
图式 1.
FLP活化氢气的反应
Scheme 1.
Activation of hydrogen by FLPs

路易斯酸碱理论是化学学科最基本的理论之一, 早在1923年, Lewis[1]就提出了将物质根据能够接受或者提供一对电子进行分类, 将具有最低未占分子轨道(LUMO)空轨道可以接受外来电子对的分子、离子或原子团定义为酸; 具有孤对电子并可以提供电子对的分子、离子或原子团定义为碱.路易斯酸与路易斯碱通常情况下会形成热力学惰性的加合物, 很长一段时间里, 这种路易斯酸碱加合物并未得到科学家的足够重视[2].
在早期的研究中, 就有科学家注意到并非所有的路易斯酸和路易斯碱均能够形成稳定加合物. 1942年, Brown课题组[3]报道了当使用2, 6-二甲基吡啶与BF3反应时, 能够形成稳定加合物; 若将BF3换成酸性较弱且具有较大空间位阻的B(CH3)3时, 两者则不能发生反应, 即使将温度升高至80 ℃两者仍未反应.遗憾的是他们并没有对这种现象进行更深入的研究.随后, Wittig课题组[4]的报道也证实了具有较大位阻的路易斯酸碱对不利于热力学稳定加合物的形成, 这些发现为后续的研究提供了一种新思路.
之后, 一些课题组分别报道了分子内同时含有路易斯酸性基团和路易斯碱性基团的例子, 其中一些化合物(1和2)被用作两性分子配体与金属形成配合物[5].也有些化合物(3)表现出了能够与多种极性分子反应的特性, 比如HCl, H2O, LiH等分子[6].
|
|
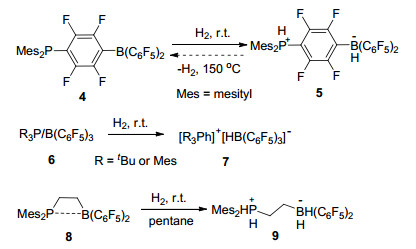
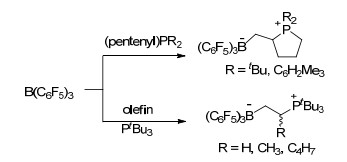
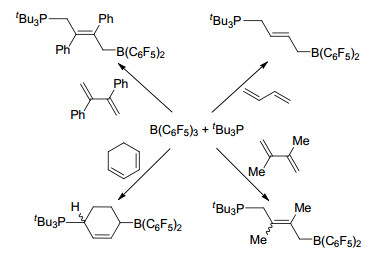
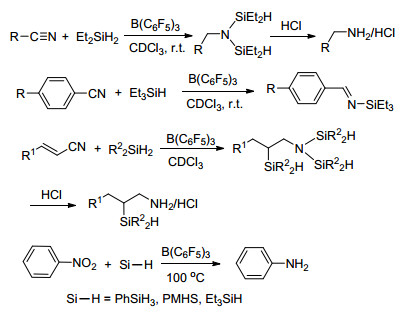
2006年, Stephan课题组[7]首次发现并报道了存在于同一分子内的路易斯酸碱对4具有在常温常压下活化氢气的能力, 这极大地推进了路易斯酸碱对化学的发展.紧接着, Stephan课题组[8]发现了大位阻膦化物与B(C6F5)3形成的分子间路易斯酸碱对体系同样具有在常温下活化氢气的能力, 并能与氢气生成磷氢/硼氢盐7 (Scheme 1).同年, Erker课题组[9]报道的分子内磷/硼路易斯酸碱对8也具有类似的活化氢气的能力(Scheme 1).这三例报道是最早关于路易斯酸碱对活化小分子的报道, 同时也为化学家们打开了一扇非金属体系催化的崭新的大门. Stephan等[10]将这种由于较大的空间位阻作用而导致路易斯酸和路易斯碱不能形成稳定加合产物的现象称之为“frustrated Lewis pairs (FLPs)”, 国内文献将其译为“受阻路易斯酸碱对”[11].
Stephan教授课题组获得的突破性研究工作进展使“受阻路易斯酸碱对(FLP)”迅速成为研究的热点.经过最近十多年的研究和发展, 许多种“受阻路易斯酸碱对”体系被发现, 并在小分子活化方面得到了广泛的应用, 比如能够催化活化氢气、二氧化碳、氮氧化物、二氧化硫、烯烃、炔烃、四氢呋喃、硅烷等小分子的活化[12~17].本文将从“受阻路易斯酸碱对”活化小分子入手, 分类介绍“受阻路易斯酸碱对”的催化反应.
Repo和Rieger等[18]于2011年报道了氮/硼FLP(如10)活化氢气的例子, 并用核磁、中子衍射等测试手段对其产物进行表征.结果表明, 这种分子内同时含有氮/硼路易斯酸碱对的两性分子能够将氢气异裂并在分子内形成N-H+/B-H-离子对.其空间结构中, 能够观察到H+与H-之间存在联系, 它们之间的键长变为0.167 nm.之后不久, Ashley和O'Hare等[19]报道了一种与之类似的FLP中子衍射结构11.同样的, 在该FLP中也能够观察到H+与H-之间的键长为0.1805 nm, 显然这种联系比化合物10中弱了很多.这两例关于氮/硼结构FLP的报道, 将反应中的路易斯酸碱对前体究竟是如何与小分子发生作用这个问题呈现在了人们面前.
|
|
许多科学家对分子间受阻路易斯酸碱对的作用机理进行研究, Autrey课题组[20]用热力学方法对路易斯酸碱对2, 6-二甲基吡啶(Lut)/B(C6F5)3加合物进行了测定.结果表明, 在反应Lut+B(C6F5)3⇔Lut/B(C6F5)3中, ΔH=(-74.9±4.2) kJ/mol, ΔS=(-205.9±10.5) kJ·mol-1.同时, 他们研究了其它几种受阻路易斯酸碱对.
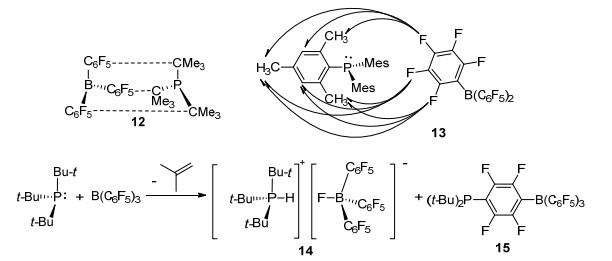
Rhee等[21]的研究表明, 酸碱对tBu3P/B(C6F5)3 (12)体系里B(C6F5)3中的C6F5基团与tBu3P中的tBu基团之间形成了一种非共价键的相互作用.而路易斯酸碱对tBu3P/B(C6F5)3与H2的反应是H2进入硼和磷之间的空间内, 与加合物12进行的双分子反应.在这之后Pápai课题组[22]对路易斯酸碱对tBu3P/B(C6F5)3进行了化学动力学计算.结果表明, 室温下在路易斯酸碱对tBu3P/B(C6F5)3的甲苯溶液中, 只有大约2%的tBu3P与B(C6F5)3产生了上述的这种非共价键作用形成加成产物12.当对路易斯酸碱对Mes3P/B(C6F5)3 (13)溶液进行类似的研究时[23], 并未检测到酸碱对之间的直接成键关系, 它们之间更可能是通过分散力相结合而非硼/磷之间的直接相互作用(Scheme 2).
2011年, Piers等[24]报道了关于溶液中FLP稳定性的研究.在缺少H2或者其他反应底物的磷化氢溶液中, 能够观察到路易斯酸碱对tBu3P/B(C6F5)3之间发生反应, 脱去一分子异丁烯, 并生成受阻路易斯酸碱对14和产物15.若从产物的动力学特性考虑, 产物14和15可能是受阻路易斯酸碱对tBu3P/B(C6F5)3活化氢气的必经过程(Scheme 2).
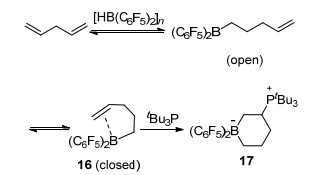
随后, Stephan课题组[25]报道了烯烃类小分子与硼通过范德华力相互作用并发生FLP加成反应的可能机理, 他们利用核磁证明了低温下通过范德华力作用形成的加合物16的存在.之后, 16再与tBu3P进一步反应生成FLP与烯烃的加成产物17 (Scheme 3).
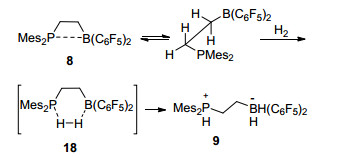
分子内受阻路易斯酸碱对与小分子进行的通常为双分子反应, 分子内路易斯酸碱对的相互作用在反应中通常起到了重要的作用.在最初报道的关于通过乙烯桥联的P/B分子内受阻路易斯酸碱对8的报道中[26], 通过密度泛函方法计算可知, 分子内P/B受阻路易斯酸碱对通过磷与硼之间较弱的相互作用, 形成了一个四元环结构(Scheme 4, 8).在之后与H2的反应中继续形成过渡态18, 并最终形成产物9.
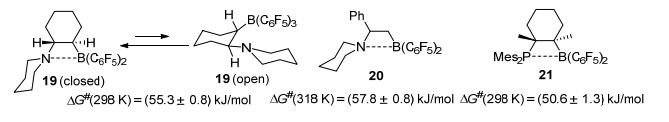
Erker课题组[27]报道了一系列分子内邻位氮/硼受阻路易斯酸碱对, 并利用单晶衍射确定其结构, 通过实验计算了其活化能垒.环己烯吡咯烷酮与HB(C6F5)2反应可得到分子内FLP 19, 其单晶结构显示氮原子与硼原子之间具有微弱的相互作用力, 键长为0.1824(6) nm, 核磁硼谱也证明了在溶液中这种相互作用的存在.这种结构的存在导致了连接在硼原子上的两个C6F5为非对映异构结构, 因此在19F NMR图谱上会显示出两组C6F5的信号峰.而如果打断B…N键后, 则连在硼原子上的两个C6F5在结构上变为等同的, 因此在氟谱上将会得到一组峰.基于此, 可以利用核磁监测来计算出B…N键断裂前后增加的吉布斯自由能ΔG#(298 K)=(55.3±0.8) kJ/mol.同样的, 可以计算出手性氮/硼受阻路易斯酸碱对20的吉布斯自由能ΔG#(318 K)=(57.8±0.8) kJ/mol.与之类似, 邻位磷/硼受阻路易斯酸碱对21显示出与之类似的结构特征, 单晶中P…B键长为0.2188(5) nm.[28]利用19F NMR可测得到P…B键解离的活化能垒为ΔG#(298 K)=(50.6±1.3) kJ/mol.与分子内氮/硼FLP相比, 磷/硼路易斯酸碱之间的相互作用显得更弱(Scheme 5).上述这些数据表明, 在这些邻位受阻路易斯酸碱对中, 氮/硼与磷/硼之间的相互作用很弱, 而在动力学上氮/硼与磷/硼之间的断键步骤为快反应, 这使得反应中开链结构的分子内受阻路易斯酸碱对更容易产生, 虽然这种构型在热力学上处于不利地位.
固态核磁同样是研究FLP的有利工具[29], 尤其是研究磷/硼类FLP中路易斯酸与路易斯碱之间相互作用的重要手段.可以对比固态核磁与其在溶剂中的11B NMR化学位移的变化或31P-11B偶合常数的变化得到相关结构信息.特别是在固态核磁中, 11B NMR四极偶合常数(CQ)和11B电场张量提供了硼的协同作用的信息.另外, 通过31P退偶11B REDOR(旋转回波双共振)实验可以准确计算出不同硼/磷受阻路易斯酸碱对硼/磷之间的距离.
与之相对应的是, 具有C4桥结构的二茂铁磷/硼FLP 22不论在溶液还是晶体中, 均为开环结构, 而在低温核磁中才发现了其闭环产物的信号峰(Eq. 1).这是为数不多的能够分别直接检测到分子内磷/硼加合物以及处于解离的自由态的磷/硼路易斯酸碱对的例子之一(Eq. 1)[30].
|
|
(1) |
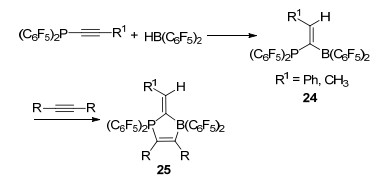
如果分子内的路易斯碱基的碱性被削弱, 那么分子内P…B之间则不易产生相互作用力.炔烃取代的P(C6F5)2与HB(C6F5)2反应得到偕磷/硼FLP体系24, 当R基团为苯基取代时, 其X射线单晶衍射表明磷/硼之间没有直接相互作用力, 磷硼之间的距离为0.2951 nm.固态核磁和溶剂中核磁的数据也都表明磷硼之间没有直接联系[31].有趣的是, 化合物24不具有活化氢气的能力, 不能与氢气发生反应, 却能与一分子炔烃继续反应, 生成化合物25 (Scheme 6).
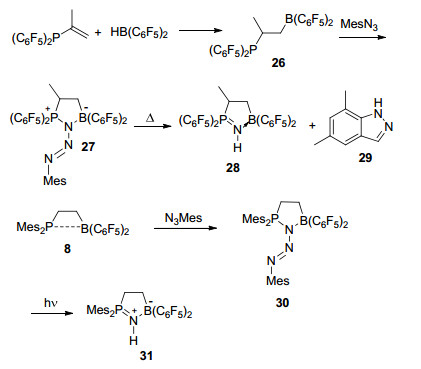
化合物(C6F5)2PC(H)=CHCH3发生硼氢化反应后生成相应的饱和偕磷/硼FLP体系(C6F5)2PCH(C2H5)B(C6F5)2, 而化合物(C6F5)2PC(Me)=CH2的硼氢化反应则会生成邻位磷/硼FLP体系26[32](Scheme 7).与Scheme 4中的8相比较, 化合物26的核磁中没有观察到磷路易斯碱与硼路易斯酸的任何相互作用.尽管如此, 化合物8和化合物26都能够与MesN3发生反应.其中26反应生成1, 1-加成产物27, 若将化合物27加热, 则会发生Staudinger反应生成产物28和29.而2, 4, 6-三甲苯基叠氮与化合物8的加合物30, 在光照条件下则会发生不规则Staudinger反应生成产物31[33](Scheme 7).
有时候, 有些路易斯酸和路易斯碱形成的看起来稳定的“不活泼的”路易斯酸碱对加合产物, 也能够与小分子发生反应.这个概念最初于2009年, 由Stephan等[34]在2, 6-二甲基吡啶与B(C6F5)3的反应中提出, 这两种物质既能够生成稳定加合产物, 又能够与氢气作用发生异裂反应, 生成[C6H3Me2NH][HB(C6F5)3].另一个类似的隐性FLP的例子是脒基硼与CO2或者CO的反应[35].
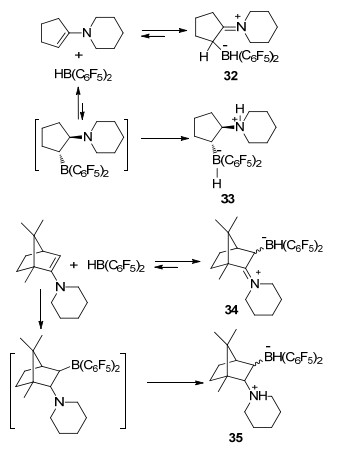
最近的一些报道进一步研究了这一现象.例如, 在烯胺哌啶环戊烯与HB(C6F5)2的反应中, 能够检测到其碳/硼加合物32, 并通过单晶进行了结构表征[36].当化合物32暴露在2.5×105 Pa氢气氛围30 min后, 能够完全反应并生成氮/硼FLP活化氢气后的产物33, 而这一过程表明了在化合物32中存在着互变异构的平衡, 正是这个平衡使得体系中能够短暂存在具有活化氢气能力的分子内氮/硼FLP.与之类似, 在樟脑烯胺类化合物与HB(C6F5)2的反应中生成exo/endo异构的碳/硼加合物混合物34, 将其在氢气或者氘气氛围下充分反应, 能够生成还原的非对映异构体混合物35[37].再次证明了反应体系中的硼酸亚胺离子化合物的互变异构化平衡, 产生了一种隐性的氮/硼FLP (Scheme 8).
大位阻膦类化合物与B(C6F5)3的加合物具有活化氢气的能力(Scheme 1), 受阻路易斯酸碱对的概念最初即是由此发展而来[38, 8].这种无金属存在条件下活化氢气的能力得到了广大科研工作者的关注, 随后便有一系列关于FLP活化氢气性质的研究见诸报道.
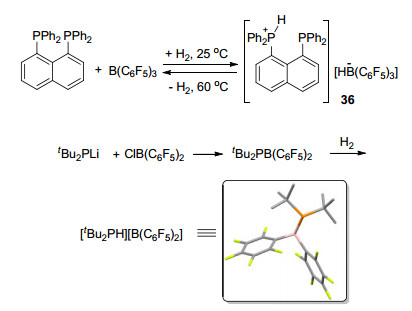
Erker课题组[39]研究并报道了1, 8-双(二苯基膦)-萘酚与B(C6F5)3形成FLP的反应.该二苯基膦是一种具有较大空间位阻的膦电子给体, 就像一个“质子海绵”, 具有捕获质子的能力, 其性质类似于路易斯碱.在B(C6F5)3的存在下, 形成FLP, 能够与氢气发生反应生成磷硼二氢盐[C10H6(PPh2)2H][HB(C6F5)3] (36) (Scheme 9).他们课题组[9]还报道了分子内FLP Mes2PCH2CH2B(C6F5)2 (8)快速可逆地活化氢气的例子(Scheme 1), 并对该类化合物活化氢气的机理进行了进一步研究[40]. 2008年, Stephan课题组[41]报道了一种磷锂类化合物与ClB(C6F5)3生成的含磷FLP, 该FLP也具有活化氢气的能力并能够生成[tBu2PH][HB(C6F5)2], 文献中还得到了其单晶结构(Scheme 9). Blagg等[42]又以P(tBu)3与不同的大位阻氟代苯硼酸形成的FLP体系为例, 讨论了FLP中的不同结构和电子效应对活化氢气所带来的影响.
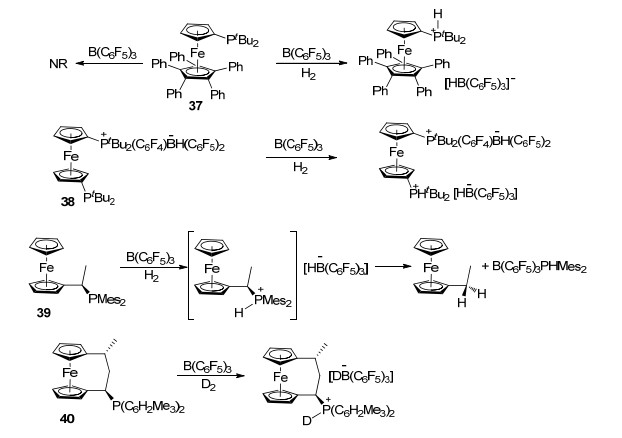
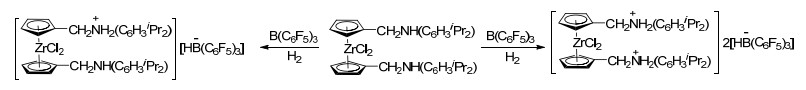
随后, Stephan课题组[43]报道了含膦二茂铁类化合物与B(C6F5)3形成FLP, 并能够活化氢气的例子.在没有氢气的存在下, 含膦二茂铁37与B(C6F5)3并不发生反应, 而若将体系内充入氢气, 则能够以73%的产率直接生成红色固体两性离子(Scheme 10).类似的含膦二茂铁化合物38、39也显示出类似的化学性质, 并具有与B(C6F5)3形成FLP活化氢气的能力[43, 44]. Erker课题组[44b]报道了含膦二茂铁化合物40在B(C6F5)3存在下活化D2的例子(Scheme 10).进一步研究表明, 含膦二茂化锆类化合物也具有类似的性质, 能够与B(C6F5)3形成FLP并活化氢气(Scheme 11)[45].
B(C6F5)3的衍生物B(p-C6F4H)3也能够与膦类路易斯碱发生反应形成受阻路易斯酸碱对, 并且具有活化氢气的能力, 这一发现极大地扩展了FLP的种类.实验表明, 由于相对B(C6F5)3而言, B(p-C6F4H)3的路易斯酸性较弱, 因此导致了在25 ℃的真空条件下, 活化氢气后生成的两性分子[(o-C6H4Me)3PH][HB(p-C6F4H)3] (41)会缓慢分解释放氢气变成游离状态的B(p-C6F4H)3和(o-C6H4Me)3P (Eq. 2).实验表明, 该逆反应在室温下真空条件中反应9 d, 转化率达到85%, 加热则能够明显增加两性分子41释放氢气的反应速度, 在80 ℃的真空中, 仅需过夜反应(12 h)即可达到同样的转化率[46].
|
|
(2) |
O’Hare课题组[47]发展了固相FLP活化异裂氢气的报道, 他们利用≡SiOB(C6F5)2与tBu3P组成的FLP体系[≡SiOB(C6F5)2][tBu3P]完成了在温和条件下异裂H—H键并生成[≡SiOB(H)(C6F5)2][tBu3PH]的反应.
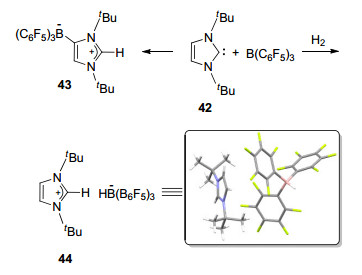
随后, Tamm课题组和Stephan课题组[48]分别报道了较大位阻氮杂卡宾与B(C6F5)3形成的FLP活化氢气的例子(Scheme 12).实验中, 氮杂卡宾42与B(C6F5)3形成的FLP体系的甲苯溶液呈黄色.室温下, 该溶液于2 h内变为无色, 并以95%的产率生成稳定的加合产物43, 丧失活化氢气的能力.而若将42与B(C6F5)3的甲苯溶液直接置于氢气氛围下并于20 ℃搅拌10 min, 则能够以82%的产率生成两性离子44, 并对其单晶结构进行了表征.之后, Stephan等[49]实现了氮杂卡宾与B(C6F5)3类化合物所组成的FLP体系催化不对称还原亚胺类化合物的反应.
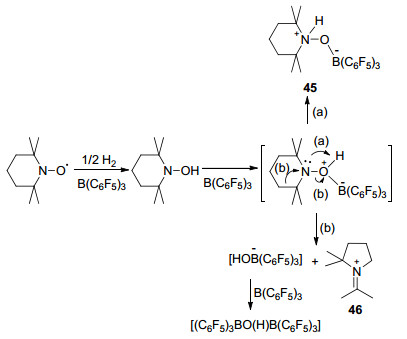
随后的研究发现, 四甲基哌啶氮氧化物(TEMPO)自由基与B(C6F5)3形成的FLP也具有活化氢气的能力, 并生成产物TEMPOH.而TEMPOH在B(C6F5)3的进一步催化下, 能够进一步发生反应, 并生成产物45和46, 分别得到了产物的单晶结构, 并对反应机理进行了一定的研究(Scheme 13)[50].
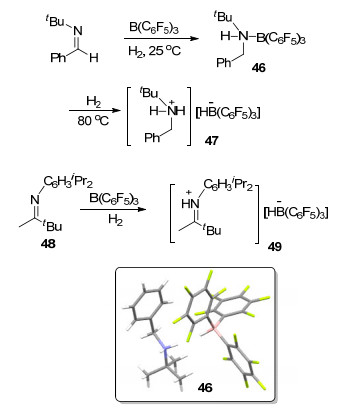
将含氮路易斯碱引入到受阻路易斯酸碱对体系之中极大地扩充了FLP体系的适用范围.早在2008年, Stephan等[51]报道的关于B(C6F5)3催化亚胺类化合物与氢气发生还原反应的文献中, 研究了室温下等物质的量的B(C6F5)3与tBuN=CPh(H)在氢气氛围下的反应, 得到硼-胺加合产物tBu(PhCH2)NH(B(C6F5)3) (46), 并得到了其单晶结构.在404 kPa氢气氛围下将加合物46加热至80 ℃并保持1 h, 则能够进一步得到异裂氢气的两性离子[tBuNH2(CH2Ph)][HB(C6F5)3] (47).之后, 他们又尝试了更大位阻的(C6H3iPr2)N=CMe(tBu) (48)在相同条件下进行类似的反应, 结果表明48与B(C6F5)3能够活化氢气并生成产物[(C6H3iPr2)N(H)=CMe(tBu)][HB(C6F5)3] (49) (Scheme 14).之后, Paradies等[52]进一步发展了B(C6F5)3类化合物催化氢气还原亚胺的反应.与此同时, 许多其它含氮FLP活化氢气的例子也见诸报道[53].
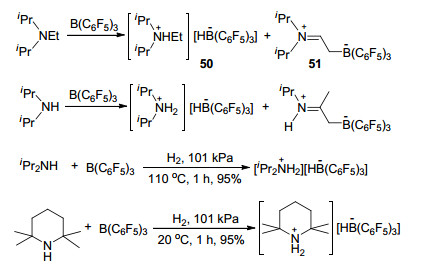
Repo和Rieger等[54]报道了iPr2NEt与B(C6F5)3活化氢气的反应.反应中, 他们发现了一个有趣的现象, iPr2NEt与B(C6F5)3在溶液中能够1:1的生成[iPr2NHEt][HB(C6F5)3] (50)和iPr2N=CHCH2B(C6F5)3 (51)的混合物, iPr2NH(或TMP)也具有同样的反应性质.但是当体系中有氢气存在时, iPr2NH(或TMP)则能与B(C6F5)3以很高产率生成对应的两性离子盐[iPr2NH2][HB(C6F5)3](或[TMPH][HB(C6F5)3]) (Scheme 15).之后, 不同课题组对更多含氮碱基与硼烷构成的FLP活化氢气的反应性质和机理做了更加详细的研究, 进一步扩展了FLP活化氢气反应的适用范围[55].
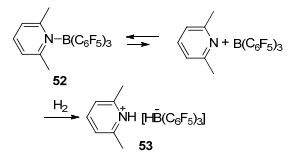
研究表明较大位阻吡啶类化合物在FLP中作为碱基端时, 具有良好的活化氢气的能力[56, 34]. 2, 6-二甲基吡啶与B(C6F5)3在溶液中存在游离态的路易斯酸碱对和其加合态52之间的平衡, 正是这个平衡的存在使体系表现出了FLP的性质.当把该体系暴露在101 kPa氢气氛围2 h后, 能以87%的产率得到活化氢气后的固体两性离子53 (Scheme 16).随后, 化学家们研究报道了更多关于吡啶类化合物所形成的FLP活化氢气的例子, 并对其机理进行了更进一步研究[57].
杜海峰课题[58a~58f]组在FLP催化活化氢气不对称氢化反应的研究中做出了杰出的贡献.他们课题组报道了若干种使用FLP途径活化氢气还原不同底物的研究, 并在手性配体的作用下得到较高的空间选择性, 对FLP活化氢气的应用作出了进一步发展(Scheme 17).王华冬课题组[58g]也对FLP催化活化氢气还原烯烃的反应进行了详细的研究, 并通过计算对其反应机理进行了详细的研究.
在Stephan最初提出“受阻路易斯酸碱对”概念的相关报道中便有关于磷-硼FLP与烯烃的反应研究.报道中研究了分子间FLP与烯烃反应的机理并尝试了一些反应(Scheme 18)[10].
他们利用2D 1H-19F NOESY等表征手段, 对硼烷与烯烃之间的相互作用进行了研究, 确认了烯烃与硼之间可能存在范德华作用力(Scheme 3)[25].与上述结论相同, Pápai课题组和黎书华课题组等[59]的研究报道也分别证明了烯烃与硼之间范德华作用力的存在.之后, Stephan课题组[60]又报道了亲电烯氧基硼与tBu3P、Me3P、2, 6-二甲基吡啶、四甲基哌啶、吡咯、卡宾和苄基三苯基磷类等多种试剂的反应及其反应性质(Scheme 19).
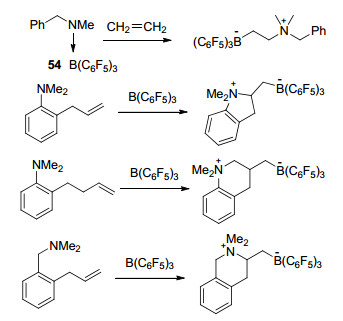
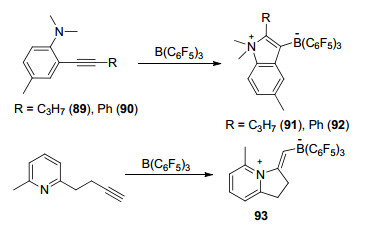
氮/硼类受阻路易斯酸碱对PhCH2NMe2B(C6F5)3 (54)与乙烯可以发生加合反应并生成两性离子PhCH2NMe2CH2CH2B(C6F5)3 (Scheme 20)[61].利用这一反应特性可以完成链状烯烃的环化反应, 邻(2-丙烯基)-N, N-二甲基苯胺、邻(3-丁烯基)-N, N-二甲基苯胺和邻(2-丙烯基)-N, N-二甲基苄胺都能够与B(C6F5)3发生反应, 并得到对应的氮杂环两性离子(Scheme 20)[62].
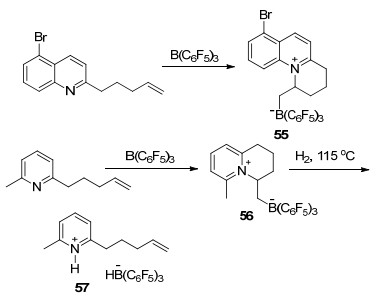
这一环化策略更是被利用到了稠环类化合物的合成之中.例如, 带有链状烯烃取代的喹啉或者吡啶类化合物与B(C6F5)3反应后可以分别得到对应的稠环类化合物55和56.有趣的是, 若将56在氢气氛围下升温至115 ℃, 则能得到活化氢气后的开环产物57 (Scheme 21)[63].
Stephan课题组还对FLP与二烯类化合物的反应进行了研究.他们研究了tBu3P和B(C6F5)3形成的FLP体系与丁二烯类衍生物生成对应的1, 4-加成硼磷类化合物的反应.这些反应中的分离产率通常为50%~60%, 这可能是由于反应中生成了1, 2-加成的副产物, 遗憾的是他们并没有对可能生成的副产物做出详细的研究和表征(Scheme 22)[64].
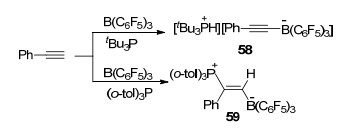
FLP与端炔的反应类型主要分为两类:去质子化反应和加成反应, 而具体发生的反应类型则与路易斯碱的碱性有关.其中最典型的一个例子便是B(C6F5)3/tBu3P体系与苯乙炔反应时能够生成化合物[tBu3PH]- [PhC≡CB(C6F5)3] (58), 而B(C6F5)3和(o-tol)3P体系与苯乙炔反应时则只能生成化合E-(o-tol)3P(Ph)C=C(H)B(C6F5)3 (59) (Scheme 23)[16].
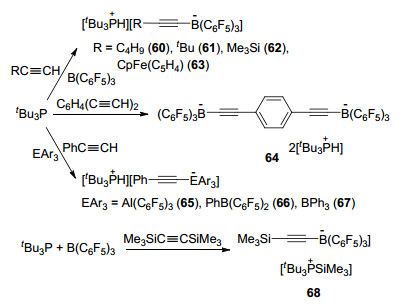
许多关于端炔与FLP的去质子化反应已经见诸报道.不同种类的端炔可以和B(C6F5)3/tBu3P体系发生去质子化反应并形成[tBu3PH][RC≡CB(C6F5)3] [R=C4H9 (60), tBu (61), Me3Si (62), CpFe(C5H4) (63)等].类似的, 1, 4-二乙炔基苯也能够形成两性离子[tBu3PH]2[(C6F5)3-BC≡C(C6H4)C≡CB(C6F5)3] (64).同时, 其它几种不同的路易斯酸也能够与[tBu3PH]和PhC≡CH发生去质子化反应并形成[tBu3PH][PhC≡CEAr3]结构, 其中EAr3可以为Al(C6F5)3 (65), PhB(C6F5)2 (66)和BPh3 (67)等.而含硅炔烃Me3SiC≡CSiMe3则与B(C6F5)3和tBu3P生成[tBu3PSiMe3][Me3SiC≡CB(C6F5)3] (68) (Scheme 24)[65].
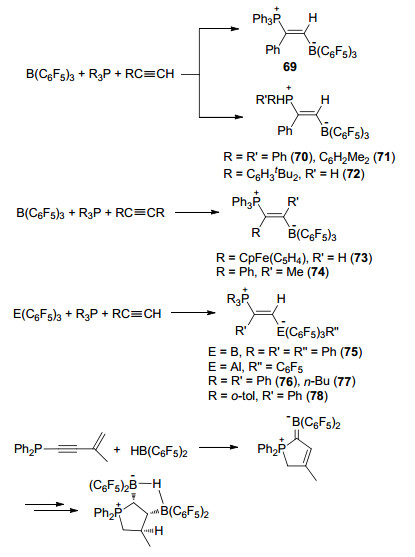
磷/硼FLP与端炔的加成反应同样也有诸多报道, 利用该类型的反应能够合成如E-Ph3P(Ph)C=C(H)- B(C6F5)3 (69), E-Ph3P(CpFe(C5H4))C=C(H)B(C6F5)3 (70), Ph3P(Ph)C=C(Me)B(C6F5)3 (71), E-R2PH(Ph)C=C(H)B(C6F5)3 [R=Ph (72), C6H2Me3 (73)], E-(C6H3tBu2)- PH2(Ph)C=C(H)B-(C6F5)3 (74)等两性离子.其中路易斯酸又能够进行各种衍生或者替换, 比如E-Ph3P(Ph)C=C(H)EAr3 [EAr3=PhB(C6F5)2 (75), Al(C6F5)3 (76)], E- Ph3P-(C4H9)C=C(H)Al(C6F5)3 (77)和E-(o-tol)3P(Ph)C=C(H)Al(C6F5)3 (78)等(Scheme 25)[65].与之类似, Erker教授[66]报道了磷/硼FLP与分子内炔烃加成, 并进一步环化的反应.得到了产物的单晶结构, 对反应机理做了详细研究(Scheme 25).
上述加成反应可以用来合成大环化合物, 比如可以由苯炔和Mes2PC6F4B(C6F5)2合成大环化合物[(H)C=C(Ph)Mes2PC6F4B(C6F5)2]2 (79) (Eq. 3)[65].
|
|
(3) |
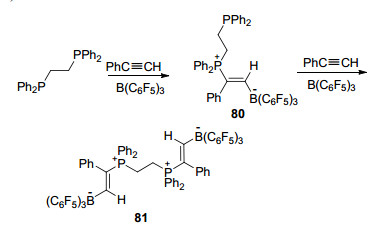
利用Ph2PCH2CH2PPh2类的二磷类化合物进行加成反应时, 可以通过控制B(C6F5)3和PhC≡CH的量来选择性生成E-Ph2PCH2CH2PPh2(Ph)C=C(H)B(C6F5)3 (80)或E-[CH2PPh2(Ph)C=C(H)B(C6F5)3]2 (81) (Scheme 26)[65].
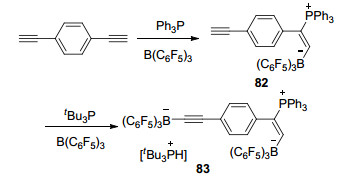
1, 4-二乙炔基苯能够与Ph3P/B(C6F5)3 FLP体系发生加成反应生成E-HC≡C≡C6H4C(PPh3)=C(H)B(C6F5)3 (82), 若紧接着再将化合物82与tBu3P/B(C6F5)3体系进一步反应, 则能够生成两性离子[tBu3PH][(C6F5)3BC≡ C≡C6H4C(PPh3)=C(H)B(C6F5)3] (83).在该反应中, 同一分子先后发生了加成反应和去质子化反应(Scheme 27)[53].
之后, Yamaguchi课题组[67]对P/B类FLP与炔烃的加成反应的应用做了相应的研究.并利用分子内的磷基团和硼基团的加成反应, 形成了一系列两性离子, 其产物在光敏材料或者电化学材料的应用中具有广阔前景(Eq. 4).
|
|
(4) |
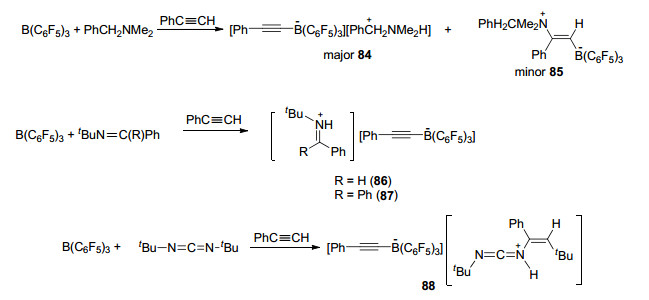
Stephan课题组[65a]对含氮受阻路易斯酸碱对与炔烃的反应也做了相关研究和报道. PhCH2NMe2和B(C6F5)3组成的FLP体系与PhC≡CH反应时, 产物以84:16的比例形成去质子化产物[PhCH2NMe2H][PhC≡C-B(C6F5)3] (84)和加成产物PhCH2NMe2(Ph)C=C(H)B-(C6F5)3 (85)的混合物.相反的, 当使用亚胺类化合物tBuN=C(R)Ph代替PhCH2NMe2时, 只有去质子化产物[tBuHN=C(R)Ph][PhC≡CB(C6F5)3] [R=H (86)或Ph (87)]生成(Scheme 28).同时, 他们还考察了碳化二亚胺tBuNCNtBu, B(C6F5)3和2 equiv. PhC≡CH发生的反应.出乎意料的是, 产物为[tBuNCN(H)C(Ph)=C(H)tBu]- [PhC≡CB(C6F5)3] (88), 也就是说在该反应中同时发生了去质子化反应和加成反应(Scheme 28).王华冬课题组[65b]报道了大位阻有机硼化物与有机碱1, 4-二氮杂二环[2.2.2]辛烷(DABCO)所组成的受阻路易斯酸碱对体系, 能够活化端炔发生去质子化反应, 并能够进一步脱去碱基形成端炔的硼氢化产物(Scheme 29).
与链状烯烃类似, 苯胺上的炔基基团也可以被用来进行分子内环化反应[62].例如, 邻戊炔基-N, N-二甲基苯胺89或者其类似物90均可以发生环化并分别生成91和92.含氮杂环如吡啶、喹啉等的衍生物也能够与分子内炔基发生环化反应, 生成对应的环化两性离子93 (Scheme 30)[63].类似的, 也可以用同样的手段构筑磷杂环戊二烯类化合物[68].
含硫FLP能够与炔烃发生加成反应, 以苯乙炔和R2S为例, 与B(C6F5)3反应可生成E-R2S(Ph)C=C(H)- B(C6F5)3 [R=Me (96), PhCH2 (97)[65].二聚分子内含硫FLP (PhSCH2B(C6F5)2)2在室温下存在单体与二聚体的互变平衡, 其单体能够与端位炔烃或分子中间位置的炔烃发生加成反应, 并得到相应的产物98~102.比较特殊的是, 该反应中所生成的加成产物均为cis构型(Scheme 31)[69].
当使用氮杂卡宾作为FLP中的碱基时, 由于其具有较强的碱性, 因此与炔烃发生反应的产物通常为去质子化产物.例如在氮杂卡宾ItBu与B(C6F5)3组成的受阻路易斯酸碱对与苯乙炔发生的反应中, 产物为去质子化产物[ItBuH][Ph反应.B(C6F5)3] (Eq. 5)[65].
|
|
(5) |
当使用化合物1-吗啉环己烯(103)与B(C6F5)3形成的FLP和苯乙炔在甲苯溶液中反应时, 产物为去质子化产物[C6H10N(CH2CH2)2O][PhC≡CB(C6F5)3] (104)和加成产物C6H9(2-PhC=C(H)B(C6F5)3)(N(CH2CH2)2O (105)的混合物, 两者比例为1:1.其中产物105的生成为形成C—C键提供了一种新思路.当把苯乙炔换为二茂铁乙炔时, 则只有去质子化产物[C6H10N(CH2CH2)2O]-[CpFe(C5H4)C≡CB(C6F5)3] (106)生成(Scheme 32)[70].
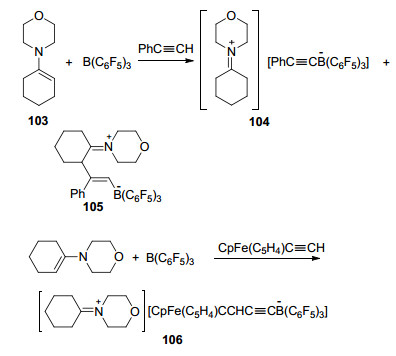
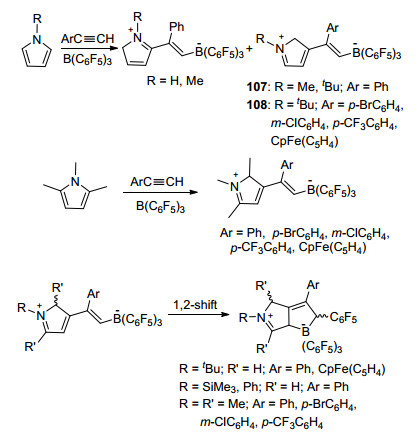
这篇报道中, 还研究了吡咯类衍生物、B(C6F5)3和苯乙炔类化合物发生的加成反应(Scheme 33).结果表明, 吡咯作为路易斯碱时产物为单一的HNC4H4(2-Ph- C=C(H)B(C6F5)3), N-甲基吡咯作为路易斯碱时产物以3:2的比例生成MeNC4H4(2-PhC=C(H)B(C6F5)3)和MeNC4H4(3-PhC=C(H)B(C6F5)3), 而当使用N-叔丁基吡咯时产物则为单一的tBuNC4H4(3-Ph-C=C(H)B(C6F5)3), 使用1, 2, 5-三甲基吡咯时的产物为对应的3-位取代的加成产物.其它构型的苯乙炔类化合物也能够发生相同反应.值得注意的是, 其中107、108在溶液中静置, 即可得到重排产物tBuNC4H3(3-ArC=C(H)(C6F5)B(C6F5)2), 进一步的研究表明, 在吡咯与炔烃的反应中, 能够直接得到产物RNC4H3(3-PhC=C(H)(C6F5)B(C6F5)2) (R=SiMe3, Ph)和MeNC4H(2, 5-Me2)(3-PhC=C(H)(C6F5)B-(C6F5)2).
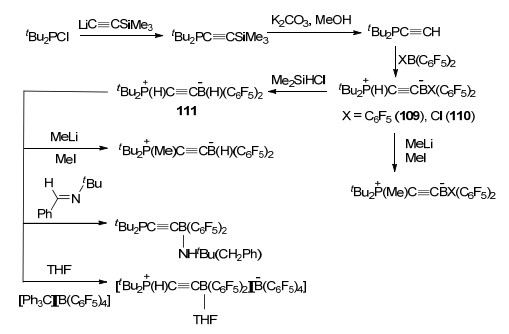
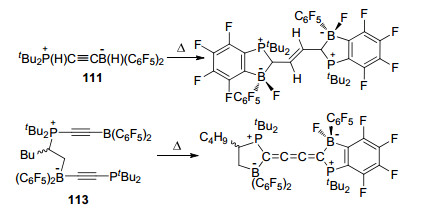
在关于连接炔基的FLP的研究中, Stephan课题组[71]报道了tBu2PC≡CH与B(C6F5)3或ClB(C6F5)2反应得到两性离子tBu2P(H)C≡CB(C6F5)3 (109)或tBu2P(H)- C≡CB(Cl)(C6F5)2 (110)的反应, 并对产物109和110的反应性质做了详细的研究.结果显示, 当使用Me2SiHCl对上述产物分别进行处理时, 均可得到氢化产物tBu2P(H)C≡CB(H)(C6F5)2 (111).若将产物111继续与[Ph3C][B(C6F5)4]或tBuN=CHPh反应仍能进一步得到对应产物(Scheme 34).
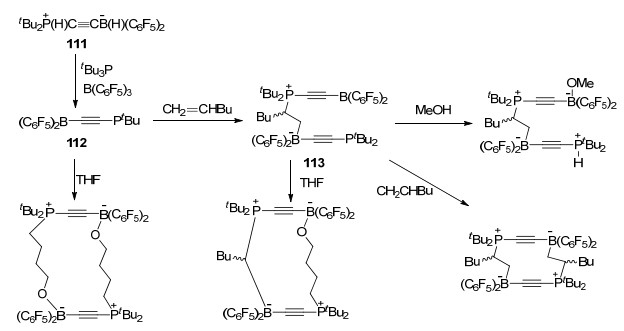
若将化合物111继续与tBu3P和B(C6F5)3混合, 则能够得到磷/硼产物tBu3PC≡CB(C6F5)2 (112), 尽管报道中未能成功分离得到该化合物, 但是112仍能够继续与1-己烯发生反应, 得到FLP与烯烃的加成反应产物113.而113能够与MeOH或者CH2=CHBu继续发生反应.同时, 化合物112和113均能使四氢呋喃(THF)发生开环反应, 并与之形成大环化合物(Scheme 35)[72].
有趣的是, 化合物111和113在受热时会发生分解, 并分别能够生成C—C偶联的C4-烯烃衍生物tBu2PCH- BF(C6F4)(C6F5)CH)2和累积多烯烃衍生物(tBu2P)C6F4BF-(C6F5)C4B(C6F5)2(BuCHCH2)(PtBu2) (Scheme 36)[72].
化合物111还能够与AlX (X=Cl, Br), Zn(C6F5)2·C7H8和Al(C6F5)3·C7H8等金属化合物发生加成产物.同时, 111或112均能够与金属Ni衍生物发生反应, 其中金属Ni能够与炔烃产生相互作用力, 在该力的作用下金属Ni能够进一步与分子内的硼基产生相互作用力(Scheme 37)[71].
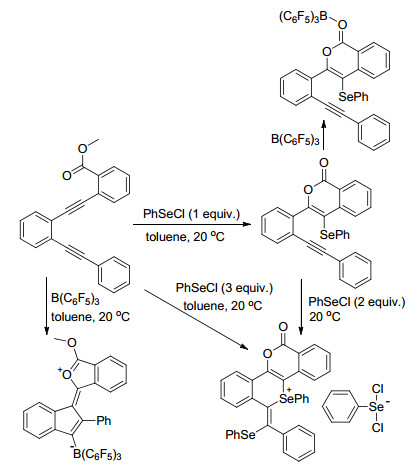
Melen课题组[73]通过对比研究了二炔烃类化合物分别与PhSeCl和B(C6F5)3的反应, 发现在FLP活化二炔烃类化合物的反应中, 分别以PhSeCl和B(C6F5)3为代表的软硬路易斯酸显示出了截然不同的反应性质, 并利用单晶等手段对结果进行了佐证(Scheme 38).而在以往的研究中, 通常更偏重于对硬路易斯酸B(C6F5)3的研究, 对于更柔软的路易斯酸体系则研究较少. Melen教授[73]对软路易斯酸在FLP反应中的研究进一步拓展了FLP的应用范围.
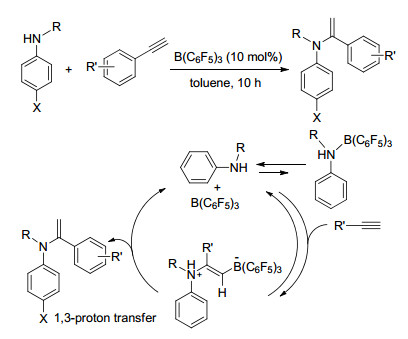
FLP还具有催化端炔与胺类化合物发生氢胺化反应的能力.在催化量硼烷存在的苯胺类化合物甲苯溶液中, 室温或低温下缓慢滴加苯炔类衍生物, 能够得到一系列芳香胺类化合物, 产率在62%~84%之间, 并对反应的机理进行了详细的研究(Scheme 39)[74].
受阻路易斯酸碱对在环醚类化合物、内脂类化合物以及环丙烷类化合物的开环反应中得到了广泛的应用.早在1950年Wittig等[75]就报道了类似的反应, 尽管当时还没有发现FLP体系.报道中, 他们使用PH3CNa与THF(BPh3)发生反应, 得到了硼酸盐阴离子[Ph3C(CH2)4OBPh3]-, 这可以看作是最初的关于FLP促进四氢呋喃开环反应.到了1992年, Stephan等[76]报道的ZrCl4(THF)2与PCy3在四氢呋喃溶液中生成两性离子二聚体[Cl4Zr(m-O(CH2)4PCy3)]2的反应, 同样也可以看作是一例FLP促进的THF开环反应.之后, 许多金属路易斯酸与N-或P-路易斯碱组成的FLP体系促进THF开环反应见诸报道, 报道中所提及的金属有铀[77]、钐[78]、钛[79]、锆[80]、碳硼烷[81]、碲[82]、和AlH3[83]等.
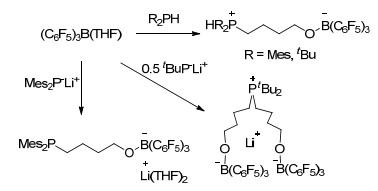
2006年, Stephan课题组[38]报道了硼烷和膦类化合物促进的四氢呋喃开环反应, 这是最初的典型FLP促进四氢呋喃开环反应的报道.报道中(THF)B(C6F5)3能够与大位阻磷酸如tBu2PH或Mes2PH等生成THF开环后的两性离子R2PH(CH2)4OB(C6F5)3 (R=tBu或Mes).同时(THF)B(C6F5)3与较大位阻的磷酸锂盐也能够促进THF的开环反应, 并生成相对应的盐(Scheme 40).
2009年, Stephan课题组[34]又报道了2, 6-二甲基吡啶/B(C6F5)3组成的FLP体系与THF的开环反应, 他们利用核磁等手段对其进行了确认和表征, 并得到了产物2, 6-Me2C5H3N(CH2)4OB(C6F5)3 (114)的单晶结构(Eq. 6).
|
|
(6) |
我们课题组[84]发现并报道了苯胺类化合物与B(C6F5)3形成的FLP具有活化四氢呋喃类环醚开环的能力.在路易斯酸的催化下, 其开环产物能够进一步脱水并发生环化反应, 并能够以较高产率得到N-芳基吡咯.我们通过单晶以及核磁等手段, 对反应机理进行了研究, 认为在该反应过程中经历了FLP促进THF开环的反应过程, 开环产物在对甲苯磺酸的作用下脱去一分子水并进一步发生环化反应, 最终生成目标产物N-芳基吡咯(Scheme 41).
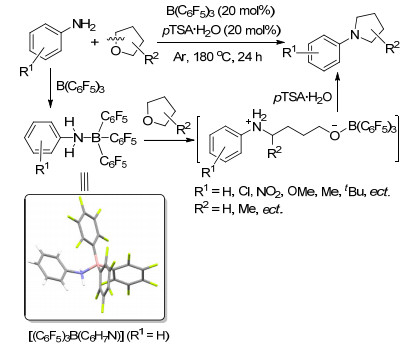
卡宾/B(C6F5)3 FLP也是活化THF开环的良好体系, Tamm课题组[48c]报道的氮杂卡宾42与B(C6F5)3形成的FLP体系能够与THF发生开环反应生成[ItBu(CH2)4-OB(C6F5)3].于此同时, 许多其他路易斯碱基与硼烷类化合物形成的FLP体系也被发现能促进THF的开环反应, 如tBu2PH, C6H5CH2NMe2, Me2NC6H4NMe2, Me3N, Et3N, Me2N(CH2)2NMe2, tBuHN(CH2)2NHtBu2等(Scheme 42)[71, 85].
THF与含有炔基的硼/磷FLP体系反应能够生成大环产物, 如前文中提到的112、113等(Scheme 35).
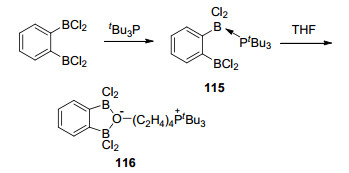
2012年, Stephan课题组[86]报道了二硼类化合物1, 2-C6H4(BCl2)2与tBu3P所形成的化合物1, 2-C6H4-(BCl2)2·(PtBu3) (115)与THF发生开环反应, 并生成特殊的THF开环产物1, 2-C6H4(BCl2)2(O(CH2)4PtBu3) (116), 该分子内氧-硼之间存在一种独特的桥联结构(Scheme 43).
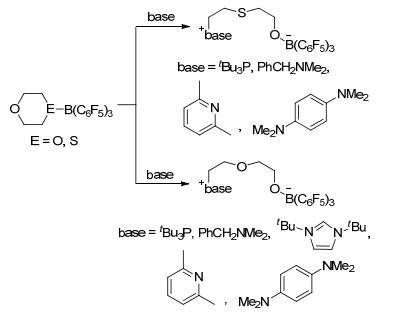
B(C6F5)3与多种路易斯碱形成的FLP体系也能够与1, 4-二氧六环或者1, 4-噻噁烷发生开环反应并生成对应产物.文献中报道的碱tBu3P, C6H5CH2NMe2, ItBu, Me2NC6H4NMe2, 2, 6-二甲基吡啶(C5H3Me2N)等与B(C6F5)3形成的FLP体系能够与1, 4-二氧六环反应生成开环产物(base)(CH2)2- O(CH2)2OB(C6F5)3, 同时tBu3P, C6H5CH2NMe2, ItBu, Me2NC6H4NMe2, C5H3Me2N等路易斯碱也能够促进1, 4-噻噁烷的开环反应并生成(base)(CH2)2S(CH2)2OB(C6F5)3 (Scheme 44)[85].
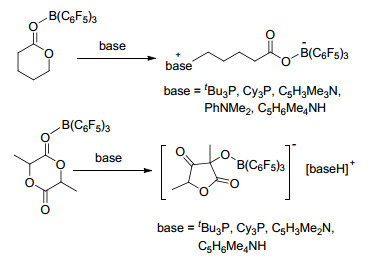
当使用tBu3P, Cy3P, C5H3Me2N, PhNMe2, C5H6Me4- NH作为碱基时, 与B(C6F5)3能够促进内脂的开环反应, 并生成两性离子(base)(CH2)4CO2B(C6F5)3.而当使用tBu3P, Cy3P, C5H3Me2N, 2, 2, 6, 6-四甲基哌啶(C5H6Me4- NH)作为碱基时, 还能够与rac-丙交酯发生去质子化反应并引发环收缩, 最终生成五元环产物[(base)H][O-C≡CHMeCO2(CMe)O(C6F5)3] (Scheme 45)[87].
2010年, Stephan课题组[88]报道, 环丙烯PhC3H5能在tBu3P/B(C6F5)3 FLP体系中发生开环反应, 并以69%的产率得到开环后的磷-硼两性离子tBu3PCH-(Ph)CH2CH2B(C6F5)3.还研究了分子内含有烯键的环己烯类化合物在FLP体系中发生的开环反应(Scheme 46).
由于CO2是一种重要的温室气体, 并且具有廉价易得、原料丰富和无毒等特点, 因此如何合理利用CO2一直是化学家的热点研究问题. CO同样也是一种廉价易得的C1源, 因此利用FLP体系活化CO2和CO具体重大意义.
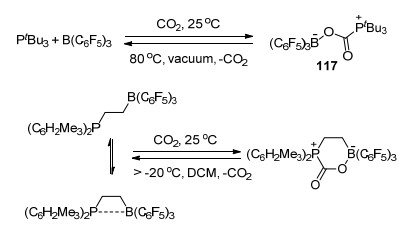
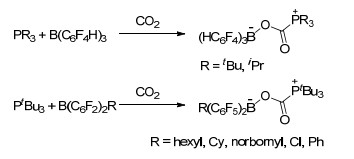
2009年, Stephan和Erker等[12a]发现了tBu3P和B(C6F5)3组成的磷/硼FLP体系具有捕捉CO2的能力, 将tBu3P和B(C6F5)3的溴苯溶液在室温下暴露于CO2中, 能够以87%的产率得到白色固体加成产物tBu3PCO2B(C6F5)3 (117), 并得到了其单晶结构.若将化合物117的溴苯溶液在密封的真空体系中加热至80 ℃以上5 h, 则能够释放出CO2并重新得到tBu3P/B(C6F5)3 FLP溶液.有趣的是若将该密封体系重新在室温下静置若干小时后, 则能够重新观察到化合物117的生成.文献中还研究了分子内FLP Mes2PCH2CH2B(C6F5)2对CO2的固定作用, 结果表明其与CO2生成的加成产物的稳定性弱于化合物117, 其二氯甲烷溶液在高于-20 ℃的条件下即可释放出CO2 (Scheme 47)[12a].
之后, Stephan和Erker等[12b]又报道了分子间FLP体系活化CO2的反应, 比如R3P(CO2)B(C6F4H)3 (R=iPr, tBu)和tBu3P(CO2)BR(C6F5)2 (R=hexyl, Cy, norbornyl, Cl、Ph)等(Scheme 48).
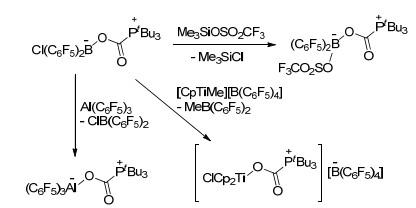
Stephan课题组[89]对FLP与CO2加成后产物的性质做了进一步研究.他们尝试用tBu3P(CO2)B(C6F5)2Cl分别与Me3SiOSO2CF3, Al(C6F5)3或[Cp2TiMe][B(C6F5)4]进行反应, 分别得到了对应产物tBu3P(CO2)B(C6F5)2-(OSO2CF3), tBu3P(CO2)Al(C6F5)3和[tBu3P(CO2)TiCp2Cl]-[B(C6F5)4] (Scheme 49).
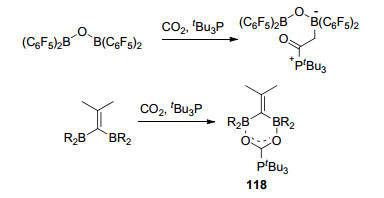
二硼化合物1, 1-bis-(C6F5)2BOB(C6F5)2也能够与CO2发生反应, O(B(C6F5)2)2能够与CO2和PtBu3反应生成产物O(B(C6F5)2)2(O2CPtBu3), 并得到了该产物的单晶结构[90].该文中还报道了二硼化合物Me2C=C(BR2)2 (R=Cl, C6F5)与CO2和PtBu3的反应, 得到对称的环化产物118, 尽管在分子118内硼与CO2形成了螯合作用, 但是仍能够在较低的温度下(低于15 ℃)分解并释放出CO2, (Scheme 50)[90].
2009年, O'Hare和Ashley等[91]发表的文章中报道了选择性氢化CO2得到CH3OH的反应, 反应中使用了FLP无金属体系, 仅需要101~202 kPa的低压即可使反应完成.报道中将四甲基哌啶与B(C6F5)3等物质的量的混合, 在氢气氛围下于160 ℃反应6 d, CO2能够转化为CH3OB(C6F5)2, 经过蒸馏分离, 能够以17%~25%的收率得到MeOH.这是关于FLP体系在无金属条件下活化还原CO2的一个重要例子, 报道中并对可能的反应机理进行了详细的研究.随后, Piers等[92]报道了四甲基哌啶与B(C6F5)3在硅氢的存在下活化氢化CO2生成甲烷的例子, 文章中同时报道了其可能的反应机理, 实现了由FLP催化的从CO2生成CH4的反应路径.
2012年, Stephan等[93]报道了甲苯中((CH3)3Si)3P和B(C6F4H)3在氢气氛围下与等物质的量的CO2的反应, 生成硅甲基二氧化碳产物((Me3Si)2PC(OSiMe3)O)- B(p-C6F4H)3.而此时若将溶剂更换为二氯甲烷, 则能进一步与另一分子CO2生成双插入产物((Me3SiO)2C=P-C(OSiMe3)=O)B(p-C6F4H)3, 这可能是由于分子内的Me3Si基团迁移引起的(Scheme 51).

受阻路易斯酸碱对B(C6F5)3/tBu3P则能够与CO2和H2在室温下生成甲酰基硼化物119, 将产物119加热至90 ℃时则会发生重排反应生成120, 而120则能继续跟CO和H2反应, 最终以3:2的比例得到还原产物121和羰基化产物122 (Scheme 52)[94].
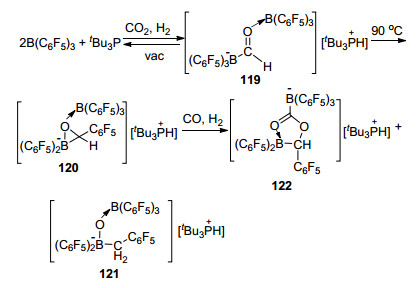
我们课题组[95]发现, 邻苯二胺与B(C6F5)3组成的氮/硼FLP体系也具有活化CO2的能力, 我们利用原位核磁对反应机理进行了研究.在体系中存在硅烷的条件下, 活化CO2后形成的产物能够进一步被还原生成甲酰胺类产物, 并继续脱去一分子水发生环化反应, 生成环化产物苯并咪唑(Eq. 7).于此同时, 其他研究组报道了关于含氮碱基与B(C6F5)3组成的FLP活化CO2的机理的研究[96].
|
|
(7) |
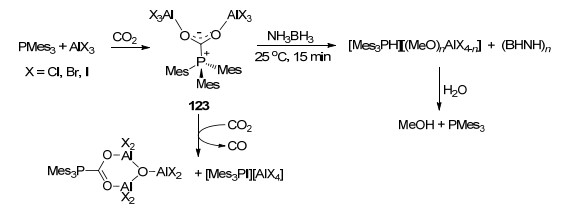
铝基FLP也是活化氢化CO2的良好试剂, AlX3 (X=Cl, Br, I)与PMes3形成的FLP体系能够有效与CO2反应生成加合产物Mes3P(CO2)(AlX3)2 (123), 若继续使用NH3BH3对产物进行处理, 则能够在室温下15 min内生成铝基甲醇盐, 将其进一步水解即能够以50%产率得到甲醇[97].后续的研究中发现, 若将化合物123继续暴露在CO2中18 h, 能够进一步生成Mes3P(CO2)(AlX2)2-(OAlX3)和[Mes3PX][AlX4], 并释放出一分子CO (Scheme 53)[98].
最近, 一系列关于FLP活化CO2的表面化学反应见诸报道, 这进一步扩展了FLP在CO2活化上的应用[99].
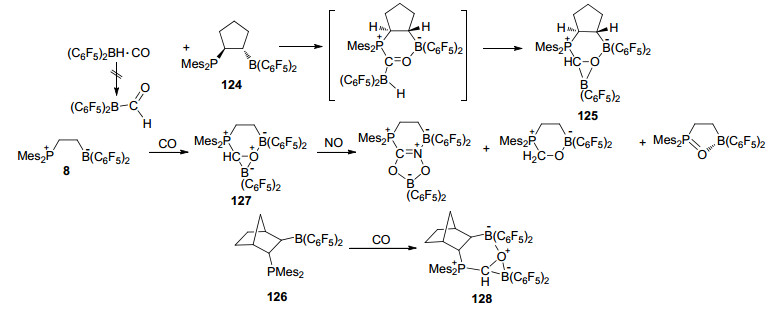
Erker课题组[100]报道了FLP与CO的反应.他们首先使用[HB(C6F5)2]与CO反应, 得到了加成产物[(C6F5)2BH·CO], 并得到了其单晶结构.在该反应中并没有发生CO的还原.然而, 当将[HB(C6F5)2]/CO用原位生成的FLP 124处理时, CO被还原为甲酰基, 并进一步与硼烷发生作用, 最终形成产物125 (Scheme 54).类似的, FLP 8和126与CO能够形成产物127和128[101].进一步的研究表明, 127还能够继续与一分子NO进行反应, 并生成CO/NO偶合产物(Scheme 54)[102].
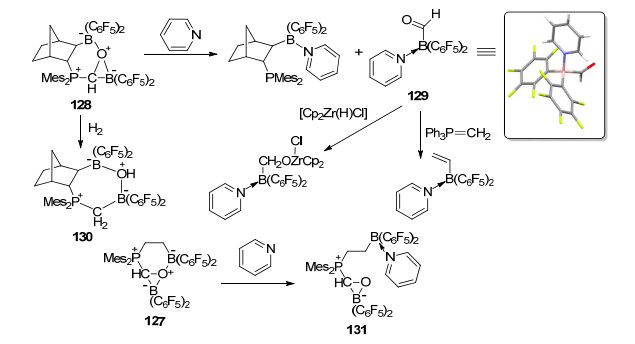
之后, Erker课题组还对FLP与CO形成的加成产物128的性质进行了详细研究.将化合物128与过量的吡啶进行反应, 能够得到稳定的甲酰基硼化物吡啶配合物129, 通过重结晶能够将其分离并得到其单晶结构.化合物129具有与醛类化合物类似的反应性质, 比如能发生Wittig反应或者被铬(VI)酸氧化, 并分别生成乙烯基硼烷和羧基硼烷.化合物128还能够与氢气发生反应, 生成进一步氢化的产物130.与之类似, 化合物127也能够与吡啶发生类似反应, 生成硼-氧键断裂产物131 (Scheme 55)[100].
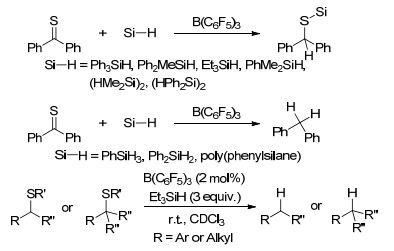
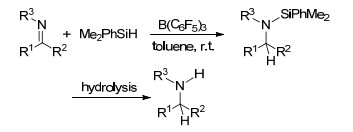
近年, FLP活化硅烷的反应也被广泛地研究和报道[103].在B(C6F5)3活化硅烷的反应中, 通常是B(C6F5)3先与硅烷中的Si—H键形成[R3Si…H…B(C6F5)3]中间体, 之后被活化的硅烷能够继续与多种底物发生进一步反应[104]. 1996年, Piers课题组[105]报道了B(C6F5)3催化活化Ph3SiH与C=O键发生的硅氢化反应, 其中C=O键可以为酮基、醛基、酯基等, 并通过动力学实验对其反应机理进行了探索(Scheme 56).后续的相关报道中, 对B(C6F5)3活化硅烷发生羰基硅氢化的机理进行了更进一步的研究(Scheme 57)[104a, 106]. Piers课题组[107]还研究了B(C6F5)3催化活化三取代硅烷与共轭烯酮发生的硅氢化反应.反应具有很好的化学选择性, 倾向于生成1, 4-加成产物而非1, 2-加成产物.文章中还对不同种类硅烷的反应活性进行了探索, 结果表明当Si上具有较大位阻取代基时, 不利于反应的进行.他们并对其反应机理进行了研究(Scheme 56).
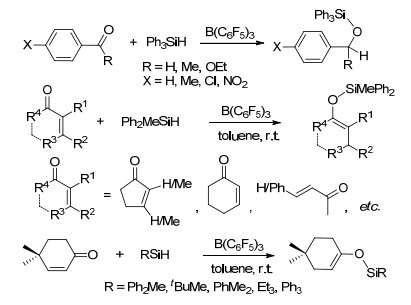
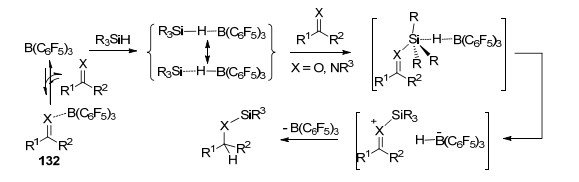
他们还报道了B(C6F5)3活化三取代硅烷(Ph3SiH或Et3SiH)与醇发生的硅烷基取代反应, 并对其机理进行了研究(Eq. 8)[108].
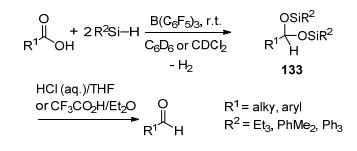
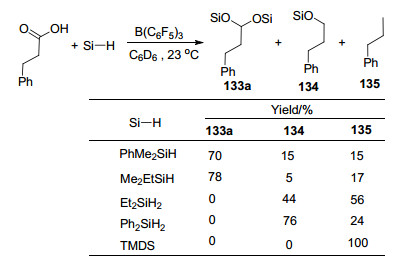
B(C6F5)3也具有催化硅烷与羧基发生硅氢化反应生成二硅缩醛类产物133的能力, 产物133则能在酸性环境中进一步水解生成对应的醛(Scheme 58)[109b].在该报道中, 当使用硅烷Et2SiH2, Ph2SiH2, PhMe2SiH或者Me2EtSiH时, 产物中能够观察到脱氧还原产物134和135的生成; 当使用1, 1, 3, 3-四甲基二硅氧烷(TMDS)时, 则产物完全为产物135 (Scheme 59).此前, Gevorgyan和Yamamoto等[109a]也对这一现象进行了系统的报道, 在他们的报道中不仅实现了羧基的脱氧还原, 还实现了醛基、酰氯以及脂基的脱氧还原反应, 并最终生成甲基.
|
|
(8) |
在此之后, 更多的关于B(C6F5)3活化硅烷进行脱氧还原的反应见诸报道, 酰胺基、酮基、醇羟基等在内的基团还原成对应的烷基化产物也得以实现[110].最近, Oestreich等[111]报道了B(C6F5)3催化Et3SiH脱氧还原砜或者亚砜生成硫醚类产物的反应(Eq. 9).
|
|
(9) |
更有趣的是, Gagné等[112]实现了利用催化量的B(C6F5)3催化Et2SiH2脱氧还原单糖或者多糖化合物生成对应的饱和或不饱和烷烃化合物.这为将可再生的植物纤维转化为可被直接利用的化学原料或者燃料提供了一个新思路(Eq. 10).
|
|
(10) |
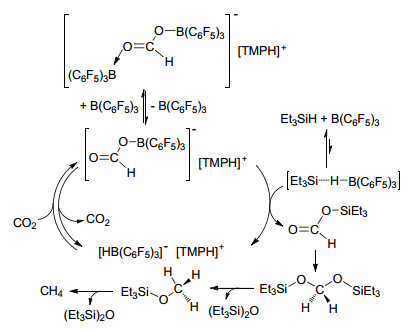
Piers等[113]报道了2, 2, 6, 6-四甲基哌啶(TMP)与B(C6F5)3组成的FLP体系在硅烷存在的条件下还原CO2成甲烷的反应.反应中CO2首先被TMP/B(C6F5)3 FLP体系活化, 之后进一步与被B(C6F5)3活化的硅烷反应, 最终生成产物CH4 (Scheme 60). Chen和Falivene等[114]报道了首例[Al(C6F5)3+B(C6F5)3]协同催化CO2硅氢化反应, 并最终生成甲烷.反应中铝和硼这两种催化剂的协同作用至关重要.前文中还提到了FLP活化的硅烷还原氢化CO2生成甲烷, 或者在邻苯二胺存在的条件下生成苯并咪唑的反应, 在此便不再赘述[92, 95].
Rosenberg课题组[115a]报道了B(C6F5)3催化三取代硅烷或者二聚硅烷与二苯基硫酮发生的硅氢化反应, 该反应具有与Scheme 57相类似的反应机理.不同的是, 当底物为硫酮时类似的加合物132相比较底物为羰基酮时更难生成, 反应速率也比对应的羰基酮较慢(Scheme 61).几年之后, 他们课题组又研究了相似条件下聚苯硅烷与二苯基硫酮的反应, 形成同时含有Si—H键和Si—SR键的多聚硅烷化合物.同时他们还发现若当把反应中的三取代硅烷更换为单取代硅烷、二取代硅烷或者聚苯硅烷时, 反应中的二苯基硫酮会被“过度还原”发生脱硫反应, 最终生成二苯基甲烷(Scheme 61)[115b]. 2015年, Akiyama等[115c]报道了B(C6F5)3活化硅烷与硫化物的反应, 实现了利用B(C6F5)3催化硅烷进行的氢化脱硫反应(Scheme 61).
2000年, Piers课题组[116a]报道了亚胺类产物的还原反应, 反应中首先形成了亚胺的硅氢化产物, 之后该产物进一步发生了水解反应, 最终形成生成胺类化合物(Scheme 62).不久之后, Oestreich等[116b, 111c]利用手性硼烷, 将该方法用于不对称氢化还原亚胺的反应之中, 并对其机理进行了详细的研究.
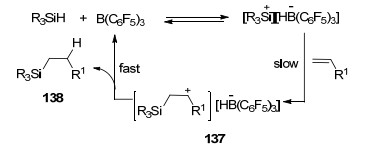
烯烃类底物也能够通过类似的方法发生硅氢化反应, Gevorgyan等[117a]首先报道了B(C6F5)3活化后的Si—H与烯烃的加成反应, 生成了β-硅基碳正离子中间体137, 再经过一步与硼氢化物的氢转移反应最终生成目标产物138 (Scheme 63).该报道中尝试了若干种硅烷均能够以较高产率得到对应产物, 但当使用较大位阻硅烷iPr3SiH时反应则不能发生, 该反应具有较宽的底物适用范围.近年, Oestreich等[117b]实现了B(C6F5)3催化3-硅甲烷环己基-1, 4-二烯与烯烃的硅氢化反应, 反应最终产物为硅甲基取代烷烃和苯.最近, 烯烃的硅氢化还原反应更是被运用到了高分子化合物的修饰之中[117c].同时, 炔烃类化合物也能够通过类似的方法进行还原[117d].
此外, 腈基和硝基也能够在类似的条件下发生硅氢化反应, 最终生成胺或亚胺类产物(Scheme 64)[118].
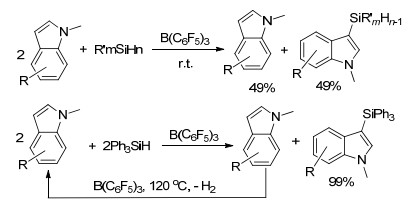
最近, 张越涛课题组[119]报道了B(C6F5)3催化硅烷与N-甲基吲哚在室温下发生的歧化反应, 反应能够1:1生成吲哚的氢化产物和C3-硅烷基取代产物.若将反应温度升高至120 ℃, 则单一生成C3-硅烷基取代产物(Scheme 65).该反应具有较高的原子经济性, 无溶剂条件下能够将反应催化剂用量降低至0.01 mol%, 并且在循环了10次之后仍然保持很高转化率.文章中并对其反应机理进行了详细研究.
含硫杂环化合物也能够被B(C6F5)3和硅烷还原, Ingleson课题组[120]报道了在体系中加入2, 6-二取代吡啶的条件下, 还原噻吩类化合物的反应(Eq. 11).报道中并对其反应机理进行了详细的研究.
|
|
(11) |
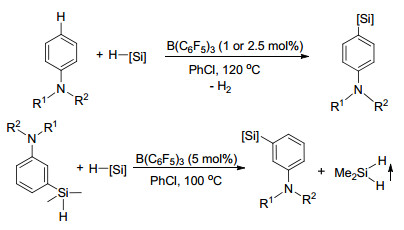
最近, 侯召民课题组[121a]报道了一系列关于B(C6F5)3催化的N, N-二取代苯胺4-位C—H键硅烷基化反应, 其中B(C6F5)3对硅烷的活化在反应中起到了关键的作用.该反应具有非常广泛的底物适用范围, 同时不论是单取代硅烷还是二取代、三取代硅烷都能够以较高产率得到目标化合物, 这也是首例关于FLP催化芳基硅基化的报道(Scheme 66).之后, 他们课题组[121b]又实现了B(C6F5)3催化N, N-二取代苯胺-3-硅烷类化合物的重组反应(Scheme 66).
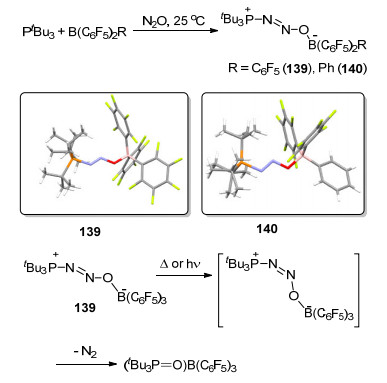
Stephan等[122]在2009年报道了FLP与N2O的反应, 室温下tBu3P和B(C6F5)2R (R=C6F5, Ph)与N2O反应生成化合物tBu3P(NNO)B(C6F5)2R (R=C6F5 139, Ph 140), 并分别得到了其单晶结构.将化合物139加热至135 ℃并保持44 h, 或利用光催化, 则能释放出氮气并生成(tBu3P=O)B(C6F5)3 (Scheme 67).
之后, 他们[13b]又对FLP体系与氮氧化物的反应进行了更为详细的研究, 深入探讨了一系列氟硼烷如B(C6F5)3, PhB(C6F5)2, MesB(C6F5)2, (C6F5)2BOC6F5, B(C6F4-p-H)3, B(C6H4-p-F)3和1, 4-(C6F5)2BC6F4B(C6F5)2等与大位阻磷酸tBu3P和N2O的反应, 并利用核磁、单晶等手段详细的研究了生成产物的结构及性质.
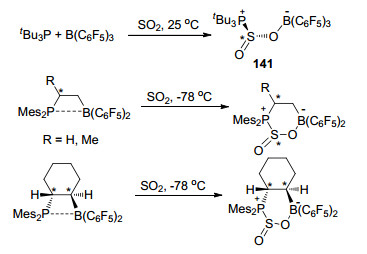
2013年, 关于FLP与SO2的反应见诸报道[14]. tBu3P/B(C6F5)3能够与SO2生成两性离子tBu3P—S(O)—OB(C6F5)3 (141), 该化合物结构与前文中提到的tBu3P/B(C6F5)3与CO2生成的117 (Scheme 47)结构类似, 不同之处在于117中CO2加成后的碳原子为平面结构, 而141中的硫原子则为三角锥形, 文献中报道了141的单晶结构.报道中还研究了其它几种类似的FLP体系与SO2的反应, 如Scheme 68所示, 同时详细报道了其产物的结构和性质.
FLP还具有活化氧气的能力, Erker课题组[123]报道了N/B FLP在室温下与O2反应生成四氢异噁唑/硼酸盐, 并通过单晶结构以及密度泛函(DFT)计算给出了可能的机理(Eq. 12).
|
|
(12) |
FLP也具有异裂二硫化物的能力, 与FLP异裂氢气的反应类似, 二硫化物(PhSSPh)与FLP tBu2P(C6F4)- B(C6F5)2反应生成两性离子[tBu2P(SPh)(C6F4)B(SPh)-(C6F5)2].同样tBu3P/B(C6F5)3与RSSR反应生成[tBu3P(SR)][(RS)B(C6F5)3] (R=Ph, p-tolyl, iPr).令人出乎意料的是, 当使用tBu3P/B(C6F5)3与BnSSBn反应时, 则能够以1:1:1的比例得到tBu3P=S、Bn2S和B(C6F5)3的混合物, 这可能是由于反应过程中生成了类似的两性离子[tBu3P(SBn)][(BnS)B(C6F5)3]所导致(Scheme 68)[124].
氮杂卡宾ItBu与(NH3)B(C6F5)3或者(RR'NH)B-(C6F5)3 (R=H, R'=Ph或R'=R=Ph)具有活化N—H的作用, 反应生成咪唑阳离子和硼胺阴离子盐类化合物[(tBuN)2C3H3][RR’NB(C6F5)3] (Eq. 13)[125, 48a].
|
|
(13) |
2016年, 杜海峰课题组[126]报道了吡啶类化合物与B(C6F5)3所组成的FLP体系活化氨硼烷中N—H键和B—H键, 并能够进一步还原吡啶成为哌啶的报道.其中氨硼烷作为氢源, 反应中避免了氢气的使用, 并能够以较高收率(44%~88%)选择性得到cis-构型的哌啶产物.同年, 他们课题组[127]还发表了利用类似的体系完成不对称还原亚胺类化合物的报道, 取得了较高的转化率和选择性(Scheme 69).
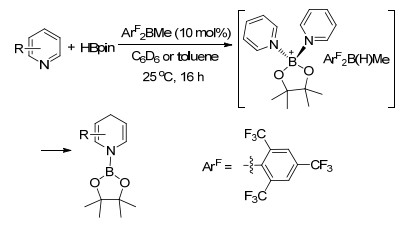
2015年, 王华冬课题组[128]报道了大位阻有机硼试剂高效催化吡啶类化合物与频那醇硼烷所发生的1, 4-硼氢化反应, 反应具有化学选择性高和区域选择性高等特点, 他们并利用NMR, 密度泛函理论计算等手段对反应中间体[Py2Bpin][ArF2B(H)Me]进行了研究(Scheme 70).
B(C6F5)3与2, 6-二甲基吡啶组成的FLP体系还具有活化C—H键的能力, 并能够形成对应的B(C6F5)3取代的硼化产物.反应体系中加入了10 mol%的儿茶酚硼烷作为催化剂, 能够在室温下以较高产率得到目标产物, 文献中还报道了产物的单晶结构, 并通过计算给出了可能的反应机理(Eq. 14)[129].
|
|
(14) |
张俊良等[130]报道了B(C6F5)3催化苯酚与重氮乙酸酯类化合物发生的邻位取代的反应.以5 mol%的催化剂量在室温下于0.5 h内以较高的收率和化学选择性得到目标产物, 他们并通过氘代控制实验对反应机理进行了深入的研究(Eq. 15).
|
|
(15) |
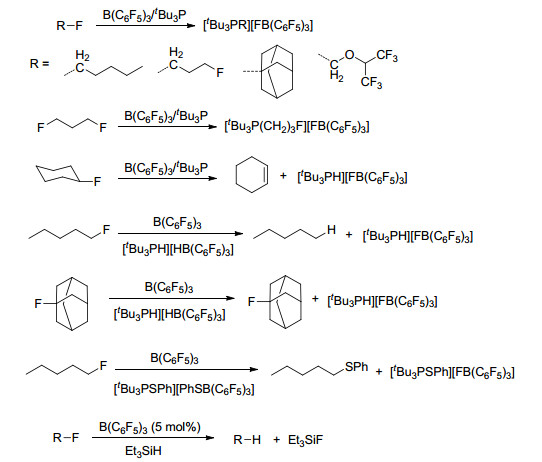
B(C6F5)3与较大空间位阻的磷化物组成的FLP具有活化C—F键的能力, 例如, 将B(C6F5)3/烷基氟化物与[tBu3PH][HB(C6F5)3]或[tBu3PSPh][PhSB(C6F5)3]进行反应, 能够得到烷烃与[tBu3PH][FB(C6F5)3]或[tBu3PSPh]-[FB(C6F5)3]的混合物.在Et3SiH存在的条件下, B(C6F5)3也能够与烷基氟化物发生类似的反应(Scheme 71)[131].
除了上述化合物外, B(C6F5)3类化合物所形成的FLP体系还具有活化多种其它小分子或基团的能力, 比如能够活化卤素[132]、碳卤键[133]、羰基[134], C(sp2)—H[135]、P—H键[136]和硝基[137]等.
“受阻路易斯酸碱对”化学是一片崭新而充满活力的领域.得益于化学家对未知世界的积极探索, 受阻路易斯酸碱对化学在近几年得到了迅速的发展. FLP体系为活化小分子提供了一个新方法和新思路, 包括氢气、烯烃、炔烃、二氧化碳/一氧化碳、环醚等在内的小分子都可以利用FLP体系进行活化并参与反应.同时FLP体系通常为无金属体系, 相比较而言更加绿色环保, 更重要的是这些发现打破了人们通常认为的活化小分子离不开过渡金属的固有思维, 也使化学家重新对主族元素产生了浓厚的兴趣.未来几年关于FLP的研究可能会更加倾向于这些新方法的应用, 尤其是在天然产物合成、生物活性中间体合成等领域可能有突飞猛进的发展.
Lewis, G. N. Valence and the Structure of Atoms and Molecules, Chemical Catalogue Company, New York, 1923.
Shore, S. G.; Parry, R. W. J.Am.Chem. Soc. 1955, 77, 6084. doi: 10.1021/ja01627a103
Brown, H. C.; Schlesinger, H. I.; Cardon, S. Z. J. Am.Chem.Soc. 1942, 64, 325. doi: 10.1021/ja01254a031
(a) Wittig, G. ; Benz, E. Chem. Ber. 1959, 92, 1999.
(b) Tochtermann, W. Angew. Chem., Int. Ed. Engl. 1966, 5, 351.
(a) Bontemps, S. ; Bouhadir, G. ; Miqueu, K. ; Bourissou, D. J. Am. Chem. Soc. 2006, 128, 12056.
(b) Grobe, J. ; Lütke-Brochtrup, K. ; Krebs, B. ; Läge, M. ; Niemeyer, H. -H. ; Würthwein E. -U. Z. Naturforsch. B 2006, 61, 882.
(c) Hudnall, T. W. ; Kim, Y. M. ; Bebbington, M. W. P. ; Bourissou, D. ; Gabba, F. P. J. Am. Chem. Soc. 2008, 130, 10890.
(d) Chiu, C. -B. ; Gabba, F. P. Dalton Trans. 2008, 814.
(e) Lin, T. -P. ; Gualco, P. ; Ladeira, S. ; Amgoune, A. ; Bourissou, D. ; Gabba, F. P. C. R. Chim. 2010, 13, 1168.
(f) Courtemanche, M. A. ; Legare, M. A. ; Maron, L. ; Fontaine, F. G. J. Am. Chem. Soc. 2013, 135, 9326
Roesler, R.; Piers, W. E.; Parvez, M. J.Organomet. Chem. 2003, 680, 218. doi: 10.1016/S0022-328X(03)00384-X
Welch, G. C.; Juan, R. R. S.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124. doi: 10.1126/science.1134230
Welch, G. C.; Stephan, D. W. J.Am. Chem.Soc. 2007, 129, 1880. doi: 10.1021/ja067961j
Spies, P.; Erker, G.; Kehr, G.; Bergander, K.; Fröhlich, R.; Grimme, S.; Stephan, D. W. Chem.Commun. 2007, 5072. http://www.ncbi.nlm.nih.gov/pubmed/18049757
McCahill, J. S. J.; Welch, G. C.; Stephan, D. W. Angew.Chem., Int.Ed. 2007, 46, 4968. doi: 10.1002/(ISSN)1521-3773
(a) Peng, B. ; Nie, Y. China Terminol. 2010, 12, 44(in Chinese).
(彭斌, 聂永, 中国科技术语, 2010, 12, 44. )
(b) Liu, Y. ; Du, H. Acta Chim. Sinica 2014, 72, 771(in Chinese).
(刘勇兵, 杜海峰, 化学学报, 2014, 72, 711. )
(a) Möming, C. M. ; Otten, E. ; Kehr, G. ; Frölich, R. ; Grimme, S. ; Stephan, D. W. ; Erker, G. Angew. Chem., Int. Ed. 2009, 48, 6643.
(b) Peuser, I. ; Neu, R. C. ; Zhao, X. ; Ulrich, M. ; Schirmer, B. ; Tannert, J. A. ; Kehr, G. ; Froehlich, R. ; Grimme, S. ; Erker, G. ; Stephan, D. W. Chem. -Eur. J. 2011, 17, 9640
(a) Cardenas, A. J. P. ; Culotta, B. J. ; Warren, T. H. ; Grimme, S. ; Stute, A. ; Frölich, R. ; Kehr, G. ; Erker, G. Angew. Chem., Int. Ed. 2011, 50, 7567.
(b) Neu, R. C. ; Otten, E. ; Lough, A. ; Stephan, D. W. Chem. Sci. 2011, 2, 170
Sajid, M.; Klose, A.; Birkmann, B.; Liang, L.; Schirmer, B.; Wiegand, T.; Eckert, H.; Lough, A. J.; Fröhlich, R.; Daniliuc, C. G.; Grimme, S.; Stephan, D. W.; Kehr, G.; Erker, G. Chem.Sci. 2013, 4, 213. doi: 10.1039/C2SC21161K
Möming, C. M.; Fröel, S.; Kehr, G.; Frölich, R.; Grimme, S.; Erker, G. J.Am.Chem.Soc. 2009, 131, 12280. doi: 10.1021/ja903511s
Dureen, M. A.; Stephan, D. W. J.Am.Chem.Soc. 2009, 131, 8396. doi: 10.1021/ja903650w
(a) Erker, G. ; Stephan, D. W. Frustrated Lewis Pairs I Uncovering and Understanding, Springer-Verlag, Berlin, Heidelberg, 2013.
(b) Stephan, D. W. ; Erker, G. Angew. Chem., Int. Ed. 2010, 49, 46.
(c) Stephan, D. W. ; Erker, G. Angew. Chem., Int. Ed. 2015, 54, 6400.
(d) Lu, Z. ; Ye, H. ; Wang, H. Top. Curr. Chem. 2013, 334, 59.
(e) Wang, H. ; Zheng, Y. ; Pan, Z. ; Fu, H. ; Ling, F. ; Zhong, W. Chin. J. Org. Chem. 2017, 37, 301(in Chinese).
(王辉, 郑亿, 潘振涛, 傅鸿樑, 凌飞, 钟为慧, 有机化学, 2017, 37, 301. )
(f) Stephan, D. W. Acc. Chem. Res. 2015, 48, 306.
(g) Stephan, D. W. Org. Biomol. Chem. 2012, 10, 5740
Schulz, F.; Sumerin, V.; Heikkinen, S.; Pedersen, B.; Wang, C.; Atsumi, M.; Leskel, M.; Repo, T.; Pyykkö, P.; Petry, W.; Rieger, B. J.Am. Chem.Soc. 2011, 133, 20245. doi: 10.1021/ja206394w
Zaher, H.; Ashley, A. E.; Irwin, M.; Thompson, A. L.; Gutmann, M. J.; Krämera, T.; O'Hare D. Chem. Commun. 2013, 49, 9755. doi: 10.1039/c3cc45889j
Karkamkar, A.; Parab, K.; Camaioni, D. M.; Neiner, D.; Cho, H. M.; Nielsen, T. K.; Autrey, T. Dalton Trans. 2013, 42, 615. doi: 10.1039/C2DT31628E
Kim, H. W.; Rhee, Y. M. Chem.-Eur.J. 2009, 15, 13348. doi: 10.1002/chem.200902322
Bakó, I.; Stirling, A.; Bálint, S.; Pápai, I. Dalton Trans. 2012, 41, 9023. doi: 10.1039/c2dt30370a
Rocchigiani, L.; Ciancaleoni, G.; Zuccaccia, C.; Macchioni, A. J. Am.Chem.Soc. 2014, 136, 112. doi: 10.1021/ja4119169
Marwitz, A. J. V.; Dutton, J. L.; Mercier, L. G.; Piers, W. E. J. Am.Chem.Soc. 2011, 133, 10026. doi: 10.1021/ja203214f
Zhao, X.; Stephan, D. W. J.Am.Chem.Soc. 2011, 133, 12448. doi: 10.1021/ja205598k
(a) Rokob, T. A. ; Hamza, A. ; Stirling, A. ; Soós, T. ; Pápai, I. Angew. Chem., Int. Ed. 2008, 47, 2435.
(b) Grimme, S. ; Kruse, H. ; Goerigk, L. ; Erker, G. Angew. Chem., Int. Ed. 2010, 49, 1402
Schwendemann, S.; Fröhlich, R.; Kehr, G.; Erker, G. Chem.Sci. 2011, 2, 1842. doi: 10.1039/c1sc00124h
(a) Axenov, K. ; Mömming, C. ; Kehr, G. ; Fröhlich, R. ; Erker, G. Chem. -Eur. J. 2010, 16, 14069.
(b) Spies, P. ; Kehr, G. ; Bergander, K. ; Wibbeling, B. ; Fröhlich, R. ; Erker, G. Dalton Trans. 2009, 1534
Wiegand, T.; Eckert, H.; Ekkert, O.; Fröhlich, R.; Kehr, G.; Erker, G.; Grimme, S. J.Am.Chem.Soc. 2012, 134, 4236. doi: 10.1021/ja210160k
Wang, X.; Kehr, G.; Daniliuc, C. G.; Erker, G. J.Am.Chem. Soc. 2014, 136, 3293. doi: 10.1021/ja413060u
(a) Rosorius, C. ; Daniliuc, C. G. ; Fröhlich, R. ; Kehr, G. ; Erker, G. J. Organomet. Chem. 2013, 744, 149.
(b) Rosorius, C. ; Kehr, G. ; Fröhlich, R. ; Grimme, S., Erker, G. Organometallics 2011, 30, 4211
(a) Stute, A. ; Kehr, G. ; Fröhlich, R. ; Erker, G. Chem. Commun. 2011, 47, 4288.
(b) Stute, A. ; Kehr, G. ; Daniliuc, C. G. ; Fröhlich, R. ; Erker, G. Dalton Trans. 2013, 42, 4487
Stute, A.; Heletta, L.; Fröhlich, R.; Daniliuc, C. G.; Kehr, G.; Erker, G. Chem.Commun. 2012, 48, 11739. doi: 10.1039/c2cc36782c
Geier, S. J.; Stephan, D. W. J.Am.Chem.Soc. 2009, 131, 3476. doi: 10.1021/ja900572x
Dureen, M. A.; Stephan, D. W. J.Am.Chem.Soc. 2010, 132, 13559. doi: 10.1021/ja1064153
Xu, B.-H.; Bussmann, K.; Fröhlich, R.; Daniliuc, C. G.; Brandenburg, J. G.; Grimme, S.; Kehr, G.; Erker, G. Organometallics 2013, 32, 6745. doi: 10.1021/om4004225
(a) Schwendemann, S. ; Oishi, S. ; Saito, S. ; Fröhlich, R. ; Kehr, G. ; Erker, G. Chem. -Asian J. 2013, 8, 212.
(b) Lindqvist, M. ; Axenov, K. ; Nieger, M. ; Raeisaenen, M. ; Leskelä, M. ; Repo T. Chem. -Eur. J. 2013, 19, 10412
Welch, G. C.; Masuda, J. D.; Stephan, D. W. Inorg.Chem. 2006, 45, 478. doi: 10.1021/ic051713r
Wang, H., Fröhlich, R.; Kehr, G.; Erker, G. Chem.Commun. 2008, 5966. http://www.mendeley.com/research/heterolytic-dihydrogen-activation-18bisdiphenylphosphinonaphthalenebc6f53-pair-application-metalfree/
(a) Oezguen, T. ; Ye, K. -Y. ; Daniliuc, C. G. ; Wibbeling, B. ; Liu, L. ; Grimme, S. ; Kehr, G. ; Erker, G. Chem. -Eur. J. 2016, 22, 5988.
(b) Oezguen, T. ; Bergander, K. ; Liu, L. ; Daniliuc, C. G. ; Grimme, S. ; Kehr, G. ; Erker, G. Chem. -Eur. J. 2016, 22, 11958
Geier, S. J.; Gilbert, T. M.; Stephan, D. W. J.Am.Chem. Soc. 2008, 130, 12632. doi: 10.1021/ja805493y
Blagg, R. J.; Lawrence, E. J.; Resner, K.; Oganesyan, V. S.; Herrington, T. J.; Ashley, A. E.; Wildgoose, G. G. Dalton Trans. 2016, 45, 6023. doi: 10.1039/C5DT01918D
Ramos, A.; Lough, A. J.; Stephan, D. W. Chem.Commun. 2009, 1118. http://onlinelibrary.wiley.com/resolve/reference/XREF?id=10.1039/b819525k
(a) Liptau, P. ; Neumann, M. ; Erker, G. ; Kehr, G. ; Fröhlich, R. ; Grimme, S. Organometallics 2004, 23, 21.
(b) Huber, D. P. ; Kehr, G. ; Bergander, K. ; Fröhlich, R. ; Erker, G. ; Tanino, S. ; Ohki, Y. ; Tatsumi, K. Organometallics 2008, 27, 5279
(a) Axenov, K. V. ; Kehr, G. ; Fröhlich, R. ; Erker, G. J. Am. Chem. Soc. 2009, 131, 3454.
(b) Unverhau, K. ; Lübbe, G. ; Wibbeling, B. ; Fröhlich, R. ; Kehr, G. ; Erker, G. Organometallics 2010, 29, 5320
(a) Ullrich, M. ; Lough, A. J. ; Stephan, D. W. J. Am. Chem. Soc. 2009, 131, 52.
(b) Ullrich, M. ; Lough, A. J. ; Stephan, D. W. Organometallics 2010, 29, 3647
Xing, J.-Y.; Buffet, J.-C.; Rees, N. H.; Norby, P.; O'Hare, D. Chem. Commun. 2016, 52, 10478. doi: 10.1039/C6CC04937K
(a) Chase, P. A. ; Stephan, D. W. Angew. Chem., Int. Ed. 2008, 47, 7433.
(b) Geier, S. J. ; Chase, P. A. ; Stephan, D. W. Chem. Commun. 2010, 46, 4884.
(c) Holschumacher, D. ; Bannenberg, T. ; Hrib, C. G. ; Jones, P. G. ; Tamm, M. Angew. Chem., Int. Ed. 2008, 47, 7428
Lam, J.; Guenther, B. A. R.; Farrell, J. M.; Eisenberger, P.; Bestvater, B. P.; Newman, P. D.; Melen, R. L.; Crudden, C. M.; Stephan, D. W. Dalton Trans. 2016, 45, 15303. doi: 10.1039/C6DT02202B
Tao, X.; Kehr, G.; Wang, X.; Daniliuc, C. G.; Grimme, S.; Erker, G. Chem.-Eur. J. 2016, 22, 9504. doi: 10.1002/chem.201602058
Chase, P. A.; Jurca, T., Stephan, D. W. Chem.Commun. 2008, 1701. http://europepmc.org/abstract/MED/18368170
(a) Tussing, S. ; Paradies, J. Dalton Trans. 2016, 45, 6124.
(b) Tussing, S. ; Kaupmees, K. ; Paradies, J. Chem. -Eur. J. 2016, 22, 7422
Courtemanche, M.-A.; Rochette, E.; Legare, M.-A.; Bi, W.; Fontaine, F.-G. Dalton Trans. 2016, 45, 6129. doi: 10.1039/C5DT03916A
Sumerin, V.; Schulz, F.; Nieger, M.; Leskela, M.; Repo, T.; Rieger, B. Angew.Chem., Int.Ed. 2008, 47, 6001. doi: 10.1002/anie.200800935
(a) Lu, Z. P. ; Cheng, Z. H. ; Chen, Z. X. ; Weng, L. H. ; Li, Z. H. ; Wang, H. D. Angew. Chem., Int. Ed. 2011, 50, 12227.
(b) Yepes, D. ; Jaque, P. ; Fernandez, I. Chem. -Eur. J. 2016, 22, 18801
Geier, S. J.; Gille, A. L.; Gilbert, T. M.; Stephan, D. W. Inorg. Chem. 2009, 48, 10466. doi: 10.1021/ic901726b
Zheng, J.; Lin, Y.-J.; Wang, H. Dalton Trans. 2016, 45, 6088. doi: 10.1039/C5DT03815D
(a) Liu, Y. ; Du, H. J. Am. Chem. Soc. 2013, 135, 6810.
(b) Liu, Y. ; Du, H. J. Am. Chem. Soc. 2013, 135, 12968.
(c) Wei, S. ; Du, H. J. Am. Chem. Soc. 2014, 136, 12261.
(d) Wang, W. ; Meng, W. ; Du, H. Dalton Trans. 2016, 45, 5945.
(e) Wang, W. ; Feng, X. ; Du, H. Org. Biomol. Chem. 2016, 14, 6683.
(f) Ren, X. ; Du, H. J. Am. Chem. Soc. 2016, 138, 810.
(g) Wang, Y. W. ; Chen, W. Q. ; Lu, Z. P. ; Li, Z. H. ; Wang, H. D. Angew. Chem., Int. Ed. 2013, 52, 7496
(a) Stirling, A. ; Hamza, A. ; Rokob, T. A. ; Pápai, I. Chem. Commun. 2008, 3148.
(b) Guo, Y. ; Li, S. Eur. J. Inorg. Chem. 2008, 2501
Zhao, X.; Stephan, D. W. Chem.Sci. 2012, 3, 2123. doi: 10.1039/c2sc20262j
Voss, T.; Mahdi, T.; Otten, E.; Fröhlich, R.; Kehr, G.; Stephan, D. W.; Erker, G. Organometallics 2012, 31, 2367. doi: 10.1021/om300017u
Voss, T.; Chen, C.; Kehr, G.; Nauha, E.; Erker, G.; Stephan, D. W. Chem.-Eur. J. 2010, 16, 3005. doi: 10.1002/chem.v16:10
Dornan, P. K.; Longobardi, L. E.; Stephan, D. W. Synlett 2014, 1521. http://www.chem.utoronto.ca/staff/DSTEPHAN/pubs.html
Ullrich, M.; Seto, K. S.-H.; Lough, A. J.; Stephan, D. W. Chem. Commun. 2009, 2335. http://onlinelibrary.wiley.com/resolve/reference/XREF?id=10.1039/b901212e
(a) Dureen, M. A. ; Brown, C. C. ; Stephan, D. W. Organometallics 2010, 29, 6594.
(b) Ye, H. Y. ; Lu, Z. P. ; You, D. ; Chen, Z. X. ; Li, Z. H. ; Wang, H. D. Angew. Chem., Int. Ed. 2012, 51, 12047
Yu, J.; Kehr, G.; Daniliuc, C. G.; Erker, G. Chem.Commun. 2016, 52, 1393. doi: 10.1039/C5CC07954C
Fukazawa, A.; Yamada, H.; Yamaguchi, S. Angew.Chem., Int. Ed. 2008, 47, 5582. doi: 10.1002/anie.v47:30
Klose, A.; Kehr, G.; Daniliuc, C. G.; Erker, G. Dalton Trans. 2016, 45, 2023. doi: 10.1039/C5DT03055B
Tanur, C. A.; Stephan, D. W. Organometallics 2011, 30, 3652. doi: 10.1021/om200336t
Dureen, M. A.; Brown, C. C.; Stephan, D. W. Organometallics 2010, 29, 6422. doi: 10.1021/om1008346
Zhao, X.; Lough, A.; Stephan, D. W. Chem.-Eur.J. 2011, 17, 6731. doi: 10.1002/chem.v17.24
Zhao, X.; Gilbert, T. M.; Stephan, D. W. Chem.-Eur.J. 2010, 16, 10304. doi: 10.1002/chem.201001575
Wilkins, L. C.; Gunther, B. A. R.; Walther, M.; Lawson, J. R.; Wirth, T.; Melen, R. L. Angew.Chem., Int.Ed. 2016, 55, 11292. doi: 10.1002/anie.201605239
Mahdi, T.; Stephan, D. W. Angew.Chem., Int.Ed. 2013, 52, 12418. doi: 10.1002/anie.201307254
Wittig, G.; Ruckert, A. Liebigs Ann.Chem. 1950, 566, 101. doi: 10.1002/(ISSN)1099-0690
Breen, T. L.; Stephan, D. W. Inorg.Chem. 1992, 31, 4019. doi: 10.1021/ic00045a032
(a) Avens, L. R. ; Barnhart, D. M. ; Burns, C. J. ; McKee, S. D. Inorg. Chem. 1996, 35, 537.
(b) Campello, M. P. C. ; Domingos, A. ; Santos, I. J. Organomet. Chem. 1994, 484, 37
Evans, W. J.; Leman, J. T.; Ziller, J. W.; Khan, S. I. Inorg. Chem. 1996, 35, 4283. doi: 10.1021/ic951627z
Mommertz, A.; Leo, R.; Massa, W.; Harms, K.; Dehnicke, K. Z.Anorg. Allg.Chem. 1998, 624, 1647. doi: 10.1002/(ISSN)1521-3749
(a) Breen, T. L. ; Stephan, D. W. Inorg. Chem. 1992, 31, 4019.
(b) Guo, Z. Y. ; Bradley, P. K. ; Jordan, R. F. Organometallics 1992, 11, 2690.
(c) Polamo, M. ; Mutikainen, I. ; Leskela, M. Acta Crystallogr. C 1997, 53, 1036
GomezSaso, M.; Mullica, D. F.; Sappenfield, E.; Stone, F. G. A. Polyhedron 1996, 15, 793. doi: 10.1016/0277-5387(95)00328-7
(a) Chivers, T. ; Schatte, G. Eur. J. Inorg. Chem. 2003, 3314.
(b) Kunnari, S. M. ; Oilunkaniemi, R. ; Laitinen, R. S. ; Ahlgren, M. J. Chem. Soc., Dalton. Trans. 2001, 3417
Campbell, J. P.; Gladfelter, W. L. Inorg.Chem. 1997, 36, 4094. doi: 10.1021/ic970171d
Zhang, Z.; Miao, C.; Xia, C.; Sun, W. Org.Lett. 2016, 18, 1522. doi: 10.1021/acs.orglett.6b00157
Birkmann, B.; Voss, T.; Geier, S. J.; Ullrich, M.; Kehr, G.; Erker, G.; Stephan, D. W. Organometallics 2010, 29, 5310. doi: 10.1021/om1003896
Sgro, M. J.; Dömer, J.; Stephan, D. W. Chem.Commun. 2012, 48, 7253. doi: 10.1039/c2cc33301e
Kreitner, C.; Geier, S. J.; Stanlake, L. J. E.; Caputo, C.; Stephan, D. W. Dalton Trans. 2011, 6771. http://www.ncbi.nlm.nih.gov/pubmed/21614351
Morton, J. G. M.; Dureen, M. A.; Stephan, D. W. Chem.Commun. 2010, 46, 8947. doi: 10.1039/c0cc02862b
Neu, R. C.; Ménard, G.; Stephan, D. W. Dalton Trans. 2012, 41, 9016. doi: 10.1039/c2dt30206c
Zhao, X.; Stephan, D. W. Chem.Commun. 2011, 47, 1833. doi: 10.1039/c0cc04791k
Ashley, A. E.; Thompson, A. L.; OÏHare, D. Angew.Chem., Int.Ed. 2009, 48, 9839. doi: 10.1002/anie.v48:52
Berkefeld, A.; Piers, W. E.; Parvez, M. J.Am.Chem. Soc. 2010, 132, 10660. doi: 10.1021/ja105320c
Takeuchi, K.; Stephan, D. W. Chem.Commun. 2012, 48, 11304. doi: 10.1039/c2cc36470k
Dobrovetsky, R.; Stephan, D. W. J.Am.Chem.Soc. 2013, 135, 4974. doi: 10.1021/ja401492s
Zhang, Z.; Sun, Q.; Xia, C.; Sun, W. Org.Lett. 2016, 18, 6316. doi: 10.1021/acs.orglett.6b03030
Liu, L.; Vankova, N.; Heine, T. Phys.Chem.Chem. Phys. 2016, 18, 3567. doi: 10.1039/C5CP06925D
Ménard, G.; Stephan, D. W. J.Am.Chem.Soc. 2010. 132, 1796. doi: 10.1021/ja9104792
Ménard, G.; Stephan, D. W. Angew.Chem., Int.Ed. 2011, 50, 8396. doi: 10.1002/anie.v50.36
(a) Ghoussoub, M. ; Yadav, S. ; Ghuman, K. K. ; Ozin, G. A. ; Singh, C. V. ACS Catal. 2016, 6, 7109.
(b) Ghuman, K. K. ; Hoch, L. B. ; Szymanski, P. ; Loh, J. Y. Y. ; Kherani, N. P. ; El-Sayed, M. A. ; Ozin, G. A. ; Singh, C. V. J. Am. Chem. Soc. 2016, 138, 1206.
(c) Ghuman, K. K. ; Hoch, L. B. ; Wood, T. E. ; Mims, C. ; Singh, C. V. ; Ozin, G. A. ACS Catal. 2016, 6, 5764
Sajid, M.; Kehr, G.; Daniliuc, C. G.; Erker, G. Angew.Chem., Int.Ed. 2014, 53, 1118. doi: 10.1002/anie.201307551
Sajid, M.; Elmer, L. M.; Rosorius, C.; Daniliuc, C. G.; Grimme, S.; Kehr, G.; Erker, G. Angew.Chem., Int.Ed. 2013, 52, 2243. doi: 10.1002/anie.201208750
Ye, K.-Y.; Kehr, G.; Daniliuc, C. G.; Liu, L.; Grimme, S.; Erker, G. Angew.Chem., Int.Ed. 2016, 55, 9216. doi: 10.1002/anie.201603760
(a) Oestreich, M. ; Hermeke, J. ; Mohr, J. Chem. Soc. Rev. 2015, 44, 2202.
(b) Fernandez-Alvarez, F. J. ; Aitani, A. M. ; Oro, L. A. Catal. Sci. Technol. 2014, 4, 611
(a) Houghton, A. Y. ; Hurmalainen, J. ; Mansikkamaki, A. ; Piers, W. E. ; Tuononen, H. M. Nat. Chem. 2014, 6, 983.
(b) Nikonov, G. I. ; Vyboishchikov, S. F. ; Shirobokov, O. G. J. Am. Chem. Soc. 2012, 134, 5488.
(c) Mathew, J. ; Eguchi, K. ; Nakajima, Y. ; Sato, K. ; Shimada, S. ; Choe, Y. K. Eur. J. Org. Chem. 2017, 4922
Parks, D. J.; Piers, W. E. J.Am.Chem.Soc. 1996, 118, 9440. doi: 10.1021/ja961536g
(a) Parks, D. J. ; Blackwell, J. M. ; Piers, W. E. J. Org. Chem. 2000, 65, 3090.
(b) Sakata, K. ; Fujimoto, H. J. Org. Chem. 2013, 78, 12505.
(c) Keess, S. ; Simonneau, A. ; Oestreich, M. Organometallics 2015, 34, 790
Blackwell, J. M.; Morrison, D. J.; Piers, W. E. Tetrahedron 2002, 58, 8247. doi: 10.1016/S0040-4020(02)00974-2
Blackwell, J. M.; Foster, K. L.; Beck, V. H.; Piers, W. E. J. Org.Chem. 1999, 64, 4887. doi: 10.1021/jo9903003
(a) Gevorgyan, V. ; Rubin, M. ; Liu, J. -X. ; Yamamoto, Y. J. Org. Chem. 2001, 66, 1672.
(b) Bézier, D. ; Park, S. ; Brookhart, M. Org. Lett. 2013, 15, 496
(a) Bajracharya, G. B. ; Nogami, T. ; Jin, T. ; Matsuda, K. ; Gevorgyan, V. ; Yamamoto, Y. Synthesis 2004, 308.
(b) Nimmagadda, R. D. ; McRae, C. Tetrahedron Lett. 2006, 47, 5755.
(c) Tan, M. ; Zhang, Y. Tetrahedron Lett. 2009, 50, 4912
Porwal, D.; Oestreich, M. Synthesis 2017, 49, 4698. doi: 10.1055/s-0036-1588476
Adduci, L. L.; McLaughlin, M. P.; Bender, T. A.; Becker, J. J.; Gagné, M. R. Angew.Chem., Int.Ed. 2014, 53, 1646. doi: 10.1002/anie.201306864
Berkefeld, A.; Piers, W. E.; Parvez, M. J.Am.Chem. Soc. 2010, 132, 10660. doi: 10.1021/ja105320c
Chen, J. W.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E. Y. X. J.Am.Chem.Soc. 2016, 138, 5321. doi: 10.1021/jacs.6b01497
(a) Harrison, D. J. ; McDonald, R. ; Rosenberg, L. Organometallics 2005, 24, 1398.
(b) Lee, P. T. K. ; Skjel, M. K. ; Rosenberg, L. Organometallics 2013, 32, 1575.
(c) Saito, K. ; Kondo, K. ; Akiyama, T. Org. Lett. 2015, 17, 3366-3369
(a) Blackwell, J. M. ; Sonmor, E. R. ; Scoccitti, T. ; Piers, W. E. Org. Lett. 2000, 2, 3921.
(b) Mewald, M. ; Oestreich, M. Chem. -Eur. J. 2012, 18, 14079.
(c) Hermeke, J. ; Mewald, M. ; Oestreich, M. J. Am. Chem. Soc. 2013, 135, 17537
(a) Rubin, M. ; Schwier, T. ; Gevorgyan, V. J. Org. Chem. 2002, 67, 1936.
(b) Simonneau A. ; Oestreich, M. Angew. Chem., Int. Ed. 2013, 52, 11905.
(c) Lee, P. T. K. ; Rosenberg, L. Dalton Trans. 2017, 46, 8818.
(d) Zhang, L. W. ; Wen, Z. G. ; Borzov, M. ; Nie, W. L. Acta Chim. Sinica 2017, 75, 819(in Chinese).
(张露文, 温志国, Borzov Maxim, 聂万丽, 化学学报, 2017, 75, 819. )
(a) Gandhamsetty, N. ; Jeong, J. ; Park, J. ; Park, S. ; Chang, S. J. Org. Chem. 2015, 80, 7281.
(b) Gandhamsetty, N. ; Park, J. ; Jeong, J. ; Park, S. W. ; Park, S. ; Chang, S. Angew. Chem., Int. Ed. 2015, 54, 6832
Han, Y. X.; Zhang, S. T.; He, J. H.; Zhang, Y. T. J.Am. Chem.Soc. 2017, 139, 7399. doi: 10.1021/jacs.7b03534
Curless, L. D.; Clark, E. R.; Dunsford, J. J.; Ingleson, M. J. Chem. Commun. 2014, 50, 5270. doi: 10.1039/C3CC47372D
(a) Ma, Y. H. ; Wang, B. L. ; Zhang, L. ; Hou, Z. M. J. Am. Chem. Soc. 2016, 138, 3663.
(b) Ma, Y. H. ; Zhang, L. ; Luo, Y. ; Nishiura, M. ; Hou, Z. M. J. Am. Chem. Soc. 2017, 139, 12434
Otten, E.; Neu, R. C.; Stephan, D. W. J.Am.Chem. Soc. 2009, 131, 9918. doi: 10.1021/ja904377v
Wang, T.; Kehr, G.; Liu, L.; Grimme, S.; Daniliuc, C. G.; Erker, G. J. Am.Chem.Soc. 2016, 138, 4302. doi: 10.1021/jacs.6b00325
Dureen, M. A.; Welch, G. C.; Gilbert, T. M.; Stephan, D. W. Inorg. Chem. 2009, 48, 9910. doi: 10.1021/ic901590s
Chase, P. A.; Gille, A. L.; Gilbert, T. M.; Stephan, D. W. Dalton Trans. 2009, 7179. http://www.ncbi.nlm.nih.gov/pubmed/20449161
Zhou, Q.; Zhang, L.; Meng, W.; Feng, X.; Yang, J.; Du, H. Org. Lett. 2016, 18, 5189. doi: 10.1021/acs.orglett.6b02610
Li, S.; Li, G.; Meng, W.; Du, H. J.Am.Chem.Soc. 2016, 138, 12956. doi: 10.1021/jacs.6b07245
Fan, X. T.; Zheng, J. H.; Li, Z. H.; Wang, H. D. J.Am. Chem.Soc. 2015, 137, 4916. doi: 10.1021/jacs.5b03147
Zheng, J.; Fan, X.; Zhou, B.; Li, Z. H.; Wang, H. Chem.Commun. 2016, 52, 4655. doi: 10.1039/C6CC00347H
Yu, Z.; Li, Y.; Shi, J.; Ma, B.; Liu, L.; Zhang, J. Angew.Chem., Int.Ed. 2016, 55, 14807. doi: 10.1002/anie.201608937
Caputo, C. B.; Stephan, D. W. Organometallics 2012, 31, 27. doi: 10.1021/om200885c
Froemel, S.; Daniliuc, C. G.; Bannwarth, C.; Grimme, S.; Bussmann, K.; Kehr, G.; Erker, G. Dalton Trans. 2016, 45, 19230. doi: 10.1039/C6DT04206F
Samigullin, K.; Georg, I.; Bolte, M.; Lerner, H.-W.; Wagner, M. Chem.-Eur. J. 2016, 22, 3478. doi: 10.1002/chem.201504791
Fasano, V.; Radcliffe, J. E.; Ingleson, M. J. ACS Catal. 2016, 6, 1793. doi: 10.1021/acscatal.5b02896
Chernichenko, K.; Lindqvist, M.; Kotai, B.; Nieger, M.; Sorochkina, K.; Papai, I.; Repo, T. J.Am.Chem.Soc. 2016, 138, 4860. doi: 10.1021/jacs.6b00819
Liu, Y. Z.; Fan, X. T.; Li, Z. H.; Wang, H. D. Chem.Commun. 2017, 53, 10890. doi: 10.1039/C7CC05028C
Chen, G.-Q.; Kehr, G.; Daniliuc, C. G.; Erker, G. Dalton Trans. 2016, 45, 6820. doi: 10.1039/C6DT00857G

图式 4 分子内P/B受阻路易斯酸碱对8活化氢气的可能机理
Scheme 4 Proposed mechanism for activation of hydrogen by intermolecular P/B FLP

图式 14 B(C6F5)3与tBuN=CPh(H)在氢气氛围下的反应
Scheme 14 Reaction between B(C6F5)3and tBuN=CPh(H) under H2 atmosphere

图式 16 2, 6-二甲基吡啶与B(C6F5)3活化氢气
Scheme 16 Activation of hydrogen by 2, 6-lutidine/B(C6F5)3

图式 32 1-吗啉环己烯、B(C6F5)3和炔烃的反应
Scheme 32 Reactions of 1-morpholinocyclohexene, B(C6F5)3 and alkynes

图式 41 苯胺和环醚生成N-芳基吡咯
Scheme 41 Synthesis of N-aryl-substituted azacycles from anilines and cyclic ethers

图式 51 ((CH3)3Si)3P和B(C6F4H)3与CO2的反应.
Scheme 51 Reaction of CO2 with ((CH3)3Si)3P and B(C6F4H)3

图式 52 B(C6F5)3/tBu3P FLP与CO2和H2的反应
Scheme 52 Reaction of B(C6F5)3/tBu3P FLP with CO2 and H2

图式 56 B(C6F5)3活化硅烷与C=O发生的硅氢化反应
Scheme 56 Hydrosilylation of C=O with hydrosilylation activated by B(C6F5)3

图式 57 B(C6F5)3催化酮类(X=O)和亚胺类(X=NR3)化合物硅氢化反应的机理
Scheme 57 Mechanism of the B(C6F5)3-catalysed hydrosilylation of ketones (X=O) and imines (X=NR3)

图式 58 B(C6F5)3催化的还原羧酸成醛的反应
Scheme 58 Reduction of carboxylic acids to aldehydes catalyzed by B(C6F5)3

图式 59 B(C6F5)3催化羧酸的脱氧还原
Scheme 59 B(C6F5)3-catalysed deoxygenation of carboxylic acids

图式 60 硅烷存在条件下B(C6F5)3催化还原CO2生成CH4
Scheme 60 B(C6F5)3-catalyzed reduction of CO2 to CH4 in the presence of hydrosilane

图式 61 B(C6F5)3催化硅烷与二苯基硫酮的硅氢化反应
Scheme 61 B(C6F5)3-catalyzed hydrosilylation of thioketones

图式 65 B(C6F5)3催化硅烷与N-甲基吲哚的歧化反应
Scheme 65 B(C6F5)3‑catalyzed disproportionation reaction of indoles

图式 66 B(C6F5)3催化的芳基C—H硅烷基化反应
Scheme 66 B(C6F5)3-catalyzed aromatic C—H bond silylation

图式 70 有机硼催化吡啶类化合物1, 4-硼氢化反应
Scheme 70 Organoborane catalyzed 1, 4-hydroboration of pyridines

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




















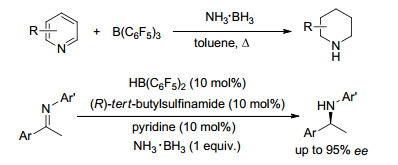



 下载:
下载:























































 下载:
下载: