Received Date:
25 December 2017 Revised Date:
29 January 2018 Available Online:
01 June 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21402129, 21572142, 21372165) and the National Key Research and Development Program of China (No. 2017YFA0505903)
Abstract:
Triplet-triplet annihilation (TTA) upconversion, a unique technique that converting low-energy photons into higher-energy photons, has attracted much attention owing to its potential applications in various fields, such as solar cells, bioimaging, photocatalysis and photoelectric device. TTA upconversion has several advantages over other upconversion methods, such as allowing for the use of low excitation power density, readily tunable excitation/emission wavelength and high upconversion quantum yield. Both triplet-triplet energy transfer (TTET) and TTA processes in TTA upconversion follow the Dexter energy transfer mechanism. The components involved have to diffuse in the media and collide within the lifetime of their excited states to complete the energy transfer. Thus, most efficient TTA-based upconversion has been achieved with donor-acceptor pairs that are molecularly dissolved in deaerated organic solvents, which however significantly limited their practical applications. In recent years, more and more efforts have beendevoted to explore high-efficient TTA upconversion under aerated conditions by employing specific solid materials or viscous liquids as matrices to block oxygen. The recent advance of research of TTA upconversion in aggregated systems is summarized, including rubbery polymers, gels, molecular crystals, nanoparticles and supramolecular self-assemblies.
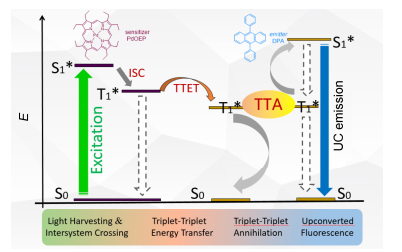
Figure 1.
Jablonski diagram illustrating the triplet ernergy transfer between triplet sensitizer and acceptor and triplet-triplet annihilation between the acceptors
Figure 6.
(a) Chemical structures of CDCB-derived heavy-atom-free sensitizers for TTA-UC in PU films and (b) schematic representation of the polymer TTA-UC system
Adopted with permission from Ref. [28]. Copyright The Royal Society of Chemistry 2016
Figure 7.
(a) Rendering of the materials morphology, (b) chemical structures of the constituents forming the glassy polymer, the surfactant, and the liquid phase and (c) emission pictures of the A-1/S-1 doped polymer under 543 nm laser irradiation
Adopted with permission from Ref. [29]. Copyright 2017 Wiley-VCH
Figure 9.
(a) Chemical structures of acceptors A-1 and donor S-6, solvents tetralin, gelator DMDBS and (b) photographs of a gelated material performing green-to-blue upconversion in an inverted cuvette
Adopted with permission from Ref. [35]. Copyright The Royal Society of Chemistry 2015
Figure 11.
(a) Molecular structures of the Organogelator FF and (b) upconversion photoluminescence intensity of S-5/A-1/FF with varied incident power density of 532 nm laser under ambient conditions
Inset: photograph of the upconversion emission in semitransparent gels. Adopted with permission from Ref. [39]. Copyright 2016 Wiley-VCH
Figure 12.
(a) Chemical structures of acceptors A-8/A-9 and donors S-7/S-8 and (b) schematic illustration of the host-guest complexation facilitated TTA-UC of S-8@A-9
Adopted with permission from Ref. [41]. Copyright 2016 American Chemical Society
Figure 14.
(a) Chemical structures of liquid acceptor A-10 and donor S-9, (b) photoluminescence spectra of the doped liquid (S-9/A-10=0.01 mol%) with different incident power intensities, and (c) schematic illustration of donor-to-acceptor TTET and energy migration among the acceptor liquid molecules
Inset: photographs of the S-9-doped liquid upon being exposed to white light (up) and a 532 nm green laser (bottom). Adopted with permission from Ref. [42]. Copyright 2013 American Chemical Society
Figure 15.
Chemical structures of A-11 and S-5, and schematic representation of nonvolatile TTA-UC systems using triplet energy migration in acceptor ion liquids
Adopted with permission from Ref. [43]. Copyright 2015 Wiley-VCH
Figure 16.
(a) align="center"hemical structure of lipophilic acceptor A-12, (b) schematic illustration of acceptor monolayer membrane A-12 formed in chloroform and (c) picture of chloroform dispersion showing upconverted emission upon 532 nm excitation under an aerated condition
Adopted with permission from Ref. [44]. Copyright 2015 Nature Publishing Group
Figure 17.
Chemical structures of water-soluble amphiphilic acceptor A-13 and donor S-10 and schematic illustration of aqueous molecular self-assembly and 10 bound to the assembly by electrostatic and hydrophobic interactions
Adopted with permission from Ref. [45]. Copyright 2016 The Royal Society of Chemistry
Figure 20.
Schematic representation of a dual-doped nanoparticle with S-5 (red sphere) and A-1 (blue spheres), showing the shielding from the external environment exerted by the NPs
Adopted with permission from Ref. [48]. Copyright 2012 Wiley-VCH
Figure 23.
Schematic illustration of TTA-UCL process of the upconversion nanocapsules, and chemical structures of Sensitizers (S-12) and Annihilators (A-16, and A-17)
Adopted with permission from Ref. [15]. Copyright 2013 American Chemical Society
Figure 24.
A procedure to fabricate bioprobe-attached SNC-B and SNC-G (A~E) and the schematic illustration of the cancer-specific, dual color imaging of (F) breast and (G) colorectal-cancer cells
Adopted with permission from Ref. [51]. Copyright 2016 American Chemical Society
Figure 25.
(a) Photocleavage of c[R]GDfK. DEACM-OH and intact cRGDfK peptide are released upon irradiation at 400 nm and (b) schematic of the phototriggering of the polymeric micellar nanoparticle by TTA-UC and FRET
Adopted with permission from Ref. [52]. Copyright 2015 American Chemical Society
(a) Islangulov, R. R.; Kozlov, D. V.; Castellano, F. N. Chem. Commun. 2005, 3776. (b) Chen, H. C.; Hung, C. Y.; Wang, K. H.; Chen, H. L.; Fann, W. S.; Chien, F. C.; Chen, P.; Chow, T. J.; Hsu, C. P.; Sun, S. S. Chem. Commun. 2009, 4064. (c) Schulze, T. F.; Czolk, J.; Cheng, Y. -Y.; Fückel, B.; MacQueen, R. W.; Khoury, T.; Crossley, M. J.; Stannowski, B.; Lips, K.; Lemmer, U.; Colsmann, A.; Schmidt, T. W. J. Phys. Chem. C2012, 116, 22794. (d) Singh-Rachford, T. N.; Castellano, F. N. J. Phys. Chem. A2008, 112, 3550. (e) Singhrachford, T. N.; Haefele, A.; Ziessel, R.; Castellano, F. N. J. Am. Chem. Soc. 2008, 130, 16164.
[2]
Zou, W.; Visser, C.; Maduro, J. A.; Pshenichnikov, M. S.; Hummelen, J. C. Nat. Photonics. 2012, 6, 560. doi: 10.1038/nphoton.2012.158
[3]
(a) Qian, L.; Yang, T.; Wei, F.; Li, F. J. Am. Chem. Soc. 2012, 134, 5390. (b) Wohnhaas, C.; Turshatov, A.; Mailänder, V.; Lorenz, S.; Baluschev, S.; Miteva, T.; Landfester, K. Macromol. Biosci. 2011, 11, 772. (c) Kim, J. H.; Kim, J. H. J. Am. Chem. Soc. 2012, 134, 17478.
[4]
(a) Islangulov, R. R.; Castellano, F. N. Angew. Chem., Int. Ed. 2006, 45, 5957. (b) Wu, W.; Wu, W.; Ji, S.; Guo, H.; Zhao, J. J. Organomet. Chem. 2011, 696, 2388. (c) Yang, C.; Nakamura, A.; Fukuhara, G.; Origane, Y.; Mori, T.; Wada, T.; Inoue, Y. J. Org. Chem. 2006, 71, 3126. (d) Yang, C.; Nakamura, A.; Wada, T.; Inoue, Y. Org. Lett. 2006, 8, 3005. (e) Yao, J.; Yan, Z.; Ji, J.; Wu, W.; Yang, C.; Nishijima, M.; Fukuhara, G.; Mori, T.; Inoue, Y. J. Am. Chem. Soc. 2014, 136, 6916.
(a) Zhao, J.; Ji, S.; Guo, H. RSC Adv. 2011, 1, 937. (b) Zhao, J.; Xu, K.; Yang, W.; Wang, Z.; Zhong, F. Chem. Soc. Rev. 2015, 44, 8904.
[7]
Baluschev, S.; Miteva, T.; Yakutkin, V.; Nelles, G.; Yasuda, A.; Wegner, G. Phys. Rev. Lett. 2006, 97, 143903. doi: 10.1103/PhysRevLett.97.143903
[8]
(a) Amemori, S.; Sasaki, Y.; Yanai, N.; Kimizuka, N. J. Am. Chem. Soc. 2016, 138, 8702. (b) Cheng, Y. Y.; Fuckel, B.; Khoury, T.; Clady, R. G.; Ekins-Daukes, N. J.; Crossley, M. J.; Schmidt, T. W. J. Phys. Chem. A2011, 115, 1047.
[9]
(a) Wu, W.; Zhao, J.; Sun, J.; Huang, L.; Yi, X. J. Mater. Chem. C2013, 1, 705. (b) Wu, W.; Ji, S.; Wu, W.; Guo, H.; Wang, X.; Zhao, J.; Wang, Z. Sens. Actuators, B: Chem. 2010, 149, 395. (c) Wu, W.; Zhao, J.; Guo, H.; Sun, J.; Ji, S.; Wang, Z. Chemistry2012, 18, 1961. (d) Wu, W.; Sun, J.; Ji, S.; Wu, W.; Zhao, J.; Guo, H. Dalton Trans. 2011, 40, 11550. (e) Wu, W.; Liu, L.; Cui, X.; Zhang, C.; Zhao, J. Dalton Trans. 2013, 42, 14374. (f) Wu, W.; Sun, J.; Cui, X.; Zhao, J. J. Mater. Chem. C2013, 1, 4577. (g) Wu, W.; Guo, H.; Wu, W.; Ji, S.; Zhao, J. J. Org. Chem. 2011, 76, 7056.
Monguzzi, A.; Tubino, R.; Meinardi, F. J. Phys. Chem. A2009, 113, 1171. doi: 10.1021/jp809971u
[12]
Murakami, Y.; Kikuchi, H.; Kawai, A. J. Phys. Chem. B2013, 117, 2487. doi: 10.1021/jp3124082
[13]
(a) Zhao, J.; Wu, W.; Sun, J.; Guo, S. Chem. Soc. Rev. 2013, 42, 5323. (b) Cui, X.; Zhao, J.; Mohmood, Z.; Zhang, C. Chem. Rec. 2016, 16, 173. (c) Singh-Rachford, T. N.; Castellano, F. N. Coord. Chem. Rev. 2010, 254, 2560.
[14]
(a) Khnayzer, R. S.; Blumhoff, J.; Harrington, J. A.; Haefele, A.; Deng, F.; Castellano, F. N. Chem. Commun. 2012, 48, 209. (b) Kim, H. I.; Weon, S.; Kang, H.; Hagstrom, A. L.; Kwon, O. S.; Lee, Y. S.; Choi, W.; Kim, J. H. Environ. Sci. Technol. 2016, 50, 11184.
Islangulov, R. R.; Lott, J.; Weder, C.; Castellano, F. N. J. Am. Chem. Soc. 2007, 129, 12652. doi: 10.1021/ja075014k
[20]
Singhrachford, T. N.; Lott, J.; Weder, C.; Castellano, F. N. J. Am. Chem. Soc. 2009, 131, 12007. doi: 10.1021/ja904696n
[21]
(a) Merkel, P. B.; Dinnocenzo, J. P. J. Phys. Chem. A2008, 112, 10790. (b) Merkel, P. B.; Dinnocenzo, J. P. Luminescence2009, 129, 303. (c) Lee, S. H.; Lott, J. R.; Simon, Y. C.; Weder, C. J. Mater. Chem. C2013, 1, 5142.
[22]
Wu, W.; Guo, H.; Wu, W.; Ji, S.; Zhao, J. J. Org. Chem. 2011, 76, 7056. doi: 10.1021/jo200990y
[23]
Kim, J.-H.; Deng, F.; Castellano, F. N.; Kim, J.-H. Chem. Mater. 2012, 24, 2250. doi: 10.1021/cm3012414
[24]
Baluschev, S.; Jacob, J.; Avlasevich, Y. S.; Keivanidis, P. E.; Miteva, T.; Yasuda, A.; Nelles, G.; Grimsdale, A. C.; Müllen, K.; Wegner, G. ChemPhysChem2005, 6, 1250. doi: 10.1002/(ISSN)1439-7641
[25]
Boutin, P. C.; Ghiggino, K. P.; Kelly, T. L.; Steer, R. P. J. Phys. Chem. Lett. 2013, 4, 4113. doi: 10.1021/jz402311j
[26]
(a) Wu, W.; Cui, X.; Zhao, J. Chem. Commun. 2013, 49, 9009. (b) Wu, W.; Zhao, J.; Sun, J.; Guo, S. J. Org. Chem. 2012, 77, 5305.
[27]
Wu, T. C.; Congreve, D. N.; Baldo, M. A. Appl. Phys. Lett. 2015, 107, 031103. doi: 10.1063/1.4926914
Hosoyamada, M.; Yanai, N.; Ogawa, T.; Kimizuka, N. Chemistry2016, 22, 2060. doi: 10.1002/chem.201503318
[35]
Sripathy, K.; MacQueen, R. W.; Peterson, J. R.; Cheng, Y. Y.; Dvořák, M.; McCamey, D. R.; Treat, N. D.; Stingelin, N.; Schmidt, T. W. J. Mater. Chem. C2015, 3, 616. doi: 10.1039/C4TC02584A
Figure 1
Jablonski diagram illustrating the triplet ernergy transfer between triplet sensitizer and acceptor and triplet-triplet annihilation between the acceptors
Figure 6
(a) Chemical structures of CDCB-derived heavy-atom-free sensitizers for TTA-UC in PU films and (b) schematic representation of the polymer TTA-UC system
Adopted with permission from Ref. [28]. Copyright The Royal Society of Chemistry 2016
Figure 7
(a) Rendering of the materials morphology, (b) chemical structures of the constituents forming the glassy polymer, the surfactant, and the liquid phase and (c) emission pictures of the A-1/S-1 doped polymer under 543 nm laser irradiation
Adopted with permission from Ref. [29]. Copyright 2017 Wiley-VCH
Figure 9
(a) Chemical structures of acceptors A-1 and donor S-6, solvents tetralin, gelator DMDBS and (b) photographs of a gelated material performing green-to-blue upconversion in an inverted cuvette
Adopted with permission from Ref. [35]. Copyright The Royal Society of Chemistry 2015
Figure 11
(a) Molecular structures of the Organogelator FF and (b) upconversion photoluminescence intensity of S-5/A-1/FF with varied incident power density of 532 nm laser under ambient conditions
Inset: photograph of the upconversion emission in semitransparent gels. Adopted with permission from Ref. [39]. Copyright 2016 Wiley-VCH
Figure 12
(a) Chemical structures of acceptors A-8/A-9 and donors S-7/S-8 and (b) schematic illustration of the host-guest complexation facilitated TTA-UC of S-8@A-9
Adopted with permission from Ref. [41]. Copyright 2016 American Chemical Society
Figure 14
(a) Chemical structures of liquid acceptor A-10 and donor S-9, (b) photoluminescence spectra of the doped liquid (S-9/A-10=0.01 mol%) with different incident power intensities, and (c) schematic illustration of donor-to-acceptor TTET and energy migration among the acceptor liquid molecules
Inset: photographs of the S-9-doped liquid upon being exposed to white light (up) and a 532 nm green laser (bottom). Adopted with permission from Ref. [42]. Copyright 2013 American Chemical Society
Figure 15
Chemical structures of A-11 and S-5, and schematic representation of nonvolatile TTA-UC systems using triplet energy migration in acceptor ion liquids
Adopted with permission from Ref. [43]. Copyright 2015 Wiley-VCH
Figure 16
(a) align="center"hemical structure of lipophilic acceptor A-12, (b) schematic illustration of acceptor monolayer membrane A-12 formed in chloroform and (c) picture of chloroform dispersion showing upconverted emission upon 532 nm excitation under an aerated condition
Adopted with permission from Ref. [44]. Copyright 2015 Nature Publishing Group
Figure 17
Chemical structures of water-soluble amphiphilic acceptor A-13 and donor S-10 and schematic illustration of aqueous molecular self-assembly and 10 bound to the assembly by electrostatic and hydrophobic interactions
Adopted with permission from Ref. [45]. Copyright 2016 The Royal Society of Chemistry
Figure 20
Schematic representation of a dual-doped nanoparticle with S-5 (red sphere) and A-1 (blue spheres), showing the shielding from the external environment exerted by the NPs
Adopted with permission from Ref. [48]. Copyright 2012 Wiley-VCH
Figure 23
Schematic illustration of TTA-UCL process of the upconversion nanocapsules, and chemical structures of Sensitizers (S-12) and Annihilators (A-16, and A-17)
Adopted with permission from Ref. [15]. Copyright 2013 American Chemical Society
Figure 24
A procedure to fabricate bioprobe-attached SNC-B and SNC-G (A~E) and the schematic illustration of the cancer-specific, dual color imaging of (F) breast and (G) colorectal-cancer cells
Adopted with permission from Ref. [51]. Copyright 2016 American Chemical Society
Figure 25
(a) Photocleavage of c[R]GDfK. DEACM-OH and intact cRGDfK peptide are released upon irradiation at 400 nm and (b) schematic of the phototriggering of the polymeric micellar nanoparticle by TTA-UC and FRET
Adopted with permission from Ref. [52]. Copyright 2015 American Chemical Society

 下载:
下载:


























 下载:
下载:


























 下载:
下载:
