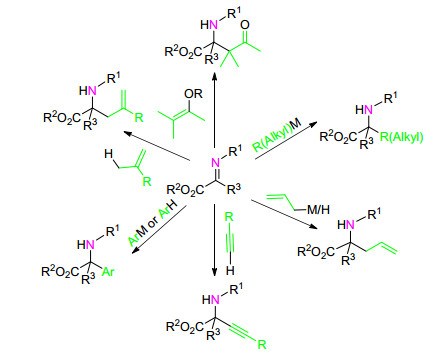
图式 1.
α-亚胺酯不对称加成合成手性α-氨基酸衍生物的方法
Scheme 1.
Synthesis of chiral α-amino acid derivatives by asymmetric addition of α-imino ester

手性α-氨基酸衍生物的应用价值广泛, 是许多药物分子、生物活性分子及天然产物结构的重要组成砌块, 也可作为手性催化剂用于多肽、蛋白质合成及全合成等领域[1].因此, 合成有机化学家开发了多种多样的合成手性α-氨基酸衍生物的方法, 包括亲核试剂对α-亚胺酯的不对称亲核加成反应、α-烯胺及亚胺酯不对称氢化[2]、α-酮酸及其派生物的不对称还原胺化[3]、亚胺的不对称环加成反应[4]等, 虽然针对α-亚胺酯的不对称亲核加成反应研究较多[5], 但相关综述目前还比较少, 所以本文对此进行总结和归纳, 以期为后续研究作以参考.
亲核加成反应底物α-亚胺酯是一类重要的前手性源[6], 由于该化合物中的C=N双键在结构上与C=O双键类似, 表现出一定的亲电性, 可以发生高立体选择性亲核加成反应.然而, 由于氮原子相对于氧原子电负性较弱, 造成C=N双键在亲核加成反应中的活性较C=O双键要弱一些.目前解决该问题的方法主要有三种:第一, 改变氮原子的保护基, 通过电性的影响增强其活性; 第二, 使用亲核性更强的亲核试剂; 第三, 利用Lewis酸等添加剂活化氮原子以增强其亲电性.
本文从反应类型、手性控制策略及亲核试剂类型的角度对α-亚胺酯不对称亲核加成反应合成手性α-氨基酸衍生物的方法进行了较为详细的介绍.总结了该类反应的研究现状, 具体介绍了α-亚胺酯的不对称烯丙基化、芳基化、烯基化、炔基化、烷基化和Mannich反应(Scheme 1).对相应的反应机理进行了阐述, 并对合成手性α-氨基酸衍生物的发展方向进行了展望.
α-亚胺酯的不对称烯丙基化反应是合成烯丙基取代氨基酸衍生物的有效途径.烯丙基化反应中, 亲核试剂对α-亚胺酯的C=N双键加成, 得到高选择性的烯丙基氨基酸衍生物.其结构中的烯丙基易于官能团化, 经特定转化可得到种类丰富、价值多样手性含氮化合物.
2010年, 林国强课题组[7]报道了金属促进的溴取代苯丙烯与叔丁基磺酰亚胺的烯丙基化反应, 通过手性诱导获得带有两个相邻手性中心且β-位芳基取代的含氮化合物.该反应巧妙地使用LiCl添加剂, 在烯丙基锌试剂插入的过程中对立体构型的翻转起到了关键性的作用.通过手性辅基导作用在N, N-二甲基甲酰胺(DMF)溶剂中获得(1R, 2R)立体构型产物, 避免了致癌物六甲基磷酰三胺的使用.该工作主要以烷基亚胺底物为主, 虽然涉及的亚胺酯底物较少, 但对实现烯丙基金属化合物作为亲核试剂对α-亚胺酯的亲核加成反应研究具有重要启发性意义(Eq. 1).
|
|
(1) |
|
|
α-烯丙基取代氨基酸衍生物可通过α-亚胺酯与多种烯丙基金属试剂(如烯丙基硅烷、烯丙基锡烷、烯丙基硼烷等)加成反应得到. 2002年, Lectka课题组[8]以Binap为配体实现了烯丙基硅试剂与N-对甲苯磺酰醛亚胺的亲核加成反应, 在手性Binap类型配体的调控作用下合成了一系列α-烯丙基氨基酸衍生物.该工作是较早利用α-亚胺酯烯丙基化反应合成手性氨基酸衍生物的成功例子(Eq. 2).
|
|
(2) |
|
|
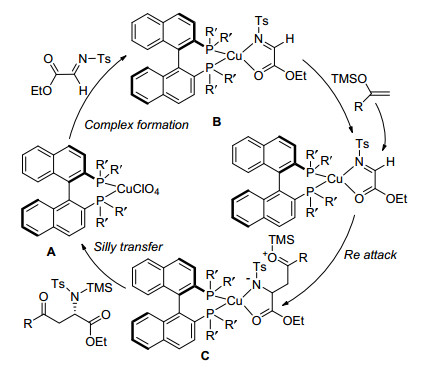
同时, Lectka课题组通过动力学同位素效应(KIE)实验、红外光谱分析、单晶衍射等方法确定了金属与配体形成络合物的结构, 从而对该反应的机理做出了解释(Scheme 2), 在四氢呋喃(THF)溶液中, Cu(ClO4)2与手性配体形成金属络合物A, 金属络合物A与α-亚胺酯的羰基氧原子、C=N双键氮原子配位形成五元环状中间体B, 随后亲核试剂与B中C=N双键发生选择性的顺式加成, 双键转移得到中间体C, 最后, 硅基转移得到目标产物和金属络合物A, 完成催化循环.该工作是金属催化、手性配体控制合成手性氨基酸衍生物的先驱性工作, 为该类反应的进一步研究和发展奠定了基础.
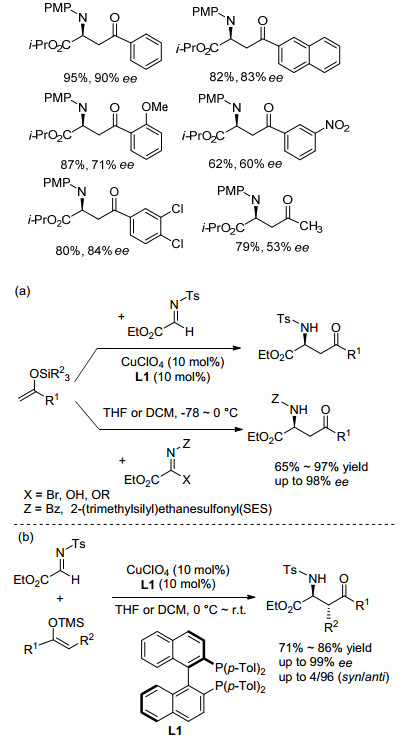
手性氨基酸衍生物的氮原子保护基多为强吸电子基团(Ts、Cbz等). 2003年, Kobayashi课题组[9]首次以酰腙为亚胺底物、烯丙基三甲氧基硅为烯丙基化试剂, 合成了一系列手性α-烯丙基氨基酸衍生物.由于反应中使用了稳定性较强的酰腙, 使得该反应可以在四氢呋喃与水的混合溶剂发生, 并保持较高的对映选择性(85% ee).同时, 与其他的催化模式相比, 该反应催化剂ZnF2具有双重特点:一是Zn2+作为Lewis酸活化亚胺底物; 二是F-作为Lewis碱活化烯丙基硅试剂.在此作用下有效地提高了目标产物收率(Eq. 3).该工作的重要意义在于拓宽了合成手性α-氨基酸衍生物的底物范围, 并为该类反应催化模式的筛选提供了借鉴.
之后, Kobayashi课题组[10]再次报道了腙作为亚胺底物与取代的烯丙基三氯硅烷的烯丙基化反应(Eq. 4).该反应通过手性双膦-氧配体与烯丙基三氯硅烷的配位作用形成了高价硅化合物, 该高价硅化合物可有效地与亲电试剂反应, 从而以高达99% ee值得到目标产物.该反应不仅为合成生物活性多肽前体D-alloisoleucin提供了方法基础, 也为烯丙基化反应提供了一种新的催化模式.
烯丙基锡试剂在α-亚胺酯的不对称烯丙基化反应中也有所应用, 较早关于该方面工作报道的有Jørgensen课题组[11]利用铜催化烯丙基锡试剂与α-亚胺酯的烯丙基化反应.从实验结果来看, 该反应具有较好的收率(94%)和对映选择性(83% ee), 并且适用于环状和非环状的烯丙基锡试剂, 底物适用范围更广.此外, 烯丙基硅试剂也可作为该反应的亲核试剂, 但实验结果不如烯丙基锡试剂理想(Eq. 5).
|
|
(3) |
|
|
(4) |
|
|
2007年, Benaglia课题组[12]报道了一例手性亚胺与一价银络合物促进的烯丙基化反应.作者设计合成了一系列亚胺双齿配体, 最终发现含有噻吩基团的双齿亚胺配体可以给出较为满意的效果(90%~99%收率, 71% ee) (Eq. 6).该工作丰富了α-亚胺酯不对称烯丙基化反应的配体类型, 为该类反应配体的开发和设计提供了新的思路.
|
|
(5) |
|
|
(6) |
除烯丙基硅和烯丙基锡试剂以外, 烯丙基硼试剂也是α-亚胺酯烯丙基化反应常见的亲核试剂, 烯丙基硼试剂普遍具有活性高、底物范围广、用量少等特点, 而受到很大关注.
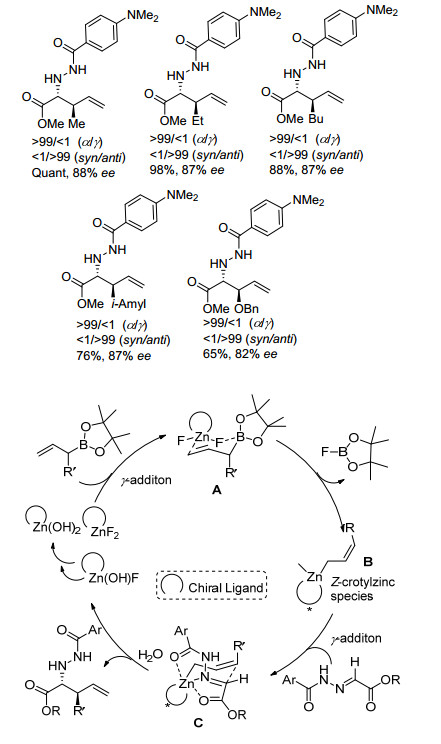
2008年, Kobayashi课题组[13]在之前工作基础上发现以烯丙基硼试剂代替烯丙基硅试剂可以降低烯丙基试剂用量, 并能够以良好的收率和优良的对映选择性得到不同烯丙基取代的α-氨基酸衍生物(Eq. 7).从反应的机理来看, 烯丙基硼酸酯与Zn的络合物通过γ-加成形成六元环状椅式过渡态A, 经配体交换得到手性烯丙基锌化合物B, 随后与腙发生立体选择性γ-加成产生中间体C, C水解后释放出目标产物和Zn(OH)F, 完成催化循环(Scheme 3).值得注意的是, 水参与了目标产物的形成过程, 是反应发生的必要条件.此外, 该研究表明, 若以Zn(OH)2为催化剂, 反应亦可获得80%收率和85% ee值, 首次实现金属氢氧化物催化的烯丙基化反应, 对金属氢氧化物催化体系的研究具有一定启发性意义.
α-亚胺酯不对称芳基化反应的亲核试剂包括芳基金属试剂、富电子芳环及杂芳环化合物等.通过亲核试剂与α-亚胺酯的亲核加成反应可制备α-芳基取代的甘氨酸骨架结构, 该结构是许多药物分子的重要骨架.通过α-亚胺酯的不对称芳基化反应, 为合成具有该结构的不同芳基甘氨酸衍生物提供了一种简洁有效的途径.
|
|
(7) |
2006年, Ellman课题组[14]利用手性辅基诱导策略实现芳基硼酸与磺酰基亚胺酯的不对称芳基化反应, 成功制备了一系列手性α-芳基氨基酸衍生物(Eq. 8).该反应在金属铑催化、膦配体的促进作用下, 经手性磺酰基诱导以69%~90%收率及优异的非对映选择性得到目标产物(>98/2 dr).该方法底物适用范围广, 取代基兼容性好, 可用于不同电性及位阻的芳基硼酸底物.此外, 作者发现将产物N-磺酰基取代的氨基酸衍生物进行脱保护, 进一步衍生化还原成醇, 均不影响产物的对映选择性.
|
|
(8) |
|
|
金属催化的亲核试剂与α-亚胺酯的芳基化反应多以金属铑络合物为催化剂, 而金属钯络合物催化的芳基化反应鲜有报道, 其原因在于金属钯络合物亲核性较弱, 难以实现与α-亚胺酯的亲核加成反应. 2007年, 陆熙炎课题组[15]发现金属钯为催化剂的反应体系中使用联吡啶配体可以有效增强钯络合物的亲核性.基于此, 该课题组通过手性辅基诱导, 成功实现了阳离子钯催化的芳基硼酸与α-亚胺酯的芳基化反应(Eq. 9).在机理方面, 作者认为阳离子钯络合物与芳基硼试剂经转金属化, 钯通过配位作用活化α-亚胺酯, 实现芳基对亚胺双键的顺式加成.该方法的重要价值在于发现了联吡啶配体对金属钯催化性能的重要影响, 使得对金属钯催化体系在该类型反应中的研究进一步深化.
2010年, 徐明华课题组[16]报道了利用手性辅基诱导的Friedel-Crafts反应, 该反应以吲哚和N-叔丁基亚磺酰亚胺为底物合成了一系列高光学活性α-吲哚取代的氨基酸衍生物(Eq. 10).经过对反应过渡态的分析, 作者认为Lewis酸与亚胺配位存在两种模式, 一是Lewis酸与亚胺结构中C=N双键氮原子和C=O双键氧原子配位(TS-1); 二是Lewis酸与亚胺结构中C=N双键氮原子和S=O双键氧原子配位(TS-2), 其中前者由于磺酰基的S=O键与吲哚位于双键同侧, 在活化亚胺的同时也便于吲哚从位阻较小的一侧进攻亚胺.该方法实现了α-(3-吲哚)甘氨酸的高效不对称合成.
|
|
(9) |
|
|
芳基金属试剂与α-亚胺酯通过亲核加成反应可制备手性α-氨基酸衍生物, 在众多芳基金属试剂中, 由于芳基硼试剂具有种类多样和兼容性良好等特点, 被广泛用于α-亚胺酯的加成反应中.
2012年, 曾伟课题组[17]在Pd(Ⅱ)和手性联噁唑啉配体的催化体系中, 实现了芳基硼酸对N-芳基保护α-亚胺酯的不对称加成反应, 成功制备了多种芳基取代的α-氨基酸酯衍生物(Eq. 11).在底物的普适性方面, 亚胺底物的电子效应对反应影响不大, 均可以较高的收率及对映选择性得到相应的目标产物; 相反, 由于硼酸底物受取代基电性影响较大, 使其在底物选择上具有一定限制, 表现为芳基硼酸可以发生反应, 烷基硼酸不反应, 若为含有杂原子的芳基硼酸则对反应的化学选择性及对映选择性存在不利影响.另外, 作者对所得的α-氨基酸衍生物进行脱保护基、水解酯基处理获得手性氨基酸, 又将手性氨基酸还原为氨基醇化合物, 实现了芳基取代手性氨基酸、氨基醇的制备.
|
|
(10) |
|
|
目前关于合成高对映选择性的α, α-二芳基取代氨基酸衍生物报道较少, 且主要以不对称Strecker反应为主, 此方法需水解氰基, 操作复杂且底物选择具有局限性. 2016年, 徐明华课题组[18]实现了铑催化芳基硼酸与五元环状亚胺酯底物的不对称芳基化反应.该反应以手性膦-烯化合物为配体, 成功制备了含季碳的手性二芳基化合物(84%~99%收率, 83%~97% ee), 在此基础上产物经开环得到了α, α-二芳基取代的直链氨基酸衍生物(Eq. 12).该方法为BACE1(淀粉蛋白前β-分解酶1)抑制剂的合成提供了一种简单有效的合成途径.
六元环状亚胺酯底物也可通过不对称芳基化反应制得α, α-二芳基取代氨基酸衍生物. Nishimura等[19]将其设计的手性双烯类配体成功用于铑催化芳基硼酸对六元环状亚胺酯的不对称加成反应中, 获得了较高收率(84%~99%)和中等较好立体选择性(83%~97% ee)的加成产物(Eq. 13).体系中的添加剂K3PO4可以避免形成副产物芳基羧酸, 但K3PO4形成的碱性环境对反应体系有不利影响.为了避免该影响, 作者选择兼容性良好的六元环状亚胺叔丁酯作为反应底物, 发现苯环无取代的亚胺酯具有很好的反应活性, 而当亚胺酯苯环氧原子对位带有吸电子基(F、Br)时, 反应的对映选择性会有所下降.另外, 在芳基硼酸底物拓展方面, 该课题组主要对芳基硼酸的取代基进行了考察, 发现吸电子基和给电子基取代的芳基硼酸均适用于该反应, 说明取代基的电性对反应的收率及对映选择性无明显影响.
|
|
(11) |
|
|
(12) |
|
|
|
|
(13) |
|
|
2017年, Hayashi等[20]报道了钯催化膦-亚胺配体促进环状磺酰基取代醛亚胺的不对称芳基化反应(Eq. 14), 通过该方法可以得到良好的收率(84%~99%)和对映选择性(83%~97%).研究发现该体系在底物筛选方面具有良好普适性, 不仅适用于C=N键上甲基、酯基、苯基取代的五元环状醛、酮亚胺底物, 也适用于甲基、不同酯基取代的六元环状醛、酮亚胺底物.由此可见, 此类膦-亚胺配体在合成α, α-二芳基取代氨基酸衍生物的不对称芳基化反应中有着良好的催化活性和应用前景.
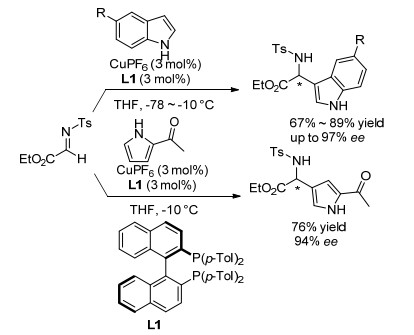
Friedel-Crafts反应是合成手性α-芳基取代氨基酸衍生物的经典方法之一. Friedel-Crafts反应(氮杂FriedelCrafts反应)可通过富电子芳环(杂环化合物)对亚胺C=N键的对映选择性加成获得手性氨基酸衍生物.较早关于α-亚胺酯的氮杂Friedel-Crafts反应是由Johannsen课题组[21]报道的, 该课题组以CuPF6和(R)-p-Tol-Binap形成的络合物为催化剂, 通过富电子吲哚、2-乙酰基吡咯与N-对甲苯磺酰亚胺酯的对映选择性加成反应, 分别以良好的收率和对映选择性获得了目标产物(Scheme 4).
|
|
(14) |
|
|
此外, Jørgensen课题组[22]用此催化体系实现了多种乙醛酸亚胺酯的芳基化反应.研究过程中, 作者发现亚胺氮原子上不同位阻取代基对反应的收率和立体选择性存在影响, 体积较大的N-叔丁氧基羰基保护的亚胺收率略低, 产物为S构型; 体积较小的N-乙氧基、N-甲氧基, N-苄氧羰基保护的亚胺, 产物为R构型.构型的差异源于手性还原的过程中存在着相反的两个方向所致(Eq. 15).
|
|
(15) |
|
|
经典的Mannich反应是含有活泼氢的化合物与甲醛、二级胺(或氨)的缩合反应.随着对反应认识的不断深化, 醛亚胺与α-亚甲基羰基化合物、烯醇硅醚化合物、席夫碱等亲核试剂的加成反应现也被视为Mannich反应, 并且该类反应在手性氨基酸衍生物合成领域已广泛应用.
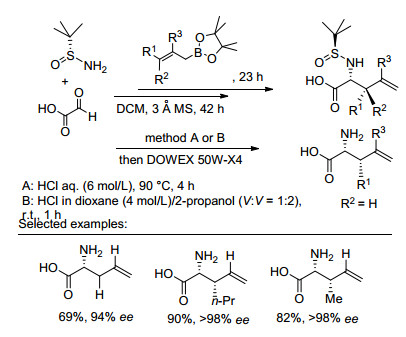
2011年, Hutton课题组[23]采用手性辅基诱导策略以取代的苯乙烯基硼酸、叔丁基亚磺酰亚胺、乙醛酸为底物实现了三组分的Mannich反应, 获得了系列α, β-不饱和的手性氨基酸衍生物.底物叔丁基亚磺酰亚胺选择巧妙:一是手性诱导效果理想, 获得较高的非对映选择性; 二是脱保护条件温和, 对不饱和双键无影响.此外, 研究发现, 反应底物浓度决定着目标产物的收率, 在高浓度环境下, 二阶动力学效应和溶解度效应的共同作用会促使产物析出, 使反应趋于完全、收率最大化(Eq. 16).
2012年, 徐明华课题组[24]进一步报道了以叔丁基亚磺酰亚胺为底物诱导合成手性氨基酸衍生物的反应体系.与以往研究不同之处在于该体系增加了Lewis酸催化剂, 利用Lewis酸与亚胺配位形成五元环状过渡态, 叔丁亚磺酰基较大的空间位阻使烯烃在re-面进攻C=N键, 形成R构型的产物, 大大提高了烯丙基转移的立体选择性, 获得一系列高光学活性α, β-不饱和的手性氨基酸衍生物(96%~99% de)(Eq. 17).同时, 该反应底物普适性广, 不仅适用于苯乙烯基硼酸还适用于杂环芳基硼酸和直链的烯基硼酸, 且均以较高的非对映选择性得到目标产物.
基于同样的手性辅基诱导策略, Keitaro Ishii等[25]在Lewis酸促进作用下以烯丙基频哪醇硼酯为亲核试剂与手性叔丁基亚磺酰胺、乙醛酸发生Mannich反应(Scheme 5).反应底物烯丙基频哪醇硼酯的C=C键可连接三个不同取代基, 反应后获得含有末端双键及两个手性中心的产物, 可用于制备含有两个手性中心的氨基酸.对于反应机理, 作者认为烯丙基硼酯被亚胺中间体的羧基活化, 使烯丙基进攻亚胺, 从而发生了Petasis borono- Mannich的反应.
|
|
(16) |
|
|
(17) |
|
|
手性调控的两种策略各具特点, 辅基诱导策略选择性高、普适性好, 但需要大量的手性辅基和脱保护操作; 手性催化剂策略所用催化剂用量少、经济环保, 通过设计合成与反应体系匹配的催化剂可有效提高反应的催化效率.
较早利用Mannich反应合成手性氨基酸衍生物的工作是由Sodeoka课题组[26]报道的Pd(Ⅱ)与(S)-p-Tol-Binap络合物促进三甲基取代烯醇硅醚与乙醛酸亚胺的反应.在反应条件的筛选过程中, 作者发现碱性溶剂DMF可以中和反应过程中产生的HBF4, 从而有效地促进了反应进行, 并获得最优的对映选择性.此外, 为明确配体对反应对映选择性的调控作用, 作者通过对核磁和质谱数据的分析研究发现:反应过程中形成了烯醇-钯中间体, 由于配体的位阻作用, 使得烯醇中间体从位阻较小的一侧对α-亚胺酯加成, 从而获得构型单一的产物(Eq. 18).
2001年, Lectka课题组[27]以芳基甲基酮衍生的二取代烯醇硅醚和α-亚胺酯为底物, 通过Mannich反应制得γ-羰基-α-氨基酸衍生物(Sheme 6a).该工作以CuClO4为催化剂, (R)-Tol-Binap为配体获得了最佳的对映选择性.与此同时, 该反应体系也适用于三取代烯醇硅醚底物(Sheme 6b)[28], 不同的是, 后者的反应温度(0 ℃~r.t.)较低, 顺、反构型的烯醇硅醚加成后均得到反式构型产物.究其原因, 作者认为金属铜催化剂起到了手性Lewis酸的作用:铜通过与亚胺配位活化亚胺, 利用空间位阻效应仅获得反式构型产物.此外, 金属铜催化剂对氮原子脱保护也具有促进作用.由此可见, 反应体系中的铜催化剂不仅可以促进缩醛胺上X基团的游离, 还可以通过配位作用活化亚胺.
|
|
(18) |
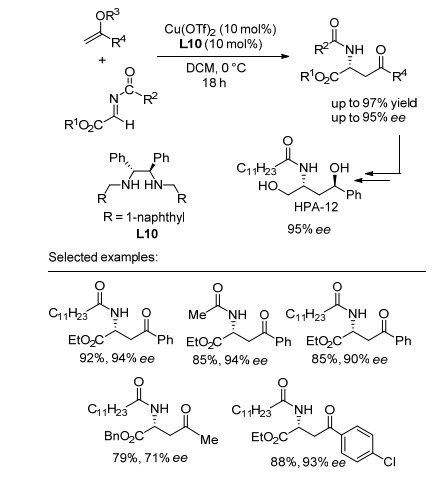
在此之后, Kobayashi课题组[29]报道了N-羰基取代亚胺酯与烯醇硅醚发生的Mannich反应(Sheme 7), 并取得了良好的对映选择性和较高的收率.同时, 该反应体系底物普适性良好, 在Cu(OTf)2和手性二胺配体络合物催化作用下, 酯、内酯、硫酯衍生的烯醇硅醚于-78 ℃可顺利地与N-苯甲酰亚胺发生反应; 于0 ℃可与N-乙酰基亚胺酯、N-羰基取代的亚胺酯反应.并且该反应也适用于烷基烯基醚(R3=Me), 可取得与烯醇硅醚(R3=TMS)同样令人满意的结果.此外, 该方法可以用于合成具有较高生物活性的手性氨基二醇化合物HPA-12, 有着重要的应用价值.
α-肼酯也可以与烯醇硅醚发生此类Mannich反应, Kobayashi课题组[30]率先以α-肼酯和烯醇硅醚为底物在手性锌络合物的催化下, 以较好的收率和对映选择性得到α-氨基酸衍生物(Eq. 19).该反应的特殊之处在反应溶剂为水, 相比于其他溶剂而言, 水具有清洁友好、无污染的特点, 但底物在水中的溶解度低, 反应速度慢.为解决此问题, 作者通过向体系中加入阳离子表面活性剂或三氟甲磺酸添加剂可有效地加快反应速率.作者通过XRD单晶衍射实验对手性控制的机理进行了研究, 发现配体L中连在手性碳原子上的两个苯基可以将手性传递到与苄基相连的两个氮原子上, 从而实现手性控制.此外, 作者进一步研究发现烯醇硅醚的几何构型和反应产物几何构型之间的关系有如下规律: Z-烯醇硅醚得到顺式立体异构的产物, 为主要反应产物; 而E-烯醇硅醚主要得到反式立体异构的产物.
|
|
(19) |
|
|
除上述研究外, 烯醇硅醚参与的Mannich类型反应还有较多报道[31], 在这里不再详述.
在Mannich反应中, 亲核试剂种类多种多样.除上述介绍的烯醇硅醚外, 烯胺、α-羰基化合物、席夫碱、硝基甲烷等也作为亲核试剂用于Mannich反应[32~40].
2004年, Kobayashi课题组[41]使用烯胺作为Mannich反应的亲核试剂, 实现了不同烯胺对多种亚胺酯加成制备手性α-氨基酸衍生物的反应(Eq. 20).该反应同样使用Cu(OTf)2和手性二胺配体的催化体系, 目标产物的对映选择性最高可达到94% ee.同时, 由于底物烯胺与α-亚胺酯取代基的多样性, 可获得带有不同官能团取代的产物, 该方法可用于合成某些天然产物、药物以及手性配体, 应用价值广泛.
α-酮酸酯作为亲核试剂也用于Mannich反应, 通过与α-亚胺酯发生Mannich反应可以合成系列手性γ-位羰基取代氨基酸衍生物. Jørgensen课题组[42]率先进行了报道:在室温条件下, Cu(OTf)2与(R, R)-Ph-Box配体的络合物催化α-酮酸酯与α-亚胺酯发生Mannich反应, 得到高对映选择性(>98% ee)的顺式构型产物(Scheme 8).手性铜络合物可起到促进烯醇形成的作用, 同时, 铜络合物与亚胺中的氮、氧原子配位形成的五元环状中间体同样对反应进行具有促进作用.另外, 通过该方法得到的光学纯手性氨基酸衍生物, 可以进一步还原和脱保护基转换成高度官能团化、高生物活性的α-氨基-γ-内酯化合物.
|
|
(20) |
|
|
除以上列举的亲核试剂外, 席夫碱也可用作Mannich反应的亲核试剂. 2003年, Jørgensen等[43]实现了CuClO4催化的席夫碱对N-对甲苯磺酰亚胺酯的加成反应.该反应利用手性氮-膦配体(Eq. 21)取得了良好的收率(最高84%)和中等的非对映选择性(syn:anti=60:40).通过席夫碱与亚胺酯的加成得到带有两个氨手性中心的产物, 可以用于制备光学纯α, β-双氨基酸衍生物, 此反应的出现为光学纯α, β-双氨基酸的合成提供了一种有效的新方法.
|
|
(21) |
|
|
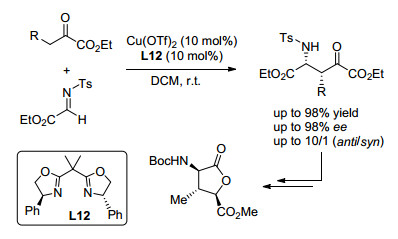
另外, 双羰基化合物作为一类重要的亲核试剂(包括丙二酸酯、β-酮酸[44]等)也可与α-亚胺酯发生Mannich反应制备手性α-氨基酸衍生物. 2003年, Jørgensen课题组[45]首次报道了Cu(OTf)2与手性噁唑啉配体催化丙二酸酯与α-亚胺酯的Mannich反应(Sheme 9).在研究反应对映选择性过程中, 作者发现丙二酸酯所连接的酯基类型对反应选择性具有重要影响, 体现为二、三级醇形成的丙二酸酯获得较高选择性的产物; 除选择合适的底物类型, 作者还根据底物的不同, 通过针对性使用添加剂六氟异丙苯(HFIP)及控制反应温度的策略, 有效地提高了反应产物的对映选择性(最高96% ee).
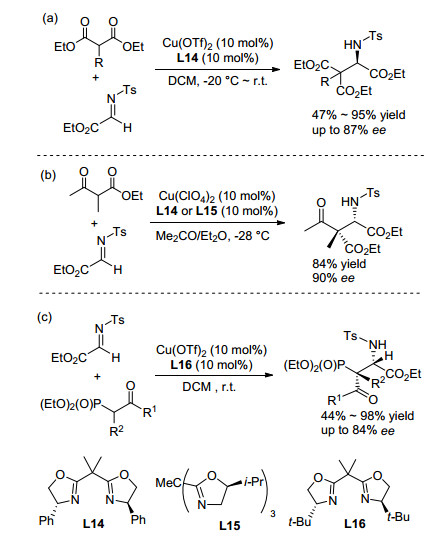
随后, Jørgensen等在以上研究的基础上, 再次报道了铜与手性联噁唑啉或三噁唑啉构成的金属络合物催化的β-酮酸酯[46]和β-羰基磷酸酯[47]作为亲核试剂与亚胺酯的Mannich反应.两项工作所得产物都以非对映异构体为主.在β-酮酸酯参与的Mannich反应中(Sheme 9), 高氯酸铜和三噁唑啉或联噁唑啉的络合物为催化剂均可以得到最高90% ee. β-羰基磷酸酯发生的Mannich反应(Scheme 9), 以Cu(OTf)2和联噁唑啉的络合物为催化剂, β-羰基磷酸酯经烯醇化对亚胺酯加成, 以中等偏上的收率以及良好的立体选择性(4.6/1 dr, 84% ee)得到含磷氨基酸衍生物.
α-羟基苯乙酮的α-位碳原子具有一定亲核性也被用作Mannich反应的亲核试剂. 2003年, Trost课题组[48]将设计合成的手性双核锌催化剂用于α-羟基苯乙酮与α-亚胺酯参与的Mannich反应(Eq. 22), 取得了良好的非对映选择性(15/1 dr)和极高的对映选择性(均大98% ee).同时, 将得到的β-羟基-α-氨基酸衍生物经BaeyerVilliger氧化和氧化脱羧转化成β-羟基-α-氨基酸, 在转化的过程中含给电子的α-羟基苯乙酮的对应产物更易于发生Baeyer-Villiger氧化.此外, 作者通过向体系中加入添加剂Ph3PS, 在不影响转化率和选择性控制的前提下提高反应的原子经济性.该方法为合成高光学纯的β-羟基-α-氨基酸衍生物提供了一种有效的方法.
|
|
(22) |
除上述工作使用的金属络合物催化剂外, 有机小分子催化剂因其来源丰富、不含金属、高效稳定且环境友好的优点使其也常被用在不对称Mannich反应中.
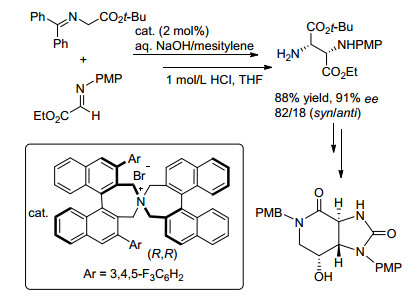
手性季铵盐作为相转移催化剂具有反应条件温和、选择性好、副反应少等特点, 已用于催化不对称Mannich反应. 2004年, Maruoka课题组[49]报道了手性季铵盐催化的α-亚胺酯与甘氨酸席夫碱发生的不对称Mannich反应.该反应在碱性溶液中, 利用对称的联萘手性铵盐催化, 合成了含有两个手性中心的氨基酸衍生物, 且产物经盐酸酸化后仍具有较高的对映选择性以及良好的非对映选择性.在条件优化的过程中, 作者发现较低的温度有利于稳定亚胺酯, 较强的碱性环境有利于反应发生, 并且溶剂效应对反应的对映选择性和非对映选择性都有重要的影响.经过条件的优化选择, 发现使用氢氧化钠溶液和均三甲苯形成的混合溶剂可以使反应的对映选择性和收率均处于较高水平.在此条件下, 具有酒石酸骨架结构的氨基酸衍生物经系列转化可高效合成streptolidine lactam的前体(Sheme 10).
手性脯氨酸是一种结构简单、廉价易得的有机小分子催化剂, 在不对称催化反应中表现出良好的催化性能. 2002年, Barbrs Ⅲ课题组[50]率先报道了L-脯氨酸催化酮羰基α-碳原子对亚胺酯的对映选择性加成反应.羰基α-碳原子上的取代基影响着反应产物的构型, 羰基α-碳原子上无取代基时得到构型单一的产物(95%~99% ee); 反之, 主要得到含有季碳的顺式构型产物(>99% ee), 二者的非对映体比例均高于19/1 (Eq. 23).反应机理方面, 作者认为L-脯氨酸与酮作用产生烯胺, 再与亚胺酯形成的中间体经历船式或椅式构象后分别得到E-式或Z-式构型的产物.
此外, 结构多样的手性硫脲分子在催化过程中表现出良好的催化活性、宽广的底物适用性及优异的对映选择性, 使其广泛应用于包括Mannich反应在内的各种不对称催化反应中. 2014年, Jacobsen课题组[51]报道了手性硫脲催化α-卤代甘氨酸酯与双羰基化合物的Mannich反应.作者认为硫脲通过攫取α-卤代甘氨酸酯上的卤原子形成高活性的N-酰基甘氨酸亚胺正离子, 随后富电性的双羰基化合物对亚胺进行亲核加成, 从而实现“一锅法”制备一系列手性α-氨基酸衍生物的反应.此外, 该条件可以在保证较高收率和对映选择性的前提下实现克级规模的反应(Eq. 24).
|
|
(23) |
|
|
|
|
(24) |
|
|
含氟氨基酸衍生物具有重要应用价值, 但合成方法有限, 2016年, 周剑课题组[52]通过手性有机脲分子催化α-亚胺酯与氟代烯醇硅醚的Mannich反应, 以高对映选择性(94%)得了双氟原子取代的手性氨基酸衍生物(Eq. 25).对于苯环上带有不同电性及位阻的环状亚胺, 在该体系下均能取得较高的对映选择性(>84% ee), 对于二氟代烯醇硅醚, 芳基取代底物选择性和收率良好, 但脂肪族取代基底物效果并不理想.
|
|
(25) |
|
|
除此之外, 有机小分子催化剂手性二酚也可用于催化不对称Mannich反应. 2008年, Schaus课题组[53]报道了手性二酚催化烯基硼酸酯、二级胺、乙醛酸酯进行Mannich反应合成系列手性氨基酸衍生物的工作.研究发现手性二酚催化剂取代基的空间位阻效应和电子效应对反应的收率和选择性具有重要影响, 表现在以联萘酚为配体时, 3, 3'位连有大位阻取代基对反应对映选择性和收率都有不利影响, 若联萘酚一侧单取代时收率和选择性会略有提高.通过对反应的分析及对催化剂的筛查, 最终以(S)-Vapol为催化剂, 体系获得了良好的收率(>77%)和选择性(>87/13 er).另外, 值得一提的是该手性二酚催化剂可以重复使用, 回收的催化剂活性和构型不会改变, 使该反应具有清洁友好, 节约环保的特点(Eq. 26).
α-亚胺酯的不对称烯基化反应也是合成手性α-氨基酸衍生物的方法之一, 此类反应中, 烯烃与C=N键加成可得到α-烯基取代氨基酸衍生物; 产物若经电子转移则得到α-烯丙基取代的氨基酸衍生物.
|
|
(26) |
|
|
Lectka课题组[54]和Jørgensen课题组[55]率先报道了α-亚胺酯的烯基化反应.两篇工作的相同之处在于选择相同的亚胺酯底物(N-对甲苯磺酰亚胺酯)和手性配体(联萘骨架的手性双齿膦化合物); 不同之处在于溶剂、催化剂种类以及底物选择范围.首先, 在溶剂选择上, 前者以不常用的三氟甲苯为溶剂, 通过该溶剂的使用有效地溶解催化剂与配体络合物的同时提高反应的对映选择性(高达99% ee) (Eq. 27).后者选择二氯甲烷为溶剂, 经CuPF6或CuClO4催化需在较低温度下(-20 ℃或0 ℃)进行(Eq. 28), 可获得高达95% ee.其次, 在底物范围选择上, 前者以芳基取代烯烃为主, 后者以烷基取代烯烃为主; 两者的工作互相补充, 为含不同末端双键的氨基酸衍生物的合成提供了方法.
2004年, Hutchings课题组[56]发现了一种新的催化模式, 通过将铜与双噁唑啉的螯合物固定在沸石上形成square-planar complex(正方形平面络合物), 用于催化α-亚胺酯烯基化反应.实验表明室温下反应20 h, 该多相催化剂可以有效地催化N-苄基取代α-亚胺酯与α-甲基苯乙烯的加成反应, 并以87%的收率和高达99%的对映选择性得到目标产物(Eq. 29).
2015年, 贾义霞课题组[57]实现了Ni(Ⅱ)催化手性Binap配体促进六元环状磺酰亚胺酯的不对称烯基化反应.与之前工作不同, 该反应得到烯基化产物(Eq. 30).该催化体系底物适用性良好, 苯并磺酰亚胺的苯环取代基电子效应对反应选择性影响不大, 所得产物均可以保持良好的对映选择性(88%~99% ee).同时, 研究进一步发现由于亲核性不同, 苯环带有给电子基的苯乙烯在收率上优于带有吸电子基的苯乙烯底物(35%~83%收率).该工作为烯基取代α-氨基酸衍生物的合成提供了一种有效的新方法.
|
|
(27) |
|
|
(28) |
|
|
(29) |
|
|
(30) |
|
|
α-亚胺酯不对称炔基化反应是合成手性α-炔基氨基酸衍生物的重要方法.此处对手性催化剂调控末端炔烃与α-亚胺酯的亲核加成反应做简要概括.
2007年, Chan课题组[58]报道了CuOTf•0.5C6H6催化Pybox配体促进的不对称炔基化反应[57](Eq. 31).在研究中作者发现使用相同的手性配体, 通过改变手性配体与金属催化剂的比例可以很好地控制对映选择性, 且ee值比例相差不大.经研究表明, 手性配体和金属催化剂的比例与产物构型之间存在的非线性效应关系[59].在机理方面, 作者认为手性催化剂、亚胺酯底物与炔烃形成络合物中间体, 该中间体有利于末端炔烃H原子的活化和亚胺的质子化, 为炔基对亚胺C=N双键的加成奠定了基础.该研究说明了不对称催化过程中配体与金属比例对反应的选择性具有重要影响.
同年, Rueping课题组[60]报道了AgOAc和手性磷酸共同催化的不对称炔基化反应(Eq. 32).该反应中的双催化剂构成了两个平行的催化循环, 其一为手性磷酸活化亲电试剂亚胺酯的过程; 其二为金属盐活化亲核试剂末端炔烃的过程(Sheme 11).由于炔基银衍生物在质子性溶剂中具有良好的稳定性且易于水解, 所以选择醋酸银作为共催化剂.该催化体系以较好的收率和对映选择性合成不同苯乙炔基取代的手性氨基酸衍生物.
|
|
(31) |
|
|
(32) |
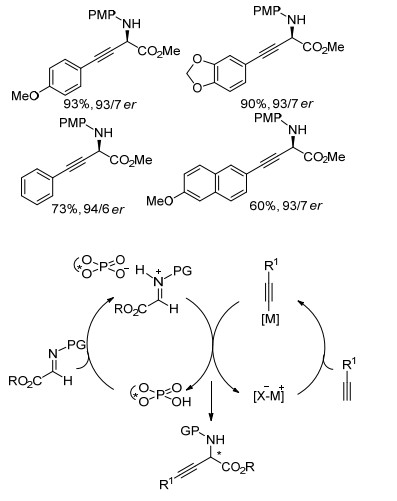
2011年, 张新刚课题组[61]报道了手性联萘酚配体与二烷基锌促进亚胺酯的炔基化反应(Eq. 33).研究发现, 配体分子中联萘骨架3, 3'-位的取代基对反应的对映选择性有明显影响, 使用3, 3'-位被三甲基硅基(TMS)取代的配体可以得到对映选择性高达97.3%的炔基化产物.该反应具有良好的底物适用性, 无论是芳基炔烃还是烷基炔烃, 都能得到较高的收率和对映选择性的目标产物(95%收率, 99% ee).
|
|
(33) |
|
|
随后, 该催化体系经进一步优化改造被成功用于1, 3-二炔对亚胺酯的不对称亲核加成反应中, 通过适当增加催化剂和配体的用量, 可适用于不同芳香族、脂肪族和甲硅烷基二炔, 并高效地获得目标炔基化产物(97%收率, 97% ee)(Eq. 34)[62].
|
|
(34) |
2013年, Ohshima课题组[63]报道了铑与噁唑啉的络合物催化亚胺酯炔丙基化反应(Eq. 35), 该催化体系避免了大量金属试剂的使用且具有良好的底物普适性, 适用于不同芳香族、脂肪族和甲硅烷基炔烃, 在脂肪族、甲硅烷基炔烃与亚胺的炔基化反应中, 炔烃底物和催化剂的用量是芳香族炔烃炔基化反应中的2倍.此外, 为确保反应具有良好的对映选择性, 需将炔基底物与铑络合物在较低温度下反应一段时间再加入亚胺底物.
|
|
(35) |
|
|
α-亚胺酯的烷基化反应中, 以格氏试剂与α-亚胺酯的亲核加成反应最为常见, 然而, 由于亚胺酯结构中含有三个亲电中心, 使反应通常获得多种加成产物, 所以区域选择性控制是实现α-亚胺酯不对称烷基化反应的关键.
2002年, Roland等[64]在详细考察手性α-亚胺酯底物、催化剂及溶剂类型的基础上, 发现具有(R)-1-苯基乙胺骨架的手性辅基的底物, 在溴化锌的促进作用下, 可以实现区域选择性和对映选择性α-烷基氨基酸衍生物的合成(Eq. 36).该反应是不对称烷基化反应的先驱性工作, 为此类反应的研究和发展提供了借鉴和参考.
|
|
(36) |
|
|
2015年, Ishihara课题组[65]报道了ZnCl2促进格氏试剂与α, β-不饱和手性亚胺酯的加成反应, 以59%~99%收率及高达99/1的对映选择性得到了一系列光学活性化合物(Eq. 37).反应过程中, 格氏试剂与氯化锌形成复杂锌盐化合物([R3Zn]-[MgX]+[{MgX2}2])可有效增加反应活性, 通过[MgX]+与C=N双键氮原子及C=O双键氧原子作用, 使[R3Zn]-的烃基具有选择性地进攻C=N双键碳原子, 从而实现手性亚胺酯的不对称烷基化.
|
|
(37) |
|
|
目前, 关于亲核试剂和α-亚胺酯加成反应制备手性α-氨基酸衍生物的研究已取得较大进展, 主要体现在以下两点:首先, 发展了不同的反应类型, 包括α-亚胺酯不对称烯丙基化反应、芳基化反应、Mannich反应、烯基化反应、炔基化反应和烷基化反应等.其中Mannich反应的亲核试剂种类极其丰富, 而且部分亲核试剂性能优良、结构稳定、易于保存, 为合成多样的手性氨基酸衍生物提供了极具价值的方法. 与其相比, α-亚胺酯的不对称烷基化和烯基化的反应模式报道较少, 处在研究的初级阶段, 尚有较大的研究空间.其次, 丰富了手性催化剂调控策略和手性辅基诱导策略的方法, 尤其是手性催化剂调控策略中一系列简洁高效手性配体的研发和应用, 为丰富手性氨基酸衍生物的合成方法做出了重要贡献.但是该策略在个别体系中仍存在着催化剂用量大、手性配体类型单一等问题, 有待进一步研究和探索.鉴于以上分析, 通过设计结构优良的手性配体, 探究简捷高效的催化体系, 寻找新的催化模式, 开发多样的亲核试剂等途径必定有助于手性氨基酸衍生物合成方法的进一步完善.
(a) Salih, N.; Adams, H.; Jackson, R. J. Org. Chem. 2016, 81, 8386.
(b) Chen, Y.-Y.; Chang, L.-T.; Chen, H.-W.; Yang, C.-Y.; Hsin, L.-W. ACS. Comb. Sci. 2017, 19, 131.
(c) Vanda, D.; Jorda, R.; Lemrová, B.; Volná, T.; Krytšof, V.; McMaster, C.; Soural, M. ACS Comb. Sci. 2015, 17, 426.
(d) Jarvo, E. R.; Miller, S. J. Tetrahedron 2002, 58, 2481.
(e) Kazmaier, U. Angew. Chem., Int. Ed. 2005, 44, 3509.
(f) Prabhu, G.; Narendra, N.; Pandurangaa, V.; Sureshbabu, V. V.; RSC Adv. 2015, 5, 48331.
Wei, Q.-L.; Zhang, F.; Zhao, X.-F.; Wang, C.; Xiao, J.-L.; Tang.W.-J. Org. Biomol. Chem. 2017, 15, 5468. doi: 10.1039/C7OB01329A
(a) Tararov, V. I.; Bőrner, A. Synlett 2005, 203.
(b) Tararov, V. I.; Kadyrov, R.; Riermeier, T. H.; Bőrner, A. Chem. Commun. 2000, 1867.
(c) Kadyrov, R.; Riermeier, T. H.; Dingerdissen, U.; Tararov, V.; Bőrner, A. J. Org. Chem. 2003, 68, 4067.
(a) Avenoza, A.; Busto, J. H.; Peregrina, J. M.; Pérez-Fernández, M. Tetrahedron 2005, 61, 4165.
(b) Avenoza, A.; Busto, J. H.; Canal, N.; Peregrina, J. M.; Pérez-Fernández, M. Org. Lett. 2005, 7, 3597.
(c) Akiyama, T.; Daidouji, K.; Fuchibe, K. Org. Lett. 2003, 5, 3691.
(d) Yao, S.; Saaby, S.; G.Hazell, R.; Johannsen, K. A. Chem.-Eur. J. 2000, 6, 2435.
(a) Chen, D.; Xu, M.-H. Chin. J. Org. Chem. 2017, 37, 1589(in Chinese).
(陈雕, 徐明华, 有机化学, 2017, 37, 1589.)
(b) Bian, Q.-H.; Zhong, J.-C.; Hou, S.-C.; Wang, M. Chin. J. Org. Chem. 2010, 30, 1261(in Chinese).
(边庆花, 钟江春, 侯士聪, 王敏, 有机化学, 2010, 30, 1261.)
(c) Cheng, Y.-H.; Zou, X.-M.; Wu, C.; Yang, H.-Z. Chin. J. Org. Chem. 2001, 30, 1(in Chinese).
(程永浩, 邹小毛, 吴超, 杨华铮, 有机化学, 2001, 30, 1.)
(d) Li, Z.-J.; Wan, G.-H.; Wei, P.; Shi, Y.-H.; Ou-yang, P.-K. Chin. J. Org. Chem. 2005, 25, 881(in Chinese).
(李振江, 万红贵, 韦萍, 石玉瑚, 欧阳平凯, 有机化学, 2005, 25, 881.)
(e) Lin, J.; Fan, H.-D.; Yan, S.-J. Chin. J. Org. Chem. 2007, 27, 925(in Chinese).
(林军, 樊会丹, 严胜骄, 有机化学, 2007, 27, 925.)
(f) Lin, J. Chin. J. Org. Chem. 2009, 24, 33(in Chinese).
(刘静, 有机化学, 2009, 24, 33.)
Eftekhari-Sis, B.; Zirak. M. Chem. Rev. 2017, 117, 8326. doi: 10.1021/acs.chemrev.7b00064
Liu, M.; Shen, A.; Sun, X.-W.; Deng, F.; Xu, M.-H.; Lin, G.-Q. Chem. Commun. 2010, 46, 8460. doi: 10.1039/c0cc03230a
Ferraris, D.; Young, B.; Cox, C.; Dudding, T.; Drury, W. J.; Ryzhkov, L.; Taggi, A. E.; Lectka, T. J. Am. Chem. Soc. 2002, 124, 67. doi: 10.1021/ja016838j
Hamada, T.; Manabe, K.; Kobayashi, S. Angew. Chem., Int. Ed. 2003, 42, 3927. doi: 10.1002/(ISSN)1521-3773
Ogawa, C.; Sugiura, M.; Kobayashi, S. Angew. Chem., Int. Ed. 2004, 43, 6491. doi: 10.1002/(ISSN)1521-3773
Fang, X.-M.; Johannsen, M.; Yao, S.; Gathergood, N.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 1999, 64, 4844. doi: 10.1021/jo990238+
Colombo, F.; Annunziata, R.; Benaglia, M. Tetrahedron. Lett. 2007, 48, 2687. doi: 10.1016/j.tetlet.2007.02.071
Fujita, M.; Nagano, T.; Schneider, U.; Hamada, T.; Ogawa, C.; Kobayashi, S. J. Am. Chem. Soc. 2008, 130, 2914. doi: 10.1021/ja710627x
Beenen, M. A.; Weix, D. J.; Ellman, J. A. J. Am. Chem. Soc. 2006, 128, 6304. doi: 10.1021/ja060529h
Dai, H.-X.; Lu, X.-Y. Org. Lett. 2007, 9, 3077. doi: 10.1021/ol0711220
Ji, D.-M.; Xu, M.-H. Chem. Commun. 2010, 46, 1550. doi: 10.1039/b914687c
Chen, J.-Y.; Lu, X.-X.; Lou, W.-Y.; Ye, Y.; Jiang, H.-F.; Zeng, W. J. Org. Chem. 2012, 77, 8541. doi: 10.1021/jo301423e
Li, Y.; Yu, Y.-N.; Xu, M.-H. ACS Catal. 2016, 6, 661. doi: 10.1021/acscatal.5b02403
Takechi, R.; Nishimura, T. Org. Biomol. Chem. 2015, 13, 4918. doi: 10.1039/C5OB00431D
Zhou, B.; Li, K.-Z.; Jiang, C.-H.; Lu, Y.-X.; Hayashi, T. Adv. Synth. Catal. 2017, 359, 1. doi: 10.1002/adsc.v359.1
Johannsen, M. Chem. Commun. 1999, 2233.
Saaby, S.; Fang, X.-M.; Gathergood, N.; Johannsen, K. A. Angew. Chem., Int. Ed. 2000, 39, 4114. doi: 10.1002/(ISSN)1521-3773
Churches, Q. I.; White, J. M.; Hutton, C. A. Org. Lett. 2011, 13, 2900. doi: 10.1021/ol200917s
Li, Y.; Xu, M.-H. Org. Lett. 2012, 14, 2062. doi: 10.1021/ol300581n
Sugiyama, S.; Imai, S.; Ishii, K. Tetrahedron:Asymmetry 2013, 24, 1069. doi: 10.1016/j.tetasy.2013.07.026
Hagiwara, E.; Fujii, A.; Sodeoka, M. J. Am. Chem. Soc. 1998, 120, 2474. doi: 10.1021/ja973962n
Ferraris, D.; Young, B.; Cox, C.; Dudding, T.; Drury Ⅲ, W. J.; Ryzhkov, L.; Taggi, A. E.; Lectka, T. J. Am. Chem. Soc. 2002, 124, 67. doi: 10.1021/ja016838j
Ferraris, D.; Young, B.; Dudding, T.; Lectka, T. J. Am. Chem. Soc. 1998, 120, 4548. doi: 10.1021/ja9802450
Kobayashi, S.; Matsubara, R.; Nakamura, Y.; Kitagawa, H.; Sugiura, M. J. Am. Chem. Soc. 2003, 125, 2507. doi: 10.1021/ja0281840
Hamada, T.; Manabe, K.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 7768. doi: 10.1021/ja048607t
(a) Xie, L.; Ma, H.-L.; Li, J.-Q.; Yu, Y.; Qin, Z.-H.; Fu, B. Org. Chem. Front. 2017, 4, 1858.
(b) Yu, J.-S.; Zhou, J. Org. Chem. Front. 2016, 3, 298.
Zhang, H.; Syed, S.; Barbas Ⅲ, C. F. Org. Lett. 2010, 12, 708. doi: 10.1021/ol902722y
Perera, S.; Sinha, D.; Rana, N. K.; Trieu-Do, V.; Zhao, C.-G. J. Org. Chem. 2013, 78, 10947. doi: 10.1021/jo4019304
Valero, G.; León, C. M.; Moyano, A. Asymmetric Catal. 2015, 2, 7.
Liu, X.-D.; Deng, L.-J.; Jiang, X.-X.; Yan, W.-J.; Liu, C.-L.; Wang, R. Org. Lett. 2010, 12, 876. doi: 10.1021/ol902916s
Wu, L.; Li, G.-X.; He, M.-G.; Wang, Y.-W.; Zhao, G.; Tang, Z. Can. J. Chem. 2016, 94, 769. doi: 10.1139/cjc-2016-0089
Tao, Z.-L.; Adele, A.; Wu, X.; Gong, L.-Z. Chin. J. Chem. 2014, 32, 969. doi: 10.1002/cjoc.201400453
Veverková, E.; Liptáková, L.; Veverka, M.; Šebesta, R. Tetrahedron:Asymmetry 2013, 24, 548. doi: 10.1016/j.tetasy.2013.03.016
Hernández-Toribio, J.; Arrayás, R. G.; Carretero, J. C. Chem.-Eur. J. 2010, 16, 1153. doi: 10.1002/chem.v16:4
(a) Veverková, E.; Štrasserová, J.; Šebesta, R.; Toma, S. Tetrahedron: Asymmetry 2010, 21, 58.
(b) Lu, N.; Fang, Y.-H.; Gao, Y.; Wei, Z.-L.; Cao, J.-G.; Liang, D.-P.; Lin, Y.-J.; Duan, H.-F. J. Org. Chem. 2018, 83, 1486.
(c) Wang, Y.-H.; Liu, Y.-L.; Cao, Z.-Y.; Zhou, J. Asian J. Org. Chem. 2014, 3, 429.
Matsubara, R.; Nakamura, Y.; Kobayashi, S. Angew. Chem., Int. Ed. 2004, 43, 1679. doi: 10.1002/(ISSN)1521-3773
Juhl, K.; Gathergood, N.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2001, 40, 2995. doi: 10.1002/(ISSN)1521-3773
Bernardi, L.; Gothelf, A. S.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 2003, 68, 2583. doi: 10.1021/jo026766u
Yang, C.-F.; Shen, C.; Wang, J.-Y.; Tian, S.-K. Org. Lett. 2012, 14, 3092. doi: 10.1021/ol301180z
Marigo, M.; Kjærsgaard, A.; Juhl, K.; Gathergood, N.; Jørgensen, K. A. Chem.-Eur. J. 2003, 9, 2359. doi: 10.1002/chem.200204679
Foltz, C.; Stecker, B.; Marconi, G.; Bellemin-Laponnaz, S.; Wadepohla, H.; Gade, L. H. Chem. Commun. 2005, 5115. http://www.ncbi.nlm.nih.gov/pubmed/16220189
Kjærsgaard, A.; Jørgensen, K. A. Org. Biomol. Chem. 2005, 3, 804. doi: 10.1039/B416294C
Trost, B. M.; Terrell, L. R. J. Am. Chem. Soc. 2003, 125, 338. doi: 10.1021/ja028782e
Ooi, T.; Kameda, M.; Fujii, J.-I.; Maruoka, K. Org. Lett. 2004, 6, 2397. doi: 10.1021/ol049215u
Córdova, A.; Notz, W.; Zhong, G.-F.; Betancort, J. M.; Barbas Ⅲ, C. F. J. Am. Chem. Soc. 2002, 124, 1842. doi: 10.1021/ja017270h
Wasa, M.; Liu, R. Y.; Roche, S. P.; Jacobsen, E. N. J. Am. Chem. Soc. 2014, 136, 12872. doi: 10.1021/ja5075163
Yu, J.-S.; Zhou, J. Org. Chem. Front. 2016, 3, 298. doi: 10.1039/C5QO00407A
Lou, S.; Schaus, S. E. J. Am. Chem. Soc. 2008, 130, 6922. doi: 10.1021/ja8018934
Drury Ⅲ, W. J.; Ferraris, D.; Cox, C.; Young, B.; Lectka, T. J. Am. Chem. Soc. 1988, 120, 11006. doi: 10.1002/chin.199849128/full
Yao, S.; Fang, X.-M.; Jørgensen, K. A. Chem. Commun. 1998, 2547.
Caplan, N. A.; Hancock, F. E.; Bulman Page, P. C.; Hutchings, G. J. Angew. Chem., Int. Ed. 2004, 43, 1685. doi: 10.1002/(ISSN)1521-3773
Liu, R.-R.; Wang, D.-J.; Wu, L.; Xiang, B.; Zhang, G.-Q.; Gao, J.-R.; Jia, Y.-X. ACS Catal. 2015, 5, 6524. doi: 10.1021/acscatal.5b01793
Shao, Z.-H.; Wang, J.; Ding, K.; Chan, A. S. C. Adv. Synth. Catal. 2007, 349, 2375. doi: 10.1002/(ISSN)1615-4169
Peng, F.-Z.; Shao, Z.-H.; Chan, A. S. C. Tetrahedron:Asymmetry 2010, 21, 465. doi: 10.1016/j.tetasy.2010.02.020
Rueping, M.; Antonchick, A. P.; Brinkmann, C. Angew. Chem., Int. Ed. 2007, 46, 6903. doi: 10.1002/(ISSN)1521-3773
Huang, G.-C.; Yang, J.; Zhang, X.-G. Chem. Commun. 2011, 47, 5587. doi: 10.1039/c1cc10403a
Zhang, F.-G.; Ma, H.; Zheng, Y.; Ma, J.-A. Tetrahedron 2012, 68, 7663. doi: 10.1016/j.tet.2012.05.086
Morisaki, K.; Sawa, M.; Nomaguchi, J.-Y.; Morimoto, H.; Takeuchi, Y.; Mashima, K.; Ohshima, T. Chem.-Eur. J. 2013, 19, 8417. doi: 10.1002/chem.v19.26
Chiev, K.-P.; Roland, S.; Mangeney, P. Tetrahedron:Asymmetry 2002, 13, 2205. doi: 10.1016/S0957-4166(02)00587-6
Hatano, M.; Yamashita, K.; Mizuno, M.; Ito, O.; Ishihara, K. Angew. Chem., Int. Ed. 2015, 127, 2745. doi: 10.1002/ange.201408916

图式 1 α-亚胺酯不对称加成合成手性α-氨基酸衍生物的方法
Scheme 1 Synthesis of chiral α-amino acid derivatives by asymmetric addition of α-imino ester

图式 6 (R)-Tol-Binap/铜催化α-亚胺酯的Mannich反应
Scheme 6 (R)-Tol-Binap/Cu-catalyzed Mannich reacton of α-imino ester

图式 7 Mannich反应合成手性氨基二醇化合物
Scheme 7 Synthesis of chiral amino glycols by Mannich reaction

图式 9 双羰基化合物、β-羰基磷酸酯参与的Mannich反应
Scheme 9 Mannich reaction of dicarbonyl compounds and β-ketophosphonates

图式 10 Streptolidine lactam前体的合成
Scheme 10 Synthesis of precursor of streptolidine lactam.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




























































 下载:
下载:






 下载:
下载: