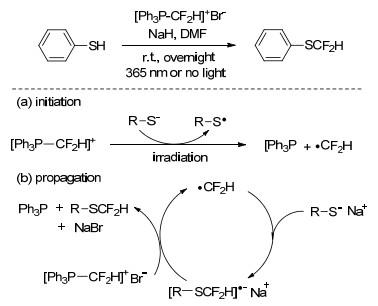
对于自由基反应类型的巯基二氟甲基化, 人们也在尝试使用一些已有的二氟甲基化试剂为前体生成二氟甲基自由基的研究.二氟甲基三苯基溴化磷([Ph3P-CF2H]+Br-)等磷盐化合物已被应用于包括硫醇在内的各种有机分子的二氟甲基化研究[27], 反应大多经过磷盐化合物产生二氟卡宾中间体的过程, 但对于以该类化合物为前体得到碳自由基用于二氟甲基化的研究报道相对较少.基于此, 2017年Studer等[28]报道了在温和条件下, 以[Ph3P-CF2H]+Br-为二氟甲基试剂, 对各种硫醇化合物进行基于自由基反应的二氟甲基化研究, 反应采用光引发产生二氟甲基自由基中间体, 避免了过渡金属的使用, 反应体系可适用于含OH、NH2、酰胺、酯基等基团的硫醇化合物的二氟甲基化(Scheme 9).
最近, 由于可见光光催化的有机反应具有条件温和的突出优势, 也被应用于二氟甲基化研究. 2017年, 付华课题组[29]报道了光催化还原方法的硫酚化合物的二氟甲基化研究.反应使用二氟溴乙酸作为二氟卡宾的前驱体, 以[fac-Ir(ppy)3]为光催化剂, 室温条件下经23 W的荧光灯照射, 即可获得高收率的二氟甲基化产物.苯环上含有卤素、硝基、氰基、甲氧基、酯基等取代基的硫酚化合物均可适用于该反应体系(Eq. 1).
|
|
(1) |
目前, 在众多合成含氟有机化合物的方法中, 亲核的氟烷基化反应受到许多研究人员的关注[30], 通过对醛、酮类化合物的亲核二氟甲基化是高效合成含α-CF2H醇化合物的重要途径.
2016年, 肖吉昌课题组[31]报道了一种在Cs2CO3存在下, 使用二氟甲基三苯基溴化磷(DFPB)对α-芳基醛或酮进行二氟甲基化的新方法, 反应可以高收率地得到含α-CF2H的醇化合物, 同时反应过程中基于Wittig反应的烯烃副产物可被有效抑制.该反应体系适用于含不同取代基的苯甲醛的二氟甲基化, 但含强吸电子基团的苯甲醛的二氟甲基化反应需在相对较低的温度下进行, 因为该类底物结构中的醛基易于发生安息香缩合副反应, 将降低目标产物收率.对于芳香酮的二氟甲基化, 底物结构中含强吸电子基团有利于二氟甲基化反应的进行, 产物收率相对较高.研究认为, 芳基醛或酮在该反应体系下的反应过程是通过DFPB中的二氟甲基的直接亲核转移来进行的(Scheme 10).

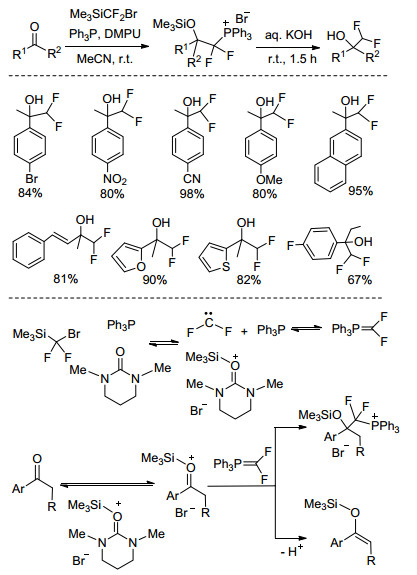
硅基试剂作为一种重要的氟烷基化试剂, 具有易于活化及反应条件温和的特点.在羰基化合物的二氟甲基化研究领域, 报道的硅基二氟甲基化试剂主要有Me3SiCF3、Me3SiCF2H、Me3SiCF2Cl、Me3SiCF2Br等[32]. 2016年, Prakash等[33]在三苯基磷存在下使用Me3SiCF3对芳香醛或酮进行二氟甲基化, 较高收率的合成了含二氟亚甲基的1, 1-二氟烯烃, 但该方法对芳基酮的二氟甲基化反应效果较差. Me3SiCF2H具有和Me3SiCF3相似的结构, 可作为亲核二氟甲基化的HCF2-来源.然而, CF2H与CF3相比吸电子性较弱, 早期的研究表明[34], 采用Me3SiCF2H的亲核氟烷基化反应通常需要苛刻的反应条件使得Si—CF2H键断裂, 同时反应仅适用于一些芳香醛的二氟甲基化, 对芳香酮的二氟甲基化产物收率仅为20%~35%.
2011年, 胡金波课题组在Me3SiCF2H的亲核二氟甲基化反应研究中发现, 通过选择合适的路易斯碱及溶剂对Si—CF2H的活化具有重要的作用.研究表明, Me3SiCF2H为二氟甲基试剂条件下, 以CsF为活化剂、DMF为溶剂, 室温下即可实现对芳香醛的二氟甲基化, 获得高收率的目标产物; 以t-BuOK为活化剂、THF为溶剂, —78 ℃即可高效实现Me3SiCF2H对二芳基酮的二氟甲基化, 但对于苯乙酮等化合物, 由于底物的可烯醇化特性及碱性条件下相应发生的醛醇缩合副反应, 难以得到相应的目标产物[35].
为解决Me3SiCF2H在可烯醇化酮类化合物二氟甲基化反应中存在的问题, 有必要对高价态的硅中间体化合物作深入研究, 同时探究该中间体结构中二氟甲基的转移性能. 2016年, 胡金波课题组[36]在以Me3SiCF2H为二氟甲基化试剂对醛、酮类化合物的二氟甲基化大量研究的基础上, 尝试在反应体系中引入冠醚作为助剂以降低HCF2-和碱土金属阳离子间的相互作用, 从而实现高价态硅中间体的稳定.研究表明, Me3SiCF2H经CsF/18-crown-6活化后可实现对可烯醇化酮类化合物的亲核二氟甲基化.反应体系中的双(二氟甲基)三甲基硅酸盐负离子[Me3Si(CF2H)2]-为重要的中间体, [(18-crown-6)Cs]+对该中间体的稳定具有重要的作用, 同时可以提高其亲核活性(Eq. 2).
|
|
(2) |
2016年, Dilman等[37]报道了在温和条件下, 以溴二氟甲基三甲基硅烷(Me3SiCF2Br)、三苯基膦(Ph3P)为原料, 经1, 3-二甲基丙撑脲(DMPU)催化后生成二氟化的磷内鎓盐的新方法, 该反应体系可应用于芳香甲基酮或杂环的芳香甲基酮化合物的亲核二氟甲基化, 均可较高收率地获得二氟甲基醇产物.该反应体系下的二氟甲基化反应过程为, Me3SiCF2Br和Ph3P在路易斯碱DMPU催化下生成二氟卡宾和相应的甲硅烷盐, 其中, 二氟卡宾和三苯基膦可以生成一定平衡浓度的磷内鎓盐.羰基经甲硅烷盐活化后生成阳离子化合物, 磷内鎓盐经亲核进攻该阳离子得到加成产物, 该加成产物经氢氧化钾处理后便得到二氟甲基醇化合物(Scheme 11).
在有机化合物中引入各种二氟烷基(CF2R)成为氟化学领域中的研究热点, CF2R不仅可改变分子化合物的性能, 还可通过所带烷基的官能团实现进一步改性[38].以烯烃为底物进行二氟甲基化是制备二氟烷基化合物的重要途径, 但现有文献报道的烯烃二氟烷基化的许多方法存在底物结构适用性窄的缺点, 且需要多步预处理过程[39].
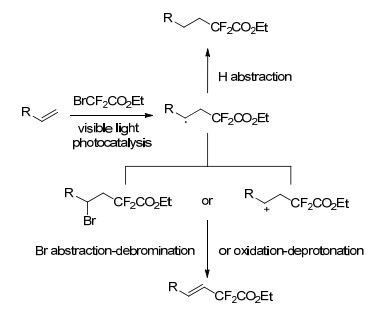
可见光光氧化还原的最新进展引起了人们的广泛关注, 2014年, Cho等[40]设计了一种新的可选择性光氧化催化烯烃的二氟烷基化反应, 该方法避免了繁琐的预处理过程, 同时反应条件温和、工艺环境友好.在光氧化催化剂存在下, 烯烃、2-溴-2, 2-二氟乙酸乙酯(BrCF2CO2Et)和碱的混合物经可见光照射后可实现烯烃的二氟烷基化.研究发现, 以二氯乙烷为溶剂, 四甲基乙二胺(TMEDA)和1, 8-二氮杂二环十一碳-7-烯(DBU)为碱的条件下可生成二氟烷基化的烷烃, 而以DMF为溶剂, K2CO3和DBU为碱, 反应将生成二氟烷基化的烯烃产物.因此, 反应体系中碱化合物种类的选择对于选择性生成二氟烷基化的烯烃或者烷烃产物至关重要.
光催化的烯烃二氟烷基化反应过程如Scheme 12所示, 首先BrCF2CO2Et在可见光催化下生成二氟烷基自由基(·CF2R), 该自由基通过与烯烃的加成反应生成二氟烷基取代的自由基中间体, 根据反应体系中碱的不同, 该中间体可通过夺得氢生成二氟烷基化的烷烃产物, 亦可直接通过氧化反应或者从BrCF2CO2Et中夺去溴, 再通过脱溴化氢生成二氟烷基化的烯烃.
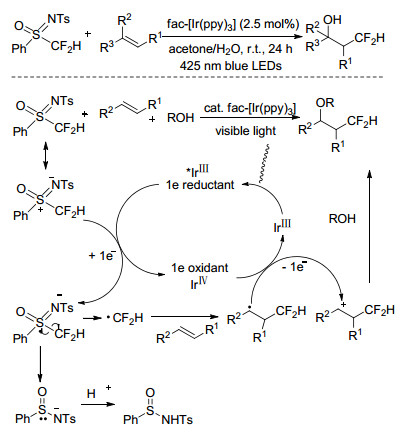
2016年, Akita等[41]首次报道了用N-甲苯磺酰基-S-二氟甲基-S-苯基亚磺酰亚胺为二氟甲基化试剂对苯乙烯衍生物进行光催化的二氟甲基化的研究, 设计的氧化还原中性反应体系可以获得系列β-CF2H取代的醇、醚及酯类化合物.该反应体系下, 首先光催化剂IrⅢ在可见光激发下得到激发态的*IrⅢ, 该物质作为强的单电子还原剂可将电子转移到二氟甲基化试剂, 进一步生成高氧化态的IrⅣ, 同时生成二氟甲基自由基, 二氟甲基自由基通过和烯烃的加成反应得到稳定的自由基中间体, 再经IrⅣ氧化后得到含β-CF2H的碳正离子, 最后经水解或醇解可得到相应的醇或醚类化合物(Scheme 13).
铜催化剂在烯烃二氟烷基化反应中得到了大量应用. 2016年, Poisson等[42]对铜催化的杂环烯烃的二氟烷基化反应进行了研究, 反应体系所用的催化剂主要有Cu(OTf)2, CuPF6·(MeCN)4, Cu2O, CuI等.二氢吡喃及其衍生物等杂环烯烃在上述铜催化剂作用下与BrCF2CO2Et、BrCF2CONR2等反应, 均可生成β位取代的二氟烷基化烯烃产物.铜催化杂环烯烃的二氟烷基化过程见Scheme 14.适用于该反应体系的杂环烯烃还包括苯并呋喃和呋喃衍生物以及少量的烯酰胺化合物, 经铜催化的杂环烯烃的二氟烷基化烯烃产物结构见Scheme 15.
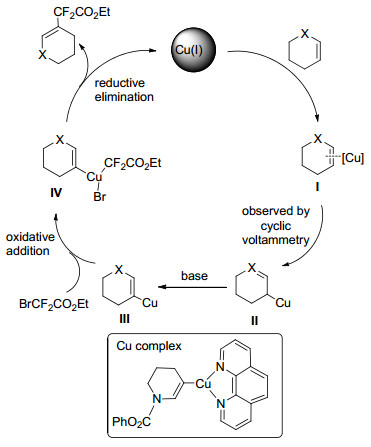

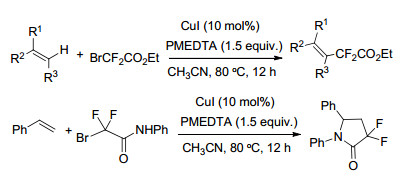
2017年, 唐向阳课题组[43a]报道了基于铜-有机胺催化体系的烯烃化合物二氟烷基化的高效方法, 反应体系优选的铜催化剂为CuI, 配体为五甲基二乙烯三胺(PMDETA).以BrCF2CO2Et为二氟烷基化试剂, 苯乙烯衍生物、1, 1-取代的烯烃等化合物在该条件下均可较高收率地获得二氟烷基化产物, 反应对于未活化的脂肪烯烃也可获得具有立体和空间选择性的相应产物(Scheme 16).该课题组在上述催化体系下拓展使用N-芳基的溴二氟乙酰胺对苯乙烯类衍生物进行二氟烷基化时得到了α, α-二氟-γ-内酰胺产物, 研究发现碱助剂的选择对于反应过程非常关键, 在碳酸钾、碳酸铯、氢氧化钠、三乙胺等条件下几乎不能得到该产物[43b].
针对强吸电子基团取代的烯烃化合物的二氟甲基化, Dilman等[37]以溴二氟甲基三甲基硅烷(Me3Si-CF2Br)、三苯基膦(Ph3P)为原料, 通过1, 3-二甲基丙撑脲(DMPU)催化生成二氟化的磷内鎓盐, 再经碱处理得到二氟甲基化的硝基烷烃.该反应体系下, 含硝基取代基的芳香烯烃、杂环的烯烃及烷基烯烃均可较高收率的获得硝基烷烃产物(Scheme 17).
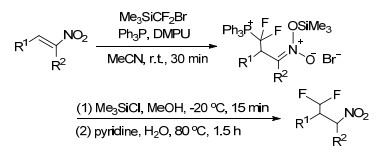
2016年, Jacobsen等[44]报道了一种经β-CO2Y (Y可为NH2、CH3)取代的苯乙烯的二氟甲基化反应合成各种含有叔碳或季碳立体中心的二氟甲基化产物的方法.反应使用手性芳基碘(Ar’Ⅰ)为催化剂, 以HF为氟源, 在间氯过苯甲酸和吡啶存在下经过苯鎓离子中间体的重排可生成1, 1-二氟取代的产物.反应中存在的取代基效应和对映选择性的温度效应表明, Cation-π相互作用在催化剂的立体控制中具有重要作用(Scheme 18).
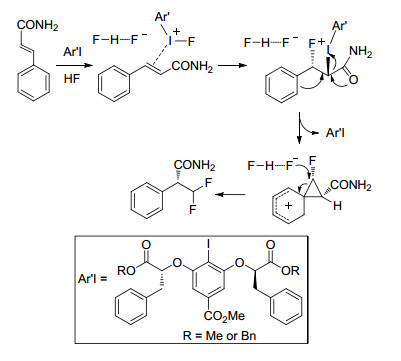
卤代苯在过渡金属络合物催化下发生羟基化反应可生成苯酚, 基于此, Hartwig等[15]设计了卤代苯经过羟基化、二氟甲基化两步反应制备二氟甲基苯基醚化合物的反应工艺, 氯苯或溴苯在该反应体系下均可较高收率的得到二氟甲基醚产物(Eq. 3).
2014年, 沈其龙课题组[45a]开展了在卤代苯化合物中直接引入二氟甲基的研究, 该方法使用钯和银两种催化剂, 以Me3SiCF2H为二氟甲基化试剂, 同时使用叔丁醇钠作为活化剂(Eq. 4). 2016年, 该课题组使用相同的钯催化剂, 选用DPPF为配体, [(SIPr)Ag(SCF2H)]为二氟甲硫基试剂, 进行了卤代杂环芳香烃化合物的二氟甲硫基化研究, 溴或碘取代的杂环芳烃在该催化体系下均可高收率的获得二氟甲基产物[45b].由于反应使用昂贵的钯金属催化剂, 同时二氟甲基化试剂的用量较大, 因此, 继续寻求开发高效催化剂和降低二氟甲基化试剂用量很有必要. 2017年, 该课题组报道了Pd催化卤代杂环芳香烃化合物二氟甲基化的最新进展, 以Pd(dba)2为催化剂, [(SIPr)Ag(CF2H)]为二氟甲基试剂, 优选出DPEPhos为二齿双膦配体, 实现了溴或碘取代的吡啶、吡咯、呋喃、噻吩、喹啉、吡嗪、噻唑、噁唑、吡唑等卤代杂环芳香烃化合物的高效二氟甲基化, 目标产物收率可达90%.由于C—Cl键相对于C—Br键作用较强, 氯取代的杂环芳香烃化合物二氟甲基化在上述反应体系下相对困难, 仅适用于杂原子邻位氯取代化合物的二氟甲基化, 而间位或对位氯取代化合物的二氟甲基化难以发生[45c].
|
|
(3) |
|
|
(4) |
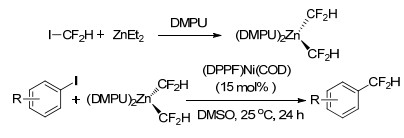
2016年, Vicic等[46]以ICF2H、二乙基锌为原料, 在DMPU存在下制备了新型二氟甲基化试剂[(DMPU)2Zn(CF2H)2], 对该试剂在金属诱导的卤代芳烃二氟甲基化反应中的应用进行了研究.反应体系优选的催化剂为[(dppf)Ni(COD)], 溶剂选用二甲基亚砜, 在25 ℃下反应24 h即可较高收率地获得二氟甲基取代的芳烃产物.碘或溴取代的芳烃以及含氧、硫、氮原子的杂环芳烃在该催化反应条件下均可生成相应的目标产物, 但烷基、甲氧基等给电子基团取代的卤代芳烃在该催化体系下反应活性较低, 部分烷基取代的卤代芳烃的二氟甲基化反应甚至难以进行(Scheme 19).
2016年, 针对碘取代芳烃的二氟甲基化, Mikami等[47]报道了以[(DMPU)2Zn(CF2H)2]为二氟甲基试剂, 碘化亚铜为催化剂的反应体系, 研究发现该反应体系对含强吸电子基团的碘取代芳烃的二氟甲基化具有较高的反应活性, 如邻硝基碘苯的二氟甲基产物收率可达75%, 而4-叔丁基碘苯的二氟甲基产物收率仅为14%.此外, 反应体系表现出明显的溶剂效应, 除在DMPU、DMF溶剂中具有较好的反应效果, 在NMP、DMSO、MeCN、THF、甲苯等溶剂中目标产物收率均低于5% (Eq. 5).
|
|
(5) |
为解决[(DMPU)2Zn(CF2H)2]试剂在含供电子取代基的碘苯的二氟甲基化反应中活性较低的问题, 2016年, Mikami等[48]制备了[(TMEDA)Zn(CF2H)2], 以此为二氟甲基化试剂开发了基于钯催化的Negishi交叉偶联反应的碘苯化合物的二氟甲基化新方法.反应过程中P配体的性质对目标产物的收率具有较大影响, Pd(dba)2/2DPPF相比P(1-Ad)nBu2具有好的催化效果, 目标产物收率可达97%, 而P(1-Ad)nBu2条件下目标产物收率仅为42%.对位含有硝基、氰基、酯基、氯的碘苯均可获得收率大于80%的目标产物, 对位含叔丁基、甲氧基等供电子取代基的碘苯也可获得收率大于85%的二氟甲基化产物.
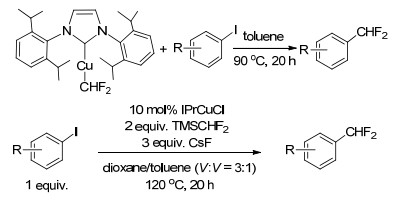
最近, Pd, Ni, Cu催化的亲电芳基化合物和亲核二氟甲基化试剂的交叉偶联反应的研究有较多报道[42, 43, 49], 其中, Cu基催化体系是最为常用的方法, 但对反应体系下的中间体[Cu(CHF2)]缺乏深入认识. 2017年, Sanford等[50]以(NHC)CuCl和TMSCHF2为原料, 首次合成了基于氮杂环卡宾配体(NHCs)的可稳定分离的(NHC)Cu(CHF2)络合物.通过选择合适的NHCs, 该类络合物可在室温下稳定存在至少24 h.当选用的NHCs为较大位阻的IPr时, 可高收率地获得稳定的(IPr)Cu(CHF2)络合物.应用该络合物对碘苯类化合物进行二氟甲基化, 含吸电子取代基团的碘苯可高收率地获得二氟甲基产物, 相反, 含有供电子取代基团的碘苯相应的二氟甲基产物收率较低(Scheme 20).此外, 研究探讨了原位生成(IPr)Cu(CHF2)络合物进行碘苯二氟甲基化的方法, 结果表明, 反应体系对含供电子取代基团的碘苯化合物的二氟甲基化具有较好的催化活性, 分析认为IPr配体使得铜催化中心具有充分的稳定性, 以致于可以耐受该类底物氧化加成所需要的高温条件.该研究有助于进一步开发基于(IPr)CuCl催化剂的含供电子基团的碘苯化合物的高效二氟甲基化方法(Scheme 20).
利用卤代苯的格氏试剂进行二氟甲基芳香化合物的制备是一种新的合成策略. 2018年, 胡金波课题组[51]首次报道了Fe基催化的苯基溴化镁(PhMgBr)的二氟甲基化, 反应以二氟甲基杂环砜(2-PySO2CF2H)为二氟甲基试剂, 通过C—S键的断裂生成二氟甲基自由基, 借助交叉偶联反应进行二氟甲基苯化合物的制备.由于格氏试剂活性较高, 需要将其缓慢滴加到—40 ℃的2-PySO2CF2H和Fe(acac)3的溶液中, 优选四甲基1, 4-丁二胺为配体后, 二氟甲基苯收率最高仅为44%.研究发现, 在无Fe(acac)3存在下, 格氏试剂也易于进攻2-PySO2CF2H发生其他副反应, 该结果为反应体系无法高效获得二氟甲基产物提供了合理解释(Eq. 6).然而, 当以活性较为温和的二苯基锌(Ph2Zn)取代PhMgBr后, 二氟甲基苯的收率达到了95%, 同时反应也适用于众多含取代基的二芳基锌的二氟甲基化.
|
|
(6) |
针对卤代烷烃的二氟甲基化研究相关报道较少, 吕剑课题组[16]基于氯代烷烃或氯代烯烃和HF的反应, 开展了气相氟化反应催化剂的结构设计和制备技术研究, 合成了CF3CHF2、CH2F2、CH3CHF2、CH3CH2CHF2等化合物.同时, 反应可以获得氯代的含二氟甲基化合物的中间体, 以此为原料也可以合成多种结构的二氟甲基化合物.
近年来, 通过C—H键的二氟甲基化制备二氟甲基取代芳烃引起人们的持续关注. 2012年, Baran等[52]报道了在t-BuOOH存在下, CH2Cl2/H2O为溶剂, 以Zn(SO2CF2H)2(DFMS)为二氟甲基化试剂对含氮杂环芳香烃化合物进行二氟甲基化的研究, 吡啶、吡唑、喹啉、黄碱等衍生物均可在该反应体系下实现芳香环C—H键的二氟甲基化.同时研究发现, 通过改变溶剂的极性可调变4-乙酰基吡啶的二氟甲基化产物的选择性(Eq. 7).
|
|
(7) |
2013年, 针对芳香烃的二氟甲基化, Hartwig等[15]采用C—H键的硼化、氧化及二氟甲基化三步反应, 得到二氟甲基芳香醚产物, 含酰胺基、酯基、卤素取代的芳香烃均适用于该反应体系(Eq. 8).
|
|
(8) |
2017年, Nielsen等[53]开发了使用二氟乙酸为二氟甲基化试剂, 对杂环芳香化合物进行C—H活化的二氟甲基化方法, 具有工艺简单, 成本相对较低的优点.以吡啶衍生物为反应底物, 在二氟乙酸存在下, 经Ag+/ K2S2O8催化后可在吡啶环上引入二氟甲基.当催化体系中AgNO3和K2S2O8的物质的量比为1/10时, 目标产物收率最高, 优选的反应溶剂为乙腈, 在乙醇、甲醇、二氯甲烷、二氧六环、二甲基甲酰胺等溶剂中反应难以进行.使用tBuOOH、H2O2、(NH4)2S2O8等氧化剂替代K2S2O8, 目标产物收率均低于10%.该反应体系可以拓展应用到吡啶、嘧啶、吡嗪、喹啉、喹喔啉和某些天然产物的二氟甲基化, 研究发现, 即使底物结构中具有多个反应活性位点, 二氟甲基均可选择性的引入到杂环结构中氮原子的邻位(Scheme 21).
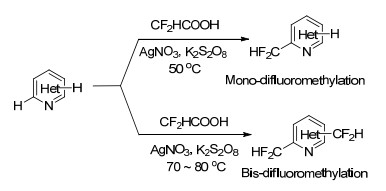
人们针对双二氟甲基取代的芳烃化合物的制备也开展了较多研究, 如在DMFS为二氟甲基试剂的反应体系中, 喹啉经二氟甲基化后会得到双二氟甲基取代的产物.基于此, Sanford等[50]尝试在AgNO3/K2S2O8催化体系下, 以二氟乙酸为二氟甲基化试剂对喹啉化合物进行二氟甲基化, 通过强化反应条件, 如提高反应温度, 可以获得双二氟甲基取代为主的产物(双取代产物与单取代产物比高达16/10).该反应体系下, 一些其他结构的杂环芳香化合物也可制备双取代的二氟甲基产物.
在众多二氟取代基团中, CF2CO2Et具有非常重要的地位, 其可以转化为各种含二氟亚甲基的其他官能团.近年来, 人们在基于自由基反应的芳香烃C—H键官能化领域开展了一些研究, 该方法可以在芳香环上直接引入CF2CO2Et, 避免了起始原料的官能化预处理过程, 但是由于反应使用昂贵的贵金属或光催化剂, 以及反应底物仅适用于富电子的芳烃化合物等不足[54], 因此, 有必要开展更加高效的芳香烃C—H键官能化引入CF2CO2Et的新方法.
2016年, 郝健课题组等[55]以AgOTf为催化剂, KF为助剂, 二氯乙烷为反应溶剂, TMSCF2COOEt为二氟烷基化试剂, 对苯、甲苯、二甲苯及三甲苯等芳香烃及其衍生物进行芳香环C—H键的官能化研究.研究发现, 增加AgOTf、KF用量, 可显著提高产物的收率, 但增加TMSCF2COOEt的用量将导致产物收率大幅下降.对于烷基取代的芳香烃的二氟烷基化, 烷基的邻位、对位的C—H键更易官能化, 而对于烷氧基取代的芳香烃, 邻位C—H键的二氟烷基化具有高的选择性(Eq. 9).
|
|
(9) |
该自由基反应的历程如Scheme 22所示, 反应首先生成AgCF2COOEt, 通过分解生成·CF2COOEt, 该中间体与芳香环发生加成反应生成新的自由基, 随后通过氧化反应生成阳离子中间体, 最后经过脱质子化得到最终产物.由于氟原子和乙氧羰基的吸电特性导致·CF2COOEt具有亲电性, ·CF2COOEt更加容易使烷基、烷氧基等给电子取代基的邻位或对位的C—H键官能化, 因为反应过程更容易生成稳定的自由基及阳离子中间体.
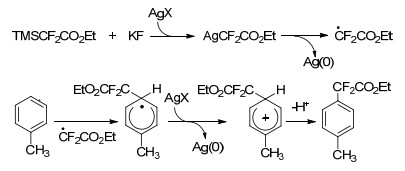
2017年, Shibata等[56]报道了在HF2CSO2Na/Ph2PCl/ Me3SiCl的反应体系下进行苯酚和萘酚的C—H键的直接二氟甲硫基化, 相应的SCF2H化合物具有较好的收率和区域选择性.其它Csp2和Csp3亲核体如吲哚、吡咯、吡唑生成的相应SCF2H产物也具有较高的产率(Scheme 23).
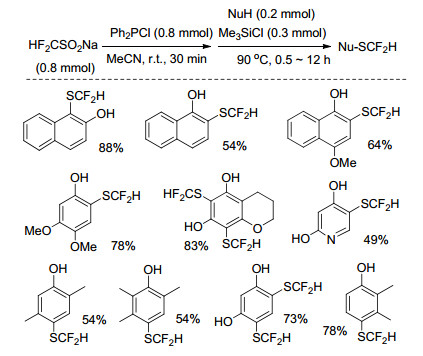
2017年, Billard等[57]设计合成了一种新的氟烷基甲磺酰胺试剂, 在三甲基氯硅烷(ClSiMe3)催化下, 该试剂可以在芳香环或杂环芳香环化合物中引入SCF2CO2Me基团, 对于亲电性较弱或位阻较大的芳香化合物, 反应需在较高的温度和其它强质子型催化剂下(如对甲基苯磺酸、三氟甲基磺酸)进行才能获得较高的产物收率(Eq. 10).
|
|
(10) |
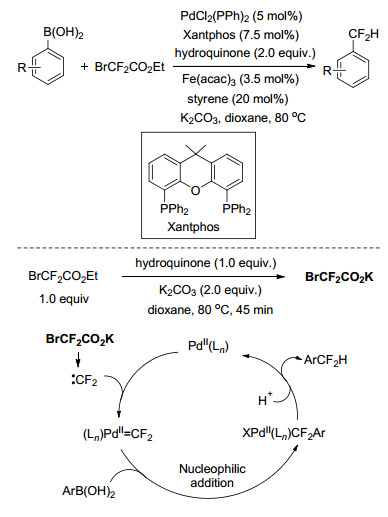
过渡金属催化的芳基硼酸和二氟甲基试剂的反应也是制备二氟甲基芳香化合物的有效方法. 2016年, 张新刚课题组[58]报道了Pd催化的芳基硼酸的二氟甲基化, 以廉价的溴代二氟乙酸乙酯为二氟甲基试剂, 对苯二酚和醋酸铁为助剂, 反应具有高效、底物适用范围广的突出特点.初步研究表明, 反应为二氟卡宾参与的钯催化循环过程, 首先PdⅡ(Ln)和原位生成的二氟卡宾反应得到(Ln)PdⅡ=CF2, 该物质被芳基硼酸捕获后生成关键中间体XPdⅡ(Ln)CF2Ar, 其经质子交换反应得到二氟甲基化的芳香化合物以及PdⅡ(Ln).对于整个反应过程而言, 助剂对苯二酚对于反应的促进必不可少, 但其具体作用仍需要进一步研究(Scheme 24).同年, 肖吉昌课题组[59]对芳基硼酸和二氟卡宾的偶联反应涉及的Pd催化的二氟卡宾转移过程从实验方面提供了有价值的证据, 证实了Pd=CF2络合物在该转移过程中的重要作用.
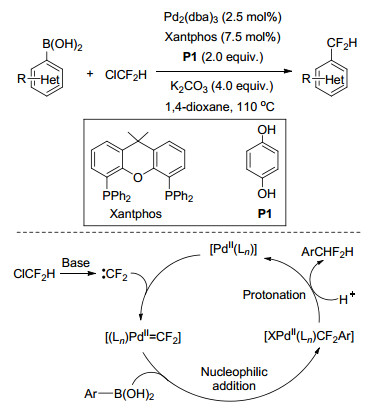
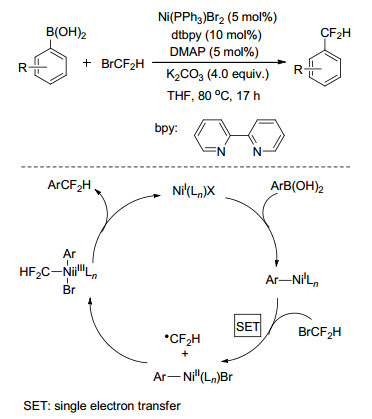
2017年, 张新刚课题组[60]从廉价的一氯二氟甲烷(ClCF2H)出发, 以钯为催化剂, Xantphos为配体、对苯二酚为添加剂、K2CO3作为碱, 高效地实现了系列芳基硼酸(酯)的二氟甲基化.反应高效简洁、底物普适性广、具有良好的官能团兼容性, 反应对含氮杂环化合物以及复杂生物活性的分子均适用.初步的机理研究表明, 反应经历了二氟卡宾的途径.通过反应中间体捕获也观察到了二氟卡宾钯中间体, 表明催化循环中可能存在二氟卡宾钯[Pd=CF2]物种.氘代实验和一系列对比实验表明该反应中芳基硼酸、ClCF2H以及对苯二酚都可以作为质子源形成最终产物, 其中添加剂对苯二酚对反应的启动至关重要, 它可能扮演了质子穿梭载体和配体的作用(Scheme 25).在此研究基础上, 2018年, 该课题组[61]选用ClCF2H的同系物BrCF2H作为氟源, 对非贵金属Ni催化下的芳基硼酸和BrCF2H的偶联反应进行了研究, 结果表明, 在NiBr2/bpy/PPh3体系下反应可以高效得到二氟甲基芳香化合物.尽管BrCF2H在碱性条件下倾向生成二氟卡宾, 但通过自由基抑制实验、自由基Clock实验等研究证明二氟甲基自由基参与了反应过程, 表明Ni催化下有效避免了该副反应, 使得芳基硼酸的二氟甲基化可以在二氟甲基自由基路径下进行(Scheme 26).同年, 王细胜课题组[62]也对Ni催化下的芳基硼酸和BrCF2H的偶联反应进行了报道, 重点通过不同种类的含氮、含膦配体与各种Ni催化剂组合, 以获得高催化活性的催化体系, 优选的催化体系为Ni(OTf)2/dtbpy/PPh3, 该反应体系下4-苯基硼酸等底物的二氟甲基化产物收率与张新刚课题组报道的数据相近.
以含二氟甲基砌块的化合物为原料, 借助普通有机化学反应对该类化合物进行改性是制备二氟甲基化合物的重要途径.该类官能化反应具有反应温和、选择性好以及产率较高的优点.
在含二氟甲基化合物的合成中, 2-氯-1, 1-二氟乙烷、二氟乙酸酯及二氟乙酸衍生物类、二氟乙腈、3, 3-二氟- 1, 2-二氯-1-丙烯等砌块可工业化生产, 而且其含有的卤素、羰基、烯基等官能团可以作为反应位点, 使二氟甲基高效地引入至其他有机分子中, 因而在含二氟甲基的医药、农药开发中具有重要的地位.一些含二氟甲基砌块的化合物如Scheme 27所示.
二氟乙醇、二氟乙胺为重要的含氟精细化学品, 是二氟甲基C2化合物的典型代表, 广泛应用于新型医药、农药的合成.针对该类化合物的合成, 国内外众多科研机构均开发了采用含二氟甲基砌块化合物为原料的工艺技术, 可用于上述化合物的规模化制备[63].吕剑课题组[64a, 64b]开展了以1, 1-二氟-1-氯乙烷为原料合成2, 2-二氟乙醇及2, 2-二氟乙胺的研究, 重点研究了反应涉及的酯化、醇解及氨解等过程的高效转化问题, 得到了纯度≥99%的2, 2-二氟乙醇和2, 2-二氟乙胺.针对经二氟乙酸乙酯加氢合成二氟乙醇的反应过程, 制备了Cu-Al-Zn催化剂, 研究发现, 适量的Zn助剂有助于提高反应转化率和选择性.在温度为200 ℃, 压力为9 MPa, 原料溶剂体积比为1:3的条件下, 反应8 h, Cu-Al-Zn催化剂性能最佳, 转化率和选择性分别可达99.5%和97.3%, 在此条件下, 催化剂重复使用12次活性无明显下降[64c].
含二氟甲基砌块的化合物应用于多官能团化合物[65]或复杂有机分子[66](如氟烷基取代的吡唑衍生物)的合成也受到研究人员的关注, 并建立了该类化合物合成的一些方法. 2015年, Mykhailiuk[67]在氯仿溶剂中加入二氟乙胺和亚硝酸叔丁酯以及催化量的乙酸, 成功制备了二氟甲基重氮甲烷(CF2HCH=N2). CF2HCH=N2可在室温下可与各种单取代和双取代的炔烃发生[3+2]环加成反应得到含二氟甲基取代的吡唑类化合物(Scheme 28).然而, 该方法仅适用于炔烃类化合物, 但商品化的炔烃类化合物种类相对较少, 导致该方法具有一定的局限性.

随后, Mykhailiuk等[68]在2017年研究了CF2HCH=N2与来源更为广泛的烯烃化合物的反应活性.通过原位生成CF2HCH=N2与马来酸酐发生[3+2]环加成反应, 得到了二氟甲基取代的吡唑啉化合物.对于含单取代基团的马来酸酐(当取代基为苯环, 或当苯环上含有卤素、甲基、甲氧基、硝基、三氟甲基等取代基时), 反应均可较高收率的获得目标产物.对于含单取代基团的烯烃, 当结构中有CO2Me、CO2Et、CN等吸电子基团时, 产物收率相对较高; 当烯烃为乙氧基乙烯、苯乙烯等底物时, 反应不能得到目标产物.此外, 合成的二氟甲基取代的吡唑啉化合物可作为起始原料合成其他化合物, 如经二氧化锰氧化处理后可得含二氟甲基的吡唑化合物(Scheme 28).
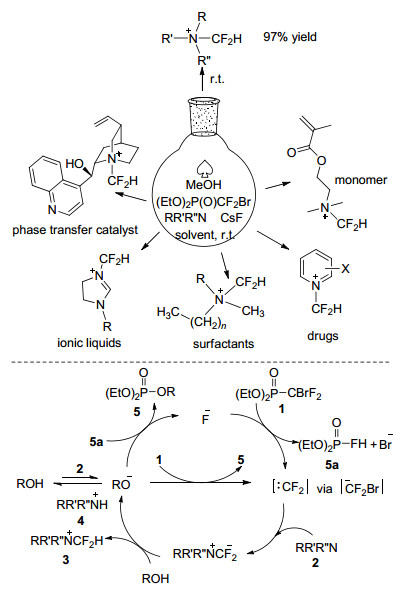
近年来, 人们尝试将二氟甲基引入到季铵盐结构中以获得独特的性能.但现有研究大多存在使用的二氟甲基试剂昂贵、不易得或者反应条件苛刻, 此外, 应用于医药、相转移催化剂等领域的季铵盐大多含有其他活性或敏感性基团, 这类基团在上述反应条件下可能不稳定[18].因此, 开发叔胺化合物实用、选择性的二氟甲基化方法仍然是挑战性的课题. 2007年, Jonczyk等[69]采用HCF2Cl和叔胺反应得到含二氟甲基的季铵盐化合物, 但反应仍需在质量分数为50% NaOH的强碱性条件下得到二氟卡宾中间体. 2016年, Zafrani等[70]报道了一种使用溴二氟甲基膦酸二乙酯和氟化物对叔胺进行二氟甲基化的新方法, 膦酸酯与氟离子可以稳定地生成二氟卡宾中间体, 该中间体在氢质子和叔胺存在下可生成相应的二氟甲基季铵盐化合物.反应尽管涉及二氟卡宾中间体, 但二氟甲基化几乎完全发生在分子结构中的氮原子上, 同时氟化物的引入使得反应在相对温和的条件下进行, 可避免由于分子结构中含有羟基、烯烃、炔基、酯基等官能团而导致的副反应(Scheme 39).
将含氟基团选择性地引入分子化合物在有机氟化学领域中具有重要的意义.最近十几年, 二氟甲基化反应得到了快速的发展, 其中包括不同类型的亲电、亲核二氟甲基试剂的开发以及各种自由基类型的反应和过渡金属催化的反应体系的建立.归纳起来, 有机分子化合物的二氟甲基化主要有三种策略.首先, 用二氟甲基化试剂官能化是非常有吸引力的合成途径, 该类转化通常是官能团耐受的, 并且可使用市售和容易制备的试剂, 适用于含OH, SH, NH2, C=O等基团的分子.最近人们已经致力于将CF2H直接引入各种分子, 如基于芳香烃化合物C—H键活化、烯烃加成反应的二氟甲基化, 该方法作为一种新的途径, 其避免使用含氧、硫、氮分子或额外的合成步骤来获得含氧、硫、氮分子前体.上述研究工作为科研工作者方便简洁地引入二氟甲基基团提供了一个较为丰富的手段.此外, 利用含二氟甲基切块的商业含氟化学品进行下游产品的开发或对现有分子进行改性, 也是二氟甲基化途径的一个有效手段.
尽管现有的二氟甲基化方法满足了简单和一些相对复杂分子的官能化, 也为更加复杂分子的官能化提供了经验技术积累, 但与此同时, 该领域仍然存在着一些问题与挑战需要我们去解决.首先, 对于直接二氟甲基化反应而言, 通常都需要用到Ru、Ir、Ag、Pd、Cu的络合物, 成本比较昂贵.其次, 具有普适性的二氟甲基化方法仍然是一个具有挑战性的目标, 特别是复杂(生物活性)分子的后期二氟甲基化.因此, 发展高效、高选择性、低成本的二氟甲基引入手段也必将随之成为有机氟化学研究的热点.
(a) Vulpetti, A. ; Dalvit, C. Drug Discovery Today 2012, 17, 890.
(b) Wang, J. ; Sánchez-Roselló, M. ; Acea, J. L. ; Pozo, C. D. ; Sorochinsky, A. E. ; Fustero, S. ; Soloshonok, V. A. ; Liu, H. Chem. Rev. 2014, 114, 2432.
(a) Mìller, K. ; Faeh, C. ; Diederich, F. Science 2007, 317, 1881.
(b) Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester (UK), 2009.
(a) Tomashenko, O. A. ; Grushin, V. V. Chem. Rev. 2011, 111, 4475.
(b) Hollingworth, C. ; Gouverneur, V. Chem. Commun. 2012, 48, 2929.
(c) Koike, T. ; Akita, M. J. Fluorine Chem. 2014, 167, 30.
(d) Merino, E. ; Nevado, C. Chem. Rev. Soc. 2014, 43, 6598.
(e) Ni, C. ; Hu, M. ; Hu, J. Chem. Rev. 2015, 115, 765.
(a) Toulgoat, F. ; Alazet, S. ; Billard, T. Eur. J. Org. Chem. 2014, 2415.
(b) Barata-Vallejo, S. ; Bonesi, S. ; Postigo, A. Org. Biomol. Chem. 2016, 14, 7150.
(c) Shi, J. ; Ren, G. ; Wu, N. ; Liu, X. ; Xu, T. ; Tan, C. Chin. J. Org. Chem. 2017, 37, 2131 (in Chinese).
(史建俊, 任贵华, 吴宁捷, 刘幸海, 许天明, 谭成侠, 有机化学, 2017, 37, 2131.)
(d) Hu, C. ; Liu, J. ; Du, X. Chin. J. Org. Chem. 2016, 36, 1051 (in Chinese).
(胡崇波, 刘建华, 杜晓华, 有机化学, 2016, 36, 1051.)
(a) Kirsch, P. Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim (Germany), 2013.
(b) Besset, T. ; Poisson, T. ; Pannecoucke, X. Eur. J. Org. Chem. 2015, 2765.
(a) Xiong, H. ; Pannecoucke, X. ; Besset, T. Chem. Eur. J. 2016, 22, 16734.
(b) Bos, M. ; Poisson, T. ; Pannecoucke, X. ; Charette, A. B. ; Jubault, P. Chem. Eur. J. 2017, 23, 4950.
(c) Pan, X. ; Xia, H. ; Wu, J. Org. Chem. Front. 2016, 3, 1163.
(d) Yerien, D. E. ; Barata-Vallejo, S. ; Postigo, A. Chem. Eur. J. 2017, 23, 14676.
(e) Zhang, X. ; Cao, S. Tetrahedron Lett. 2017, 58, 375.
(f) Ni, C. ; Zhu, L. ; Hu, J. Acta Chim. Sinica 2015, 73, 90 (in Chinese).
(倪传法, 朱林桂, 胡金波, 化学学报, 2015, 73, 90.)
(a) Manteau, B. ; Pazenok, S. ; Vors, J. -P. ; Leroux, F. R. J. Fluorine Chem. 2010, 131, 140.
(b) Huang, C. -H. ; Liang, T. ; Harada, S. ; Lee, E. ; Ritter, T. J. Am. Chem. Soc. 2011, 133, 13308.
(c) Hojczyk, K. N. ; Feng, P. ; Zhan, C. ; Ngai, M. -Y. Angew. Chem., Int. Ed. 2014, 53, 14559.
(d) Shen, X. ; Zhou, M. ; Ni, C. -F. ; Zhang, W. ; Hu, J. -B. Chem. Sci. 2014, 5, 117.
(e) Huang, R. ; Huang, Y. ; Lin, X. ; Rong, M. ; Weng, Z. Angew. Chem. 2015, 127, 5828.
(f) Liu, J. -B. ; Xu, X. -H. ; Qing, F. -L. Org. Lett. 2015, 17, 5048.
(g) Feng, P. ; Lee, K. N. ; Lee, J. W. ; Zhan, C. ; Ngai, M. -Y. Chem. Sci. 2016, 7, 424.
Chauret, N.; Guay, D.; Li, C.; Day, S.; Silva, J.; blouin, M.; Ducharme, Y.; Yergey, J. A.; Nicoli-Griffith, D. A. Bioorg. Med. Chem. Lett. 2002, 12, 2149. doi: 10.1016/S0960-894X(02)00349-9
Mizukado, J.; Matsukawa, Y.; Quan, H.-D.; Tamura, M.; Sekiya, A. J. Fluorine Chem. 2006, 127, 400. doi: 10.1016/j.jfluchem.2005.12.034
Flynn, R. M.; Burton, D. J. J. Fluorine Chem. 2011, 132, 815. doi: 10.1016/j.jfluchem.2011.05.034
(a) Fedorov, O. V. ; Struchkova, M. I. ; Dilman, A. D. J. Org. Chem. 2016, 81, 9455.
(b) Song, X. ; Tian, S. ; Zhao, Z. ; Zhu, D. ; Wang, M. Org. Lett. 2016, 18, 3414.
Mizukado, J.; Matsukawa, Y.; Quan, H.-D.; Tamura, M.; Sekiya, A. J. Fluorine Chem. 2005, 126, 365. doi: 10.1016/j.jfluchem.2005.01.012
Miethchen, R.; Hein, M.; Naumann, D.; Tyrra, W. Eur. J. Org. Chem. 1995, 1995, 1717.
Levchenko, K.; Datsenko, O. P.; Serhiichuk, O.; Tolmachev, A.; Iaroshenko, V. O.; Mykhailiuk, P. K. J. Org. Chem. 2016, 81, 5803. doi: 10.1021/acs.joc.6b00628
Fier, P. S.; Hartwig, J. F. Angew. Chem. 2013, 125, 2146. doi: 10.1002/ange.v125.7
Zhang, W. ; Mao, W. ; Wang, B. ; Zeng, J. ; Yang, J. ; Lü, J. Sci. Sin. Chim. 2017, 47, 1312 (in Chinese).
(张伟, 毛伟, 王博, 曾纪珺, 杨建明, 吕剑, 中国科学: 化学, 2017, 47, 1312.)
(a) Piou, A. ; Celerier, S. ; Brunet, S. J. Fluorine Chem. 2012, 134, 103.
(b) Dolbier Jr., W. R. ; Wang, F. ; Tang, X. ; Thomoson, C. S. ; Wang, L. J. Fluorine Chem. 2014, 160, 72.
(a) Prakash, G. K. S. ; Zhang, Z. ; Wang, F. ; Ni, C. -F. ; Olah, G. A. J. Fluorine Chem. 2011, 132, 792.
(b) Prakash, G. K. S. ; Weber, C. ; Chacko, S. ; Olah, G. A. Org. Lett. 2007, 9, 1863.
Zhu, J.; Liu, Y.; Shen, Q. Angew. Chem., Int. Ed. 2016, 55, 1. doi: 10.1002/anie.201510990
(a) Wang, F. ; Zhang, W. ; Zhu, J. ; Li, H. ; Huang, K. -W. ; Hu, J. Chem. Commun. 2011, 47, 2411.
(b) Li, L. ; Wang, F. ; Ni, C. ; Hu, J. Angew. Chem. 2013, 125, 12616.
(c) Ni, C. ; Hu, J. Synthesis 2014, 46, 842.
Xie, Q.; Ni, C.; Zhang, R.; Li, L.; Rong, J.; Hu, J. Angew. Chem., 2017, 129, 3254. doi: 10.1002/ange.201611823
(a) Erickson, J. A. ; McLoughlin, J. I. J. Org. Chem. 1995, 60, 1626.
(b) Jeanmart, S. ; F. Edmunds, A. J. ; Lamberth, C. ; Pouliot, M. Bioorg. Med. Chem. 2016, 24, 317.
Thomoson, C. S.; Dolbier Jr., W. R. J. Org. Chem. 2013, 78, 8904. doi: 10.1021/jo401392f
Mehta, V. P.; Greaney, M. F. Org. Lett. 2013, 15, 5036. doi: 10.1021/ol402370f
Prakash, G. K. S.; Krishnamoorthy, S.; Kar, S.; Olah, G. A. J. Fluorine Chem. 2015, 180, 186. doi: 10.1016/j.jfluchem.2015.09.011
Ma, J.; Liu, Q.; Lu, G.; Yi, W. J. Fluorine Chem. 2017, 193, 113. doi: 10.1016/j.jfluchem.2016.11.010
Zhang, C. Adv. Synth. Catal. 2017, 359, 372. doi: 10.1002/adsc.201601011
Heine, N. B., Studer, A. Org. Lett. 2017, 19, 4150. doi: 10.1021/acs.orglett.7b02109
Yang, J., Jiang, M., Jin, Y., Yang, H., Fu, H. Org. Lett. 2017, 19, 2758 doi: 10.1021/acs.orglett.7b01118
(a) Medebielle, M. ; Dolbier, W. R., Jr J. Fluorine Chem. 2008, 129, 930.
(b) Dilman, A. D. ; Levin, V. V. Eur. J. Org. Chem. 2011, 2011, 831.
(c) Liu, X. ; Xu, C. ; Wang, M. ; Liu, Q. Chem. Rev. 2015, 115, 683.
(d) Alonso, C. ; de Marigorta, E. M. ; Rubiales, G. ; Palacios, F. Chem. Rev. 2015, 115, 1847.
Deng, Z.; Lin, J.-H.; Cai, J.; Xiao, J.-C. Org. Lett. 2016, 18, 3206. doi: 10.1021/acs.orglett.6b01425
Krishnamoorthy, S., Prakash, G. K. S. Synthesis 2017, 49, 3394. doi: 10.1055/s-0036-1588489
Krishnamoorthy, S.; Kothandaraman, J.; Saldana, J.; Prakash, G. K. S. Eur. J. Org. Chem. 2016, 29, 4965. doi: 10.1002/ejoc.201601038/pdf
Hagiwara, T.; Fuchikami, T. Synlett 1995, 717. doi: 10.1002/chin.199547114/full
Zhao, Y.; Huang, W.; Zheng, J.; Hu, J. Org. Lett. 2011, 13, 5342. doi: 10.1021/ol202208b
Chen, D.; Ni, C.; Zhao, Y.; Cai, X.; Li, X.; Xiao, P.; Hu, J. Angew. Chem., Int. Ed. 2016, 55, 12632. doi: 10.1002/anie.201605280
Trifonov, A. L.; Zemtsov, A. A.; Levin, V. V.; Struchkova, M. I.; Dilman, A. D. Org. Lett. 2016, 18, 3458. doi: 10.1021/acs.orglett.6b01641
(a) Romanenko, V. D. ; Kukhar, V. P. Chem. Rev. 2006, 106, 3868.
(b) Liang, T. ; Neumann C. N. ; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214.
(a) Sato, K. ; Omote, M. ; Ando, A. ; Kumadaki, I. J. Fluorine Chem. 2004, 125, 509.
(b) Murakami, S. ; Ishii, H. ; Tajima, T. ; Fuchigami, T. Tetrahedron 2006, 62, 3761.
(c) Leung, L. ; Linclau, B. J. Fluorine Chem. 2008, 129, 986.
(d) Nguyen, J. D. ; Tucker, J. W. ; Konieczynska, M. D. ; Stephenson, C. R. J. J. Am. Chem. Soc. 2011, 133, 4160.
(e) Wallentin, C. J. ; Nguyen, J. D. ; Finkbeiner, P. ; Stephenson, C. R. J. J. Am. Chem. Soc. 2012, 134, 8875.
(f) Belhomme, M. -C. ; Poisson, T. ; Pannecoucke, X. Org. Lett. 2013, 15, 3428.
Yu, C.; Iqbal, N.; Park S.; Cho, E. J. Chem. Commun. 2014, 50, 12884. doi: 10.1039/C4CC05467A
Arai, Y.; Tomita, R.; Ando, G.; Koike, T.; Akita, M. Chem. Eur. J. 2016, 22, 1262. doi: 10.1002/chem.201504838
Pannecoucke, X.; Poisson, T. Synlett 2016, 27, 2314. doi: 10.1055/s-0035-1562784
(a) Wang, X. ; Zhao, S. ; Liu, J. ; Zhu, D. ; Guo, M. ; Tang, X. ; Wang, G. Org. Lett. 2017, 19, 4187.
(b) Chen, H. ; Wang, X. ; Guo, M. ; Zhao, W. ; Tang, X. ; Wang, G. Org. Chem. Front. 2017, 4, 2403.
Banik, S. M.; Medley, J. W.; Jacobsen, E. N. Science 2016, 353, 51. doi: 10.1126/science.aaf8078
(a) Gu, Y. ; Leng, X. ; Shen, Q. Nat. Commun. 2014, 5, 5405.
(b) Wu, J. ; Liu, Y. ; Lu, C. ; Shen, Q. Chem. Sci. 2016, 7, 3757.
(c) Lu, C. ; Gu, Y. ; Wu, J. ; Gu, Y. ; Shen, Q. Chem. Sci. 2017, 8, 4848.
Xu, L.; Vicic, D. A. J. Am. Chem. Soc. 2016, 138, 2536. doi: 10.1021/jacs.6b00053
Aikawa, K.; Serizawa, H.; Ishii, K.; Mikami, K. Org. Lett. 2016, 18, 3690 doi: 10.1021/acs.orglett.6b01734
Serizawa, H.; Ishii, K.; Aikawa, K.; Mikami, K. Org. Lett. 2016, 18, 3686. doi: 10.1021/acs.orglett.6b01733
(a) Ge, S. ; Chaladaj, W. ; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 4149.
(b) Rong, J. ; Ni, C. ; Hu, J. Asian J. Org. Chem. 2017, 6, 139.
Bour, J. R.; Kariofillis, S. K.; Sanford, M. S. Organometallics 2017, 36, 1220. doi: 10.1021/acs.organomet.7b00025
Miao, W.; Zhao, Y.; Ni, C.; Gao, B.; Zhang, W.; Hu, J. J. Am. Chem. Soc. 2018, 140, 880. doi: 10.1021/jacs.7b11976
Fujiwara, Y.; Dixon, J. A.; Rodriguez, R. A.; Baxter, R. D.; Dixon, D. D.; Collins, M. R.; Blackmond, D. G.; Baran, P. S. J. Am. Chem. Soc. 2012, 134, 1494. doi: 10.1021/ja211422g
Tung, T. T.; Christensen, S. B.; Nielsen, J. Chem.-Eur. J. 2017, 23, 18125. doi: 10.1002/chem.201704261
(a) Ohtsuka, Y. ; Yamakawa, T. Tetrahedron 2011, 67, 2323.
(b) Lin, Q. ; Chu, L. ; Qing, F. -L. Chin. J. Chem. 2013, 31, 885.
(c) Jung, J. ; Kim, E. ; You, Y. ; Cho, E. J. Adv. Synth. Catal. 2014, 356, 2741.
Li, J.; Wan, W.; Ma, G.; Chen, Y.; Hu, Q.; Kang, K.; Jiang, H.; Hao, J. Eur. J. Org. Chem. 2016, 4916. doi: 10.1007/s11467-013-0324-x
Huang, Z.; Matsubara, O.; Jia, S.; Tokunaga, E.; Shibata, N. Org. Lett. 2017, 19, 934. doi: 10.1021/acs.orglett.7b00113
Ismalaj, E.; Glenadel, Q.; Billard, T. Eur. J. Org. Chem. 2017, 1911. doi: 10.1002/ejoc.201601526/full
Feng, Z.; Min, Q.; Zhang, X. Org. Lett. 2016, 18, 44. doi: 10.1021/acs.orglett.5b03206
Deng, X. Y.; Lin, J. H.; Xiao, J. C. Org. Lett. 2016, 18, 4384. doi: 10.1021/acs.orglett.6b02141
Feng, Z.; Min, Q.; Fu, X.; An, L.; Zhang, X. Nat. Chem. 2017, 9, 918. doi: 10.1038/nchem.2746
Fu, X. P.; Xiao, Y. L.; Zhang, X. Chin. J. Chem. 2018, 36, 143. doi: 10.1002/cjoc.201700624
Sheng, J.; Ni, H. Q.; Bian, K. J.; Li, Y.; Wang, Y. N.; Wang, X. S. Org. Chem. Front. 2018, 5, 606. doi: 10.1039/C7QO00934H
(a) Lui, N. ; Pazenok, S. ; Shermolovich, Y. G. US 2011/0060167, 2011.
(b) Antons, S. ; Lui, N. ; Gerlach, A. US 2011/0082318, 2011.
(c) Thomas, R. ; Ansgar, A. ; Christian, M. WO 2016/023773, 2016.
(d) Norbert, M. T. ; Norbert, L. ; Stefan, M. US 20140243561, 2014.
(a) Yun, J. ; Yang, J. M. ; Lu, J. ; Zhang, Q. ; Li, J. W. ; Zhao, F. W. ; Yu, Q. W. ; Mei, S. N. ; Wang, W. Q. ; Li, Y. N. ; Hui, F. CN 106831338, 2017.
(b) Yun, J. ; Yang, J. M. ; Lu, J. ; Hui, F. ; Li, Y. N. ; Zhang, Q. ; Li, J. W. ; Zhao, F. W. ; Yu, Q. W. ; Wang, W. Q. ; Mei, S. N. CN 106810421, 2017.
(c) Sun, D. A. ; Li, C. Y. ; Du, Y. M. ; Zhang, W. ; Zhang, J. W. . ; Ma, Y. B. ; Zeng J. J. ; Lu, J. J. Mol. Catal. 2013, 27, 242 (in Chinese).
(孙道安, 李春迎, 杜咏梅, 张伟, 张建伟, 马洋博, 曾纪珺, 吕剑, 分子催化, 2013, 27, 242.)
(a) Wang, Q. ; Yu, X. ; Jin, J. ; Wu, Y. ; Liang, Y. Chin. J. Chem. 2018, 36, 223.
(b) Zhou, N. ; Xu, P. ; Li, W. ; Cheng, Y. ; Zhu, C. Acta Chim. Sinica 2017, 75, 60 (in Chinese).
(周能能, 胥攀, 李伟鹏, 成义祥, 朱成建, 化学学报, 2017, 75, 60.)
(a) Fujita, T. ; Sanada, S. ; Chiba, Y. ; Sugiyama, K. ; Ichikawa, J. Org. Lett. 2014, 16, 1398.
(b) Fujita, T. ; Sugiyama, K. ; Sanada, S. ; Ichitsuka, T. ; Ichikawa, J. Org. Lett. 2016, 18, 248.
Mykhailiuk, P. K. Angew. Chem., Int. Ed. 2015, 54, 1. doi: 10.1002/anie.201410930
Li, J.; X.-L.; Yu, Cossy, J.; Lv, S.-Y.; Zhang, H.-L.; Su, F.; Mykhailiuk, P. K.; Wu, Y. Eur. J. Org. Chem. 2017, 2017, 266. doi: 10.1002/ejoc.201601462/pdf
Nawrot, E.; Jonczyk, A. J. Org. Chem. 2007, 72, 10258. doi: 10.1021/jo701735n
Zafrani, Y.; Amir, D.; Yehezkel, L.; Madmon, M.; Saphier, S.; Karton-Lifshin, N.; Gershonov, E. J. Org. Chem. 2016, 81, 9180. doi: 10.1021/acs.joc.6b01728

图式 1 不同结构底物及其二氟甲基化产物
Scheme 1 Different structural substrates and their difluoromethylated products

图式 4 氟氯交换反应合成二氟甲基芳基醚
Scheme 4 Synthesis of difluoromethoxy arenes by Cl/F exchange reaction

图式 9 巯基化合物和[PhP-CF2H]+Br-的自由基反应类型的二氟甲基化
Scheme 9 Radical difluoromethylation of thiols with [Ph3P-CF2H]+Br-

图式 10 羰基化合物的直接亲核二氟甲基化
Scheme 10 Direct nucleophilic difluoromethylation of carbonyl compounds

图式 13 IrⅢ催化下的苯乙烯衍生物的二氟甲基化
Scheme 13 Difluoromethylation of styrene derivatives catalyzed by IrⅢ

图式 14 铜催化杂环烯烃的乙氧羰基二氟烷基化
Scheme 14 Cu-catalyzed difluoroalkylation of heterocyclic alkenes

图式 15 铜催化杂环烯烃的二氟烷基化产物
Scheme 15 Difluoroalkylated product of Cu-catalyzed heterocyclic alkenes

图式 17 DMPU催化下硝基烯烃的二氟甲基化
Scheme 17 Difluoromethylation of nitro alkenes catalyzed by DMPU

图式 18 Ar’Ⅰ催化下的苯乙烯衍生物的二氟甲基化
Scheme 18 Difluoromethylation of styrene derivatives catalyzed by Ar'Ⅰ

图式 21 杂环芳香化合物的单/双二氟甲基化
Scheme 21 The mono- and bis- difluoromethylation of heteroaromatic compounds

图式 22 AgOTf催化的芳香烃的二氟烷基化反应机理
Scheme 22 Proposed mechanism of silver-mediated difluoroalkylation of arenes

图式 23 苯酚、萘酚及其衍生物的二氟甲基硫醇化
Scheme 23 Difluoromethylthiolation of phenols and related compounds

图式 24 Pd催化的芳基硼酸和BrCF2CO2Et的二氟甲基化反应
Scheme 24 Pd-Catalyzed Difluoromethylation of Arylboronic Acids with Bromodifluoroacetate

图式 25 Pd催化的芳基硼酸(或杂环芳基硼酸)和ClCF2H的偶联反应
Scheme 25 Pd-catalysed cross-coupling of ClCF2H with arylboronic and heteroarylboronic acids

图式 26 Ni催化的芳基硼酸和Pd催化的芳基硼酸和BrCF2H的二氟甲基化反应
Scheme 26 Ni-catalyzed difluoromethylation of arylboronic acids with BrCF2H

图式 28 CF2HCHN2与炔烃或烯烃的环加成反应(EWG表示吸电子基团)
Scheme 28 Cycloaddition of CF2HCHN2 with alkynes or alkenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:











 下载:
下载:













 下载:
下载: