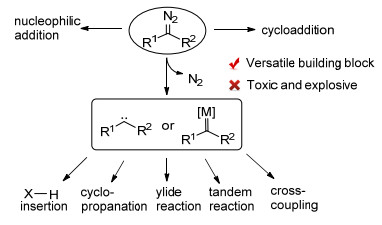
图式 1.
重氮化合物的反应活性
Scheme 1.
Reactivity of diazo compounds

重氮化合物作为一种多用途的有机砌块, 被广泛应用于各种有机分子的构建之中.除了作为1, 3-偶极子或亲核试剂直接参与反应外, 重氮分子极易作为氮气离去的N2基团使得重氮化合物能够被用作自由卡宾或金属卡宾的前体, 发生各种各样的经典转化, 例如X—H键插入反应(X=C, N, O等)、环丙烷化反应、叶立德反应以及多组分反应[1].近年来, 基于重氮化合物(或其前体N-对甲苯磺酰腙)的金属卡宾参与的偶联反应也有了长足的发展, 为各种碳碳键的构筑提供了简单易行的方法(Scheme 1)[2].然而, 高能重氮基团所带来的不稳定性以及潜在的毒性却严重限制了重氮化合物的应用.在冲击、加热以及强酸条件下, 不稳定的烷基重氮化合物会迅速分解放出氮气, 从而引发爆炸.虽然含有拉电子基团的羰基重氮化合物在实验室条件下相对稳定, 但在大批量反应以及工业生产的条件下有一定的风险.不稳定性带来的苛刻的储存条件要求也限制了重氮化合物的应用.更为重要的是, 一些重氮化合物的毒性会给实验人员带来健康隐患.以上这些问题使得一些高活性的重氮化合物在化工生产中被“遗忘”, 某些利用重氮化合物能够简单实现的转化, 却由于安全问题的考量, 只能采用原子经济性更差、合成步骤更多的方式来实现.因而发展一种全新的, 能够在保证安全的前提下大批量应用重氮化合物的反应策略成为这一领域亟待解决的问题.
近年来, 连续流动化学得到了迅速的发展[3].这种在毫米甚至微米尺度的微反应器中通过连续液流进行反应的策略成为了传统的批量反应策略的有力竞争者, 在药物中间体合成以及化学品的大量制备领域都得到了广泛的应用.与传统的批量反应策略相比, 流动化学最为显著的优势之一在于应用有害试剂时的安全性.这来源于微反应器较高的比表面积与更好的导热与传质性能, 局部过热现象受到明显抑制.同时微反应器较小的尺度也防止了反应器内部危险物质的累积.另外, 通过串联过程可以实现有害试剂在流动过程中的现制现用, 无需储存有害试剂, 既保证了实验的安全性又保证了反应的效率.在安全性得到保证的同时, 通过简单的增大流量以及延长反应时间就可以实现反应的扩大化.与之形成鲜明对比的是, 对于传统批量反应而言, 大量反应往往意味着安全隐患.因此, 将高活性重氮化合物应用于连续流动反应中能够有效降低重氮化合物的安全隐患, 从而进一步拓展重氮化合物在有机合成中的潜力.近期, 这一领域已引起了有机化学家的广泛注意[4].本文将简要介绍实验室条件下各种不同的重氮化合物在连续流动系统中实现的转化.
重氮甲烷(CH2N2, 1)是结构最为简单的重氮化合物, 作为多用途的一碳单元, 其在实验室中并没有得到充分的应用.这源于CH2N2的高毒性以及爆炸风险.由于其对DNA甲基化的能力, CH2N2具有较强的致癌性, 且由于室温下为气体(沸点为-23 ℃), 高挥发性使得其危害性更加严重.另外, CH2N2高能的分子结构使它对于振动、光、热等条件都十分敏感.实验室使用这种气体需要用到特制的无磨砂接口玻璃器皿.
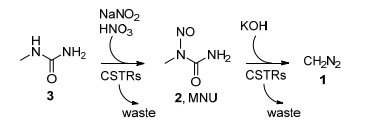
连续流动技术在CH2N2参与的反应中的应用最早来源于工业界对于大量反应安全性的考量. 1998年, 美国Aerojet公司报道了一项利用连续流动技术生产CH2N2醚溶液的专利(Scheme 2)[5].作者在连续搅拌槽式反应器(CSTR)中实现了CH2N2前体N-亚硝基-N-甲基脲(MNU, 2)以及CH2N2的连续制备. 2002年, 英国Phoenix Chemicals公司进一步改进了这一过程[6].他们采用N-甲基-N-亚硝基对甲苯磺酰胺(Diazald, 4)作为重氮甲烷前体, 经由多步连续反应合成了α-氯代酮这类重要的药物中间体, CH2N2产量达到了每年60吨.
之后, 在实验室中通过微反应器制备与应用CH2N2也得到了系统的研究.早期的工作由Löbbecke[7]以及Stark等[8]展开, 他们分别报道了将微反应器中生成的CH2N2用于苯甲酸甲基化的过程.由于Diazald需要与强碱性KOH溶液反应来产生CH2N2, 并直接通入底物溶液, 该转化的底物普适性并不强.
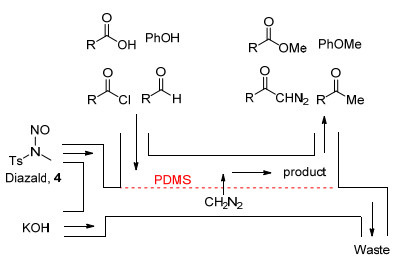
2011年, Kim等[9]采用双通道平行微反应器解决了这一问题(Scheme 3).反应器的两个通道之间采用高疏水性、可容许气体通过的聚二甲基硅氧烷(PDMS)膜隔离, 碱性水溶液中产生的CH2N2经由膜扩散进入另一个通道, 与有机相中的底物反应.这样一种连续分离手段有效避免了底物与碱性水溶液的接触, 高效实现了乙酸、苯酚、苯甲醛的甲基化以及苯甲酰氯的重氮化过程.但是, 由于微反应器容积仅有60 μL, 该装置日产量仅为0.58~2.88 mmol, 且由于PDMS的溶胀性, 四氢呋喃(THF)等低极性溶剂难应用于该体系中.
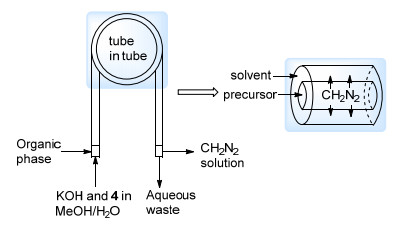
Kappe等[10a]沿用CH2N2在流动体系中原位分离的思路, 将Ley等[10b]发展的管中管(tube-in-tube)反应器应用于CH2N2的原位制备之中.这种反应器的内管以容许气体通过的Teflon AF-2400制成. CH2N2由Diazald的醇溶液与碱反应在内管中制得, 之后扩散出内管壁溶于外管中的有机相, 或与外管中的底物反应, 而水相废液从内管流出(Scheme 4).甲基化反应、环丙烷化反应以及环加成反应都能够以较高的效率实现, 且该方法中重氮甲烷的产生速率能够达到3.6 mmol/h.
之后, Kappe课题组[11]又将这一方法应用于由保护的α-氨基酸5制备α-氯代酮8的多步连续流动反应之中(Scheme 5a), 而8是制备人类免疫缺陷病毒(HIV)蛋白酶抑制剂的重要手性原料.在第一个反应线圈之中, 5被转化为酸酐6, 之后进入管中管反应器与原位生成的无水CH2N2反应得到α-重氮酮7, 最终在引入的盐酸醚溶液的作用下得到α-氯代酮8.经由这样的连续流动过程能够在4.5 h内获得最多1.84 g手性纯的α-氯代酮8.
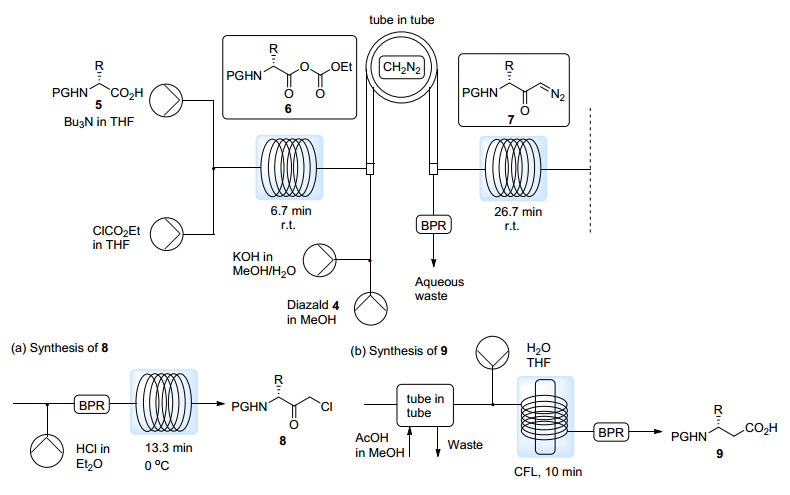
2014年, 同一课题组[12]报道了由保护的α-氨基酸5制备对应的β-氨基酸9的连续流动合成方法(Scheme 5b).经过与之前报道类似的过程得到α-重氮酮7后, 通过Arndt-Eistert增碳反应便可得到对应的β-氨基酸9.过量的CH2N2能够在外管通入醋酸醇溶液的第二个管中管反应器中被除去.之后, α-重氮酮7的Wolff重排过程既可在光化学条件下进行, 又可在填充有氧化银的填充床反应器中进行.
以上报道说明管中管反应器能够有效地实现CH2N2的原位生成、纯化与消耗, 但该体系仍存在一定的问题, 即外管中的反应速率, CH2N2的生成速率以及过膜的扩散速率难以匹配.外管溶液的流速会同时影响这三个方面, 因而难以分别研究.另外, 外管中如果有固体生成就会阻塞管路.最近, Kappe等[13]在管中管反应器的基础之上又发展了半批量化的瓶中管(tube-in-flask)反应器(Scheme 6).通过将产生CH2N2的Teflon AF-2400线圈浸入装有底物溶液的密封反应瓶中, 即可使生成的CH2N2与底物充分反应, 同时除去水相废液.该过程中, 生成CH2N2的步骤属于流动反应, 而CH2N2与底物之间的反应属于批量反应.除了CH2N2的经典转化(α-重氮酮的合成)[13b]之外, 该体系也可用于不同溶剂CH2N2溶液的制备, 产量能够达到较高的42.6 mmol/h.
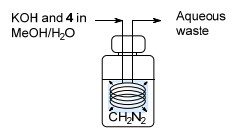
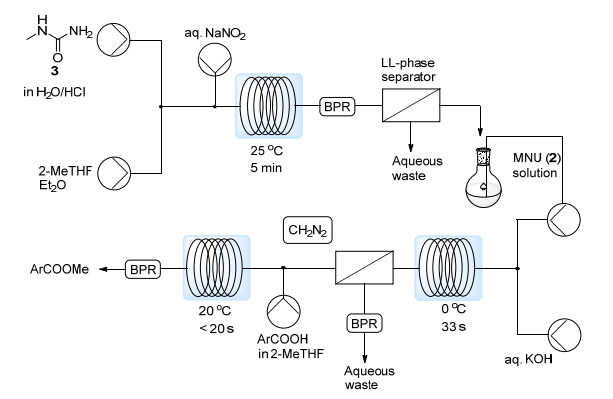
2012年, Maggini等[14]报道了一例在KOH作用下MNU分解产生CH2N2的流动相反应.利用Corning公司的Advanced-Flow反应器GEN1, CH2N2的日产量能够达到19 mol, 且能定量实现苯甲酸的甲基化.但是, 该过程并未实现CH2N2的原位分离, 需要加入过量的苯甲酸来中和KOH.最近, Novartis公司的Lehmann等[15]从无毒易处理的N-甲基脲3出发, 发展了一条先后原位生成与消耗MNU以及CH2N2的两步流动反应路线(Scheme 7). 3首先与通入的NaNO2溶液反应得到MNU, 之后经过液液双相分离器分离得到有机相. MNU溶液再与KOH水溶液反应得到CH2N2, 同样经液液分离后与通入的芳基甲酸反应能够定量获得芳基甲酸甲酯.在该体系中, 有毒害性的MNU与CH2N2均无需分离并与外界接触, 大大降低了健康风险.该过程CH2N2产量达117 mmol/h, 已被用于Novartis公司的药物研发过程之中.
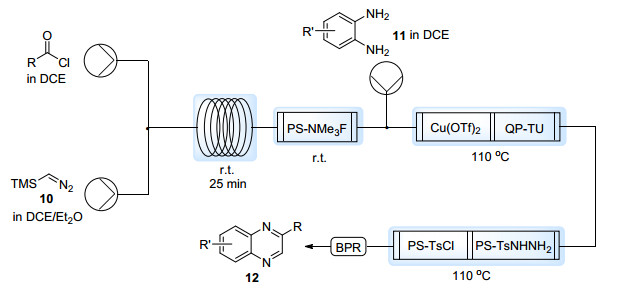
同样作为常用的一碳单元, 三甲基硅基重氮甲烷(TMSCHN2, 10)由于TMS基团的存在, 其爆炸性得到降低且沸点显著升高(沸点96 ℃), 常被用作较CH2N2更安全的替代品. 2009年, Liotta等[16]首先将TMSCHN2用于药物中间体的流动合成之中.之后, Ley等[17]报道了利用TMSCHN2制备端位重氮酮的流动过程.酰氯与TMSCHN2溶液在经过线圈反应器后, 在填充有固载氟源(PS-NMe3F)的柱子中脱除TMS基团.得到的α-重氮酮能够进一步与邻苯二胺11在Cu(OTf)2催化下缩合得到喹喔啉环系12 (Scheme 8).由于α-重氮酮与邻苯二胺的毒性, 作者希望利用线上净化的方式来避免有毒物质与外界的接触.因此, 反应混合物先后经过聚苯乙烯固载的硫脲(QP-TU), 对甲苯磺酰氯(PS-TsCl)以及对甲苯磺酰腙(PS-TsNHNH2), 从而分别除去溶解的铜盐, 未反应的邻苯二胺以及剩余的α-重氮酮.流出反应体系的溶液经简单的蒸除溶剂过程即可获得纯净的喹喔啉衍生物12.该过程体现了连续流动技术在产物分离纯化上的简便性.
Arndt-Eistert合成是向羧酸中插入一碳单元的重要手段.传统的Arndt-Eistert合成需要用到重氮甲烷以制备α-重氮酮, Kappe等将之应用于流动体系之中, 上文已给出介绍. Fuse等[18]将TMSCHN2作为重氮甲烷的替代物, 由芳基甲酸出发制备了α-芳基酯以及α-芳基酰胺.其中, 制备α-芳基酯所需的三步反应完全在流动体系中完成, 体现了流动体系在实现多步串联反应上的优势.
TMSCHN2虽然有着十分广泛的应用, 但直到最近, 其合成方法仍仅限于格氏试剂TMSCH2MgCl与叠氮磷酸二苯酯(DPPA)之间的反应, 更为简单的胺的重氮化过程无人实现.这是由于TMSCHN2具有极强的亲核性, 在生成后就会与体系中的酸直接反应. Lebel等[19]首先从TMSCH2Cl出发, 在流动体系优化了TMSCH2NH2 (13)的合成过程, 之后利用双亚硝基试剂Pr(ONO)2, 并以大位阻的AdCOOH提供酸性条件, 分别在批量反应以及流动反应中实现了TMSCHN2的合成(Eq. 1).以上条件有效地抑制了酯化副反应的发生, 保证了TMSCHN2的产率, 日产量可达2.5 mol.不同硅基取代的重氮化合物也可通过该方法合成.
|
|
(1) |
与重氮甲烷类似, 其他烃类重氮化合物也由于不稳定性与安全问题, 难以被用于实验室合成之中.其中芳基重氮化合物由于芳环的共轭作用, 稳定性较普通的烃类重氮化合物稍好, 但仍然容易分解, 故被认为是“半稳定”(semi-stabilized)的重氮化合物.例如苯基重氮乙烷虽然能够制备并存储, 但必须储存在-40 ℃的稀溶液中, 且每次使用前必须标定浓度.连续流动化学为这类活性分子的原位制备与反应提供了可能.
Bamford-Stevens反应, 即芳基磺酰腙在碱性条件下的分解, 是由酮出发制备重氮化合物的重要途径. 2009年, Barluenga等[20]报道了无过渡金属条件下N-对甲苯磺酰腙与芳基硼酸间的偶联反应, 体系中原位生成的重氮化合物参与实现了sp2-sp3碳碳键的构筑.这一过程被Kirschning等[21]在流动体系中实现(Scheme 9).酮与N-对甲苯磺酰肼14的混合溶液首先在装有钢珠的填充床反应器中经由电磁感应加热的方法反应生成N-对甲苯磺酰腙15, 之后该溶液直接被通入第二个电磁感应加热反应器与芳基硼酸反应, 并得到最终的偶联产物16.
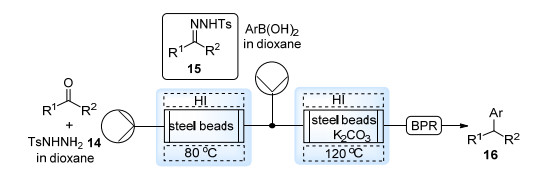
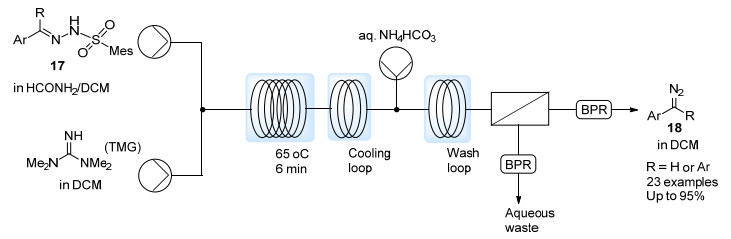
由于以上反应中重氮化合物原位参与反应, 未能分离提纯, 因此对碱性以及温度较敏感的转化无法实现.最近, Charette等[22]进一步发展了芳基磺酰腙分解产生重氮化合物的体系, 采用位阻较大的2, 4, 6-三甲基苯磺酰腙17以降低产生重氮所需温度, 同时使用NH4HCO3水溶液洗涤以除去过量的碱以及亚磺酸盐副产物, 经液液分离装置即可获得纯净的芳基重氮甲烷溶液或二芳基重氮甲烷溶液(18) (Scheme 10).得到的重氮化合物溶液能够直接用于对碱敏感的Sc(OTf)3催化的环酮扩环反应, 铜催化的醛的环氧化反应以及ZnI2催化的Simmons-Smith环丙烷化反应之中, 均取得了较好的效果.
腙类化合物的直接氧化是另一个产生重氮化合物的常用方法.将不溶的固体氧化剂填充于柱子中, 即可将液流中的腙氧化为重氮化合物, 并参与接下来的反应. Maggini等[23]于2011年以NiO2为氧化剂, 在流动体系中原位合成了芳基重氮化合物, 并将之应用于一种具有光电活性的亚甲基富勒烯的合成之中.但这种流动合成方法在当时并没有得到深入的探讨, 直到2015年起, Ley等[24~28]系统地研究了以MnO2为氧化剂的流动反应体系.
Ley课题组[24]首先以酸淬灭的方式探究了MnO2氧化腙的最佳条件.通过线上红外监测的手段, 他们发现, 应用活化过的、提前加入少量腙改性的MnO2能够起到最好的氧化效果, 且向腙溶液中加入二异丙基乙胺(i-Pr2NEt)能够有效减少副产物(Scheme 11a).他们将产生的芳基重氮溶液直接通入芳基硼酸溶液中.除了与缺电子的吡啶硼酸之间的反应需要加热外, 其余偶联反应均可在室温下高效完成, 这展现了芳基重氮化合物较高的反应活性(Scheme 11b).实验证明, 在室温条件下, 重氮化合物与硼酸偶联后发生1, 2迁移生成苄基硼酸中间体20, 之后中间体20发生脱硼质子解得到最终偶联产物.该苄基硼酸中间体具有一定稳定性, 可由过氧化氢氧化为醇.另外, 使用过的MnO2填充柱还能够通过过氧叔丁醇(TBHP)再氧化的方式循环利用.
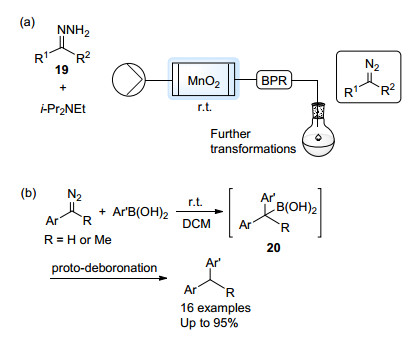
2016年, 在之前反应的基础之上, Ley等[25]继续发展了基于芳基重氮化合物的迭代碳碳键形成反应.芳基重氮化合物与烷基硼酸偶联后得到的苄基硼酸中间体能够与新加入的另一分子芳基重氮化合物继续偶联, 最终得到的硼酸能够发生脱硼质子解得到偶联产物21, 或者被转化为硼酯22.通过这种连续反应的策略, 作者最多以一种烷基硼酸与三种不同的芳基或烯基重氮化合物为砌块, 连续构建三根碳碳键, 合成了其他方法难以获得的碳链结构(Scheme 12a).烯基重氮化合物也能以类似的方式与烷基硼酸偶联得到烯丙基硼酸中间体, 实现连续碳碳键的构筑, 并最终通过对醛的烯丙基化过程终止反应, 得到高烯丙基醇23 (Scheme 12b).该过程充分展现了以流动化学为工具发现活泼重氮化合物的新反应活性的潜力.
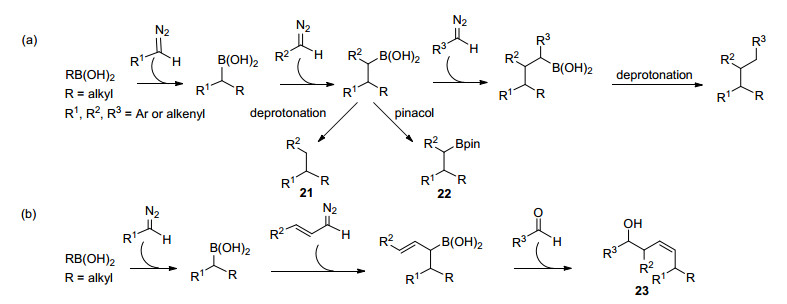
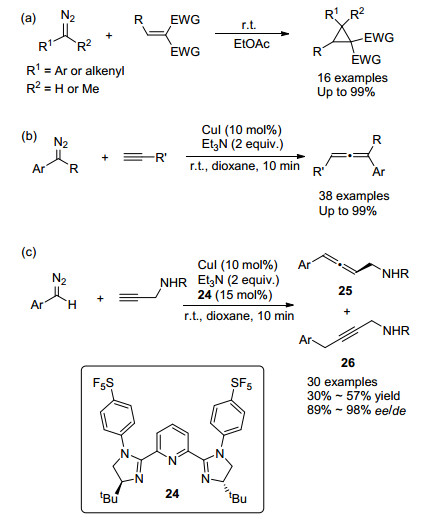
除了与硼酸的反应, Ley等[26]还利用这种连续流动产生芳基重氮化合物的方法实现了室温下无过渡金属催化的与缺电子烯烃的环丙烷化反应(Scheme 13a), 以及铜催化与端炔的偶联反应(Scheme 13b)[27].后者能够在室温下十分钟内完成转化, 提供了温和的合成二取代或三取代联烯的普适性方法.基于此方法, Ley等[28]还发展了铜催化的芳基重氮甲烷与炔丙基胺之间的不对称偶联(Scheme 13c).作者采用了手性的二咪唑啉吡啶三齿配体24, 实现了温和条件下不对称二取代联烯25的合成, ee/de值最高可达98%.这一转化为较难实现的不对称联烯合成提供了新的方法, 但该方法同时会生成炔烃副产物26, 导致化学选择性较低, 联烯产率均在60%以下.
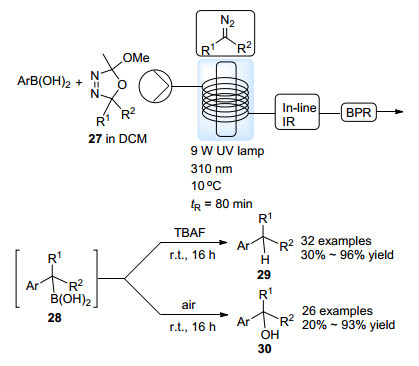
相比于半稳定的芳基重氮化合物, 不稳定的烷基重氮化合物的合成与应用更加具有挑战性.早在1989年, Warkentin等[29]发现1, 3, 4-噁二唑啉环系27能够在紫外光照射下分解为重氮化合物, 但这一发现被忽视.直到最近Ley等[30]制备了一系列稳定的噁二唑啉化合物, 并将之应用于烷基重氮化合物的连续流动制备中, 得到的重氮化合物原位与芳基硼酸反应, 从而实现了sp2-sp3碳碳键的偶联过程.烷基硼酸中间体28经由后续的脱硼质子解或者氧化过程, 得到一系列偶联产物29以及三级醇30 (Scheme 14).线上红外技术的应用使该反应更加易于监测.这一过程为不稳定的烷基重氮化合物的合成与应用提供了新的手段.
重氮乙酸乙酯(EDA, 30)是有机合成中重要的二碳合成单元之一, 也是结构最简单、最为常用的α-羰基重氮化合物.由于酯基对于重氮中心的拉电子效应能够有效稳定重氮化合物, EDA较烃类重氮化合物更加稳定, 但仍然存在爆炸的风险.
同其他重氮化合物类似, EDA在流动体系中的原位生成与消耗是十分具有实用价值的策略. Rutjes等[31]利用甘氨酸乙酯盐酸盐(31)与NaNO2在酸性介质中的反应实现了EDA的连续流动合成.作者利用实验设计算法(DoE)对反应条件进行了详尽的筛选, 最终确定保留时间为20 s时, 采用1.5 equiv. NaNO2在50 ℃下反应能够取得最高的效率以及重复性, 实现每天175 mmol EDA的合成.在加入线上液液萃取装置(FLLEX)后, 该体系能够直接获得纯净的EDA溶液.
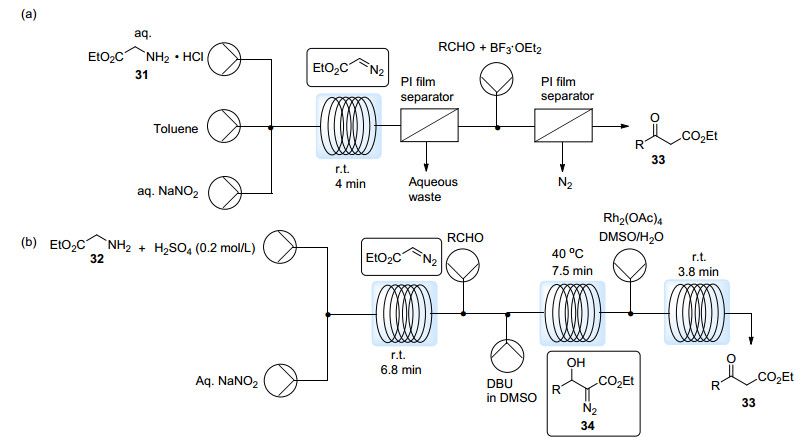
Kim课题组[32]与Wirth课题组[33]先后利用类似的方法实现EDA在流动系统中的原位合成与消耗.他们均报道了流动体系中EDA与醛在弱碱性条件下的aldol加成反应, 制备了β-羟基-α-重氮酯34. Kim等利用自制的聚酰胺(PI)膜微分离器实现了线上有机相与水相的分离.他们还探究了EDA与醛在BF3·Et2O催化下的Roskamp反应(Scheme 15a).该反应过程中会有氮气放出, 这可能会影响液流的流速, 同时也可能导致液流喷出造成安全隐患.因此作者再次引入PI微分离器作为气液分离装置, 从而保证了流速的稳定. Wirth等则研究了在流动系统中串联第三步反应的可能性, 他们在体系中继续引入Rh2(OAc)4溶液以催化β-羟基-α-重氮酯34发生1, 2氢迁移, 从而获得相同的β-酮酯产物33 (Scheme 15b).
EDA与醛的加成反应以1, 8-二氮杂二环十一碳-7-烯(DBU)为碱即可实现, 但EDA与酮或者环状内酯的加成反应则需要以LDA为碱来实现, 生成的活性中间体锂重氮乙酸乙酯(Li-EDA, 35)十分不稳定, 在-50 ℃以上即会分解得到棕色胶状不溶物.这使得批量进行的这一加成反应产率低且重复性差. Wirth等[34]将流动过程应用于该反应的优化之中(Scheme 16), 流动体系中原位产生的二异丙基氨基锂(LDA)经冷却线圈后, 在低温下与EDA反应, 经过保留时间仅12 s的反应线圈后与酮或内酯反应后得到加成产物36. EDA与LDA极短的反应时间保证了不稳定中间体35的反应活性.低温下的流动反应体系有效防止了反应管路的堵塞.该反应展示了连续流动过程易于处理不稳定金属有机中间体从而实现快速反应的优势.
连续流动化学的一大优势在于易于实现多相反应, 这其中就包括了非均相催化反应.将催化剂固载到高分子等材料上, 再置于填充床反应器中, 反应物溶液流过即发生反应, 通过提高催化剂的相对量来提升反应的效率. Martínez-Merino等[35]先后制备了高分子固载的手性双噁唑啉铜(BOX-Cu)配合物以及吡啶噁唑啉铜(pyox-Cu)配合物, 并将其用于苯乙烯与EDA的流动环丙烷化反应之中.其中, BOX-Cu配合物在非均相催化过程中不对称诱导的效率较均相催化高出20%左右.超临界CO2在这两个体系中均可用作溶剂, 并可提高反应的产率.此外, Caselli等[36]将含有吡啶的四氮杂大环配体配位的铜催化剂(Pc-L*-Cu)固载在二氧化硅上, 以超临界CO2为溶剂实现了类似的不对称转化.
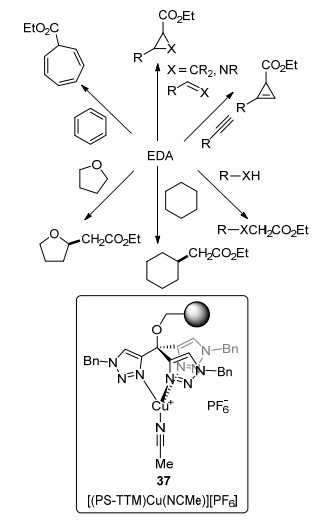
2015年, Pérez等[37]合成了一种聚乙烯连接的三(三氮唑)甲烷配位的阳离子铜催化剂[(PS-TTM)Cu(NC-Me)][PF6] (37), 并将之应用于EDA参与的一系列连续流动转化之中, 包括X—H键插入(X=C, N, O), 烯烃与亚胺的环丙烷化反应、炔烃的环丙烯化反应以及苯的Büchner扩环反应(Scheme 18).该非均相催化剂具有较高的反应效率、较好的普适性、可重复利用以及极少的催化剂滤出等优势.对于EDA与乙醇的反应, 其保留时间仅一分钟即可达到93%收率, 且可连续运转48 h, 彰显了流动化学反应规模易扩大的特点.
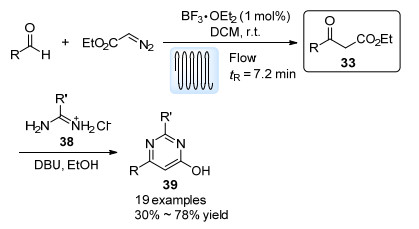
经由流动反应实现的EDA参与的杂环合成反应也有所报道. Hayes等[38]报道了一例合成2, 4, 6-三取代嘧啶环系39的方法(Scheme 19).首先, EDA与醛在BF3·Et2O催化下在线圈反应器中发生Roskamp反应获得β-酮酯33, 之后反应液不经过分离直接与脒的盐酸盐38缩合得到目标产物39.该反应展示了将两种毒害试剂(EDA与BF3·Et2O)用于同一流动体系中的例子.这两种物质在Zhang等[39]报道的哌啶酮扩环反应中也有所应用.
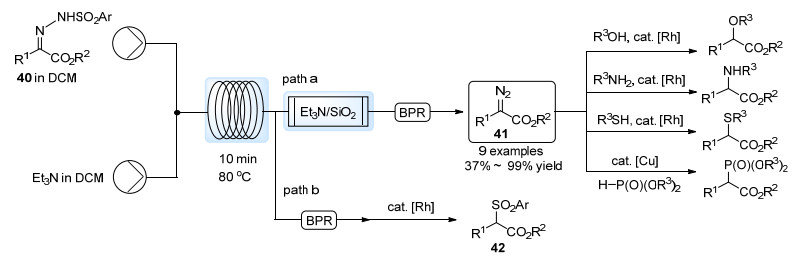
其他常用的α-羰基重氮化合物通常可根据取代基团的不同分为供体/受体类型的重氮化合物(例如芳基酯基重氮化合物)以及受体/受体类型的重氮化合物(例如双酯基重氮化合物).这类重氮化合物通常为液体, 有些为固体, 且由于拉电子基团的稳定作用, 能够较方便的制备与应用.但是重氮化合物潜在的爆炸风险以及其制备过程中有机叠氮化合物的使用都带来了安全方面的隐患, 因而在大规模应用时具有一定的风险.
为避免有机叠氮化合物的使用, Hayes和Moody等[40]利用对应的芳基磺酰腙40在流动体系中的Bamford-Stevens反应制备了α-重氮酯41 (Scheme 19).向线圈反应器中分别引入芳基磺酰腙以及Et3N的二氯甲烷溶液, 并在加热条件下反应, 经由混有Et3N的硅胶柱除去铵盐后即可几乎当量得到41的溶液.该溶液可以不经纯化直接参与铑催化的O—H键插入, N—H键插入, S—H键插入以及铜催化的P—H键插入过程(path a).此外, 若去掉流动体系中的纯化柱, 反应液中残留的亚磺酸根离子便可在醋酸铑催化下直接与α-重氮酯41反应, 得到砜类产物42 (path b).
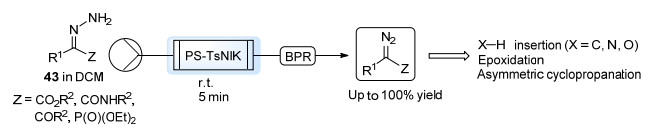
与芳基重氮化合物类似, 腙类化合物的直接氧化也是产生α-羰基重氮化合物的重要方法.与Bamford-Stevens反应相比, 氧化法无需碱性条件, 且不会产生亚磺酸根离子作为副产物. Moody等[41]将该组发展的氧化剂N-碘代对甲苯磺酰胺钾盐(TsNIK)固定在Amberlyst 15树脂上, 制备了非均相氧化剂PS-TsNIK.将腙43的二氯甲烷溶液泵入填充有PS-TsNIK的柱式反应器之中, 即可得到对应的重氮化合物, 保留时间为5 min (Scheme 20). α-重氮酯、α-重氮酰胺、α-重氮酮以及α-重氮磷酯均可以此方法高效制备, 大部分产率均在90%以上.反应后柱中氧化剂PS-TsNIK被还原为PS-TsNH2, 可通过KI3/KOH溶液重新氧化恢复活性.以此法得到的重氮化合物溶液中不含其他有机杂质, 可直接用于铑催化的O—H键插入、N—H键插入以及醛的环氧化反应之中.最近, 同一个课题组[42]将以此法产生的α-重氮酯用于分子内C—H键插入反应, 实现了一系列螺环β-或γ-内酯的合成, 将其还原后得到的螺环氧丁烷结构是药物合成的重要骨架.此外, Davies等[43]将本法应用于铑催化的不对称环丙烷化以及分子间不对称C—H键插入反应之中, 并获得了较高的ee值.获得的重氮化合物溶液中含有的微量水以及反应器中渗出的少量碘单质影响了反应的效率, 在柱式反应器之后串联装有4 Å分子筛以及Na2S2O3的吸附柱可以有效提高反应的产率.
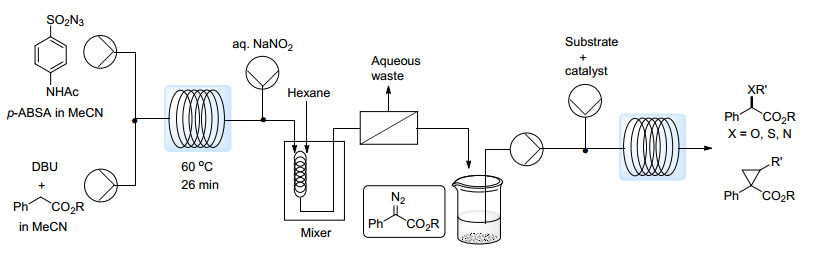
将批量反应中常用的重氮转移反应移植到流动系统中来也是降低有机叠氮化合物安全风险的有效策略.批量进行的重氮转移反应通常需要在室温下反应数个小时, 由于叠氮化合物加热风险太大, 且该反应本身放热, 不能通过升高温度来提升反应速率. Wirth等[44]在流动体系中实现了p-ABSA与苯乙酸酯之间的重氮转移反应(Scheme 21).由于流动体系中叠氮化合物爆炸风险大大降低, 作者将反应温度升至60~70 ℃, 即可在30 min内实现完全转化.加入低极性溶剂萃取后经过液液分离装置即可获得纯净的α-重氮酯溶液.通过线上红外光谱的方法, 作者还研究了重氮转移步骤反应的动力学, 并实现了X—H键(X=O, N, S)插入、环丙烷化反应等后续过程反应条件的迅速优化.这一过程被Wirth等[45]应用于治疗抑郁症的药物milnacipran类似物的流动合成之中.
Maguire等[46]也报道了类似的重氮转移过程, 与Wirth等工作的不同之处在于他们采用了对甲基苯磺酰叠氮(TsN3)为重氮转移试剂, 并将其合成也应用到流动过程之中, 提供了一个更为安全的合成方法.之后, 该课题组[47]将流动体系引入了α-重氮-β-羰基亚砜44的合成之中(Eq. 2).由于硫上孤对电子与重氮基团反键轨道的重叠作用, 这类化合物的稳定性较普通的α-羰基重氮化合物差得多, 只有环状的α-重氮-β-羰基亚砜能够被稳定分离出来.普通批量反应中, 该化合物产率通常在30%左右, 这是由于重氮转移反应中采用的碱会促进其分解. Maguire等采用固载的有机碱(PS-NMe2), 并仔细筛选流动反应中底物溶液的流速以及保留时间, 成功将产率提升为批量反应中的二到三倍.恰当的保留时间控制能够在完成底物转化的同时, 尽量减少敏感产物与碱的接触, 从而有效提高反应的效率.
|
|
(2) |
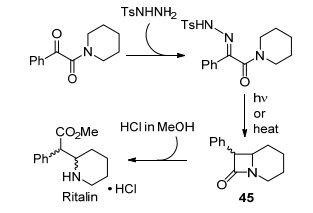
原位合成重氮化合物并消耗的策略也在大规模药物合成中得到了应用.最近, Monbaliu等[48]利用卡宾中间体的分子内C—H键插入反应作为关键步骤实现了治疗多动症的药物Ritalin的流动合成(Scheme 22).该过程已经经过中试, 关键中间体β-内酰胺45的产量已经达到了每天4.25 kg.
将较稳定的α-羰基重氮化合物直接用于流动反应之中的过程也有所报道.由于连续流动体系在实现光化学反应时具有简便高效、易扩大化的特点, 基于光引发的Wolff重排实现的串联流动反应得到了合成化学家的关注[49].经由Wolff重排, α-重氮酮被转化为活泼的烯酮中间体并参与后续反应, 实现一系列有机分子的构建.
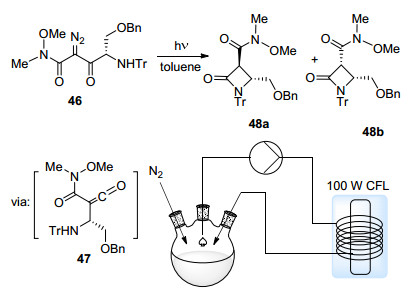
2010年, Konopelski等[50]报道了第一例连续流动体系中实现的光引发Wolff重排反应(Scheme 23). α-重氮-β-酮酰胺46经由Wolff重排得到烯酮中间体47, 之后发生分子内关环得到β-内酰胺48.该反应经由循环流动装置实现, 反应物溶液被泵入缠在光源上的FEP管路之中, 反应过后流回原容器中. FEP管路良好的透光性能以及较窄的内径保证了光在反应物溶液中的投射效率.采用100 W节能灯作为光源能够提高反式产物48a的比例.
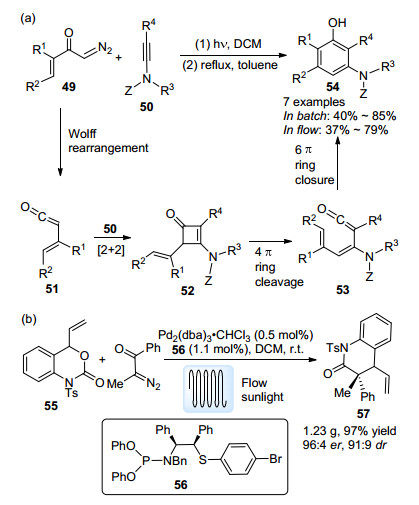
光引发的Arndt-Eistert合成是Wolff重排反应的重要应用, 上文已作出简要介绍, 此处不再赘述.烯酮中间体能够发生的另一类重要转化就是环加成反应. 2013年, Danheiser等[51]报道了α-重氮酮49与炔酰胺50之间的成环反应(Scheme 24a). α-重氮酮49发生Wolff重排后得到烯酮中间体51, 该中间体随后与炔酰胺50发生[2+2]环加成反应.得到的烯基取代环丁烯酮52在4π电环化开环后, 发生6π电环化关环, 并在芳构化后获得多取代苯酚产物54.该反应在流动条件以及批量条件下能够获得类似的产率.最近, 肖文精课题组[52]报道了一例钯催化的苯并噁嗪酮55与烯酮中间体的不对称[4+2]环加成反应(Scheme 24b), 烯酮中间体同样由光引发的Wolff重排生成.作者引入流动体系, 成功实现了目标产物57的克级制备, 且保持了优秀的er值与dr值.
基于Wolff重排的多组分串联过程也有所报道. Basso等[53]将本课题组发展的α-重氮酮、异腈以及羧酸之间的三组分反应移植到了流动体系之中(Eq. 3).该反应经由光引发的Wolff重排以及形式上的Passerini反应实现.该过程中, 形成Z式烯烃是比较有利的, 但光引发的烯烃异构化过程使得反应的顺反选择性较差, 批量反应中需加入戊间二烯或反式-1, 2-二苯乙烷作为三重态淬灭剂来抑制异构化过程[54].在流动体系之中, 产物在反应后会迅速离开反应体系, 从而有效抑制了E式烯烃的生成.本反应体现了连续流动反应能够控制产物选择性的优势.
Wolff重排除了可以由光照引发, 还能够在加热条件下实现. Ley等[55]研究了流动体系中, 利用微波加热的方法实现的α-重氮酮与亚胺之间的Wolff-Staudinger反应(Eq. 4).强热下产生的烯酮中间体会与亚胺发生[2+2]环加成反应, 得到β-内酰胺产物58.微波反应提供了一种高效的加热手段, 但微波辐射深度以及吸收效率上的限制使得微波反应很难实现扩大化.流动化学为这一问题提供了解决手段, 反应器较小的尺寸保证了微波辐射的效率.在这一例子中, 微波辐射4 min以内, 反应器内部温度就可以迅速升至165 ℃, 并保持稳定.
|
|
(3) |
|
|
(4) |
α-羰基重氮化合物也可用于非均相催化的不对称反应之中. 2011年, Hashimoto等[56]将手性铑催化剂[Rh2(S-PTTL)(S-TCPTTL)3] (60)经由共聚反应固载在高分子基底之上, 并将之利用于2-重氮-3, 6-二酮酯(59)与苯乙烯或苯乙炔之间的羰基叶立德[3+2]环加成反应之中(Scheme 25).在流动条件下该反应能达到较高的产率以及最高99%的ee值.

流动非均相催化采用的填充床反应器虽被广泛应用于各项研究之中, 但其内部较大的压力带来了泄漏等安全隐患. 2015年, Davies与Jones等[57]将一种新颖的复合高分子中空纤维用作流动反应器, 并将不对称催化中常用的[Rh2(S-DOSP)4]的类似物固载在硅基上, 在制备纤维时混入其中.当反应物溶液进入中空纤维管道后, 另一股液流垂直于多孔管壁流入, 促使反应物溶液经由管壁从另一侧流出.在此过程中, 反应物接触管壁中的催化剂并发生反应(Scheme 26).这一策略被用于α-羰基重氮化合物参与的不对称环丙烷化以及烯丙基C—H键插入过程之中, 均以与批量反应中相近的高效率以及高对映选择性实现了转化.
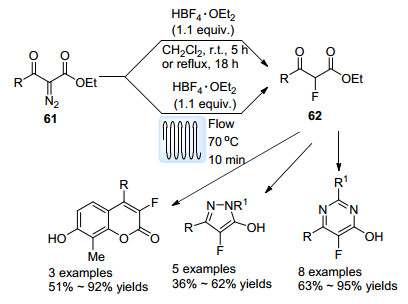
均相体系中应用流动体系能够促进一些过程的反应效率以及选择性. 2012年, Moody与Hayes等[58]探究了HBF4·Et2O对α-重氮-β-酮酯61的亲核氟化反应(Scheme 27).该反应以二氯甲烷为溶剂, 在批量以及流动体系中均可完成.流动体系中由于能够维持高压状态来抑制二氯甲烷气化, 故可通过加热缩短反应时间, 保留时间仅为10 min, 而批量反应中则需在常温下反应5 h.以此法获得的α-氟-β-酮酯62能够一步构建吡唑、嘧啶以及香豆素等杂环环系, 展现了较高的合成应用价值.
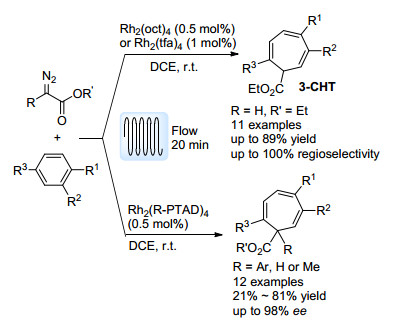
最近, Beeler等[59]报道了一个利用流动反应有效控制反应区域选择性的例证(Scheme 28).铑催化的重氮化合物与芳环之间的Buchner扩环反应是一步合成七元环系的重要途径, 然而, 对于带有取代基的苯环底物, 由于反应的区域选择性较差, 会得到多种环庚三烯(CHT)异构体.作者认为, 这是由批量反应中较差的混合效率以及散热效率引起的.当在反应物充分混合, 并能迅速散去反应放热, 体系温度稳定在室温时, 扩环反应就会选择性得到环庚三烯产物3-CHT.实验证明, 在流动反应高效的传质与传热效应的作用下, 扩环反应的区域选择性较批量反应得到了有效的控制.除了EDA之外, 其它α-重氮酯也可应用于此体系中.利用Davies等[57]发展的[Rh2(R-PTAD)4]手性催化剂, 该反应能够构建出含有手性四级碳中心的环庚三烯, ee值最高达到98%.
近年来, 含氟有机化合物的合成成为了有机化学领域的热点之一.含氟官能团的引入能够大幅度改变有机分子的生化活性, 例如三氟甲基基团能够提高化合物的新陈代谢稳定性、亲脂性以及与生物大分子的结合选择性.三氟甲基重氮甲烷(CF3CHN2)的合成早在1943年就已实现, 而二氟甲基重氮甲烷(CF2HCHN2)的合成与应用一直到2015年才被报道.在流动体系中实现含氟烷基重氮化合物的方法则直到2016年起才逐渐发展起来.
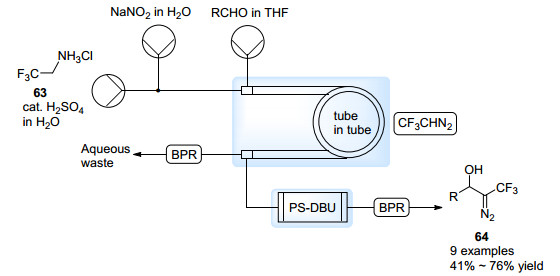
CF3CHN2的沸点仅为13 ℃, 室温下为气体. Kappe等[60]希望在管中管反应器中实现其原位制备与转化.与生成CH2N2的过程类似, 重氮化合物前体三氟乙胺盐酸盐63的酸性水溶液与NaNO2水溶液分别通入管中管反应器的内管, 逐渐释放出CF3CHN2.这一气体经由Teflon AF-2400管壁扩散至外管中.外管则通入醛的THF溶液, 溶解CF3CHN2之后进入装有固载DBU的柱式反应器之中, 并发生碱催化的aldol加成反应, 最终得到三氟甲基取代的α-羟基重氮化合物64 (Scheme 29).
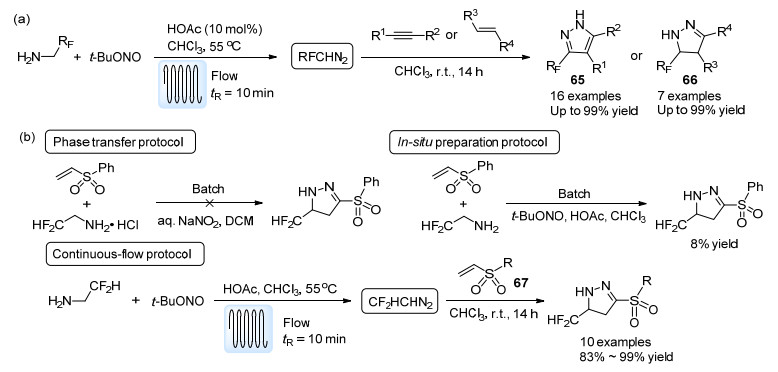
与Kappe等[61]采用的水相重氮化反应不同, Koenigs等[62]以叔丁基亚硝酸酯(t-BuONO)为重氮化试剂在有机流动相中实现了一系列含氟烷基重氮化合物的合成(Scheme 30a).该方法普适性较好, 除了CF3CHN2之外, CF2HCHN2以及含有其他含氟烷基的重氮化合物均可由此方法合成.生成的重氮化合物溶液被直接通入装有烯烃或炔烃溶液的反应瓶中, 经由室温下的[3+2]环加成反应, 即可获得药物中间体中常见的含氟吡唑(65)或吡唑啉(66)环系.之后, 该课题组又以烯基砜67为底物, 探究了CF2HCHN2与CF3CHN2在批量反应与流动反应中反应性的区别.实验发现, 以NaNO2水溶液为重氮化试剂制备的CF3CHN2直接参与批量的[3+2]环加成反应能够获得较高的产率, 但以此法获得的CF2HCHN2无法参与批量的[3+2]环加成反应, 若以t-BuONO为重氮化试剂, 在批量条件下同样产率极低.与之形成鲜明对比的是, 流动条件下产生的CF2HCHN2能够近乎定量地参与[3+2]环加成反应(Scheme 30b).作者认为相比于CF3CHN2, CF2HCHN2在批量条件下的不稳定性导致了这一差别.类似的不稳定性在同一课题组报道的铑催化的CF2HCHN2与烯烃的环丙烷化反应中也有所体现[63].以上例子均体现了流动体系在处理不稳定中间体时的优势.
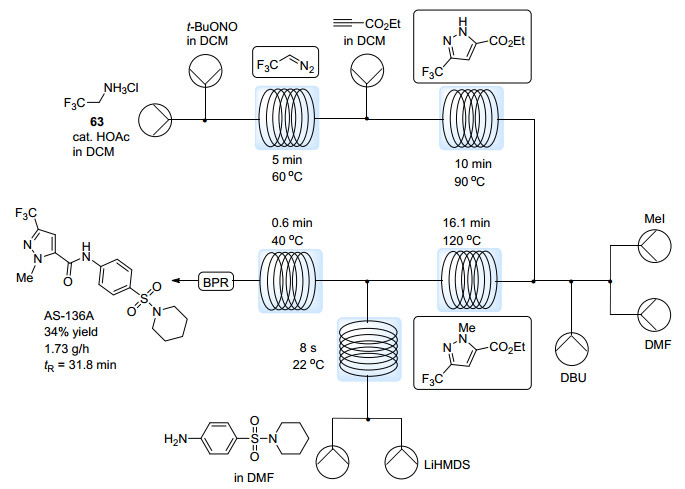
由于消耗重氮化合物的步骤是在批量条件下进行的, 以上过程严格意义上属于半批量反应.如果能够将含氟重氮化合物的产生以及后续步骤全部整合在流动体系中完成, 即可实现药物中间体的“流水线合成”, 将显著减少操作步骤并提高生产效率. Jamison等[64]最近报道了一系列具有生物活性的含氟烷基取代吡唑与吡唑啉的“流水线合成”策略.此过程的关键步骤仍是含氟重氮化合物与炔烃或烯烃的[3+2]环加成反应, 但作者通过流动体系中压力的控制, 成功实现了在二氯甲烷溶剂中的高温反应(90~132 ℃).在室温下需要14 h的[3+2]环加成反应, 在高温流动体系中仅需10 min, 且无需加入其他催化剂或添加剂.通过后续加入不同的反应模块, 能够实现氮烷基化或芳基化、脱保护、胺化反应等多种转化, 从而在流动体系中直接实现药物或农药中间体的合成.以麻疹治疗药物AS-136A为例(Scheme 31), 在完全串联的流动反应体系中能够获得34%的产率, 总保留时间约30 min.虽然由于副反应的存在, 产率与分步反应相比较低, 但仍能以1.73 g/h的速率获得该药物中间体, 并能明显简化操作.
连续流动反应作为一种新兴的反应技术, 近年来被广泛应用于天然产物全合成、药物中间体合成、有机光反应等多个领域, 其在有害化合物处理上的优势使之能够被应用于各种重氮化合物参与的反应之中.一些半稳定或不稳定的重氮化合物在连续流动条件下能够安全高效地参与转化, 从而大大扩展了重氮化合物的应用前景, 使之成为一种更加普适的合成子应用于有机合成之中.在保证安全的基础之上, 流动化学方法能够简便地实现扩大化生产, 为重氮化合物工业规模的应用提供了可能性.除此之外, 流动化学在处理高温高压反应、光化学反应、快速反应、非均相催化反应以及多步反应时所具有的优势, 也使合成化学家在应用重氮化合物时拥有除传统批量反应之外的另一个选择.
虽然经过近十几年的发展, 重氮化合物参与的流动反应已经有了相当多的报道, 逐渐形成了一定的体系, 但我们还应看到, 这一体系仍不完善.首先, 大多数报道着眼于重氮化合物的安全性以及反应的扩大化生产之上, 应用的反应往往是重氮化合物比较经典的转化, 利用连续流动方法来发现重氮化合物新的反应活性的报道比较少.实际上, 流动反应较批量反应更高的传热以及传质效率为重氮化合物全新的反应活性提供了较大的想象空间.其次, 与较稳定的α-羰基重氮化合物以及结构最为简单的重氮甲烷相比, 烃基重氮化合物在流动化学中的应用还有待于进一步的发掘.发展更为高效便捷普适的流动合成方法将大大提升这类不稳定化合物的应用潜力.类似的, 由于起步较晚, 含氟烷基重氮化合物在流动体系中的反应性仅限于环加成反应以及环丙烷化反应, 鉴于含氟基团在生物活性分子中的广泛性, 这类转化拥有较大的发展空间.我们有理由相信, 重氮化合物参与的连续流动反应将会受到更多的关注, 也会给重氮化学领域带来全新的机遇.
For selected reviews, see: (a) Ye, T. ; McKervey, M. A. Chem. Rev. 1994, 94, 1091.
(b) Doyle, M. P. ; McKervey, M. A. ; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, Wiley, New York, 1998.
(c) Zhang, Z. ; Wang, J. Tetrahedron 2008, 64, 6577.
(d) Davies, H. M. L. ; Denton, J. R. Chem. Soc. Rev. 2009, 38, 3061.
(e) Ford, A. ; Miel, H. ; Ring, A. ; Slattery, C. N. ; Maguire, A. R. ; McKervey, M. A. Chem. Rev. 2015, 115, 9981.
(f) Qiu, D. ; Qiu, M. ; Ma, R. ; Zhang, Y. ; Wang, J. Acta Chim. Sinica 2016, 74, 472(in Chinese).
(邱頔, 邱孟龙, 马戎, 张艳, 王剑波, 化学学报, 2016, 74, 472. )
(g) Liu, L. ; Zhang, J. Chin. J. Org. Chem. 2017, 37, 1117(in Chinese).
(刘路, 张俊良, 有机化学, 2017, 37, 1117. )
(a) Zhang, Y. ; Wang, J. Eur. J. Org. Chem. 2011, 6, 1015.
(b) Shao, Z. ; Zhang, H. Chem. Soc. Rev. 2012, 41, 560.
(c) Barluenga, J. ; Valdés, C. Angew. Chem., Int. Ed. 2011, 50, 7486.
(d) Xiao, Q. ; Zhang, Y. ; Wang, J. Acc. Chem. Res. 2013, 46, 236.
(e) Xia, Y. ; Zhang, Y. ; Wang, J. ACS Catal. 2013, 3, 2586.
(f) Liu, Z. ; Wang, J. J. Org. Chem. 2013, 78, 10024.
(g) Barroso, R. ; Cabal, M. P. ; Valdés, C. Synthesis 2017, 49, 4434.
(h) Xia, Y. ; Di, Q. ; Wang J. Chem. Rev. 2017, 117, 13810.
For selected reviews, see: (a) Noël, T. ; Buchwald, S. L. Chem. Soc. Rev. 2011, 40, 5010.
(b) Pastre, J. C. ; Browne, D. L. ; Ley, S. V. Chem. Soc. Rev. 2013, 42, 8849.
(c) Gutmann, B. ; Cantillo, D. ; Kappe, C. O. Angew. Chem., Int. Ed. 2015, 54, 6688.
(d) Cambié, D. ; Bottecchia, C. ; Straathof, N. J. W. ; Hessel, V. ; Noël, T. Chem. Rev. 2016, 116, 10276.
(e) Britton, J. ; Raston, C. L. Chem. Soc. Rev. 2017, 46, 1250.
(f) Plutschack, M. B. ; Pieber, B. ; Gilmore, K. ; Seeberger, P. H. Chem. Rev. 2017, 117, 11796.
(a) Müller, S. T. R. ; Wirth, T. ChemSusChem 2015, 8, 245.
(b) Deadman, B. J. ; Collins, S. G. ; Maguire, A. R. Chem. -Eur. J. 2015, 21, 2298.
(c) Movsisyan, M. ; Delbeke, E. I. P. ; Berton, J. K. E. T. ; Battilocchio, C. ; Ley, S. V. ; Stevens, C. V. Chem. Soc. Rev. 2016, 45, 4892.
Archibald, T. G. ; Barnard, J. C. ; Reese, H. F. US 5854405, 1998[Chem. Abstr. 1999, 130, 83188].
Proctor, L. D.; Warr, A. J. Org. Process Res. Dev. 2002, 6, 884. doi: 10.1021/op020049k
Ferstl, W. F.; Schwarzer, M. S.; Löbbecke, S. L. Chem. Ing. Tech. 2004, 76, 1326. doi: 10.1002/cite.200490233/full
Struempel, M.; Ondruschka, B.; Daute, R.; Stark, A. Green Chem. 2008, 10, 41. doi: 10.1039/B710554A
Maurya, R. A.; Park, C. P.; Lee, J. H.; Kim, D.-P. Angew. Chem., Int. Ed. 2011, 50, 5952. doi: 10.1002/anie.v50.26
(a) Mastronardi, F. ; Gutmann, B. ; Kappe, C. O. Org. Lett. 2013, 15, 5590.
(b) Brzozowski, M. ; O'Brien, M. ; Ley, S. V. ; Polyzos, A. Acc. Chem. Res. 2015, 48, 349.
Pinho, V. D.; Gutmann, B.; Miranda, L. S. M.; de Souza, R. O. M. A.; Kappe, C. O. J. Org. Chem. 2014, 79, 1555. doi: 10.1021/jo402849z
Pinho, V. D.; Gutmann, B.; Kappe, C. O. RSC Adv. 2014, 4, 37419. doi: 10.1039/C4RA08113G
(a) Dallinger, D. ; Pinho, V. D. ; Gutmann, B. ; Kappe, C. O. J. Org. Chem. 2016, 81, 5814.
(b) Garbarino, S. ; Guerra, J. ; Poechlauer, P. ; Gutmann, B. ; Kappe, C. O. J. Flow Chem. 2016, 6, 211.
(c) Dallinger, D. ; Kappe, C. O. Nat. Protoc. 2017, 12, 2138.
Rossi, E.; Woehl, P.; Maggini, M. Org. Process Res. Dev. 2012, 16, 1146. doi: 10.1021/op200110a
Lehmann, H. Green Chem. 2017, 19, 1449. doi: 10.1039/C6GC03066A
Pollet, P.; Cope, E. D.; Kassner, M. K.; Charney, R.; Terett, S. H.; Richman, K. W.; Dubay, W.; Stringer, J.; Eckert, C. A.; Liotta, C. L. Ind. Eng. Chem. Res. 2009, 48, 7032. doi: 10.1021/ie801885y
Martin, L. J.; Marzinzik, A. L.; Ley, S. V.; Baxendale, I. R. Org. Lett. 2011, 13, 320. doi: 10.1021/ol1027927
(a) Mifune, Y. ; Fuse, S. ; Tanaka, H. J. Flow Chem. 2014, 4, 173.
(b) Fuse, S. ; Otake, Y. ; Mifune, Y. ; Tanaka, H. Aust. J. Chem. 2015, 68, 1657.
Audubert, C.; Gamboa Marin, O. J.; Lebel, H. Angew. Chem., Int. Ed. 2017, 56, 6294. doi: 10.1002/anie.201612235
Barluenga, J.; Tomás-Gamasa, M.; Aznar, F.; Valdés, C. Nat. Chem. 2009, 1, 494. doi: 10.1038/nchem.328
Kupracz, L.; Kirschning, A. J. Flow Chem. 2012, 3, 11. https://www.uni-oldenburg.de/oc-christoffers/ehemalige/diplom-master-bachelorarbeiten/kupracz-lukas/
Lévesque, É.; Laporte, S. T.; Charette, A. B. Angew. Chem., Int. Ed. 2017, 56, 837. doi: 10.1002/anie.201608444
Rossi, E.; Carofiglio, T.; Venturi, A.; Ndobe, A.; Muccini, M.; Maggini, M. Energy Environ. Sci. 2011, 4, 725. doi: 10.1039/C0EE00314J
Tran, D. N.; Battilocchio, C.; Lou, S.-B.; Hawkins, J. M.; Ley, S. V. Chem. Sci. 2015, 6, 1120. doi: 10.1039/C4SC03072A
Battilocchio, C.; Feist, F.; Hafner, A.; Simon, M.; Tran, D. N.; Allwood, D. M.; Blakemore, D. C.; Ley, S. V. Nat. Chem. 2016, 8, 360. doi: 10.1038/nchem.2439
Roda, N. M.; Tran, D. N.; Battilocchio, C.; Labes, R.; Ingham, R. J.; Hawkins, J. M.; Ley, S. V. Org. Biomol. Chem. 2015, 13, 2550. doi: 10.1039/C5OB00019J
Poh, J.-S.; Tran, D. N.; Battilocchio, C.; Hawkins, J. M.; Ley, S. V. Angew. Chem., Int. Ed. 2015, 54, 7920. doi: 10.1002/anie.201501538
Poh, J.-S.; Makai, S.; vonKeutz, T.; Tran, D. N.; Battilocchio, C.; Pasau, P.; Ley, S. V. Angew. Chem., Int. Ed. 2017, 56, 1864. doi: 10.1002/anie.201611067
Majchrzak, M. W.; Bekhazi, M.; Tse-Sheepy, I.; Warkentin, J. J. Org. Chem. 1989, 54, 1842. doi: 10.1021/jo00269a019
Greb, A.; Poh, J.-S.; Greed, S.; Battilocchio, C.; Pasau, P.; Blakemore, D. C.; Ley, S. V. Angew. Chem., Int. Ed. 2017, 56, 16602. doi: 10.1002/anie.201710445
Delville, M. M. E.; van Hest, J. C. M.; Rutjes, F. P. J. T. Beilstein J. Org. Chem. 2013, 9, 1813. doi: 10.3762/bjoc.9.211
Maurya, R. A.; Min, K.-I.; Kim, D.-P. Green Chem. 2013, 16, 116. http://onlinelibrary.wiley.com/resolve/reference/XREF?id=10.1039/c3gc41226a
Müller, S. T. R.; Smith, D.; Hellier, P.; Wirth, T. Synlett 2014, 25, 871. doi: 10.1055/s-00000083
Müller, S. T. R.; Hokamp, T.; Ehrmann, S.; Hellier, P.; Wirth, T. Chem.-Eur. J. 2016, 22, 11940. doi: 10.1002/chem.201602133
(a) Burguete, M. I. ; Cornejo, A. ; García-Verdugo, E. ; García, J. ; Gil, M. J. ; Luis, S. V. ; Martínez-Merino, V. ; Mayoral, J. A. ; Sokolova, M. Green Chem. 2007, 9, 1091.
(b) Aranda, C. ; Cornejo, A. ; Fraile, J. M. ; García-Verdugo, E. ; Gil, M. J. ; Luis, S. V. ; Mayoral, J. A. ; Martinez-Merino, V. ; Ochoa, Z. Green Chem. 2011, 13, 983.
Castano, B.; Gallo, E.; Cole-Hamilton, D. J.; Santo, V. D.; Psaro, R.; Caselli, A. Green Chem. 2014, 16, 3202. doi: 10.1039/C4GC00119B
Maestre, L.; Ozkal, E.; Ayats, C.; Beltrán, Á.; Díaz-Requejo, M. M.; Pérez, P. J.; Pericìs, M. A. Chem. Sci. 2015, 6, 1510. doi: 10.1039/C4SC03277B
Bartrum, H. E.; Blakemore, D. C.; Moody, C. J.; Hayes, C. J. J. Org. Chem. 2010, 75, 8674. doi: 10.1021/jo101783m
Zhang, X.; Stefanick, S.; Villani, F. J. Org. Process Res. Dev. 2004, 8, 455. doi: 10.1021/op034193x
(a) Bartrum, H. E. ; Blakemore, D. C. ; Moody, C. J. ; Hayes, C. J. Chem. -Eur. J. 2011, 17, 9586.
(b) Bartrum, H. E. ; Blakemore, D. C. ; Moody, C. J. ; Hayes, C. J. Tetrahedron 2013, 69, 2276.
Nicolle, S. M.; Hayes, C. J.; Moody, C. J. Chem.-Eur. J. 2015, 21, 4576. doi: 10.1002/chem.201500118
Nicolle, S. M.; Nortcliffe, A.; Bartrum, H. E.; Lewis, W.; Hayes, C. J.; Moody, C. J. Chem.-Eur. J. 2017, 23, 13623. doi: 10.1002/chem.201703746
Rackl, D.; Yoo, C.-J.; Jones, C. W.; Davies, H. M. L. Org. Lett. 2017, 19, 3055. doi: 10.1021/acs.orglett.7b01073
Müller, S. T. R.; Murat, A.; Maillos, D.; Lesimple, P.; Hellier, P.; Wirth, T. Chem.-Eur. J. 2015, 21, 7016. doi: 10.1002/chem.201500416
Müller, S. T. R.; Murat, A.; Hellier, P.; Wirth, T. Org. Process Res. Dev. 2016, 20, 495. doi: 10.1021/acs.oprd.5b00308
Deadman, B. J.; O'Mahony, R. M.; Lynch, D.; Crowley, D. C.; Collins, S. G.; Maguire, A. R. Org. Biomol. Chem. 2016, 14, 3423. doi: 10.1039/C6OB00246C
(a) McCaw, P. G. ; Deadman, B. J. ; Maguire, A. R. ; Collins, S. G. J. Flow Chem. 2016, 6, 226.
(b) McCaw, P. G. ; Buckley, N. M. ; Eccles, K. S. ; Lawrence, S. E. ; Maguire, A. R. ; Collins, S. G. J. Org. Chem. 2017, 82, 3666.
Gérardy, R.; Winter, M.; Vizza, A.; Monbaliu, J.-C. M. React. Chem. Eng. 2017, 2, 149. doi: 10.1039/C6RE00184J
Fuse, S.; Otake, Y.; Nakamura, H. Eur. J. Org. Chem. 2017, 44, 6466.
Vaske, Y. S. M.; Mahoney, M. E.; Konopelski, J. P.; Rogow, D. L.; McDonald, W. J. J. Am. Chem. Soc. 2010, 132, 11379. doi: 10.1021/ja1050023
Willumstad, T. P.; Haze, O.; Mak, X. Y.; Lam, T. Y.; Wang, Y.-P.; Danheiser, R. L. J. Org. Chem. 2013, 78, 11450. doi: 10.1021/jo402010b
Li, M.-M.; Wei, Y.; Liu, J.; Chen, H.-W.; Lu, L.-Q.; Xiao, W.-J. J. Am. Chem. Soc. 2017, 139, 14707. doi: 10.1021/jacs.7b08310
Garbarino, S.; Protti, S.; Basso, A. Synthesis 2015, 47, 2385. doi: 10.1055/s-00000084
Basso, A.; Banfi, L.; Garbarino, S.; Riva, R. Angew. Chem., Int. Ed. 2013, 52, 2096. doi: 10.1002/anie.v52.7
Musio, B.; Mariani, F.; Śliwiński, E. P.; Kabeshov, M. A.; Odajima, H.; Ley, S. V. Synthesis 2016, 48, 3515. doi: 10.1055/s-0035-1562579
Takeda, K.; Oohara, T.; Shimada, N.; Nambu, H.; Hashimoto, S. Chem.-Eur. J. 2011, 17, 13992. doi: 10.1002/chem.v17.50
Moschetta, E. G.; Negretti, S.; Chepiga, K. M.; Brunelli, N. A.; Labreche, Y.; Feng, Y.; Rezaei, F.; Lively, R. P.; Koros, W. J.; Davies, H. M. L.; Jones, C. W. Angew. Chem., Int. Ed. 2015, 54, 6470. doi: 10.1002/anie.201500841
Pasceri, R.; Bartrum, H. E.; Hayes, C. J.; Moody, C. J. Chem. Commun. 2012, 48, 12077. doi: 10.1039/c2cc37284c
Fleming, G. S.; Beeler, A. B. Org. Lett. 2017, 19, 5268. doi: 10.1021/acs.orglett.7b02537
Pieber, B.; Kappe, C. O. Org. Lett. 2016, 18, 1076. doi: 10.1021/acs.orglett.6b00194
Mertens, L.; Hock, K. J.; Koenigs, R. M. Chem.-Eur. J. 2016, 22, 9542. doi: 10.1002/chem.201601707
Hock, K. J.; Mertens, L.; Metze, F. K.; Schmittmann, C.; Koenigs, R. M. Green Chem. 2017, 19, 905. doi: 10.1039/C6GC03187K
Hock, K. J.; Mertens, L.; Koenigs, R. M. Chem. Commun. 2016, 52, 13783. doi: 10.1039/C6CC07745E
Britton, J.; Jamison, T. F. Angew. Chem., Int. Ed. 2017, 56, 8823. doi: 10.1002/anie.v56.30

图式 2 Aerojet公司发展的CH2N2连续流动生产技术
Scheme 2 Aerojet continuous flow technique for CH2N2 production

图式 3 CH2N2在双通道平行微反应器中的原位生成与反应
Scheme 3 In situ generation and reaction of CH2N2 in dual-channel microreactor

图式 5 CH2N2在管中管反应器中实现的多步连续流动反应
Scheme 5 Multi-step continuous flow reactions of CH2N2 in tube-in-tube microreactor

图式 9 N-对甲苯磺酰腙与芳基重氮的偶联反应
Scheme 9 Coupling reaction of N-tosylhydrazone with aryl boronic acid

图式 10 通过腙热分解实现的芳基重氮甲烷的流动合成与纯化
Scheme 10 Flow synthesis and purification of aryldiazomethanes via hydrazone fragmentation

图式 11 腙氧化法制重氮化合物及后续偶联反应
Scheme 11 Diazo compound formation by hydrazone oxidation and subsequent coupling

图式 12 基于芳基重氮化合物的迭代碳碳键形成反应
Scheme 12 Iterative C—C bond formation reaction of diazo compounds

图式 13 芳基重氮化合物参与的环丙烷化反应与联烯合成
Scheme 13 Cyclopropanation and allene synthesis with aryl diazo compounds

图式 14 烷基重氮化合物的流动合成与转化
Scheme 14 Flow synthesis and transformation of alkyl-substi-tuted diazo compounds

图式 17 非均相铜催化剂37在EDA参与的反应中的应用
Scheme 17 Utilization of heterogeneous Cu catalyst 37 in reactions of EDA

图式 19 通过Bamford-Stevens反应流动制备重氮酯
Scheme 19 Flow synthesis of diazoesters via Bamford-Stevens reaction

图式 20 利用固载氧化剂实现的重氮化合物流动合成
Scheme 20 Flow synthesis of diazo compounds using supported oxidant

图式 23 经由Wolff重排实现的β-内酰胺的流动制备
Scheme 23 Synthesis of β-lactams in flow via Wolff rearrangement

图式 24 流动条件下烯酮中间体的环加成反应
Scheme 24 Cycloaddition reactions of ketene intermediates under flow conditions

图式 25 非均相催化羰基叶立德[3+2]环加成反应
Scheme 25 Carbonyl ylide [3+2] cycloaddition reaction under heterogeneous catalysis

图式 26 中空纤维流动反应器的图示与原理
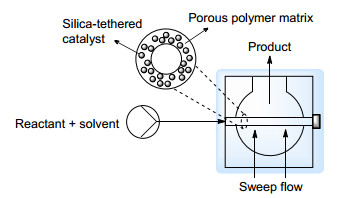
Scheme 26 Principle and schematic view of the hollow fiber flow reactor

图式 27 α-重氮-β-酮酯61的亲核氟化反应
Scheme 27 Nucleophilic fluorination of α-diazo-β-ketoester 61

图式 29 CF3CHN2在管中管反应器中的制备与应用
Scheme 29 Generation and application of CF3CHN2 in tube-in-tube reactor

图式 30 含氟烷基重氮化合物的[3+2]环加成反应
Scheme 30 [3+2] cycloaddition reactions of fluoroalkyl-substituted diazo compounds

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:






 下载:
下载:











 下载:
下载: