
图 1.
Wurster红盐和蓝盐
Figure 1.
Wurster's salts (red and blue)

自由基正离子(radical cation, RC)作为一种重要的有机活性中间体受到了广大有机化学家的广泛关注.通常来说, 自由基正离子中间体是一类既含有未成对电子又带有一个单位正电荷的活性中间体, 根据成单电子和正电荷的分布状况, 自由基正离子可分为两类: (1)成单电子和正电荷集中在同一个原子上, 称为普通自由基正离子, 这类自由基正离子会表现出不同于自由基和碳正离子的一些特殊性质; (2)成单电子和正电荷分布在不同的原子上, 这类自由基正离子称为distonic radical cation, 它的未成对电子中心表现自由基的性质, 而正离子部分通常发生经典的正离子反应.
早在1879年, 第一个自由基正离子盐(radical cation salt, RCS)就已被合成出来, 由于其带有特殊的颜色被称为Wurster蓝盐和红盐(图 1)[1]. 1926年前后, Weitz等[2]首次报道了一类三芳基胺类自由基正离子高氯酸盐的制备.随着对自由基正离子盐结构和性质的研究, 各种类型的自由基正离子盐被合成出来并用于各类有机化学转化(图 2)[3].这些自由基正离子盐通常具有较大的共轭体系或存在大位阻基团, 因此, 在室温、空气环境下都能稳定存在, 是一类具有很高实用价值的单电子氧化剂, 其中, 对溴三苯胺自由基正离子六氯锑酸盐(TBPA)还实现了商品化.自由基正离子盐除了可用于引发自由基正离子反应外, 还可作为空穴载体被广泛地应用于有机材料学的研究.本文重点综述自由基正离子盐引发的有机化学转化的最新进展及其相关的反应机理探讨.
自由基正离子盐由于其缺电子特性, 通常具有一定的氧化性, 其氧化性的强弱可以通过循环伏安法进行测定.例如:对溴三苯胺自由基正离子六氯锑酸盐(TBPA)的还原电位为1.15 V (vs. SCE), 其类似物三(2, 4-二溴三苯基)胺六氯锑酸盐(TDBPA)的还原电位可达到1.66 V (vs. SCE).
自由基正离子盐作为单电子氧化剂通常从富电子底物中夺取一个电子, 形成自由基正离子中间体(一级RC), 自身被还原(Scheme 1).生成的自由基正离子中间体发生后续的加成、重排等一系列反应产生二级自由基正离子(二级RC), 而二级自由基正离子通常具有更高的氧化性, 会从底物夺取电子生成相应产物, 并再生出一级RC完成催化循环(RC催化).这种情况下, 由于一级RC可以再生, 一般只需要催化量自由基正离子盐即可高效地引发反应.如果一级自由基正离子发生碎裂, 则会生成自由基或正离子中间体, 接着发生自由基反应或正离子反应(RC化学计量氧化).产生的自由基中间体还可以被自由基正离子盐继续氧化为正离子中间体, 从而发生相应的正离子反应.由于一级RC不能再生, 通常需要加入化学计量的自由基正离子盐来促进反应的完成.
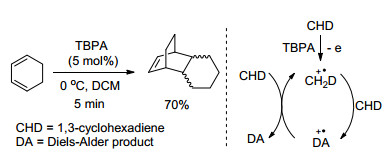
自由基正离子盐催化的Diels-Alder环加成反应可能是最著名的自由基正离子反应, 其相关机理也被充分阐明.早在二十世纪八九十年代, Bauld和Eberson等详细地研究了自由基正离子盐促进下的环己二烯Diels- Alder二聚反应, 并建立了经历自由基正离子中间体的链式反应机理(Scheme 2)[4]. 1, 3-环己二烯在5 mol% TBPA催化下, 室温反应5 min即可以70%的产率得到DA环加成产物.该反应的机理如下:环己二烯被自由基正离子盐氧化为相应的自由基正离子中间体, 接着与另一分子环己二烯发生环加成, 产生二级自由基正离子, 该自由基正离子将环己二烯氧化得到加成产物, 同时再生出环己二烯自由基正离子参与下一步环加成反应.
在此项研究的基础上, Bauld等[5]详细研究了其它富电子烯烃的DA环加成反应, 其中包括共轭二烯与环己二烯、苯乙烯衍生物与共轭二烯、乙烯基醚或乙烯基硫醚与共轭二烯的DA环加成等(Eq. 1).该反应还被成功地应用于β-芹子烯的合成中.这些研究不仅拓展了自由基正离子盐促进的反应研究, 也为类似的研究奠定了坚实的机理基础, 在此基础上, 一系列自由基正离子盐促进的环加成反应被开发出来.
|
|
(1) |
Schmittel等[6]实现了三芳基胺自由基正离子盐催化的烯酮与共轭二烯的DA环加成反应.该反应在0 ℃ 5 min即可完成, 高效地构建了樟脑酮骨架(Eq. 2).与环己二烯二聚反应类似, 该反应中三芳基自由基正离子盐首先将共轭二烯氧化为自由基正离子, 接着发生与烯酮的环加成生成二级自由基正离子中间体, 经过电子转移得到最终产物.
|
|
(2) |
在这些前人工作的启发下, 贾晓东、刘中立等[7]报道了三芳基自由基正离子盐促进的芳香亚胺1与α-甲基苯乙烯衍生物2的氮杂DA反应, 成功地用于一系列四氢喹啉衍生物的合成(Eq. 3).在该项研究中发现, α-甲基苯乙烯衍生物的氧化电位的高低会显著影响此反应的发生.当α-甲基苯乙烯的氧化电位低于相应的亚胺时, 氮杂DA反应顺利发生, 高产率地得到四氢喹啉衍生物(3a, 3b).而当亚胺的电位较低时, 反应不能有效地进行, 只得到亚胺分解产物.这是由于自由基正离子盐首先氧化苯乙烯产生相应的自由基正离子中间体, 接着与亚胺发生[4+2]环加成反应.如果亚胺电位低于苯乙烯, 亚胺被优先氧化分解, 从而阻碍了反应的正常进行.
|
|
(3) |
在这项研究的基础上, 该课题组实现了一系列富电子双键, 例如烯酰胺、烯胺和烯醇醚等, 与芳香亚胺的氮杂DA环加成反应, 构筑了各种取代类型的四氢喹啉以及喹啉骨架(Eq. 4)[8].
|
|
(4) |
由于查尔酮环氧化合物被氧化后生成自由基正离子中间体, 该中间体既可发生自由基反应, 在合适的亲核试剂存在下也可以进行相应的碳正离子反应.霍聪德课题组[12]利用富电子萘酚11、12作为亲核试剂, 实现了自由基正离子盐促进的萘酚类化合物的Friedel-Crafts类型反应(Scheme 3).有趣的是, 当使用β-萘酚12作为亲核试剂时, 会接着脱水, 得到苯并呋喃衍生物14.不仅如此, 芳香醚类也可发生类似反应得到Friedel-Crafts烷基化产物[13].
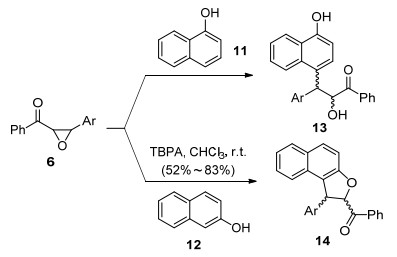
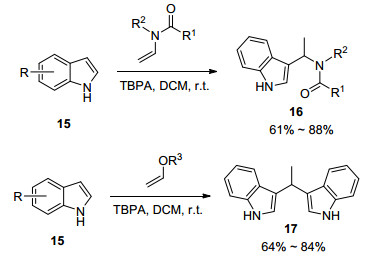
这一反应说明, 单电子氧化产生的自由基正离子中间体可以作为温和的Friedel-Crafts试剂进行芳烃的Friedel-Crafts反应, 从而避免传统Friedel-Crafts反应需要较强的酸性条件的弊端.在此策略下, 该课题组[14]选择氧化富电子烯胺以及烯醇醚作为Friedel-Crafts试剂, 实现了吲哚类化合物15的Friedel-Crafts反应(Scheme 4).值得注意的是, 当选用烯醇醚时, 会发生C—N键的断裂, 得到二吲哚取代乙烷衍生物17.
2015年曾程初和Little等[15]通过电化学氧化对溴三苯胺, 原位产生其自由基正离子盐, 同样成功地实现了烯胺与富电子芳烃如萘酚、吲哚以及三甲氧基苯的Fridel-Crafts烷基化反应, 高产率地得到了相应芳烃衍生物(Eq. 5).
|
|
(5) |
自由基正离子盐不仅能有效地氧化富电子双键, 也可使张力三元环发生单电子氧化, 从而发生一系列[3+2]环加成反应.
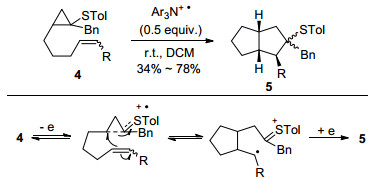
早在1995年Iwata等[9]就首次报道了三芳基胺自由基正离子盐促进的环丙基硫醚类化合物4的分子内[3+2]环加成反应(Scheme 5).在C=C双键作为自由基正离子受体存在下, 环丙基硫醚类化合物可以顺利地将三芳基胺盐氧化为相应的自由基正离子中间体, 接着与双键发生自由基加成得到自旋与电荷分离的distonic RC中间体, 经过二次电子转移, 以中等的产率和立体选择性得到苯硫基取代的并环化合物5.
2004年霍聪德、刘中立等[10]用三芳基胺自由基正离子盐实现了查尔酮环氧6的氧化断裂, 与富电子烯烃7发生[3+2]环加成反应, 合成了一系列四氢呋喃衍生物(Scheme 6).该反应使用二氯甲烷作为溶剂, 只需5 mol% TBPA在室温下反应5 min, 即可以90%产率和90%的立体选择性得到相应的四氢呋喃衍生物8, 相比于其它的呋喃环构建反应, 具有条件温和、产率高、选择性好等优势.

在三芳基胺自由基正离子盐存在下, 查尔酮环氧首先被氧化为相应的自由基正离子, 接着与烯烃发生自由基或正离子加成, 生成distonic RC中间体.该中间体将另一分子查尔酮环氧氧化得到四氢呋喃产物, 同时再生出查尔酮环氧自由基正离子参与下一个催化循环.
在此基础上, 2005年该课题组[11]选用亚胺9作为查尔酮环氧受体, 在类似的温和条件下, 又成功地实现了一系列噁唑烷10骨架的构建(Eq. 6).
|
|
(6) |
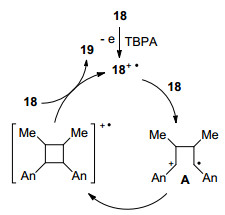
Bauld等[5]在早期的自由基正离子盐促进的DA反应研究中发现, 富电子烯烃不仅能与共轭二烯发生[4+2]环加成, 还有可能发生[2+2]环加成反应, 得到环丁烷衍生物.例如, 茴香脑18在三芳基胺盐催化下, 可以高效地发生[2+2]环加成(Scheme 7).该反应表现出头对头的区域选择性, 其立体选择性受温度的影响, 在0 ℃立体专一性地得到反式加成产物, 而在-35 ℃下得到顺反混合物.
不仅如此, 茴香脑还可以和其它富电子烯烃发生交叉[2+2]环加成. Metzger等[16]通过ESI-MS详细地研究了该反应的机理过程(Scheme 8).首先, 茴香脑被氧化为自由基正离子, 接着与另一分子茴香脑加成生成distonic自由基正离子中间体A, 再通过电子转移得到环丁烷化产物. ESI-MS检测到该distonic自由基正离子中间体A的生成, 证明该[2+2]环加成步骤是通过分步反应机理进行.
在自由基正离子盐促进的环加成反应研究中, Bauld[5]成功地实现了富电子烯烃与重氮乙酸乙酯20的[2+1]环加成反应, 在温和条件下高效构建了环丙烷衍生物骨架(Scheme 9).

受该反应启发, 霍聪德等[17]选用亚胺9作为受体, 在催化量三芳基胺自由基正离子盐诱导下, 以中等产率实现了氮杂环丙烷的合成(Eq. 7).该反应对缺电子芳香亚胺表现出良好的活性, 而富电子亚胺不能有效参与反应.值得注意的是, 该反应表现出良好的顺式立体选择性, 没有观察到相应反式产物的生成.
|
|
(7) |
Adam和Little等[18]详细研究了高张力房烷类化合物(Housane) 22的扩环重排反应, 并将该反应应用于胡萝卜烯(daucene)的合成中(Scheme 10).对该反应的一系列机理研究表明, 自由基正离子盐氧化了房烷中的张力三元碳环, 接着发生区域选择性地重排, 经过单电子回传(back electron transfer)生成了相应的扩环产物.相比于酸催化反应, 自由基正离子盐催化表现出更高的立体和区域选择性.
Jiménez和Miranda等[19]报道了三芳基自由基正离子盐可以将氮杂环丁烷氧化为其自由基正离子, 从而发生C—C键和C—N键的断裂, 得到二苯乙烯和芳香亚胺衍生物(Eq. 8).作者通过闪光光解对该碎裂反应机理的研究表明C—C键和C—N键的断裂是通过分步机理进行的.
|
|
(8) |
虽然自由基正离子盐被广泛应用于一系列单电子氧化反应, 但是, 由于常见的自由基正离子盐的氧化能力并不高, 因此, 相关的研究仅限于对富电子体系或张力环的氧化.这大大限制了自由基正离子盐催化的研究范围.另一方面, 随着C—H键活化研究的兴起, 众多有机化学家开发了多种催化体系来实现sp-, sp2-, sp3- C—H键的直接转化.这些转化通常需要过渡金属催化(如: Pd, Ni, Rh等), 或化学计量的氧化剂引发(如: DDQ、过氧化物等).而氧气作为最清洁的氧化剂由于受到其氧化能力的限制, 还不能很有效用于C—H键的直接官能团化.这就需要开发出合适的活化试剂来促进氧气参与的C—H键直接转化反应.
2012年, 贾晓东课题组[20]首次报道了三芳基胺自由基正离子盐促进下甘氨酸衍生物sp3-C—H键官能团化反应.在这项研究中, 作者发现, 三芳基胺自由基正离子盐能有效地促进氧气参与的C—H键氧化反应, 仅用催化量(5~10 mol%)对溴三苯胺自由基正离子盐, 就可以实现甘氨酸衍生物27与苯乙烯类化合物的氧化Povarov类型反应, 最高93%的产率得到相应的4-芳基喹啉-2-羧酸酯衍生物(Eq. 9).控制实验以及机理研究表明, 不加入自由基正离子盐或隔绝氧气该反应不能有效发生.这说明三芳基胺自由基正离子盐是促进C—H键需氧氧化的关键.虽然其它氧化体系也能高效的实现该反应, 但均需要加入化学计量甚至超过化学计量的氧化剂引发.很显然, 自由基正离子盐/氧气催化体系具有绿色、高效等优势, 无疑是非常理想的氧化体系.这一研究开辟了自由基正离子盐促进的C—H键活化研究的新领域, 为C—H键的直接转化提供了新方法.
|
|
(9) |
在这项研究的基础上, 该课题组进行了一系列甘氨酸酯衍生物sp3-C—H键官能团化研究, 成功实现了喹啉羧酸酯、喹啉并内酯/内酰胺以及1, 4-二氢吡啶衍生物的高效合成[21].
自由正离子盐/氧气体系促进的C—H键活化研究也启发了霍聪德课题组, 从自由基正离子盐促进的有机转化也转向C—H键活化研究. 2013年该课题组[22]报道了利用三芳基胺自由基正离子盐/氧气体系促进下甘氨酸酯衍生物27与吲哚类化合物15的氧化Fridel-Crafts反应(Eq. 10).有趣的是, 在该反应中C—N键也发生了断裂, 最终得到二吲哚取代的乙酸酯衍生物28.该反应不仅产率高、底物兼容性好, 而且能放大到20 mmol产率没有明显损失, 具有潜在的工业应用价值.
不仅如此, 该课题组[23]还将三芳基胺自由基正离子盐/氧气体系拓展到四氢异喹啉类化合物29的C—H官能团化(Scheme 11).在10 mol%三芳基胺自由基正离子促进下, 使用吲哚15或亚磷酸衍生物31可以顺利地实现相应的C—C键以及C—P键的构建.通过机理研究表明, 该反应首先将四氢异喹啉氧化为碳自由基, 接着发生单电子转移得到相应的亚胺正离子, 在亲核试剂的存在下发生亲核加成反应, 最终得到C—H键官能团化产物.

使用同样的催化氧化条件, 霍聪德等[24]又使用羰基化合物33作为亲核试剂, 进行了四氢异喹啉的氧化Mannich类型的反应, 高产率地实现了一系列β-氨基酮类衍生物34 (Eq. 11).值得注意的是, 该反应不仅适用于芳香酮, 而且对脂肪酮以及含有敏感官能团的底物(如环丙基、呋喃环等)都能以较高产率得到预期产物, 显示了良好的官能团耐受性.
由于碳中心自由基能很好的被C=C不饱和键所稳定, 贾晓东等[25]尝试了N-苄基苯胺类化合物35的需氧氧化反应(Eq. 12).使用苯乙烯类作为氧化Povarov反应的亲二烯体, 该课题组成功实现了1, 4-二芳基喹啉类化合物36的高效合成.该反应不仅兼容了甲基、卤素等惰性取代基, 对酚羟基也表现了很好的耐受性, 甚至带有强吸电子的硝基底物也能以不错的产率得到相应的产物.
|
|
(11) |
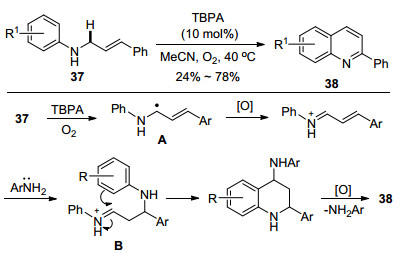
2016年贾晓东等[26]又成功地将自由基正离子盐/氧气体系用于N-肉桂基苯胺类化合物37的需氧氧化(Scheme 12).有趣的是, 该反应并未像作者预期那样生成4-芳基喹啉衍生物, 而是得到了2-芳基喹啉38.通过一系列控制实验和机理研究, 作者发现苯胺的存在对该反应的顺利进行起到了关键的作用.反应的历程如下: N-肉桂基苯胺首先被氧化为相应的自由基A, 单电子转移得到α, β-不饱和亚胺正离子中间体, 接着苯胺对该中间体进行Michael型加成, 随后中间体B经历分子内Friedel-Crafts环化建立四氢喹啉骨架.氮原子α-位活性C—H键的继续氧化促进了后续的脱氨基芳构化, 得到最终产物, 而消除的氨基继续参与下一个催化循环.
与上面的反应类似, 贾晓东等[27]又用此方法拓展到氨基巴豆酸酯39的分子内环合反应, 构筑了喹啉-2-羧酸酯40骨架(Eq. 13).
|
|
(13) |
在C—H键需氧氧化反应构筑喹啉骨架的研究中, 贾晓东等发现最终的芳构化是推动反应进行的重要驱动力.接着, 该课题组通过在底物特定位点引入取代基阻止芳构化的策略, 进行了一系列非芳香杂环骨架的构筑.
首先, 他们利用N-芳基丙氨酸酯41中氮原子α-位的甲基来阻止芳构化(Scheme 13)[28].实验结果表明, 由于最终不能形成芳香结构, 该类底物不和苯乙烯类化合物发生氧化Povarov反应, 而是与原位生成的烯胺中间体进行了自身[4+2]环加成反应, 得到了一系列1, 2-二氢喹啉类化合物42.机理研究表明, 丙氨酸酯氧化为亚胺正离子后通过互变异构产生了烯胺中间体, 相比于一般烯烃, 烯胺是更好的氮杂Diels-Alder反应亲二烯体, 其与N-芳基丙氨酸酯亚胺发生[4+2]环加成, 得到四氢喹啉, 最后消除氨基生成了1, 2-二氢喹啉.该反应不仅在喹啉骨架4-位引入了酯基官能团, 而且在2-位建立了一个季碳中心, 是一种很好的含杂环季碳中心的建立方法.

在此反应的研究中, 作者还分离得到了部分2-位酯基脱羧芳构化的产物.针对这一结果, 该课题组[29]进行了后续的研究, 成功地实现了2-甲基-4-酯基喹啉骨架的建立(Scheme 14).由于前线轨道不匹配, 丙炔酸酯并不能顺利地和芳香亚胺发生Povarov环化, 因此通过Povarov反应建立2-甲基-4-酯基喹啉骨架是完全不可行的.该课题组使用自由基正离子盐/氧气催化体系将丙氨酸酯41氧化为亚胺和烯胺的互变异构体, 利用氨基的推电子效应活化亲二烯体、酯基的拉电子效应活化氮杂二烯体, 高效地促进了该环加成的进行, 同时在芳构化的巨大能量驱动力下, 分别脱去氨基和羧基, 实现了一例双可离去活化基团促进的Povarov反应.该策略为实现热力学禁阻的Diels-Alder反应提拱了一种新思路.

2017年该课题组[30]通过在亲二烯体中引入取代基来阻止芳构化的策略, 又实现了3, 4-二氢喹啉-3-酮骨架的合成(Eq. 14).
|
|
(14) |
分子内1, 5-、1, 6-氢转移反应是一类广为人知的自由基反应, 在有机合成中有重要的应用[31].但通常需要在自由基引发剂存在下, 首先使得碳卤键或杂原子卤键发生均裂产生自由基中心, 再与分子内特定位点的C—H键发生氢原子转移反应.这样不仅需要通过复杂的合成过程制备相应的底物, 而且由于常用自由基引发剂的毒性及兼容性等问题, 大大影响了这类反应在绿色有机合成中的广泛应用.在甘氨酸酯类衍生物的催化氧化研究中, 由于推拉效应的存在, 酯基α-位的sp3-C—H键具有较高活性, 较易被氧化为相应的自由基中间体.受此启发, 贾晓东等设计了通过氧气捕获该自由基, 产生氧中心过氧自由基, 利用分子内1, 6-氢原子转移反应来活化其它惰性化学键的C—H键活化接力(C—H Activation Relay, CHAR)策略. 2016年, 该课题组[32]使用N-烃基甘氨酸酯类化合物44在自由基正离子盐的引发下, 成功实现了靛红类分子45骨架的建立(Scheme 15).该反应中不仅酰氧α-位的C—H被有效活化, 而且通过自由基β-消除反应也同时活化了非常惰性的酯基C—O键.通过一系列控制实验和自由基捕获实验证明, 该反应首先在氮原子α-位产生了自由基中心, 接着被氧气捕获生成过氧自由基, 发生分子内1, 6-氢迁移和β-碎裂得到了酰基自由基中间体, 然后经历分子内环合建立了靛红骨架.
Wittig反应是一类被广泛应用于有机合成化学的烯基化反应.一般是使用羰基化合物作为Wittig试剂的受体, 经历四元环中间体来建立碳碳不饱和键, 也可经历类似的机理用Staudinger试剂来进行碳氮不饱和键的形成[33].但用Wittig型反应建立羰基官能团尚未见报道.贾晓东课题组[34]使用α-氨基磷酸酯46通过自由基正离子盐促进的C—H键活化, 实现了用Wittig型反应构筑羰基官能团的方法(Scheme 16).该方法对氮原子芳环上各类取代基都能兼容, 分子内存在的烯丙基、苄基以及环丙基等对反应产率也没有显著影响.即使将反应放大到10 mmol规模, 也能以89%产率得到预期产物.机理研究表明, 自由基正离子盐与氧气首先将磷酸酯α-位氧化为自由基, 氧气捕获后产生的过氧中间体与磷原子生成四元环中间体, 再经过与Wittig反应类似的过程, 得到了羰基化产物.这是第一例通过Wittig反应来构筑羰基官能团的实例, 对拓展Wittig反应的合成应用具有重要的价值.
自由基正离子盐除了可以促进C—H键的需氧氧化外, 作为单电子氧化剂也可以用于sp3-C—H键的氧化官能团化. 2016年Murphy等人利用三甲基三苯胺自由基正离子盐TPTA作为单电子氧化剂实现了一系列氮原子α-位饱和碳氢键的氧化官能团化(Scheme 17)[35].在该反应中, TPTA首先将DABCO氧化为自由基正离子, 该中间体从氮原子α-位夺取氢原子产生碳中心自由基, 接着与另一分子DABCO自由基正离子发生偶联.在亲核试剂的存在下, 发生亲核取代反应完成C—H键到C—C键的转化.需要指出的是, 由于DABCO自由基正离子具有较大的位阻, 该反应表现出卓越的N-Me选择性, 即使在分子内存在众多竞争性的饱和C—H键的天然产物骨架分子中, 也能高选择性的在N-Me上发生官能团化.尤其重要的是, 通过自由基正离子盐氧化DABCO产生更为活泼的自由基正离子中间体来引发惰性C—H的活化, 这一策略为自由基正离子盐促进的C—H键活化反应提拱了新的思路, 将来有可能被用来实现更为广泛、更为惰性的C—H键活化反应.

本综述首先简述了自由基正离子盐引发的有机转化的早期研究, 重点阐述了近年来自由基正离子盐在单电子氧化反应和C—H键活化研究中的应用.自由基正离子盐作为典型的单电子氧化剂可以高效地促进一系列电子转移反应, 在此基础上, 近几年的研究结果表明, 三芳基胺类自由基正离子盐还可以有效地促进sp3-C—H键的需氧氧化, 实现C—H键的直接官能团化.通过这些研究成果可以看出, 自由基正离子盐促进的有机化学转化仍有广阔的研究空间, 特别是在C—H键的直接转化中表现出独特的优势.但是, 自由基正离子盐促进的需氧氧化机理尚未充分阐明, 制约了自由基正离子盐在有机合成中的应用.与此同时, 虽然有大量的自由基正离子盐被制备出来, 但其在促进有机转化的研究尚不充分, 目前仅三芳基胺类自由基正离子盐被广泛应用于有机合成反应中.我们相信在充分揭示自由基正离子盐促进的需氧氧化机理的基础上, 通过对自由基正离子盐的结构修饰以及制备具有更多结构多样性的自由基正离子盐, 将有望进一步拓展其在有机转化中的应用, 实现更加廉价、高效、绿色的有机合成反应.
(a) Wurster, C. ; Sendtner, R. Ber. Dtsch. Chem. Ges. 1879, 12, 1803.
(b) Wurster, C. Ber. Dtsch. Chem. Ges. 1879, 12, 2071.
(a) Weitz, E. ; Schwechten, H. W. Ber. 1926, 59, 2307.
(b) Weitz, E. ; Schwechten, H. W. Ber. 1927, 60, 545.
(a) Rathore, R. ; Kochi, J. K. J. Org. Chem. 1995, 60, 4399.
(b) Rathore, R. ; Kochi, J. K. J. Org. Chem. 1995, 60, 7479.
(c) Debroy, P. ; Shukla, R. ; Lindeman, S. V. ; Rathore, R. J. Org. Chem. 2007, 72, 1765.
(d) Schneeweis, A. ; Neidlinger, A. ; Reiss, G. J. ; Frank, W. ; Heinze, K. ; Müller T. J. J. Org. Chem. Front. 2017, 4, 839.
(e) Hiraoka, S. ; Okamoto, T. ; Kozaki, M. ; Shiomi, D. ; Sato, K. ; Takui, T. ; Okada, K. J. Am. Chem. Soc. 2004, 126, 58.
(f) Chen, X. ; Wang, X. ; Sui, Y. ; Li, Y. ; Ma, J. ; Zuo, J. ; Wang, X. Angew. Chem., Int. Ed. 2012, 51, 11878.
(a) Bauld, N. L. ; Pabon, R. J. Am. Chem. Soc. 1983, 105, 633.
(b) Eberson, L. ; Olofsson, B. Acta Chem. Scand. 1991, 45, 316.
Bauld, N. L. Tetrahedron 1989, 45, 5307. doi: 10.1016/S0040-4020(01)89486-2
(a) Schmittel, M. ; von Seggern, H. Angew. Chem., Int. Ed. 1991, 30, 999.
(b) Schmittel, M. ; von Seggern, H. J. Am. Chem. Soc. 1993, 115, 216.
Jia, X.-D.; Lin, H.-C.; Huo, C.-D.; Zhang, W.; Lü, J.-M.; Yang, L.; Zhao, G.-Y.; Liu, Z.-L. Synlett 2003, 1707.
(a) Jia, X. -D. ; Han, B. ; Zhang, W. ; Jin, X. -L. ; Yang, L. ; Liu, Z. -L. Synthesis 2006, 2831.
(b) Jia, X. -D. ; Ren, Y. ; Huo, C. -D. ; Wang, W. -J. ; Chen, X. -N. ; Xu, X. -L. ; Wang, X. -C. Tetrahedron Lett. 2010, 51, 6779.
(c) Jia, X. -D. ; Qing, C. ; Huo, C. -D. ; Peng, F. -F. ; Wang, X. -C. Tetrahedron Lett. 2012, 53, 7140.
(d) Jia, X. -D. ; Peng, F. -F. ; Qing, C. ; Huo, C. -D. ; Wang, Y. -X. ; Wang, X. -C. Tetrahedron Lett. 2013, 54, 4950.
Takemoto, Y.; Furuse, S.; Koike, H.; Ohra, T.; Iwata, C. Tetrahedron Lett. 1995, 36, 4085. doi: 10.1016/0040-4039(95)00721-N
Huo, C.; Jia, X.; Zhang, W.; Yang, L.; Lü, J.; Liu, Z. Synlett 2004, 251.
Huo, C.; Wei, R.; Zhang, W.; Yang, L. Liu, Z. Synlett 2005, 161. doi: 10.1016/j.tet.2011.10.062
Huo, C.; Xu, X.; An, J.; Jia, X.; Wang, X.; Wang, C. J. Org. Chem. 2012, 77, 8310. doi: 10.1021/jo300827s
Huo, C.; An, J.; Xu, X.; Jia, X.; Wang, X.; Kang, L. Tetrahedron Lett. 2013, 54, 1145. doi: 10.1016/j.tetlet.2012.12.078
Huo, C.; Kang, L.; Xu, X.; Jia, X.; Wang, X.; Xie, H.; Yuan, Y. Tetrahedron Lett. 2014, 55, 95. https://www.sciencedirect.com/science/article/pii/S0169409X1630151X
Li, L.-J.; Jiang, Y.-Y.; Lam, C. M.; Zeng, C.-C.; Hu, L.-M.; Little, R. D. J. Org. Chem. 2015, 80, 11021. doi: 10.1021/acs.joc.5b02222
Marquez, C. A.; Wang, H.; Fabbretti, F.; Metzger, J. O. J. Am. Chem. Soc. 2008, 130, 17208. doi: 10.1021/ja806791c
Huo, C.; Sun, C.; Hua, D.; Jia, X.; Xu, X.; Liu, Z. Tetrahedron Lett. 2011, 52, 7008. doi: 10.1016/j.tetlet.2011.10.121
(a) Adam, W. ; Librera, C. P. J. Org. Chem. 2002, 67, 576.
(b) Gerken, J. B. ; Wang, S. C. ; Preciado, A. B. ; Park, Y. S. ; Nishiguchi, G. ; Tantillo, D. J. ; Little, R. D. J. Org. Chem. 2005, 70, 4598.
(c) Park, Y. S. ; Little, R. D. J. Org. Chem. 2008, 73, 6807.
Andreu, I.; Delgado, J.; Espinós, A.; Pérez-Ruiz, R.; Jiménez, M. C.; Miranda, M. A. Org. Lett. 2008, 10, 5207. doi: 10.1021/ol802181u
Jia, X.-D.; Peng, F.-F.; Qing, C.; Huo, C.-D.; Wang, X.-C. Org. Lett. 2012, 14, 4030. doi: 10.1021/ol301909g
(a) Jia, X. -D. ; Wang, Y. -X. ; Peng, F. -F. ; Huo, C. -D. ; Yu, L. -L. ; Wang, X. -C. J. Org. Chem. 2013, 78, 9450.
(b) Wang, Y. -X. ; Peng, F. -F. ; Liu, J. ; Huo, C. -D. ; Wang, X. -C. ; Jia, X. -D. J. Org. Chem. 2015, 80, 609.
(c) Liu, J. ; Wang, Y. -X. ; Yu, L. -L. ; Huo, C. -D. ; Wang, X. -C. ; Jia, X. -D. Adv. Synth. Catal. 2014, 356, 3214.
(d) Jia, X. -D. ; Wang, Y. -X. ; Peng, F. -F. ; Huo, C. -D. ; Yu, L. -L. ; Liu, J. ; Wang, X. -C. Adv. Synth. Catal. 2014, 356, 1210.
Huo, C.; Wang, C.; Sun, C.; Jia, X.; Wang, X.; Chang, W.; Wu, M. Adv. Synth. Catal. 2013, 355, 1911. doi: 10.1002/adsc.v355.10
Huo, C.; Wang, C.; Wu, M.; Jia, X.; Wang, X.; Yuan, Y.; Xie, H. Org. Biomol. Chem., 2014, 12, 3123. doi: 10.1039/c3ob42454e
Huo, C.; Wu, M.; Jia, X.; Xie, H.; Yuan, Y.; Tang, J. J. Org. Chem. 2014, 79, 9860. doi: 10.1021/jo5017822
Liu, J.; Liu, F.; Zhu, Y.; Ma, X.; Jia, X. Org. Lett. 2015, 17, 1409. doi: 10.1021/acs.orglett.5b00244
Liu, F.; Yu, L.; Lü, S.; Yao, J.; Liu, J.; Jia, X. Adv. Synth. Catal. 2016, 358, 459. doi: 10.1002/adsc.201500574
Jia, X.; Li, P.; Shao, Y.; Yuan, Y.; Hou, W.; Liu, X.; Zhang, X.; Ji, H. Chem. Asian J. 2017, 12, 1719. doi: 10.1002/asia.v12.14
Lü, S.; Zhu, Y.; Ma, X.; Jia, X. Adv. Synth. Catal. 2016, 358, 1004. doi: 10.1002/adsc.201500885
Jia, X.-D.; Lü, S.; Yuan, Y.; Zhang, X.; Zhang, L.; Luo, L. Org. Biomol. Chem., 2017, 15, 2931. doi: 10.1039/C7OB00446J
Jia, X.; Hou, W.; Shao, Y.; Yuan, Y.; Chen, Q.; Li, P.; Liu, X.; Ji, H. Chem. Eur. J. 2017, 23, 12980. doi: 10.1002/chem.v23.53
(a) Huang, L. ; Lin, J. -S. ; Tan, B. ; Liu, X. -Y. ACS Catal. 2015, 5, 2826.
(b) Qin, Q. ; Yu, S. Org. Lett. 2015, 17, 1894.
(c) Kundu, R. ; Ball, Z. T. Org. Lett. 2010, 12, 2460.
(d) Hashimoto, T. ; Hirose, D. ; Taniguchi, T. Angew. Chem., Int. Ed. 2014, 53, 2730.
(e) Yu, P. ; Lin, J. -S. ; Li, L. ; Zheng, S. -C. ; Xiong, Y. -P. ; Zhao, L. -J. ; Tan, B. ; Liu, X. -Y. Angew. Chem., Int. Ed. 2014, 53, 11890.
Jia, X.; Zhu, Y.; Yuan, Y.; Zhang, X.; Lü, S.; Zhang, L.; Luo, L. ACS Catal. 2016, 6, 6033. doi: 10.1021/acscatal.6b01781
(a) Maryanoff, B. E. ; Reitz, A. B. Chem. Rev. 1989, 89, 863.
(b) Cristau, H. -J. Chem. Rev. 1994, 94, 1299.
(c) Rein, T. ; Pederson, T. M. Synthesis 2002, 5, 579.
(d) Hoffmann, R. W. Angew. Chem., Int. Ed. 2001, 40, 1411.
Zhu, Y.; Lü, S.; Ma, X.; Zhang, L.; Luo, L.; Jia, X. Asian J. Org. Chem. 2016, 5, 617. doi: 10.1002/ajoc.201600055
Barham, J. P.; John, M. P.; Murphy, J. A. J. Am. Chem. Soc. 2016, 138, 15482. doi: 10.1021/jacs.6b09690

Scheme 3 查尔酮环氧与萘酚的Friedel-Crafts反应
Scheme 3 Fridel-Crafts reactions between chalcone expoxides and naphthols

Scheme 5 环丙基硫醚的分子内[3+2]环加成
Scheme 5 Intramolecular [3+2] cycloaddition of cyclopropyl sulfides

Scheme 6 查尔酮环氧与烯烃的[3+2]环加成
Scheme 6 [3+2] cycloaddition between chalcone expoxides and alkenes

Scheme 9 烯烃与重氮乙酸乙酯的[2+1]环加成
Scheme 9 [2+1] cycloaddtion between alkenes and ethyl diazoacetate

Scheme 11 N-芳基四氢异喹啉的需氧氧化偶联
Scheme 11 Aerobic oxidative coupling of N-aryltetrahydro- isoquinolines

Scheme 13 通过N-芳基丙氨酸酯建立2, 3-二氢喹啉
Scheme 13 Construction of 2, 3-dihydroquinolines from N-arylalaninates

Scheme 14 通过N-芳基丙氨酸酯建立2-烷基喹啉-4-羧酸酯
Scheme 14 Construction of 2-alkylquinolin-4-carboxylates from N-arylalaninates

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



















 下载:
下载:











 下载:
下载: