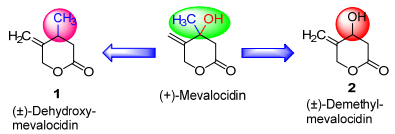
Scheme 1.
Mevalocidin and its designed analogues 1 & 2
Design and Synthesis of Natural Product Mevalocidin Chiral Center Based Analogues
Qiongyou Wu , Rui Zhang , Jinhuan Pan , John Clough , Yucheng Gu , Guangfu Yang

Although synthetic agrochemicals have played a significant role in the control of weeds, insects and fungal diseases of major agriculture crops, and their repetitive and continuous application over the years has resulted in the development of populations of resistant plants, insects and fungi.[1~4] Therefore, the search for new agrochemicals, especially those with modes of action that are different with commercial products, are of increasing importance. Although strategies for herbicide discovery are no different from those for the discovery of other agrochemicals or pharmaceuticals, herbicides with mode of action unrelated to those on the market have not been discovered during the last 20 years.[5] On the other hand, the secondary metabolites of plants and microorganisms are the result of the interaction between these species and the environment during a long period of evolution, and herbicides produced from these secondary metabolites may lead to less resistance development and fewer environmental issues.[6~8] The use of secondary metabolites directly as herbicides, or as lead compounds for further structural modifications, has been an important approach for herbicidal development. For example, the valuable commercial herbicide glufosinate, [9] is the synthetic version of the microbial natural product (phytotoxin) phosphinothricin.[10~12] Also, the widely using trike-tone class herbicides are derived from the plant-produced phytotoxin leptospermone.[13] Although these products represent only a small proportion of commercialized herbicides, they are important because each introduced a novel molecular target site for herbicides.
Mevalocidin, a phytotoxin, was discovered in 2008 from static cultures of two fungal isolates designated Rosellinia DA092917 and Fusarium DA056446.[14] The (+)-enan-tiomer of mevalocidin has broad spectrum post-emergence activity on grasses and broadleaf weeds and is currently under development for use in organic farming. Importantly, 14C-labeling experiments revealed that mevalocidin is transported via the phloem and xylem mobile, which is a highly desirable and relatively rare characteristic for herbicides to enhancing phytotoxicity by enabling the movement of a herbicide from the contact point to the growing meristems.[15] In addition, mevalocidin gives unique visual symptoms on treated plants, including stunting, meristematic inhibition and anthocyanin accumulation, which are unlike those of any known commercial herbicide, and therefore a novel mode of action is speculated.[16] However, it is should be pointed out that mevalocidin is a relatively high-dose herbicide (4 kg/ha), its potency is quite low. To achieve greater than 50% lethality, an application rate of 1 kg/ha was necessary for the control of all the weed species in the tests, showing that the mevalocidin is much weaker than modern synthetic herbicides.
Therefore, we envisioned that mevalocidin might be a useful lead for the discovery of novel herbicides. To date, no work has been published on the structural modification of this natural herbicide. In continuation of our program aimed at the discovery and development of natural product-based pesticides, [17, 18] herein to determine the role of hydroxyl and methyl groups in mevalocidin herbicidal activity, we designed two analogues (±)-deoxy-mevalocidin (1) and (±)-demethyl-mevalocidin (2) by removal of hydroxyl and methyl groups at its chiral center, respectively (Scheme 1). Further, the two compounds 1 and 2 were synthesized and herbicidal activity was evaluated. In each case, the six-membered lactone and the exocylic double bond were retained. In the two analogues described in this manuscript, the stereochemistry at the chiral center of mevalocidins was ignored, and racemic compounds were prepared.
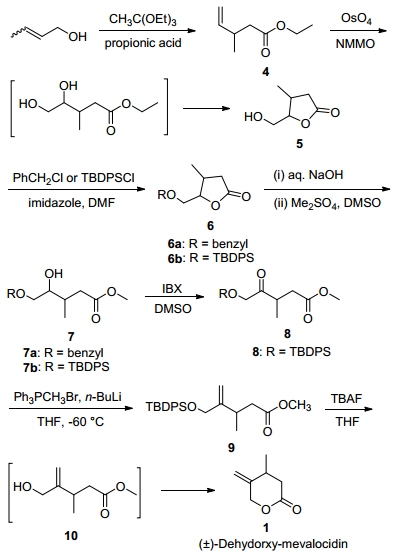
The synthetic approach for deoxy-mevalocidin 1 is depicted in Scheme 2. Treatment of 2-buten-1-ol with triethyl orthoacetate in the presence of a catalytic amount propionic acid afforded the ester 4 via a Claisen orthoester rearrangement. The olefinic bond in the ester 4 was then oxidized with OsO4 and 4-methylmorpholine N-oxide monohydrate (NMMO) to give an intermediate 1, 2-diol, which was not stable and ring-closed to give the five-membered lactone 5 in situ.[19] Selection of the protecting group for the primary hydroxyl group in lactone 5 was crucial to success of the subsequent ring-opening reaction. We initially selected benzyl as the protecting group because it can be cleanly removed by hydrogenolysis, so the benzyl ester 6a was prepared first. However, the ring opening reaction, carried out under aqueous basic conditions, failed to give the desired product 7a. The compound 7a was observed in thin-layer chromatography (TLC) but it is readily cyclized back to the lactone 6a. The bulky protection group t-butyl-diphenylsilyl (TBDPS) was chosen to protect the hydroxyl group of compound 5, and then compound 6b was synthesized. The subsequent ring-opening reaction (using aq. NaOH) and esterification (using Me2SO4 in DMSO) afforded the desired product 7b in a one-pot two-step manner in 71% yield. Ring-closure back to the lactone starting material was not observed, presumably due to the steric hindrance of the bulky TBDPS group. It is worth to mention that two chiral centers are generated in compounds 5, 6 and 7, however, the stereochemistry of these intermediates involved was ignored and the mixture of the diastereoisomers were prepared. The secondary hydroxyl group in compound 7b was then oxidized with 2-iodoxybenzoic acid (IBX) in DMSO to provide ketone 8 in a yield of 67%. Subsequent olefination of compound 8 with Wittig reagent methylenetriphenylphos-phorane (Ph3PCH2, generated in situ by treating Ph3PCH3Br with n-BuLi in THF at -60 ℃) afforded olefin 9. Finally, deprotection of TBDPS with tetrabutylammonium fluoride (TBAF) in THF gave the intermediate hydroxy-ester 10, which spontaneously cyclized to the desired racemic lactone (±)-deoxy-mevalocidin (1) in a yield of 77%.
We next investigated the synthesis of the lactone 2. The synthetic route is shown in Scheme 3. In this route, the commercially available triethyl phosphonoacetate was selected as starting material, when it reacted with an excess of formaldehyde in the presence of aqueous K2CO3 to give the allyl alcohol 11 via double hydroxymethylation and a Horner-Wadsworth-Emmons reaction.[20, 21] The allyl alcohol 11 was protected with t-butyldimethylsilyl chloride (TBSCl), its ester group 12 was reduced by using diisobu-tylaluminium hydride (DIBAL-H) to produce the singly-protected double allyl alcohol 13 in good yield. In the next step, the partial oxidation of the hydroxyl group in Swern oxidation conditions led to aldehyde 14. Reaction of aldehyde 14 with 1.2 equiv. of lithium tert-butyl acetate (in situ) gave the racemic product 15 in 88% yield after chromatography. The silyl protecting group was then removed by treatment with TBAF in THF resulted open chain form analogue of mevalocidin 16. Finally, cyclization of compound 16 achieved by treating with trifluoroacetic acid (TFA) in anisole produced lactone (±)-demethylmevalo-cidin (2).
The synthesized new mevalocidin analogues 1 and 2 were tested for herbicidal activity against two monocotyledonous and two dicotyledonous weeds at a concentration of 1000 mg/L. The results showed that the both analogues loosed herbicidal activity when compared to parent at tested concentration. In the same test, the racemic mevalocidin (in its lactone form) showed post-emergence activity against one monocotyledonous and two dicotyledonous weed species. These results show that both the hydroxyl and the methyl groups at the chiral centre of mevalocidin are crucial for its herbicidal activity.
In summary, two simplified analogues of mevalocidin were designed and successfully synthesized in an attempt to improve the potency of this herbicidal natural product. However, the herbicidal activity results showed that the analogues lost the herbicidal activity when compared to parent mevalocidin. Therefore, it can be concluded that, the methyl and/or hydroxyl groups at the chiral center of mevalocidin are crucial for its herbicidal activity and also this study provides insights into further value addition of phytotoxin mevalocidin.
Commercially available reagents and solvents were treated by standard methods before use. 1H NMR spectra were recorded on a VARIAN Mercury-Plus 600/Mercury-Plus 400 spectrometer in CDCl3 with TMS as the internal reference. 13C NMR spectra were recorded in CDCl3 or CD3OD on a VARIAN Mercury-Plus 600 (150 MHz)/ Mercury-Plus 400 (100 MHZ) spectrometer. HRMS were recorded on an Agilent 6520 Accurate-Mass Q-TOF instrument. Melting points were taken on a Buchi B-545 melting point apparatus and are uncorrected.
To a solution of 2-buten-1-ol (1.80 g, 25.0 mmol) and triethyl orthoacetate (33.7 mL) was added propionic acid (0.11 mL) at room temperature, and the mixture was heated under reflux for 11 h. The reaction mixture was poured into ice-cold sulfuric acid (5%, 44 mL), stirred for another 17 h. and then the reaction mixture was extracted with diethyl ether. The combined organic extracts were washed with aqueous sodium bicarbonate solution and brine, dried over Na2SO4, and concentrated under reduced pressure to give crude product 4 (3.52 g), which was used for the next step without further purification.
To the solution of residue 4 (11.7 mmol) dissolved in a mixed solution of acetone (53 mL), tert-butyl alcohol (12 mL) and water (12 mL), were successively added N-methylmorpholine N-oxide (NMMO, 2.38 g, 20.3 mmol) and OsO4 (2.0 mL, 2% aqueous solution). The reaction mixture was stirred at room temperature for 6 h until complete consumption of 4. The reaction was quenched by the addition of saturated aqueous sodium thiosulfate (30 mL), and stirred at room temperature for 0.5 h. The mixture was extracted with ethyl acetate, dried over Na2SO4, and concentrated under reduced pressure to yield the crude product 5 (1.64 g, yellow oil), which was used for the next step without further purification.
To a stirred solution of compound 5 (1.64 g, crude, from previous step) in dry DMF (25 mL) was added imidazole (1.11 g, 16.4 mmol) and TBDPSCl (3.92 g, 15.1 mmol), and the mixture was stirred at room temperature overnight. The reaction mixture was diluted with water (40 mL) and extracted with EtOAc. The combined organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure to give the crude product. Purification by flash chromatography on silica gel yielded compound 6b as a white solid (3.4 g, 37% in three steps). 1H NMR (600 MHz, CD3OD) δ: 7.90~7.62 (m, 4H), 7.49~7.29 (m, 6H), 4.17 (dt, J=6.0, 3.0 Hz, 1H), 3.87 (dd, J=12.0, 3.0 Hz, 1H), 3.73 (dd, J=12.0, 3.6 Hz, 1H), 3.63 (s, 1H), 2.77 (dd, J=17.4, 9.0 Hz, 1H), 2.59~2.53 (m, 1H), 2.22 (dd, J=17.4, 7.2 Hz, 1H), 1.09 (t, J=9.0 Hz, 3H), 1.05~0.98 (m, 9H); 13C NMR (150 MHz, CD3OD) δ: 179.52, 136.57, 133.92, 133.71, 133.27, 133.57, 131.25, 129.10, 88.86, 84.39, 65.50, 64.38, 49.34, 49.20, 49.05, 48.91, 38.02, 33.89, 32.82, 27.44, 20.19, 18.82, 14.35; HRMS (MALDI-TOF) calcd for C22H28NaO3Si [M+Na]+391.1705, found 391.1722.
To a solution of compound 6b (0.5 g, 1.35 mmol) in ethanol (4 mL) was added aqueous sodium hydroxide (81 mg, dissolved in 0.5 mL water), and the resulting mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure, using toluene to remove the residual water. The resulting crude carboxylic salt was dissolved in DMSO (5 mL), and dimethyl sulfate (0.15 mL) was added. The reaction mixture was stirred at room temperature for another 1 h before the addition of water (10 mL). It was extracted with Et2O, and the combined extracts were washed with brine and dried over Na2SO4, then concentrated by evaporation under reduced pressure. The residue was purified by flash chromatography on silica gel to give compound 7b as a colorless oil (438 mg, 81 %). 1H NMR (600 MHz, CDCl3) δ: 7.67 (dd, J=9.0, 7.8 Hz, 4H), 7.59~7.36 (m, 6H), 4.13~4.10 (m, 1H), 3.86 (dd, J=11.4, 3.0 Hz, 1H), 3.72 (dd, J=11.4, 3.6 Hz, 1H), 3.68 (dd, J=7.8, 4.2 Hz, 1H), 3.66~3.57 (m, 2H), 3.49 (s, 1H), 2.82 (dd, J=17.4, 9.0 Hz, 1H), 2.61~2.53 (m, 1H), 2.47 (dd, J=14.4, 4.8 Hz, 1H), 2.21~2.16 (m, 1H), 2.15 (t, J=3.0 Hz, 1H), 1.13 (d, J=7.2 Hz, 1H), 1.06 (d, J=5.4 Hz, 9H), 0.90 (d, J=6.6 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ: 176.74, 135.60, 135.50, 129.87, 127.79, 86.80, 64.17, 36.98, 31.50, 26.80, 26.71, 19.17, 18.77; HRMS (ESI) calcd for C23H32NaO4Si [M+Na]+ 423.19621, found 423.19657.
To a solution of compound 7b (385 mg, 0.96 mmol) in DMSO (3 mL) was added IBX (350 mg, 1.25 mmol). The reaction mixture was stirred at room temperature and monitored by TLC until the complete consumption of compound 7b. The reaction was quenched with water (6 mL) and filtered. The filtrate was extracted with EtOAc, and the combined extracts were washed with brine, dried over Na2SO4, concentrated under reduced pressure, and purified by flash chromatography on silica gel to give compound 8 as a colorless oil (312 mg, 82%). 1H NMR (600 MHz, CDCl3) δ: 7.68 (t, J=14.4 Hz, 4H), 7.48~7.35 (m, 6H), 4.39~4.29 (m, 2H), 3.63 (s, 3H), 3.28 (dq, J=13.8, 7.2 Hz, 1H), 2.78 (dd, J=16.8, 9.0 Hz, 1H), 2.31 (dd, J=16.8, 5.4 Hz, 1H), 1.56 (s, 1H), 1.11 (s, 9H), 1.05 (d, J=7.2 Hz, 3H); 13C NMR (150 MHz, CD3OD) δ: 213.11, 174.00, 136.61, 135.90, 131.10, 130.40, 128.74, 128.93, 128.50, 69.74, 52.13, 38.74, 37.38, 27.20, 19.94, 19.56, 18.97; HRMS (ESI) calcd for C23H30NaO4Si [M+Na]+ 421.18056, found 421.18075.
A solution of methyltriphenylphosphonium bromide (336 mg, 0.94 mmol) in THF (7 mL) was treated with n-BuLi (0.4 mL, 2.5 mol•L-1 solution in hexane) at -60 ℃ over a period of 10 min under a N2 atmosphere. The reaction mixture was warmed to room temperature, stirred for 40 min, and then cooled to -60 ℃. A solution of compound 8 (312 mg, 0.78 mmol) in THF (5 mL) was then injected dropwise, and the resulting mixture was stirred for 6 h (TLC monitored). The reaction was quenched with water (10 mL) and extracted with EtOAc. The combined organic extracts were washed with saturated brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography on silica gel to give compound 9 as a colorless oil (280 mg, 90%). 1H NMR (600 MHz, CDCl3) δ: 7.70 (m, 4H), 7.40 (m, 6H), 5.23 (s, 1H), 4.92 (s, 1H), 4.16 (s, 2H), 3.62 (s, 3H), 2.66~2.58 (m, 1H), 2.49 (dd, J=15.2, 6.4 Hz, 1H), 2.28 (dd, J=15.2, 8.3 Hz, 1H), 1.43 (s, 1H), 1.07 (s, 9H), 1.04 (d, J=6.9 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 172.97, 151.67, 135.48, 134.76, 133.50, 129.62, 127.63, 108.04, 65.49, 51.40, 40.60, 32.95, 26.76, 19.91, 19.23; HRMS (ESI) calcd for C24H33O3Si [M+H]+397.21935, found 397.21902.
A solution of compound 9 (245 mg, 0.62 mmol) in THF (3 mL) was treated with TBAF (0.93 mL, 1 mol•L-1 solution in THF) and the mixture was stirred at room temperature for 1 h (monitored by TLC). The reaction mixture was concentrated in vacuum, and purified by flash chromatography on silica gel to give compound 1 as a colorless oil (66 mg, 85 %). 1H NMR (600 MHz, CDCl3) δ: 5.13 (d, J=1.4 Hz, 1H), 5.07 (s, 1H), 4.76 (d, J=0.9 Hz, 2H), 2.79~2.74 (m, 1H), 2.71 (t, J=7.9 Hz, 1H), 2.32 (dd, J=15.8, 9.0 Hz, 1H), 1.20 (d, J=6.7 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 171.68, 143.42, 111.19, 71.69, 37.72, 30.87, 20.09; HRMS (ESI) calcd for C7H11O2 [M+H]+ 127.07536, found 127.07478.
To a mixture of triethyl phosphonoacetate (22.4 g, 100 mmol) and formaldehyde (40 mL, 400 mmol) was added K2CO3 (24.15 g, 175 mmol) in water (50 mL). The reaction mixture was stirred at room temperature for 2 h and aqueous ammonium chloride (37.5 mL) was added. The resulting mixture was extracted with EtOAc and the extracts were washed with brine and dried over Na2SO4. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatograph on silica gel to yield compound 11 (8.19 g), which was used without further purification.
Compound 11 (8.19 g, crude from previous step) and imidazole (5.13 g, 75.5 mmol) were dissolved in dry DMF (125 mL), then TBSCl (11.3 g, 75.5 mmol) was added into the solution in one portion. The reaction mixture was stirred at room temperature for 4 h (monitored by TLC) then quenched by the addition of water (150 mL). The reaction mixture was extracted with EtOAc (100 mL×3), and the combined extracts were washed with water (100 mL×2) and brine (100 mL×2), dried over Na2SO4 and concentrated under reduced pressure to give the crude compound 12 (6.91 g, 45%), which was used directly in the next step.
Compound 12 (3.36 g, crude from previous step) was dissolved in dry THF (34 mL), cooled to -78 ℃, and then DIBAL-H (33 mL, 1 mol•L-1 in THF) was added dropwise over 0.5 h. The reaction mixture was stirred at -78 ℃ for another 2 h (monitored by TLC), quenched with water (20 mL), then extracted with EtOAc (30 mL×3). The combined extracts were washed with brine (20 mL×2), concentrated under reduced pressure, then purified by flash column chromatography to give compound 13 as a colorless oil (1.6 g, 62%). 1H NMR (400 MHz, CDCl3) δ: 5.07 (m, 2H), 4.23 (s, 2H), 4.16 (s, 2H), 0.91 (s, 9H), 0.09 (s, 6H).
Dry DMSO (0.94 mL, 13.3 mmol) was added dropwise to a stirred solution of oxalyl chloride (0.6 mL, 7.07 mmol) in DCM (15 mL) at -78 ℃. After stirring for an additional 10 min, a solution of 13 (0.84 g, 4.16 mmol) in DCM (10 mL) was added slowly. After stirring for 1 h, DIPEA (4.12 mL, 24.96 mmol) was added slowly. The reaction mixture was warmed to room temperature, stirred for 10 min, and then quenched by the addition of water (10 mL). It was extracted three times with DCM and the combined extracts were washed three times with 1 mol•L-1 hydrochloric acid, three times with saturated aqueous NaHCO3, twice with brine, then dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography to give compound 14 (0.56 g, 68%) as colorless oil. 1H NMR (400 MHz, CDCl3) δ: 9.60 (s, 1H), 6.52 (d, J=1.3 Hz, 1H), 6.11 (d, J=1.4 Hz, 1H), 4.41 (t, J=2.0 Hz, 2H), 0.95 (s, 9H), 0.11 (s, 6H) (In accordance with reference 11).
Diisopropylamine (307 mg, 3.04 mmol) was dissolved in dry THF (10 mL) and cooled to -40 ℃. n-BuLi (1.46 mL, 2.5 mol/L) was added dropwise, then the reaction mixture was cooled to -78 ℃ and allowed to stir for 1 h. tert-Butyl acetate (301 mg, 2.6 mmol) and a solution of compound 14 (435 mg, 2.17 mmol) in THF (5 mL) were added to the reaction mixture. The reaction mixture was kept at -78 ℃ for 2 h (monitored by TLC) then quenched by the addition of aqueous citric acid solution (10%, 10 mL). The mixture was extracted with EtOAc (10 mL×3) and the extracts were washed with brine, concentrated under reduced pressure, and purified by flash column chromatography to give compound 15 (606 mg, 88%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 5.14 (s, 2H), 4.25~4.56 (m, 1H), 4.26 (d, J=7.8 Hz, 2H), 2.68~2.52 (m, 2H), 1.48 (s, 9H), 0.94 (s, 9H), 0.12 (s, 7H); 13C NMR (100 MHz, CD3OD) δ: 172.04, 151.07, 110.21, 81.68, 70.24, 64.43, 43.58, 28.45, 26.47, 19.25, -5.09; HRMS (ESI) calcd for C16H32NaO4Si [M+Na]+339.19621, found 339.19671.
Compound 15 (420 mg, 1.33 mmol) was dissolved in dry THF (5 mL) and cooled to 0 ℃, then TBAF (1.73 mL, 1 mol/L in THF) was added dropwise. The reaction mixture was warmed to room temperature and progress of the reaction was monitored by TLC until complete consumption of compound 15. The reaction mixture was concentrated under reduced pressure and the crude product was purified by flash column chromatography to give compound 16 (214 mg, 80%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 5.14 (s, 1H), 5.12 (s, 1H), 4.65~4.62 (m, 1H), 4.25 (d, J=13.0 Hz, 1H), 4.18 (d, J=13.0 Hz, 1H), 2.66~2.53 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CD3OD) δ: 172.13, 151.45, 110.63, 81.74, 70.40, 63.10, 43.47, 28.42; HRMS (ESI) calcd for C10H18NaO4 [M+Na]+ 225.10973, found 225.10964.
Compound 16 (135 mg, 0.67 mmol) was dissolved in anisole (142 mg, 1.31 mmol) and cooled to 0 ℃. TFA (0.74 mL) was then added dropwise at 0 ℃ over a period of 5 min. The mixture was then warmed to room temperature over another 5 min, and the reaction was complete after another 15 min (TLC monitored). The mixture was concentrated under reduced pressure and purified by flash column chromatography to give compound 2 (69 mg, 81%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 5.39 (d, J=1.1 Hz, 1H), 5.27 (d, J=1.3 Hz, 1H), 4.96 (d, J=13.4 Hz, 1H), 4.76 (dt, J=13.4, 1.2 Hz, 1H), 4.64 (dd, J=5.9, 5.0 Hz, 1H), 2.90 (dd, J=16.6, 5.0 Hz, 1H), 2.76 (dd, J=16.6, 5.9 Hz, 1H), 2.21 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 170.91, 140.55, 113.82, 70.24, 65.97, 39.83; HRMS (ESI) calcd for C6H8NaO3 [M+Na]+151.03711, found 151.03797.
Supporting Information 1H NMR and 13C NMR spectra of the target molecules and the key intermediates. This material is available free of charge via the Internet at http://sioc-journal.cn.
Guan, A. Y.; Liu, C. L.; Yang, X. P.; Dekeyser, M. Chem. Rev. 2014, 114, 7079. doi: 10.1021/cr4005605
Hashidoko, Y.; Shinano, T. J. Pest. Sci. 2011, 36, 106. doi: 10.1584/jpestics.W10-79
Jeschke, P. Pest Manage. Sci. 2010, 66, 10. doi: 10.1002/ps.v66:1
Damalas, C. A.; Eleftherohorinos, I. G. Int. J. Environ. Res. Public Health 2011, 8, 1402. doi: 10.3390/ijerph8051402
Duke, S. O. Pest Manage. Sci. 2012, 68, 505. doi: 10.1002/ps.2333
Dayan, F. E.; Owens, D. K.; Duke, S. O. Pest Manage. Sci. 2012, 68, 519. doi: 10.1002/ps.2332
Butler, M. S. J. Nat. Prod. 2004, 67, 2141. doi: 10.1021/np040106y
Dayan, F. E.; Duke, S. O. Plant Physiol. 2014, 166, 1090. doi: 10.1104/pp.114.239061
Zhang, Z.; Xing, A.; Staswick, P. Clemente, T. E. Plant Cell, Tissue Organ Cul. 1999, 56, 37. doi: 10.1023/A:1006298622969
Bayer, E.; Gugel, K. H.; Haegele, K.; Hagenmaier, H.; Esipov, S. E.; Koenig, W. A.; Zaehner, H. Helv. Chim. Acta 1972, 55, 224. doi: 10.1002/hlca.v55:1
Omura, S.; Hinotozawa, K.; Imamura, N.; Murata, M. J. Antibiot. 1984, 37, 939. doi: 10.7164/antibiotics.37.939
Hoerlein, G. Rev. Environ. Contam. Toxicol. 1994, 138, 73. doi: 10.1021/acs.est.7b04682?src=recsys
Mitchell, G.; Bartlett, D. W.; Fraser, T. E. M.; Hawkes, T. R.; Holt, D. C.; Townson, J. K.; Wichert, R. A. Pest Manage. Sci. 2001, 57, 120. doi: 10.1002/(ISSN)1526-4998
Gerwick, B. C. ; Graupner, P. R. ; Fields, S. C. ; Schmitzer, P. R. ; Brewster, W. K. US 7393812, 2008[Chem. Abstr. 2006, 485213].
Lichtner, F. Austral. J. Plant Physiol. 2000, 27, 609.
Gerwick, B. C.; Brewser, W. K.; de Bore, G. J.; Fields, S. C.; Graupner, P. R.; Hahn, D. R.; Pearce, C. J.; Schmitzer, P. R.; Webster, J. D. J. Chem. Ecol. 2013, 39, 253. doi: 10.1007/s10886-013-0238-7
Lin, L.; Mulholland, N.; Wu, Q. Y.; Beattie, D.; Huang, S. W.; Irwin, D.; Clough, J.; Gu, Y. C.; Yang, G. F. J. Agric. Food Chem. 2012, 60, 4480. doi: 10.1021/jf300610j
Lin, L.; Mulholland, N.; Huang, S. W.; Beattie, D.; Irwin, D.; Gu, Y. C.; Clough, J.; Wu, Q. Y.; Yang, G. F. Chem. Biol. Drug Des. 2012, 80, 682. doi: 10.1111/jpp.2012.80.issue-5
Larsen, C. H.; Ridgway, B. H.; Shaw, J. T.; Woerpel, K. A. J. Am. Chem. Soc. 1999, 121, 12208. doi: 10.1021/ja993349z
Villieras, J.; Rambaud, M. Synth. Commun. 1983, 300. doi: 10.1055/s-1982-29998
Suizu, H.; Shigeoka, D.; Aoyama, H.; Yoshimitsu, T. Org. Lett. 2015, 17, 126. doi: 10.1021/ol503356m

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:


 下载:
下载: