
表 1
钌配合物催化脱氢形成亚胺
Table 1.
Dehydrogenative imine formation catalyzed by ruthenium complexes

亚胺是羰基上的氧原子被氮原子取代后所形成的有机化合物, 是一类性质活泼的中间体, 能够发生还原、加成、缩合和环加成等一系列的反应.另外, 亚胺类化合物具有很好的抗菌作用, 它的药理学活性和生物学活性使其在医药和生物领域得到广泛的关注[1~3]. Yu等[4]首次由Schiff通过胺与羰基化合物共沸缩合的方法制备亚胺.在反应中首先N的孤对电子进攻羰基(醛羰基或酮羰基)基团上带有正电荷的碳原子, 发生亲核加成反应, 生成中间产物α-氨基醇, 然后进一步脱水形成亚胺.而中间产物α-氨基醇的生成是一个可逆反应, 所以要加入Lewis酸或除水剂促进中间产物的脱水.该方法原子经济性低, 操作麻烦, 对环境污染严重.另外由于醛或酮等羰基化合物较为活拨, 会导致一些副反应的发生, 使得亚胺的产率较低.因此, 迫切需要改进亚胺的合成方法是目前有机合成和化工生产领域的课题之一.近年来, 一些新兴的合成亚胺方法发展起来, 如硝基化合物加氢偶联合成亚胺[5, 6]、胺与醇的氧化缩合反应合成亚胺[7, 8]、仲胺的氧化脱氢反应合成亚胺[9]等.然而, 这些方法常常需要昂贵的等化学计量的试剂或毒性较大的反应物料, 并且会产生等化学计量的副产物, 因此难以真正地应用于工业生产.
由亚胺的结构可知, 胺-胺氧化偶联合成亚胺是较为理想方法之一.由于胺来源广泛, 理论上胺可任意组合生成多种亚胺, 且反应原子经济性高, 对环境友好.因此, 胺-胺氧化偶联合成亚胺的方法已经成为当前研究的热点.本文主要从催化剂及其催化机理两方面对胺-胺氧化偶联合成亚胺的研究进展进行综述.
1996年, Bailey等[10]首次报道了以反式[Ru4+(tmp)-(O)2] (tmp=5, 10, 15, 20-四甲基卟啉二价阴离子)配合物作为催化剂催化胺-胺氧化偶联合成亚胺.当胺为二级胺时, 产物为亚胺且产率接近100%, 而伯胺和脂肪胺氧化的产物是腈.在Bailey的开拓性工作报道不久, Albrecht等[11]用杂环卡宾(NHC)配体替代tmp制备了一系列如Eq. 1所示的“[Ru2+(η6-arene)(NHC)]”配合物, 实现了胺-胺氧化偶联合成亚胺.结果显示, 含NHC配合物的催化剂3具有最高的催化活性(95%), 主要原因在于卡宾配体表现出明显的离子特性, 可容纳正负电荷, 与金属配位影响电子轨道排布, 有效地影响金属的氧化态, 对催化效果有明显的影响, 同时, 卡宾配体对氧化反应具有很高的稳定性.含三唑基配合物的催化剂1的催化活性略低于催化剂3, 延长反应时间产率也能达到95%.与此相反, 含碳酸盐配合物的催化剂2对该反应却没有催化活性, 原因可能是碳酸配体不利于与胺形成配合物.该催化体系对芳香胺和脂肪胺均适用, 但脂肪胺的反应速率较慢且产率相对较低.
|
|
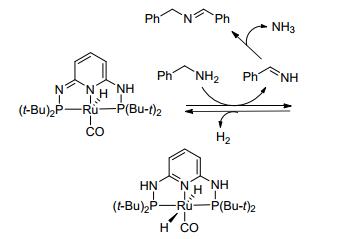
Doctorovich等[12]采用钌配合物[Ru(bpy)2(NO)Cl]2+(bpy=联二吡啶)作为催化剂成功地催化苄胺氧化偶联合成亚胺, 但大多数底物会有副产物腈的生成.为了解决腈副产物生成这一问题, Huang等[13]制备了吡啶类钳型钌配合物, 成功地催化胺-胺氧化偶联合成亚胺.这种钳型结构不仅能够提高苄胺的转化率, 避免了副产物腈的生成, 而且含取代基的苄胺也能转化为具有良好产率的亚胺(表 1).该方法提供了在无碱和无氧条件下将苄胺选择性氧化成相应的亚胺的直接策略.同时, 作者提出了如Scheme 1所示的反应机理.首先苄胺在催化剂的作用下脱氢生成中间体亚胺, 中间体亚胺与另一分子苄胺反应并释放出氨气得到目标产物亚胺.然而, 作者在文中对胺-胺之间的交叉缩合反应未作考察.而Zhang等[14]同样采用钌配合物[Et3NH]2[Ru(dipic)Cl3]作为催化剂在无溶剂条件下, 除了考察胺的自身偶联还研究了胺的交叉偶联缩合.实验结果表明, 苄胺与含给电子和弱吸电子取代基的苯胺交叉偶联取得了良好产率的不对称亚胺, 且与脂肪胺的交叉偶联能以100%的选择性获得不对称亚胺; 另外该催化剂的制备也较为简单, 可以很容易地由RuCl3、三乙胺和2, 6-吡啶二羧酸(dipicH2)为原料合成.
 下载:
导出CSV
下载:
导出CSV
 | ||
| Entry | R | Yielda/% |
| 1 | Ph | 93 |
| 2 | p-Me-C6H4 | 65 |
| 3 | m-Me-C6H4 | 80 |
| 4 | p-MeO-C6H4 | 83 |
| a 1H NMR yields. | ||
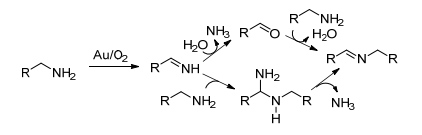
钌催化胺-胺氧化合成亚胺的进展开启了研究者对金催化胺-胺氧化偶联反应的研究. 2007年, Angelici等[15]首次报道了以金粉(粒径约103 nm)为催化剂催化胺-胺氧化偶联合成亚胺.同时, 他们还制备了氧化铝负载的金催化剂作了对比试验, 发现在相同的实验条件下, 5 mg Au/Al2O3催化剂的催化效果优于1 g金粉催化剂的催化效果(表 2), 这主要是由于Al2O3负载后可以有效地分散金颗粒, 减小金粒径(4~20 nm), 提高金的利用率.金催化剂对苄胺和含给电子基的苄胺均能以高转化率获得相应的亚胺, 但不适用于含吸电子基的苄胺和脂肪胺的氧化反应[16].同时, 作者根据实验结果推测了胺-胺氧化偶联的反应途径(Scheme 2).首先在金催化下胺氧化脱水生成中间体RCH=NH, 然后中间体进一步与第二分子伯胺或水反应, 最后生成目标产物亚胺.但作者没有给出确切的实验证据来区分这两种途径.
下载:
导出CSV
 | |||
| Entry | R | Yielda/% | |
| Au powder cat. | Au/Al2O3 cat. | ||
| 1 | Ph | 56 | 92 |
| 2 | p-Me-C6H4 | 61 | 96 |
| 3 | p-Cl-C6H4 | 7 | 59 |
| 4 | C5H11 | 5 | 29 |
| a Gold powder (1 g); b 5% Au/Al2O3. | |||
Garcia等[17]也对负载型金催化剂进行了研究, 发现金的颗粒大小显著地影响催化剂催化性能, 亚胺产率随着纳米颗粒平均粒径的减小而呈指数增长.在相同的反应条件下, 负载在TiO2上的金原子簇(3.5 nm)催化剂可以将苄胺以高转化率获得相应的亚胺, 而用金原子簇颗粒大小为25 nm的Au/TiO2催化剂却未能实现这一转化.此外, 载体也是影响催化性能的重要因素.与Au/TiO2催化剂相比, 以活性炭为载体的金催化剂(0.8% Au/C)催化该反应的反应速率提高了一个数量级, 仅需1 h转化率就可达到99%.同时, Au/C催化剂对杂环胺的自身氧化偶联和苄胺与缺α-氢原子的胺(苯胺或叔丁胺)的交叉氧化偶联均具有较好的催化效果.作者还制备了Au/CeO2和Au/PSt催化剂用于催化胺-胺氧化偶联合成亚胺, 并对比了催化活性[18].当反应进行24 h时, 两种催化剂表现出了相同的催化活性, 而反应进行4 h时, Au/CeO2催化剂的催化活性远高于Au/PSt催化剂, 表明Au/CeO2具有较短的诱导期.
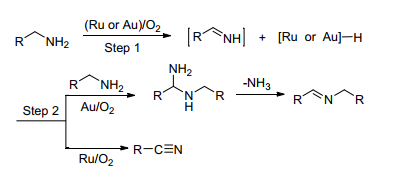
综合上述对钌和金催化剂的研究可以发现, 钌往往会有副产物腈的生成而金的主要产品是亚胺, 基于此实验结果, 研究者对钌和金的催化机理进行了总结(Scheme 3)[19].首先, 胺通过β-氢化物的消除得到中间产物亚胺, 在钌催化作用下, 亚胺中间体β-氢化物二次消除得到的主要是腈; 而在金催化作用下, 亚胺中间体的二次β-氢化物消除反应很难发生, 易与另一分子胺偶联合成主要产物亚胺.也有文献报道生成的腈还可进一步被还原为亚胺中间体, 然后亚胺中间体进一步与胺反应生成亚胺并释放出氨气[20, 21].显然, 贵金属催化体系下的催化机制并不是十分明确, 尚需进一步研究.
铜是地壳中最丰富的元素之一, 价廉易得. Patil等[22]首次报道了在无溶剂条件下, 以空气作为氧化剂, 用低负载量的CuⅠ催化剂催化胺-胺氧化偶联合成亚胺.该催化剂对于自身和交叉氧化偶联均表现出较好的催化活性(表 3和4), 底物的取代基(吸电子基和给电子基)和空间阻碍对亚胺的产率有较大的影响.然而, 在自身氧化缩合中, 大多数情况下会有5%~22%的副产物醛生成.随后, 作者又采用铜粉作为催化剂代替氯化亚铜在氧气条件下催化胺-胺氧化偶联合成亚胺[23], 发现以铜粉为催化剂可有效避免醛的生成, 且该催化剂的底物适用范围较广, 对含有给电子和吸电子取代基的苄胺、仲胺、环状仲胺、脂肪族伯胺和芳杂环胺等均有催化活性.
下载:
导出CSV
 | |||
| Entry | R | Yielda/% | 2:3c |
| 1 | Ph | 88 | 93:7 |
| 2 | p-Me-C6H4 | 94 | 85:15 |
| 3 | o-Me-C6H4 | 84 | 80:20 |
| 4 | p-MeO-C6H4 | 92 | 78:22 |
| 5 | p-F-C6H4 | 84 | 86:14 |
| 6 | o-Cl-C6H4 | 89 | 95:5 |
| 7 | m-Cl-C6H4 | 85 | 89:11 |
| 8 | p-Cl-C6H4 | 90 | 99:1 |
| 9 | o-F-C6H4 | 52b | 100:0 |
| 10 | m-F-C6H4 | 54b | 100:0 |
| 11 | C5H11 | 50 | — |
| a Yields of isolated imines. b Rest of starting benzylamine was recovered. c Selectivity determined by 1H NMR | |||
下载:
导出CSV
 | |||
| Entry | R2 | Yield/% | unsym./sym. |
| 1 | Ph | 78 | 23:77 |
| 2 | p-MeO-C6H4 | 86 | 86:14 |
| 3 | p-Me-C6H4 | 82 | 17:83 |
| 4 | p-Cl-C6H4 | 93 | 33:67 |
| 5 | m-NO2-C6H4 | 97 | 0:100 |
| 6 | o-Me-C5H3N | 94 | 0:100 |
| 7 | C6H13 | 78 | 56:44 |
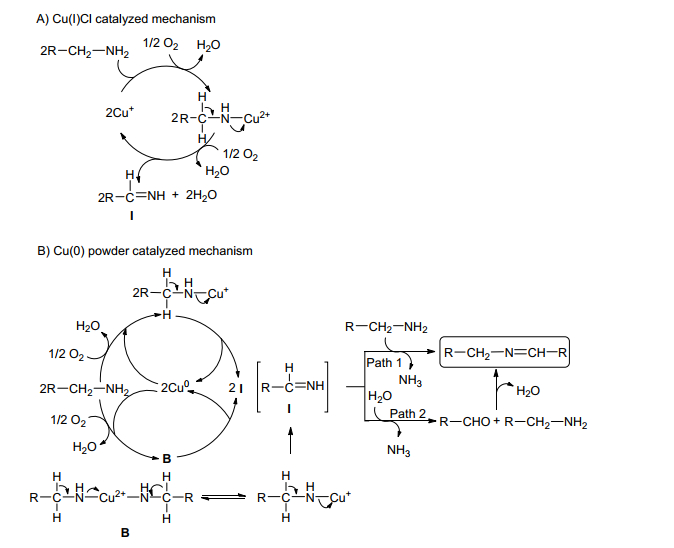
Patil课题组基于其对CuⅠ和铜粉催化剂催化胺-胺氧化偶联合成亚胺的深入研究, 提出了可能的催化机理, 如Scheme 4[24].第一步是通过铜与胺的氧化加成生成铜(Ⅱ)-胺络合物(Scheme 4A), 然后铜从铜(Ⅱ)-胺络合物中被还原脱除得到亚胺中间体Ⅰ, 如路径1 (Scheme 4B), 中间体Ⅰ与另一分子胺反应获得最终产品亚胺; 路径2是在水的作用下中间体Ⅰ部分水解生成醛, 醛和胺进一步缩合得到亚胺.
另外, Hu等[25]发展了以空气为氧化试剂, 采用碱性助剂2, 2, 6, 6-四甲基哌啶-N-氧化物(TEMPO)与溴化铜(Ⅱ)协同催化胺-胺氧化缩合生成亚胺.值得一提的是, 该催化系统除了可用于自身缩合, 还对交叉氧化反应也表现出较好的催化效果(表 5).除了CuBr2外, CuI也可与TEMPO协同作用催化胺-胺氧化偶联合成亚胺[26].作者研究发现, 该催化体系不但可催化各种取代基的苄胺以高转化率和高选择性合成亚胺, 还适用于二苄胺和一些环胺自身氧化偶联合成亚胺.
下载:
导出CSV
 | |||
| Entry | R1 | R2 | Conv.a/% |
| 1 | Ph | Ph | 48 |
| 2 | p-Me-C6H4 | Ph | 58 |
| 3 | p-MeO-C6H4 | Ph | 64 |
| 4 | o-Cl-C6H4 | Ph | 44 |
| 5 | Ph | o-Me-C6H4 | 66 |
| 6 | o-Cl-C6H4 | o-Me-C6H4 | 55 |
| 7 | Ph | p-MeO-C6H4 | 73 |
| 8 | p-Me-C6H4 | p-MeO-C6H4 | 78 |
| 9 | Ph | 2, 6-Me2-C6H3 | n.r. |
| a Conversion determined by GC using dodecane as an internal standard. n.r.=no reaction. | |||
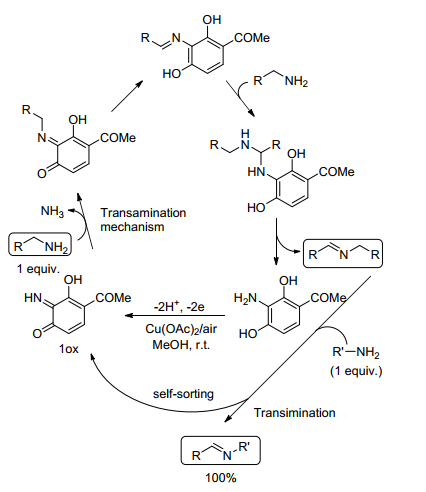
在胺-胺氧化偶联合成亚胺的反应中, 胺的交叉偶联普遍存在底物适用范围窄且产率低的现象, 针对这一问题, Largeron等[27]采用有机催化剂邻亚氨基醌(1ox)和CuⅡ金属协同催化胺-胺氧化偶联合成亚胺.邻亚氨基醌能够辅助CuⅡ激活伯胺的α-C—H键, 促进伯胺氧化为醛, 并进一步反应生成亚胺.该催化系统广泛适用于多种苄基胺及含硫和氧原子的杂环胺与1-乙基丙胺交叉氧化偶联且能够高选择性、高收率地生成相应的不对称亚胺.同时, 作者提出了可能的反应机理, 如Scheme 5.首先, 底物与邻亚氨基醌缩合生成亚氨基醌中间体并释放出氨气, 中间体异构化形成亚氨基苯醌, 接着与底物反应生成缩醛胺, 缩醛胺还原分解生成产品亚胺和氨基对苯二酚.亚胺还可与其他类型胺通过转氨作用进一步烷基化得到交叉偶联产物.
铜的络合物也被用于胺-胺氧化偶联合成亚胺, Gu等[28]就利用红铜与有机配体制备了铜络合物催化胺-胺氧化偶联反应.研究发现可以通过有机配体调控最终产物, 当1, 10-邻菲罗啉作为有机配体时获得的产品是亚胺, 而在相同条件下以异喹啉作为有机配体时获得的产品是腈.
以上报道的铜系催化剂大多为均相催化剂难以分离重复利用. Jones等[29]研究了复合型CuO/CeO2多相催化剂, 该催化剂易于分离, 可回收利用.作者研究发现CuO和CeO2单独存在时均具有催化活性胺-胺氧化偶联反应活性, 但复合型CuO/CeO2催化剂催化该反应时表现出更高的催化活性, 在整个反应中CuO起主要的催化作用, 而CeO2的存在可催化胺的氧化脱水, 从而促进反应的进行.
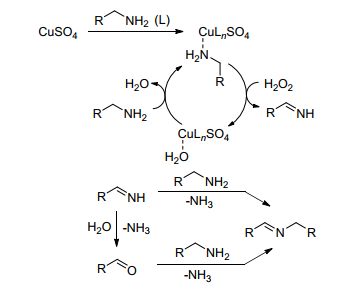
氧化试剂对胺-胺氧化偶联合成亚胺的反应有重要的影响, 空气或氧气为氧化剂, 由于方便易得, 成为研究者首选.除此之外, Marui等[30]报道了将H2O2为氧化剂用于胺-胺氧化偶联合成亚胺. H2O2氧化性强于空气使反应可在较温和的条件下进行, 该反应以CuSO4为催化剂, 水为溶剂, 环境友好.作者提出了可能的反应途径(Scheme 6).首先, 通过胺与CuSO4的反应形成胺络合物, 此络合物与H2O2反应产生中间体亚胺, 中间体亚胺和另一分子胺直接反应得到最终产物亚胺, 或通过水解放出氨气得到酮, 酮进一步与另一分子胺反应得到最终产物亚胺.
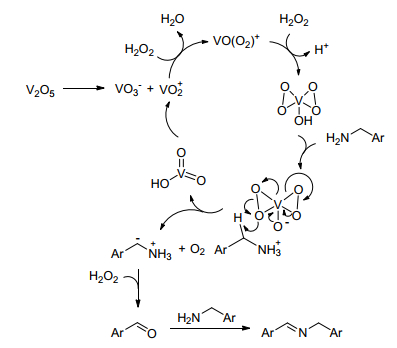
Li等[31]成功实现了以廉价而稳定的氧化钒为催化剂催化胺-胺氧化偶联合成亚胺, 但是对于含给电子基团(Me, OMe和OAc)的苄胺和1-萘甲胺未能获得相应的亚胺, 而是得到醛, 作者以此提出了如Scheme 7所示的V2O5催化的胺-胺氧化偶联合成亚胺反应的可能机理. V2O5电离后, 被H2O2氧化成黄色物质HOV(O2)2, 酸性HOV(O2)2与苄胺形成盐, 其中一个氧与苄基的氢作用使苄基去质子化生成苄型阴离子, 苄型阴离子被H2O2氧化成醛, 醛与另一分子苄胺作用得到亚胺产品.
Wang等[32]报道了钒络合物(HQ)2Vv(O)(OiPr)催化的胺-胺氧化偶联合成亚胺反应.反应可在非常温和的条件下进行且不需要任何添加剂或促进剂.该催化剂不仅适用于芳香胺而且对于合成含杂原子的亚胺也有催化活性, 但不适用于正己胺.另外, Ogawa等[33]使用氧化钒络合物VO(Hhpic)2 (H2hpic=3-羟基吡啶甲酸)作为催化剂在有氧条件下胺-胺氧化合成亚胺.该催化剂可在离子液体中显示了优异的重复利用性, 可重复使用到第8次而催化活性没有明显下降.
除了氧化钒和钒的络合物外, 一些钒和钼的混合物也被报道用于胺-胺氧化偶联合成亚胺[34~36], 其中这些混合物中, NPV6Mo6表现出最优的催化活性, 这些催化剂的一个重要优势是它们在强氧化条件下依然很稳定.
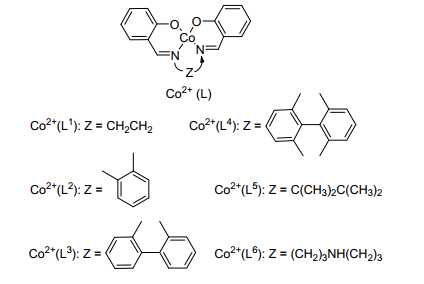
Nishinaga等[37]钴配合物催化体系催化伯胺合成亚胺.作者研究了不同配体结构的钴基催化剂的催化性能(图 1), 结果显示配体的结构对催化剂的活性有着重要的影响(表 6).
下载:
导出CSV
| Entry | Catalyst | Yielda/% |
| 1 | Co2+(L1) | 95.4 |
| 2 | Co2+(L2) | 93.8 |
| 3 | Co2+(L3) | 31.0 |
| 4 | Co2+(L4) | 16.6 |
| 5 | Co2+(L5) | 95.6b |
| 6 | Co2+(L6) | 0.90 |
| a Yields of isolated imines. b Reaction time: 1.5 h. | ||
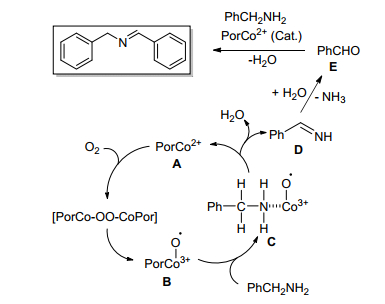
Zhao等[38]研究了CoTPP(CF3)4催化苄胺在氧气存在下氧化偶联合成亚胺, 发现该催化剂对胺自身氧化偶联可以以较高产率获得亚胺, 催化剂催化活性受配体类型和溶剂的影响较大.作者提出了可能的反应机理, 如Scheme 8. CoTPP(CF3)4与氧反应生成活性中间体B, 中间体B与苄胺配位生成中间体C, 随后将其转化为席夫碱中间体D, 在H2O的诱导下, 该亚胺中间体D水解释放出NH3分子形成苯甲醛E, E与另一分子苄胺反应得到所需的亚胺产物, 并释放一分子H2O.
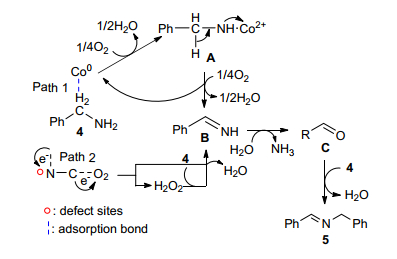
由以上文献报道可以看出, 尽管钴络合物催化剂显示了较好的催化活性, 但其制备复杂, 催化剂难以回收利用, 成本高.基于此情况, 本课题组[39]发展了一种SiO2材料负载的N掺杂碳基钴催化剂(Co-N-C/SiO2)并用于胺-胺氧化偶联合成亚胺.由X射线光电子能谱分析(XPS)表征结果显示表面掺杂的氮原子与碳结合生成石墨型氮, 吡咯型氮和吡啶型氮三种形式.该催化剂制备不使用昂贵的原料, 制备过程简单, 催化剂比表面积、孔容大, 颗粒分散均匀. Co-N-C/SiO2可在无需任何助剂, 仅在空气气氛和无溶剂条件下高产率的获得亚胺, 具有很高的催化活性.该催化剂不仅适用于胺的自身氧化缩合, 而且苄胺与脂肪胺的交叉偶联反应也具有较好的催化活性, 只是对脂肪胺的自身偶联的转化率较低(表 7). Co-N-C/SiO2通过简单的过滤即可分离, 重复使用5次催化活性无明显的降低, 环境友好.我们根据实验结果提出了较为合理的反应机理, 如Scheme 9.反应分为两种不同的途径: (1) Co为活性相:在Co和O2的作用下苄胺发生氧化反应脱水, 生成络合物A, 然后, A进一步氧化, 得到苄基亚胺B, 同时钴在该步骤中再生. (2) N—C作为活性相:苄胺和分子氧都在N—C的缺陷位点上被活化, 随后转化成苄基亚胺和H2O2中间体, H2O2可以与另一分子苄胺立即反应得到苄基亚胺B.这两种途径均通过胺的氧化脱水得到苄基亚胺B, B与初始步骤中产生的H2O反应, 得到醛C并释放出氨气, 随后醛与未反应的胺发生缩合反应, 得到最终产物5.
下载:
导出CSV
 | |||
| Entry | R | Conv.a/mol% | Selectivityb/mol% |
| 1 | C6H5 | 99 | 100 |
| 2 | p-Me-C6H4 | 96 | 100 |
| 3 | p-Cl-C6H4 | 97 | 100 |
| 4 | m-Cl-C6H4 | 95 | 100 |
| 5 | o-Cl-C6H4 | 93 | 100 |
| 6 | n-Pentyl | 32 | 28 |
| 7 | Cyclohexyl | 13 | 62 |
| a Determined by GC analysis; b Selectivity=yield/conversion. | |||
Garcia等[40]开发了一种含铁(Ⅲ)基-有机骨架的N-羟基邻苯二甲酰亚胺(NHPI)催化系统[NHPI/Fe(BTC)], 以氧气作为氧化剂, 在无溶剂条件下, 可以使各种苄胺(包括杂环胺)以高转化率和高选择性合成相应的亚胺, 但催化剂制备较复杂, 成本高且不适用于脂肪胺.作者提出了以NHPI/Fe(BTC)为催化剂催化胺-胺氧化偶联合成亚胺的催化机理.如Scheme 10所示, 首先NHPI/ Fe(BTC)在氧气的作用下生成N-氧自由基, N-氧自由基剥去苄位的H自由基形成NHPI, 从而得到苄基自由基胺, 苄基自由基胺被氧气氧化后脱氨气生成醛, 醛与另一分子胺反应得到目标产物亚胺.
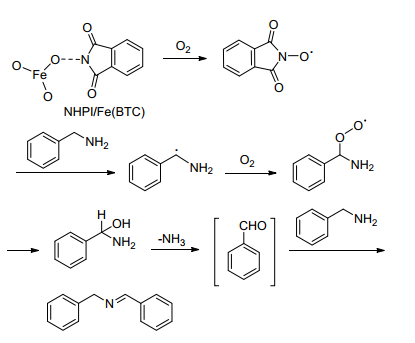
Xu等[41]采用廉价的Fe(NO3)3与添加剂TEMPO作为催化体系[Fe(NO3)3/TEMPO]用于催化胺-胺氧化偶联合成亚胺.该催化体系的通用性较强, 不仅可用于催化苄胺与苯胺交叉氧化偶联合成不对称亚胺, 而且还可用于醇与胺的有氧氧化合成亚胺.然而, 对于脂肪胺自身偶联反应, 活性不甚理想.
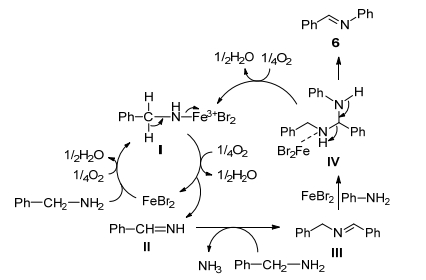
最近, Gopalaiah等[42]报道了更为简单廉价的溴化亚铁(Ⅱ)催化剂(5 mol%)成功实现了胺-胺氧化偶联合成亚胺.该催化剂不仅可以催化各种苄胺自身氧化偶联以高转化率和高选择性转化为亚胺, 而且适用于各种苄胺与各种苯胺(萘胺)和脂肪胺交叉氧化偶联合成不对称亚胺, 同时也考察正己胺和环己胺的自身氧化偶联也显示出良好的催化活性.该催化系统不需要任何助剂和溶剂且适用范围较广.
作者根据实验结果和已知的化学知识, 提出了如Scheme 11所示的反应机理.首先铁氧化与胺形成络合物Ⅰ, 进一步氧化Ⅰ得到亚胺Ⅱ并再生溴化亚铁(Ⅱ)以供循环再利用.亚胺Ⅱ与另一分子苄胺反应得到自身偶联产物Ⅲ并释放出氨气, 紧接着, 在铁催化剂的作用下苯胺与亚胺Ⅲ发生亲核加成反应得到铁-二元胺络合物Ⅳ, 最后, 二元胺络合物Ⅳ分解成交叉亚胺产物6和铁-胺络合物Ⅰ.
除了以上研究铜、钴、铁非贵金属催化剂, 锰、铬等也有报道. Kim等[43]以MnSO4为催化剂, TBHP为碱性助剂催化胺-胺氧化偶联合成亚胺.该催化体系对单取代及双取代的苄胺均具有较好的催化活性. Choudary等[44]报道了一种ZnCrCO3双金属催化剂, 也以TBHP为碱性助剂成功实现了胺-胺氧化偶联合成亚胺.该催化剂除了可以催化各种苄胺合成亚胺外, 对含杂原子的胺也适用, 但催化活性较低.锰、铬等金属催化剂虽然可以用于胺-胺氧化偶联反应, 但对环境危害较大, 相关的研究比较少.
综上, 过渡金属催化剂在胺胺氧化合成亚胺中占据重要的地位.尽管钌、金等贵金属对胺-胺氧化偶联合成亚胺反应具有优异的催化性能, 但由于钌、金等昂贵的价格限制了其发展, 因此探索廉价且性能优异的催化剂已逐渐成为研究主要方向.目前的研究已表明, 非贵金属催化剂可催化苄胺、杂环胺和部分脂肪胺, 以较高转化率和选择性合成亚胺, 且一些催化剂对不对称亚胺合成也有较好的催化效果, 但非贵金属催化体系大多需要添加专用氧化剂、碱等助剂, 反应体系复杂, 反应条件苛刻, 催化剂较难回收利用.因此, 对于过渡金属催化体系, 研究重点是继续提高非贵金属催化剂的催化活性, 简化反应体系; 同时, 从活性组分、制备方法入手发展负载型催化剂, 提高催化剂的重复利用性, 并进一步提高催化活性, 以进一步降低成本.
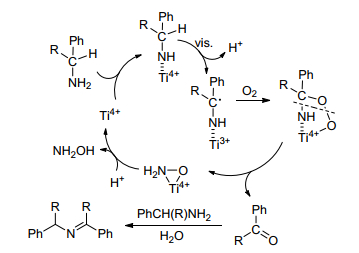
光催化是有机合成中一种反应条件温和、清洁和原子效率高的方法, 已被广泛地用于亚胺合成.在光催化剂中, 由于TiO2具有成本低、光化学稳定性高和无毒的特点, 已成为应用历史最久、最广泛的光催化材料之一. 1985年, Ohtani等[45]首次用钯负载TiO2光催化剂成功地将伯胺在乙腈中催化合成了亚胺并释放出氨气和氢气.在Ohtani的研究的基础上, Zhao等[46]在紫外光照射下, 胺可在TiO2的表面被氧化以高转化率和高选择性合成亚胺.后来作者[47]又对可见光照射下, 锐钛矿TiO2的光催化性能进行研究, 发现吸附在TiO2表面的胺可以在可见光区域吸收光, 以此考察了在乙腈溶剂中TiO2催化胺的选择性氧化性能, 结果显示各种苄胺均能以中等至优异的转化率和选择性得到相应的亚胺产品, 而且含N、O、S的杂环胺也可以获得相应的亚胺(表 8). 2013年, 他们进一步研究发现, TiO2作为光催化剂可以在水中以良好的选择性合成亚胺, 并且胺在水中选择性氧化的速率要比在乙腈中快很多, 所获得的产品与水不混溶可避免产品过氧化, 从而实现产品的轻松分离[48].显然, 该光催化体系反应条件温和, 催化剂循环利用, 以水为溶剂环保、经济.同时作者根据实验结果和已知的化学知识, 提出了如Scheme 12所示的反应机理.首先, 底物吸附在TiO2表面形成表面配合物, 在可见光照射下, 表面配合物发生光生电荷的分离, 电子从TiO2表面释放并产生电子-空穴, 所产生的电子-空穴引发胺基质子化, 并产生中性的以碳为中心的自由基, 然后, TiO2轨道中的电子转移给氧气, 氧气与自由基结合形成过氧化物中间体, 中间体分解产生醛或酮, TiO2的表面钛(Ⅵ)位点的再生完成光催化氧化循环.最后, 未反应的胺对醛或酮的亲核攻击产生相应的亚胺.
下载:
导出CSV
 | ||||
| Entry | Ar | R | Conv.a mol% | Selectivityb mol% |
| 1 | C6H5 | H | 91 | 92 |
| 2 | p-Me-C6H4 | H | 78 | 94 |
| 3 | p-t-Bu-C6H4 | H | 90 | 94 |
| 4 | p-MeO-C6H4 | H | 95 | 93 |
| 5 | p-F-C6H4 | H | 84 | 88 |
| 6 | p-Cl-C6H4 | H | 83 | 90 |
| 7 | o, p-Cl2-C6H3 | H | 92 | 91 |
| 8 | 2-Pyridyl | H | 77 | 86 |
| 9 | 2-Furyl | H | 68 | 38 |
| 10 | 2-Thienyl | H | 77 | 86 |
| 11 | C6H5 | Me | 44 | 53 |
| 12 | p-Br-C6H4 | Me | 49 | 49 |
| a Determined by GC analysis; b Selectivity=yield/conversion. | ||||
Chen等[49]通过溶剂热法合成了单晶金红石TiO2纳米棒, 在可见光照射下催化胺-胺氧化偶联合成亚胺.由于该催化剂具有3D高度有序的层次结构, 在反应中表现出了很高的光催化活性, 具有很大的应用潜力.同年, Li等[50]发现混合TiO2(B)/锐钛矿TiO2作为催化剂也可实现胺-胺氧化偶联合成亚胺.实验结果表明, 在可见光照射下, 与纯TiO2(B)和锐钛型TiO2相比, 混合TiO2(B)/锐钛矿TiO2催化剂显示出更高的光催化活性.
Zhu等[51]用制备了的锐钛型TiO2硅酸盐纳米晶体复合材料在可见光(λ>460 nm)的照射下, 催化胺-胺氧化偶联合成亚胺.作者考察了不同溶剂对催化活性的影响, 结果显示, 该催化剂可在乙腈中催化胺的氧化偶联, 以高转化率和高选择性转化为亚胺.该催化体系适用于各种苄胺及含N和S的杂环胺, 并表现出优异的催化活性, 但不适用于脂肪胺.
除了上述的二氧化钛以外, 其他类型光催化剂也被用于亚胺合成. Tanaka等[52]报道了Nb2O5作为多相光催化剂, 在常温常压下催化各种苄胺在苯中选择性氧化合成相应的亚胺.结果显示, 含给电子基团(Me和OMe)的取代苄胺比含吸电子基团(Cl和CF3)的取代苄胺更容易发生反应, 同时该催化剂也成功催化正丁胺自身偶联合成相应的亚胺, 但选择性(61%)低于苄基同系物(94%~99%)且反应时间较长.
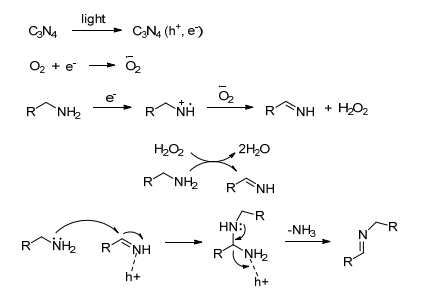
Wang等[53]报道了介孔石墨碳氮化合物(mpg-C3N4)光催化剂在可见光照射、0.5 MPa氧气下催化胺选择性氧化合成亚胺, 反应时间较短(2~3 h).该催化系统不仅适用于芳香胺, 而且对吡啶-2-基甲胺和噻吩-2-基甲胺也可以以高转化率和高选择性实现转化(95%~99%), 并且催化剂重复使用三次而不丧失催化活性.作者根据实验结果提出了合理的反应机理, 如Scheme 13所示, mpg-C3N4在光照下溢出电子产生电子空位, 氧气得电子形成超氧自由基阴离子, 然后, 胺失电子后与超氧自由基阴离子反应生成亚胺中间体, 生成的H2O2与另一分子胺反应也生成亚胺中间体, 所有的亚胺中间体与未反应的胺反应脱氨气得到目标产物亚胺.
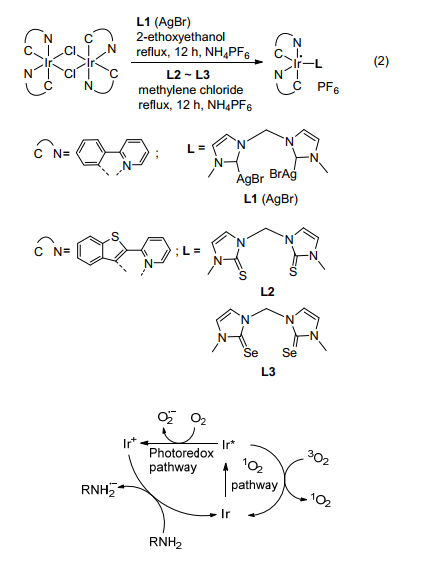
Son等[54]制备了吩噻嗪基有机染料, 用于可见光驱动下光催化有机转化.在常温常压蓝色LED可见光照射下, 0.5 mol% 3, 7-二取代吩噻嗪即可催化苄胺偶联反应, 以满意的产率得到亚胺.后来他们[55]又制备了如Eq. 2所示的含有双(N-杂环卡宾)(L1), 双(咪唑啉硫酮) (L2)和双(咪唑啉硒酮)(L3)的三种新型铱(Ⅲ)鳌合物催化胺-胺氧化偶联生成亚胺.当催化剂为带有L3的双(2-(2'-苯并噻吩基)吡啶)铱(Ⅲ)鳌合物(Ir-6)时, 在最大发射波长为460 nm处的蓝色LED照射下, 0.25 mol% Ir-6室温下催化苄胺反应5 h转化率可达到94%.该结果表明L3是调节铱(Ⅲ)鳌合物的光学性质的有效配体, 该催化剂表现出最高的催化活性.作者提出了光氧化和单线态氧两种反应路径如Scheme 14所示, 在光氧化路径中, 激发的铱络合物将电子转移到氧上形成超氧自由基阴离子, 胺在氧化铱络合物作用下得电子形成自由基阳离子, 然后, 超氧自由基阴离子和自由基阳离子反应形成亚胺.在单线态氧路径中, 三线态的激发铱络合物将能量转移到三线态氧分子上形成活性单线态氧, 然后, 诱导胺的氧化偶联合成亚胺.
Berlicka等[56]研究发现在蓝色发光二极管(LED)照射下, 2, 7, 12, 17-四丙基卟啉(H2TPrPc)为催化剂, 能有效地催化伯胺和仲胺氧化生成相应的亚胺, 但该催化体系仅适用于带有吸电子基团的芳香胺和氮原子上无烷基支链的仲胺(表 9).
下载:
导出CSV
 | |||
| Entry | Ar | R | Yielda/% |
| 1 | C6H5 | H | 42 |
| 2 | p-MeO-C6H4 | 1-Ethyl | 59 |
| 3 | C6H5 | 2-Ethyl | 99 |
| 4 | C6H5 | C6H5CH2 | 99 |
| a Determined by GC analysis. | |||
Tada等[57]研究了由金纳米颗粒和金属氧化物载体组成的等离子体光催化剂, 用于胺的选择性氧化为亚胺.考察了各种金属氧化物的使用, 其中Au/金红石TiO2在苄胺的选择性氧化中表现出很高的可见光催化活性, 可在无溶剂、室温条件下以高选择性合成相应的亚胺.
由此不难看出, 光催化具有高效、反应条件温和等优点, 且对杂环胺显示出优异的催化性能.然而, 目前使用的光催化剂制备相对复杂, 活性还需进一步提高, 且易失活.对于光催化剂可在合成方法上, 通过金属沉积、离子掺杂等方式与其他材料进行复合等延缓电子-空穴的复合, 从而提高催化活性和稳定性.
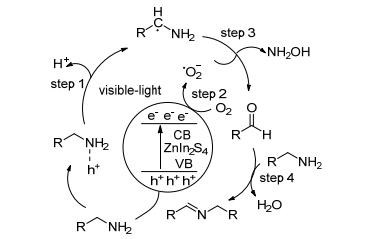
Li等[58]首次使用快速微波辅助溶剂热法制备的ZnIn2S4纳米球催化胺的选择性氧化反应.结果显示, 以空气作为氧化剂, 在可见光照射下, 对于各种取代基苄胺均以中等至高的转化率和选择性获得相应的亚胺.底物扩展实验结果表明, 催化剂的催化活性几乎不受电子效应的影响(给电子基和吸电子基), 但受空间位阻的影响较大.同时该催化剂还适用于杂环胺、仲胺和脂肪胺.
作者提出了如Scheme 15所示的一种可能的反应机理.用可见光照射ZnIn2S4时, 产生电子和空穴, 胺可以与光生空穴反应, 去质子化产生中性碳中心基团(步骤1), 然后分子氧可捕获光生电子以产生超氧自由基阴离子(步骤2), 光生碳基自由基和O2之间的相互作用将形成醛(步骤3), 该机理与在UV照射下TiO2光催化形成醛的机理类似, 而未反应的胺与醛亲核作用脱水产生相应的亚胺(步骤4).
仿生催化是集酶催化与化学催化的优点于一体, 可使常规化学反应在环境友好的反应条件下进行, 并像酶催化一样表现出高效率和高选择性的催化技术.由于仿生催化剂具有良好的稳定性, 对条件变化适应能力强等, 因而受到科学家们的广泛关注.
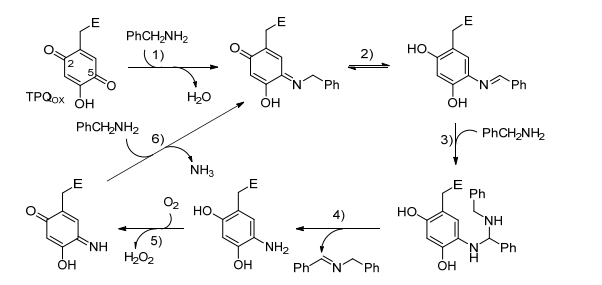
Klinman等[59, 60]对铜胺氧化酶(CuAOs)的生物活性的研究发现, 其对伯胺具有极好的底物特异性, 因此作者利用苯醌有机辅助因子(TPQox)和铜离子的协同作用, 在CuAOs和分子氧作用下催化胺-胺氧化偶联合成亚胺, 显示出良好的催化活性. Sayre等[61]也研究铜胺氧化酶催化胺-胺氧化偶联反应的催化活性, 同时通过金属酶的作用模拟CuAO的活性位点探究其反应机理, 如Scheme 16所示, 2, 4, 5-三羟基苯丙氨酸醌(TPQox)的4-羟基基团在使苯醌环免于胺环化和苄胺直接亲核加成到C-5位上起到重要作用, 在有机介质中, 生成的席夫碱中间体可以直接与苄胺加成, 得到偶联的亚胺和氨基苯酚产物, 而不是在水中形成醛, 氨基苯酚的有氧氧化生成亚氨基苯醌, 其与苄胺继续反应并释放氨气得到席夫碱中间体.除此之外, 作者[62]还研究了赖氨酰氧化酶辅因子(LTQ)在乙腈中催化苄胺氧化偶联合成亚胺, 与TPQ催化体系相比, LTQ可以有效避免副产物的生成.
最近, Wendlandt等[63]受到生物化学模型的启发, 利用4-叔丁基-2-羟基苯醌(TBHBQ)作为有机催化剂催化胺-胺氧化偶联合成亚胺, 发现在乙腈中, 101 kPa氧气压和室温条件下, 各种苄胺及杂环糠胺都能发生氧化偶联得到相应的亚胺(表 10).除了三氟甲基苄胺和间氯苄胺的反应速率较慢, 需48 h外, 其他苄胺均可在20 h内得到较高产率的亚胺, 这表明亚胺的产率不仅仅取决于苯环上的取代基, 还取决于胺的空间位阻.虽然该催化剂对脂肪胺催化活性不高, 但其对苄基伯胺的独特选择性, 允许胺以高转化率选择性合成交叉亚胺.该催化剂在催化具有醇或胺基团的伯胺与苄胺进行交叉氧化反应时, 醇或胺基团不会被氧化, 这一点要优于过渡金属催化剂.作者提出了与Largeron团队报道的类似的反应机理, 主要通过底物与邻亚氨基醌缩合生成亚氨基苯醌, 接着与底物反应生成缩醛胺, 缩醛胺还原分解生成产品亚胺.
下载:
导出CSV
 | ||
| Entry | Ar | Yielda/% |
| 1 | C6H5 | 87 |
| 2 | p-H2N-C6H4 | 76 |
| 3 | p-MeO-C6H4 | 93 |
| 4 | p-Cl-C6H4 | 90 |
| 5 | p-F-C6H4 | 91 |
| 6 | p-CF3-C6H4 | 78b |
| 7 | m-Cl-C6H4 | 72b |
| 8 | m-I-C6H4 | 70 |
| 9 | o-Me-C6H4 | 86 |
| 10 | o-MeO-C6H4 | 73 |
| 11 | Naphthyl | 81 |
| 12 | 2-Furyl | 80 |
| a Yields of imines determined by 1H NMR spectroscopy versus internal standard; b Reaction time was 48 h. | ||
Kobayashi等[64]首次报道了一种与有机辅助因子起协同作用的金属多相纳米催化剂用于胺-胺氧化偶联合成亚胺.这种多相纳米催化剂因与CuAOs催化系统有一些相似的特性, 这种协同作用可以克服反应途径上的能量障碍, 而单独使用时没有催化作用.作者用负载在苯乙烯共聚物载体的Pt/Ir双金属多相纳米催化剂和4-叔丁基-邻苯醌(TBBQ)组成的金属酶协同催化体系催化胺氧化偶联以高转化率得到亚胺.同时, 除了含吸电子基的胺以外, 该催化剂均能在室温和101 kPa氧气下催化其他胺以良好的转化率得到亚胺(表 11).而与CuAOs催化体系相比, 该催化体系最大的优点在于仲胺也可以获得较好的产率亚胺, 且催化剂易于回收, 可重复使用5次催化活性无明显下降.
下载:
导出CSV
 | |||
| Entry | Ar | GC yielda/% | Isolated yield/% |
| 1 | Ph | 77 | 74 |
| 2 | p-MeC6H4 | 80 | 75 |
| 3 | p-MeOC6H4 | 80 | 73 |
| 4 | p-ClC6H4 | 60 | 52 |
| 5 | p-CF3C6H4 | 57 | 55 |
| a Determined by GC analysis with anisole as the internal standard. | |||
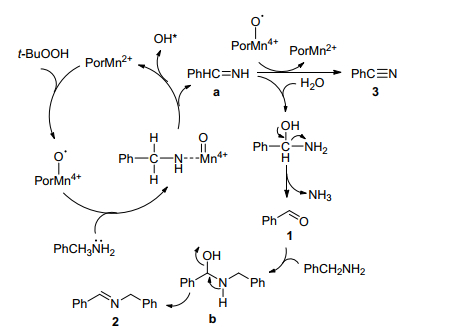
Ji等[65]报道了用内消旋氯化四苯基卟啉锰(Ⅲ)为催化剂(MnTPPCl, 0.03 mol%), 叔丁基过氧化氢(tBuOOH)为氧化剂, 催化胺-胺氧化偶联合成亚胺.研究发现在室温下, 以甲苯为溶剂催化苄胺氧化偶联合成亚胺的反应中, 用锰卟啉作为催化剂比铜、铁、钴和钌卟啉作为催化剂具有更优的催化活性.该催化系统不仅可以催化各种苄胺以高转化率和高选择性获得亚胺产物, 同时也适用于环己胺、正己胺等脂肪胺(表 12).除此之外, 锰-卟啉络合物在其他有机溶剂(二氯甲烷[66]和乙腈[67])中, 以叔丁基过氧化氢为氧化剂催化该反应也是有效的.随后, 他们[68]又提出了更环保的催化方案:用水溶性锰卟啉(MnTE4PyP, 0.05 mol%)和作为氧化剂的t-BuOOH催化苄胺的氧化偶联反应在60 ℃水中可顺利进行, 且在相同的反应条件下, 不同取代的苄胺也能转化为产率高达76%的亚胺.同时, 作者根据实验结果提出了MnTE4PyP催化剂催化胺氧化偶联合成亚胺的反应机理.如Scheme 17所示, 催化剂在t-BuOOH的作用下与胺反应生成席夫碱亚胺中间体a, 然后, 中间体a水解释放一分子氨气生成苯甲醛1, 苯甲醛1与另一分子胺反应生成中间体b, 中间体b脱水得到最终产品亚胺2.另外, 在高价锰中间体存在下, 席夫碱亚胺中间体a脱氢产生苄腈3.
下载:
导出CSV
 | ||||
| Entry | R | Time/h | Conversiona/% | Yielda/% |
| 1 | C6H5 | 0.25 | >99 | 91 |
| 2 | p-MeC6H4 | 1.0 | >99 | 88 |
| 3 | p-MeOC6H4 | 1.0 | >99 | 73 |
| 4 | p-FC6H4 | 0.5 | >99 | 96 |
| 5 | p-ClC6H4 | 0.5 | 97 | 95 |
| 6 | o, p-Cl2C6H3 | 3.0 | 70 | 57 |
| 7 | c-Hexyl | 2.0 | >99 | 91 |
| 8 | Pentyl | 1.0 | >99 | 96 |
| a Based on amines, determined by GC and GC-MS. | ||||
相对于以上类型的催化体系, 仿生催化反应条件温和, 催化效果好, 除了能够催件苄胺和杂环胺偶联反应外, 还对脂肪胺和仲胺具有优异的催化活性.对于仿生催化, 如何简化催化剂制备程序、提高催化剂对反应条件的适应性, 以及如何重复利用催化剂是下一步研究的重点.
除了上述金属催化、光催化、仿生催化外, 还有一些其它类型催化剂也被用于胺-胺氧化偶联合成亚胺.在20世纪90年代, Hirao等[69]首次以聚苯胺为催化剂, 以(NH4)2S2O8为氧化剂催化胺-胺氧化偶联合成亚胺, 显示出良好的催化活性, 但均未对底物扩展做进一步的研究.
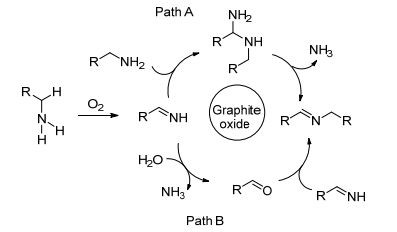
Huang等[70]报道了一种高效、低成本和可循环使用的氧化石墨(GO)多相催化剂.该催化剂具有较大的比表面积, 可在100 ℃无需任何溶剂的条件下催化胺以较高的转化率和选择性合成亚胺.同时, 该催化体系也适用于制备不对称亚胺和环状亚胺.根据实验结果并参考相关文献, 作者提出了如Scheme 18所示的反应机理.首先该反应过程经过两种不同的氧化脱氢途径, 两路径都涉及到RCH=NH中间体的形成.在路径A中, 中间体RCH=NH与第二分子伯胺作用生成缩醛胺, 缩醛胺进一步释放出NH3, 获得偶联亚胺产物RCH=NCH2R.在路径B中, 最初的中间体RCH=NH与痕量的H2O反应得到醛RCH=O, 醛随后与第二分子的伯胺反应获得最终的亚胺产物.
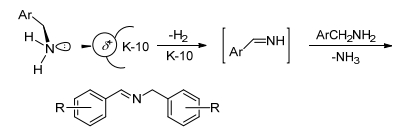
Landge等[71]制备了一种新型的多相催化剂固体酸K-10蒙脱石用于催化胺-胺氧化偶联合成亚胺.在该催化剂催化下, 各种苄胺(吸电子基和给电子基)都可在1~3 h内高选择性地转化为亚胺.同时对各种苄胺和苯胺之间的交叉氧化偶联合成不对称的交叉亚胺, 同样有催化活性.但该催化体系反应条件较为苛刻, 需要微波辅助, 且在150 ℃的高温下进行.根据已报道的文献和实验结果, 作者提出了如Scheme 19所示的可能的反应机理, K-10的酸性中心与苄胺的氮的孤对电子配位, 生成亚胺中间体, 然后此中间体与另一分子苄胺反应释放出氨气得到亚胺产物.
Fu等[72]提出了用偶氮二异丁腈(AIBN)作为催化剂, 以氧气为氧化剂催化胺-胺氧化偶联合成亚胺.实验结果显示, 该催化剂对底物适用范围较广, 对各种苄胺及含N、O、S的杂环胺具有较好的催化效果, 且几乎不受空间位阻和电子效应的影响, 但脂肪胺的转化率和选择性较低(表 13).
下载:
导出CSV
 | |||
| Entry | R | Conversiona/% | Yielda/% |
| 1 | C6H5 | 94 | 89 |
| 2 | p-Me-C6H4 | 99 | 87 |
| 3 | p-MeO-C6H4 | 97 | 76 |
| 4 | o-Me-C6H4 | 98 | 88 |
| 5 | p-Cl-C6H4 | 99 | 85 |
| 6 | p-F-C6H4 | 97 | 84 |
| 7 | 2-Thienyl | 93 | 83 |
| 8 | 2-Furyl | 90 | 46 |
| 9 | 2-Pyridyl | 92 | 86 |
| 10 | Cyclohexyl | 32 | 7 |
| a Based on amines, determined by GC. | |||
胺-胺氧化偶联是一种原子利用率高、环境友好的亚胺合成策略.通过对新型催化体系的不断探索, 反应条件趋于温和, 底物适用范围不断扩大, 同时避免了使用特定氧化剂, 有毒助剂及溶剂, 简化了制备程序.然而, 目前已发展的催化剂中, 贵金属尽管显示出优异的催化性能, 但对某些有空间位阻的胺-胺氧化偶联反应催化效果并不令人满意, 且贵金属配合物难于制备、价格昂贵.一些廉价的非贵金属催化剂的发展有效地降低了成本, 但存在着反应条件苛刻, 需要额外添加氧化试剂、碱性助剂等缺点.其他催化方法, 如光催化、仿生催化等也可在非常温和的条件下催化胺自身氧化偶联, 高转化率和高选择性获得亚胺, 但对胺-胺交叉氧化偶联反应的催化活性较低, 且催化剂制备困难.同时, 对于胺-胺氧化偶联的催化机理, 大多是基于实验结果的推测, 缺少直接的证据; 而且由于所使用催化剂类型不同, 催化机制各异, 还没有统一的定论.基于以上问题, 对胺-胺氧化偶联合成亚胺, 需进一步发展新型的、廉价的和高活性的催化体系, 降低反应条件, 简化反应步骤, 扩大底物适用范围, 特别是发展适用于胺-胺交叉氧化偶联反应高活性、高选择性的催化剂, 还要重点研究胺-胺交叉氧化偶联反应的催化机理, 理清反应路径, 从而为探索新型高效的催化剂提供理论指导.
Kobayashi, S.; Mori, Y.; Fossey, J. -S.; Salter, M. -M. Chem. Rev. 2011,111, 2626. doi: 10.1021/cr100204f
Adams, J. -P. J. Chem. Soc. ,Perkin Trans. 1 2000, 125.
Ashok, M.; Holla, B. -S.; Poojary, B. Eur. J. Med. Chem. 2007,46, 1095.
Young, J. -N.; Chang, T. -C.; Tsai, S. -C.; Yang, L.; Yu, S. -C. J. Catal. 2010,272, 253. doi: 10.1016/j.jcat.2010.04.005
Santos, L. -L.; Serna, P.; Corma, A. Chem. -Eur. J. 2009,15, 8196. doi: 10.1002/chem.v15:33
Zanardi, A.; Mata, J. -A.; Peris, E. Chem. -Eur. J. 2010,16, 10502. doi: 10.1002/chem.201000801
Maggi, A.; Madsen, R. Organometallics 2012,31, 451. doi: 10.1021/om201095m
Kwon, M. -S.; Kim, S.; Park, S.; BoscoO, W.; Chidrala, R. -K.; Park, J. J. Org. Chem. 2009,74, 2877. doi: 10.1021/jo8026609
Jiang, G.; Chen, J.; Huang, J. -S.; Che, C. -M. Org. Lett. 2009,11, 4568. doi: 10.1021/ol9018166
Bailey, A. -J.; James, B. -R. Chem. Commun. 1996,0, 2343.
Prades, A.; Peris, E.; Albrecht, M. Organometallics 2011,30, 1162. doi: 10.1021/om101145y
Doctorovich, F.; Granara, M.; Salvo, F. -D. TransitionMet. Chem. 2001,26, 505. doi: 10.1023/A:1011033312510
He, L. -P.; Chen, T.; Gong, D.; Lai, Z.; Huang, K. -W. Organometallics 2012,31, 5208. doi: 10.1021/om300422v
Zhang, Y. -C.; Lu, F.; Huang, R.; Zhang, H. -Y.; Zhao, J. -Q. Catal. Commun. 2016,81, 10. doi: 10.1016/j.catcom.2016.03.019
Zhu, B.; Angelici, R. -J. Chem. Commun. 2007,38, 2157.
Zhu, B.; Lazar, M.; Trewyn, B. -G.; Angelici, R. -J. J. Catal. 2008,260, 1. doi: 10.1016/j.jcat.2008.08.012
Grirrane, A.; Corma, A.; Garcia, H. J. Catal. 2009,264, 138. doi: 10.1016/j.jcat.2009.03.015
Pérez, Y.; Aprile, C.; Corma, A.; Garcia, H. Catal Lett. 2010,134, 204. doi: 10.1007/s10562-009-0243-1
Ishida, T.; Kawakita, N.; Akita, T.; Haruta, M. Gold Bull. 2009,42, 267. doi: 10.1007/BF03214948
Srimani, D.; Feller, M.; Ben-David, Y.; Milstein, D. Chem. Commun. 2012,48, 11853. doi: 10.1039/c2cc36639h
Lu, S. -L.; Wang, J. Q.; Cao, X. Q.; Li, X. M.; Gu, H. W. Chem. Commun. 2014,50, 3512. doi: 10.1039/C3CC48596J
Patil, R. -D.; Adimurthy, S. Adv. Synth. Catal. 2011,353, 1695. doi: 10.1002/adsc.201100100
Patil, R. -D, Adimurthy, S. RSC Adv. 2012,2, 5119. doi: 10.1039/c2ra20339a
Patil, R. -D, Adimurthy, S. Asian J. Org. Chem. 2013,2, 726. doi: 10.1002/ajoc.v2.9
Hu, Z. -Z.; Kerton, F. -M. Org. Biomol. Chem. 2012,10, 1618. doi: 10.1039/c2ob06670j
Huang, B.; Tian, H.; Lin, S.; Xie, M.; Yu, X.; Xu, Q. Tetrahedron Lett. 2013,54, 2861. doi: 10.1016/j.tetlet.2013.03.098
Largeron, M.; Fleury, M. -B. Chem. -Eur. J. 2015,21, 1.
Wang, J.; Lu, S.; Cao, X.; Gu, H. Chem. Commun. 2014,50, 5637. doi: 10.1039/c4cc01389a
Al-Hmoud, L.; Jones, C. -W. J. Catal. 2013,301, 116. doi: 10.1016/j.jcat.2013.01.027
Marui, K.; Nomoto, A.; Ueshima, M.; Ogawa, A. Tetrahedron Lett. 2015,56, 1200. doi: 10.1016/j.tetlet.2015.01.090
Chu, G. -B.; Li, C. -B. Org. Biomol. Chem. 2010,8, 4716. doi: 10.1039/c0ob00043d
Wang, L. -Y.; Chen, B.; Ren, L. -H.; Zhang, H. -Y.; Lü, Y.; Gao, S. Chin. J. Catal. 2015,36, 19. doi: 10.1016/S1872-2067(14)60196-0
Kodama, S.; Yoshida, J.; Nomoto, A.; Ueta, Y.; Yano, S.; Ueshima, M.; Ogawa, A. Tetrahedron Lett. 2010,41, 2450. http://www.sciencedirect.com/science/article/pii/S0040403910003758
Neumann, R.; Levin, M. -J. J. Org. Chem. 1991,56, 5707. doi: 10.1021/jo00019a047
Nakayama, K.; Hamamoto, M.; Nishiyama, Y.; Ishii, Y. Chem. Lett. 1993,22, 1699. doi: 10.1246/cl.1993.1699
Reddy, J. -S.; Sayari, A. Catal. Lett. 1994,28, 263. doi: 10.1007/BF00806055
Maruyama, K.; Kusukawa, T.; Highchi, Y.; Nishinaga, A. Chem. Lett. 1991,7, 1093. http://www.researchgate.net/publication/250538541_ChemInform_Abstract_Co(salen)-Catalyzed_tertButyl_Hydroperoxide_Oxidation_of_tert-_Butylphenols_Bearing_an_Unsaturated_Side_Chain
Zhao, S.; Liu, C.; Guo, Y.; Xiao, J. -C.; Chen, Q. -Y. J. Org. Chem. 2014,79, 8926. doi: 10.1021/jo5017212
Zhang, C. -H.; Zhao, P. -S.; Zhang, Z. -L.; Zhang, J. -W.; Yang, P.; Gao, P.; Gao, J.; Liu, D. RSC Adv. 2017,7, 47366. doi: 10.1039/C7RA09516C
Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. ChemCatChem 2010,2, 1438. doi: 10.1002/cctc.201000175
Zhang, E.; Tian, H. -W.; Xu, S. -D.; Yu, X. -C.; Xu, Q. Org. Lett. 2013,15, 2704. doi: 10.1021/ol4010118
Gopalaiah, K.; Saini, A. Catal. Lett. 2016,146, 1648. doi: 10.1007/s10562-016-1789-3
Kim, S. -S.; Thakur, S. -S.; Song, J. -Y.; Lee, K. -H. Bull. Korean Chem. Soc. 2005,26, 499. doi: 10.5012/bkcs.2005.26.3.499
Choudary, B. -M.; Narender, N.; Bhuma, V. Synth. Commun. 1996,26, 631. doi: 10.1080/00397919608086735
Ohtani, B.; Osaki, H.; Nishimoto, S. -I.; Kagia, T. Chem. Lett. 1985,21, 1075.
Lang, X.; Ji, H.; Chen, C.; Ma, W.; Zhao, J. Angew. Chem. ,Int. End. 2011,50, 3934. doi: 10.1002/anie.201007056
Lang, X.; Ma, W.; Zhao, Y.; Chen, C.; Ji, H.; Zhao, J. Chem. -Eur. J. 2012,18, 2624. doi: 10.1002/chem.201102779
Li, N.; Lang, X. -J.; Ma, W. -H.; Ji, H. -W.; Chen, C. -C.; Zhao, J. -C. Chem. Commun. 2013,49, 5034. doi: 10.1039/c3cc41407h
Bu, J.; Fang, J.; Leow, W. -R.; Zheng, K. -H.; Chen, X. -D. RSC Adv. 2015,5, 103895. doi: 10.1039/C5RA23428J
Dai, J.; Yang, J.; Wang, X. -H.; Zhang, L.; Li, Y. Appl. Surf. Sci. 2015,349, 343. doi: 10.1016/j.apsusc.2015.04.232
Zavahir, S.; Zhu, H. Molecules 2015,20, 1941. doi: 10.3390/molecules20021941
Furukawa, S.; Ohno, Y.; Shishido, T.; Teramura, K.; Tanaka, T. ACS Catal. 2011, 1, 1150. doi: 10.1021/cs200318n
Su, F.; Mathew, S. -C.; Möhlmann, L.; Antonietti, M.; Wang, X.; Blechert, S. Angew. Chem. ,Int. Ed. 2011,50, 657. doi: 10.1002/anie.v50.3
Park, J. -H.; Ko, K. -C.; Kim, E.; Park, N.; Ko, J. -H.; Ryu, D. -H.; Ahn, T. -K.; Lee, J. -Y.; Son, S. -U. Org. Lett. 2012,14, 5502. doi: 10.1021/ol302584y
Jin, J.; Shin, H. -W.; Park, J. -H.; Park, J. -H.; Kim, E.; Ahn, T. -K.; Ryu, D. -H.; Son, S. -U. Organometallics 2013,32, 3954. doi: 10.1021/om4004412
Berlicka, A.; König, B. Photochem. Photobiol. Sci. 2010,9, 1359. doi: 10.1039/c0pp00192a
Naya, S. -I.; Kimura, K.; Tada, H. ACS Catal. 2013,3, 10. doi: 10.1021/cs300682d
Ye, L.; Li, Z. -H. ChemCatChem 2014,6, 2540. doi: 10.1002/cctc.201402360
Mure, M.; Klinman, J. -P. J. Am. Chem. Soc. 1995,117, 8698. doi: 10.1021/ja00139a002
Mure, M.; Klinman, J. -P. J. Am. Chem. Soc. 1995,117, 8707. doi: 10.1021/ja00139a003
Lee, Y.; Sayre, L. -M. J. Am. Chem. Soc. 1995,117, 11823. doi: 10.1021/ja00153a001
Ling, K. -Q.; Kim, J.; Sayre, L. -M. J. Am. Chem. Soc. 2001,123, 9606. doi: 10.1021/ja011141j
Wendlandt, A. -E.; Stahl, S. -S. Org. Lett. 2012,14, 2850. doi: 10.1021/ol301095j
Yuan, H.; Yoo, W. -J.; Miyamura, H.; Kobayashi, S. J. Am. Chem. Soc. 2012,134, 13970. doi: 10.1021/ja306934b
Yuan, Q. -L.; Zhou, X. -T.; Ji, H. -B. Catal. Commun. 2010,12, 202. doi: 10.1016/j.catcom.2010.09.009
Tollari, S.; Fumagalli, A.; Porta, F. Inorg. Chim. Acta 1997,247, 71. http://med.wanfangdata.com.cn/Paper/Detail?id=PeriodicalPaper_JJ025202321
Kim, S. -S.; Thakur, S. -S.; Bull, K. Chem. Soc. 2005,26, 1600.
Zhou, X. -T.; Ren, Q. -G.; Ji, H. -B. Tetrahedron Lett. 2012,53, 3369. doi: 10.1016/j.tetlet.2012.04.096
Higuchi, M.; Ikeda, I.; Hirao, T. J. Org. Chem. 1997,62, 1072. doi: 10.1021/jo9617575
Huang, H.; Huang, J.; Liu, Y. -M.; He, H. -Y.; Cao, Y.; Fan, K. -N. Green Chem. 2012,14, 930. doi: 10.1039/c2gc16681j
Landge, S. -M.; Atanassova, V.; Thimmaiah, M.; Török, B. Tetrahedron Lett. 2007,48, 5161. doi: 10.1016/j.tetlet.2007.05.051
Liu, L.; Wang, Z.; Fu, X.; Yan, C. -H. Org. Lett. 2012,14, 5692. doi: 10.1021/ol302708r

图式 1 钌催化剂1催化的苄胺自偶联生成亚胺反应的机理
Scheme 1 Mechanism for the homocoupling of primary amine to imine catalyzed by ruthenium catalysts

图式 2 金催化有氧氧化伯胺合成亚胺的反应途径
Scheme 2 Proposed reaction pathways for the gold-catalyzed aerobic oxidation of primary amines to imines

图式 3 钌与金催化氧化胺的机理途径
Scheme 3 Possible mechanistic pathways for ruthenium- versus gold-catalyzed oxidation of amines

图式 4 铜催化伯胺有氧氧化生成亚胺的可能机制
Scheme 4 Possible mechanism for the copper-catalysed aerobic oxidation of primary amines to give imines

图式 5 Cu(OAc)2/1ox催化伯胺交叉偶联反应的可能两步机制
Scheme 5 Proposed overall two-step mechanism for the Cu(OAc)2/1ox-mediated cross-coupling of primary amines

图式 6 在H2O2作用下CuSO4催化伯胺氧化的可能催化机理
Scheme 6 A possible catalytic mechanism for the CuSO4-cata- lyzed oxidation of primary amines with H2O2

图式 8 苄胺氧化偶联合成亚胺的机理
Scheme 8 Proposed mechanism of the oxidative coupling of benzylamines to imines

图式 9 Co-N-C/SiO2催化胺有氧氧化成亚胺的反应机理
Scheme 9 Proposed mechanism for the aerobic oxidation of amines to imines over Co-N-C/SiO2

图式 11 铁催化伯胺氧化缩合合成亚胺的反应机理
Scheme 11 Proposed reaction mechanism for the ironcatalyzed oxidative condensation of primary amines to imines

图式 12 可见光照射下胺在TiO2表面上氧化产生亚胺的机理
Scheme 12 Proposed mechanism for the oxidation of amines to give imines on a TiO2 surface under visible-light irradiation

图式 13 胺氧化偶联合成亚胺的可能机理
Scheme 13 Proposed mechanism for aerobic oxidative coupling of amines

图式 14 光诱导的苄胺氧化偶联制亚胺的两种主要机理
Scheme 14 Two main mechanisms for the photoinduced oxidative coupling of benzylamine to imine

图式 15 在可见光照射下ZnIn2S4作用下胺的选择性有氧氧化的机理(CB=导带, VB=价带)
Scheme 15 Proposed mechanism for the selective aerobic oxidation of amines to imines over ZnIn2S4 under visible-light irradiation (CB=Conduction band, VB=valence band)

图式 16 由CuAOs苯醌模型催化伯胺有氧氧化的离子转氨酶机理
Scheme 16 Proposed ionic transamination mechanism of the aerobic oxidation of primary amines catalyzed by CuAOs quinone models

图式 17 MnTE4PyP在水中催化胺氧化偶联制亚胺的合理机理
Scheme 17 A plausible mechanism for the oxidative coupling of amine to imine catalyzed by MnTE4PyP in water

图式 18 GO催化伯胺有氧氧化制亚胺的可能机理
Scheme 18 Possible mechanisms for the GO-catalyzed aerobic oxidation of primary amines to imines

图式 19 K-10蒙脱石催化苄胺的自身氧化偶联的可能机理
Scheme 19 Possible mechanisms for the oxidative self-coupling of benzylamines in the presence of K-10 montmorillonite
表 1 钌配合物催化脱氢形成亚胺
Table 1. Dehydrogenative imine formation catalyzed by ruthenium complexes
| ||
| Entry | R | Yielda/% |
| 1 | Ph | 93 |
| 2 | p-Me-C6H4 | 65 |
| 3 | m-Me-C6H4 | 80 |
| 4 | p-MeO-C6H4 | 83 |
| a 1H NMR yields. | ||
 下载: 导出CSV
下载: 导出CSV
表 2 金催化伯胺有氧氧化合成亚胺
Table 2. Gold-catalyzed aerobic oxidation of primary amines to imines
| |||
| Entry | R | Yielda/% | |
| Au powder cat. | Au/Al2O3 cat. | ||
| 1 | Ph | 56 | 92 |
| 2 | p-Me-C6H4 | 61 | 96 |
| 3 | p-Cl-C6H4 | 7 | 59 |
| 4 | C5H11 | 5 | 29 |
| a Gold powder (1 g); b 5% Au/Al2O3. | |||
下载: 导出CSV
表 3 铜催化剂催化苄胺有氧氧化合成亚胺
Table 3. Copper-catalyzed aerobic oxidation of benzylic amines to imines
| |||
| Entry | R | Yielda/% | 2:3c |
| 1 | Ph | 88 | 93:7 |
| 2 | p-Me-C6H4 | 94 | 85:15 |
| 3 | o-Me-C6H4 | 84 | 80:20 |
| 4 | p-MeO-C6H4 | 92 | 78:22 |
| 5 | p-F-C6H4 | 84 | 86:14 |
| 6 | o-Cl-C6H4 | 89 | 95:5 |
| 7 | m-Cl-C6H4 | 85 | 89:11 |
| 8 | p-Cl-C6H4 | 90 | 99:1 |
| 9 | o-F-C6H4 | 52b | 100:0 |
| 10 | m-F-C6H4 | 54b | 100:0 |
| 11 | C5H11 | 50 | — |
| a Yields of isolated imines. b Rest of starting benzylamine was recovered. c Selectivity determined by 1H NMR | |||
下载: 导出CSV
表 4 不对称亚胺的合成
Table 4. Synthesis of unsymmetrical imines
| |||
| Entry | R2 | Yield/% | unsym./sym. |
| 1 | Ph | 78 | 23:77 |
| 2 | p-MeO-C6H4 | 86 | 86:14 |
| 3 | p-Me-C6H4 | 82 | 17:83 |
| 4 | p-Cl-C6H4 | 93 | 33:67 |
| 5 | m-NO2-C6H4 | 97 | 0:100 |
| 6 | o-Me-C5H3N | 94 | 0:100 |
| 7 | C6H13 | 78 | 56:44 |
下载: 导出CSV
表 5 CuBr2/TEMPO催化苄胺与苯胺类氧化偶联
Table 5. CuBr2-TEMPO catalyzed oxidative coupling of benzylamines with anilines
| |||
| Entry | R1 | R2 | Conv.a/% |
| 1 | Ph | Ph | 48 |
| 2 | p-Me-C6H4 | Ph | 58 |
| 3 | p-MeO-C6H4 | Ph | 64 |
| 4 | o-Cl-C6H4 | Ph | 44 |
| 5 | Ph | o-Me-C6H4 | 66 |
| 6 | o-Cl-C6H4 | o-Me-C6H4 | 55 |
| 7 | Ph | p-MeO-C6H4 | 73 |
| 8 | p-Me-C6H4 | p-MeO-C6H4 | 78 |
| 9 | Ph | 2, 6-Me2-C6H3 | n.r. |
| a Conversion determined by GC using dodecane as an internal standard. n.r.=no reaction. | |||
下载: 导出CSV
表 6 在TBHP作用下Co2+(L)催化苄胺氧化成亚胺
Table 6. Co2+(L) catalyzed oxidation of benzylic amines to imines with TBHP
| Entry | Catalyst | Yielda/% |
| 1 | Co2+(L1) | 95.4 |
| 2 | Co2+(L2) | 93.8 |
| 3 | Co2+(L3) | 31.0 |
| 4 | Co2+(L4) | 16.6 |
| 5 | Co2+(L5) | 95.6b |
| 6 | Co2+(L6) | 0.90 |
| a Yields of isolated imines. b Reaction time: 1.5 h. | ||
下载: 导出CSV
表 7 Co-N-C/SiO2催化伯苄胺氧化成亚胺
Table 7. Co-N-C/SiO2 catalyzed oxidation of primary benzylic amines to imines
| |||
| Entry | R | Conv.a/mol% | Selectivityb/mol% |
| 1 | C6H5 | 99 | 100 |
| 2 | p-Me-C6H4 | 96 | 100 |
| 3 | p-Cl-C6H4 | 97 | 100 |
| 4 | m-Cl-C6H4 | 95 | 100 |
| 5 | o-Cl-C6H4 | 93 | 100 |
| 6 | n-Pentyl | 32 | 28 |
| 7 | Cyclohexyl | 13 | 62 |
| a Determined by GC analysis; b Selectivity=yield/conversion. | |||
下载: 导出CSV
表 8 TiO2光催化伯苄胺氧化成亚胺
Table 8. TiO2-photocatalyzed oxidation of primary benzylic amines to imines
| ||||
| Entry | Ar | R | Conv.a mol% | Selectivityb mol% |
| 1 | C6H5 | H | 91 | 92 |
| 2 | p-Me-C6H4 | H | 78 | 94 |
| 3 | p-t-Bu-C6H4 | H | 90 | 94 |
| 4 | p-MeO-C6H4 | H | 95 | 93 |
| 5 | p-F-C6H4 | H | 84 | 88 |
| 6 | p-Cl-C6H4 | H | 83 | 90 |
| 7 | o, p-Cl2-C6H3 | H | 92 | 91 |
| 8 | 2-Pyridyl | H | 77 | 86 |
| 9 | 2-Furyl | H | 68 | 38 |
| 10 | 2-Thienyl | H | 77 | 86 |
| 11 | C6H5 | Me | 44 | 53 |
| 12 | p-Br-C6H4 | Me | 49 | 49 |
| a Determined by GC analysis; b Selectivity=yield/conversion. | ||||
下载: 导出CSV
表 9 不同的二级胺氧化成亚胺
Table 9. Oxidation of different secondary amines to imines
| |||
| Entry | Ar | R | Yielda/% |
| 1 | C6H5 | H | 42 |
| 2 | p-MeO-C6H4 | 1-Ethyl | 59 |
| 3 | C6H5 | 2-Ethyl | 99 |
| 4 | C6H5 | C6H5CH2 | 99 |
| a Determined by GC analysis. | |||
下载: 导出CSV
表 10 苯醌调解伯苄胺有氧氧化
Table 10. Quinone-mediated aerobic oxidation of primary benzylic amines
| ||
| Entry | Ar | Yielda/% |
| 1 | C6H5 | 87 |
| 2 | p-H2N-C6H4 | 76 |
| 3 | p-MeO-C6H4 | 93 |
| 4 | p-Cl-C6H4 | 90 |
| 5 | p-F-C6H4 | 91 |
| 6 | p-CF3-C6H4 | 78b |
| 7 | m-Cl-C6H4 | 72b |
| 8 | m-I-C6H4 | 70 |
| 9 | o-Me-C6H4 | 86 |
| 10 | o-MeO-C6H4 | 73 |
| 11 | Naphthyl | 81 |
| 12 | 2-Furyl | 80 |
| a Yields of imines determined by 1H NMR spectroscopy versus internal standard; b Reaction time was 48 h. | ||
下载: 导出CSV
表 11 负载型Pt/Ir纳米簇/TBBQ催化伯苄胺有氧氧化制亚胺a
Table 11. Aerobic oxidation of primary amines to imines catalyzed by supported Pt/Ir nanoclusters/TBBQ
| |||
| Entry | Ar | GC yielda/% | Isolated yield/% |
| 1 | Ph | 77 | 74 |
| 2 | p-MeC6H4 | 80 | 75 |
| 3 | p-MeOC6H4 | 80 | 73 |
| 4 | p-ClC6H4 | 60 | 52 |
| 5 | p-CF3C6H4 | 57 | 55 |
| a Determined by GC analysis with anisole as the internal standard. | |||
下载: 导出CSV
表 12 锰/卟啉系统催化伯苄胺氧化制亚胺
Table 12. Oxidation of primary amines to imines catalyzed by the manganese/porphyrin system
| ||||
| Entry | R | Time/h | Conversiona/% | Yielda/% |
| 1 | C6H5 | 0.25 | >99 | 91 |
| 2 | p-MeC6H4 | 1.0 | >99 | 88 |
| 3 | p-MeOC6H4 | 1.0 | >99 | 73 |
| 4 | p-FC6H4 | 0.5 | >99 | 96 |
| 5 | p-ClC6H4 | 0.5 | 97 | 95 |
| 6 | o, p-Cl2C6H3 | 3.0 | 70 | 57 |
| 7 | c-Hexyl | 2.0 | >99 | 91 |
| 8 | Pentyl | 1.0 | >99 | 96 |
| a Based on amines, determined by GC and GC-MS. | ||||
下载: 导出CSV
表 13 AIBN催化伯苄胺氧化制亚胺
Table 13. Oxidation of primary amines to imines catalyzed by AIBN
| |||
| Entry | R | Conversiona/% | Yielda/% |
| 1 | C6H5 | 94 | 89 |
| 2 | p-Me-C6H4 | 99 | 87 |
| 3 | p-MeO-C6H4 | 97 | 76 |
| 4 | o-Me-C6H4 | 98 | 88 |
| 5 | p-Cl-C6H4 | 99 | 85 |
| 6 | p-F-C6H4 | 97 | 84 |
| 7 | 2-Thienyl | 93 | 83 |
| 8 | 2-Furyl | 90 | 46 |
| 9 | 2-Pyridyl | 92 | 86 |
| 10 | Cyclohexyl | 32 | 7 |
| a Based on amines, determined by GC. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: