


图式 1.
吖啶-1, 8二酮的合成
Scheme 1.
Synthesis of acridine-1, 8-diones

吖啶由于其特殊的化学特性, 在诸多领域有着重要的应用, 因此受到科学家们的广泛关注[1~4]. 19世纪人们发现吖啶能与细胞中的核苷酸结合, 从而开始对吖啶进行了深入的研究, 在随后的研究中发现吖啶具有多种药理活性, 并且能作为抗菌[5]、抗疟疾[6]、抗肿瘤[7]、抗血吸虫[8]、多药耐药性(multidrugresistance, MDR)逆转剂[9]等药物.有些吖啶衍生物能够嵌入到DNA或RNA中[10], 吖啶作为经典的抗疟药物在疾病的治疗和防御中起着重要的作用, 在染料方面的应用也有相当长的时间[11].随着研究的继续深入, 吖啶也展现出了另一方面的特性, 在电子效应和光学性能上用作半导体材料[12]以及开关分子[13].吖啶环长的共轭结构在荧光方面也展现出不寻常的应用, 吖啶的药理活性和荧光性能作为荧光探针在医疗方面具有广泛的应用前景.人们对其合成方法进行了不断地探索与改进, 合成了一系列的吖啶类化合物. Bernthsen反应是最早合成吖啶的方法, 用二苯胺和羧酸, 与无水ZnCl2在200~270 ℃下加热合成了吖啶[14~16].近年来随着合成技术的发展, 在合成吖啶衍生物方面出现了多种简便、绿色、经济的方法.本文主要介绍2010年以来的吖啶衍生物的合成方法和其在药理活性、荧光材料、工业染料和电致发光等各方面的应用.
2011年Nawaz Khan等[17]首次报道了二氧化锡催化的以2-氨基-5-氯苯甲酮和咔唑酮为原料在微波辐射下的Friedlander缩合反应, 得到吲哚并吖啶衍生物1 (Eq. 1), 收率为47%~82.5%.并对这三种化合物做了血溶性和细胞毒性的活性测试, 其中在吲哚骨架部分的3-C位置上进行甲基取代的吖啶化合物表现出了良好的生物活性.通过分析该吖啶衍生物的化学结构, 我们可以知道该吖啶衍生物的化学结构中包含有吖啶、咔唑、吲哚三种结构片段, 而三种结构都有药理活性, 可能是这样的片段组合导致其有着别具一格的特性.
|
|
(1) |
2011年Thakur等[18]报道了经过改进的Bernthsen反应(Eq. 2), 在微波辐射下采用新的催化剂对甲苯磺酸(p-TSA)进行反应, 得到了9-H被取代的吖啶化合物2, 微波辅助的反应条件使反应时间缩短.而改进的方法具有催化剂用量较少、易得、绿色等优点.通过对催化剂的筛选和底物扩展, 收率达到了66%~88%.本课题组近期通过传统的Bernthsen反应合成9-甲基吖啶时发现产率非常低, 在20%左右, 重复性很差, 所以改进传统方法提高产率是非常必要的.
|
|
(2) |
2012年Rivero等[19]报道了无溶剂和微波辐射的条件下经过Hantzsch反应合成1, 8-二酮吖啶衍生物3 (Eq. 3), 通过对β-双酮的比例进行调整并与芳基扩展合成了8种吖啶酮衍生物, 收率达到83%~98%, 并对其做了清除DPPH (diphenyl picryl hydrazinyl radical, 二苯基苦酰肼基)自由基活性实验, 当反应原料是甲醛时所得到的产物具有清除DPPH自由基的活性.自由基是个强氧化性的物质, 对人体有各种各样的伤害, 在人体的新陈代谢和日常的饮食等活动中都会产生自由基, 因此清除自由基就显得很重要.抗氧化活性化合物也会受到空间位阻的影响, 在该1, 8-二酮吖啶衍生物的合成中使用甲醛时, 可能形成的吖啶衍生物的9-C的氢在空间上的位阻最小, 所以使用甲醛比其他任何醛类的合成的1, 8-二酮吖啶衍生物对自由基的抗氧化性高.
|
|
(3) |
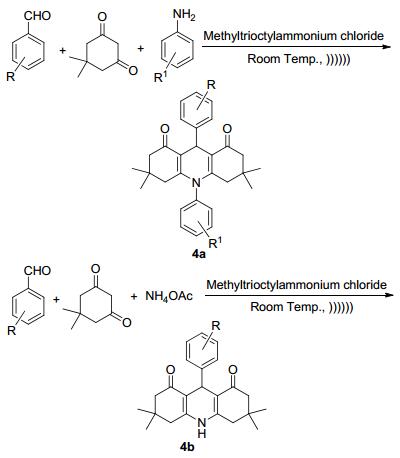
2013年Kumar等[20]在以前的工作基础上[21~23]用三辛基氯化铵作离子液体催化剂, 以苯甲醛、达米酮、苯胺或醋酸铵作为原料, 在超声辐射下绿色、简便、快捷地合成了吖啶衍生物4a和4b (Scheme 1), 收率为90%~96%.像这种芳醛和β-双酮以及胺的反应是典型的合成吖啶酮反应, 而通过对反应原料的扩展可以得到多种吖啶的衍生物.近年来发展起来的合成手段, 如微波、超声等方法使产率提高, 大大缩短了反应的时间.三辛基氯化铵作离子液体在这里起到溶剂和催化剂的双重作用, 超声的作用会使化学键的断裂更容易, 因此加快了反应的进程.
2015年Hevia等[24]通过调制不同配比的单金属络合物得到联合双金属络合物的芳基化试剂, 用于吖啶环特定位置的芳基化, 得到9-C芳基化的吖啶化合物5 (Eq. 4).在125 ℃下微波辐射20 min就能得到9-H被取代的吖啶衍生物, 收率高达95%.与以往的单金属络合物促进的反应相比, 相比于反应时间为20 h以上传统的方法, 微波辅助的方法降低了反应时间和能源的消耗并且提高了产率.如果说双金属络合物起到一个定位作用, 那么高活性的氧化剂2, 3-二氯-5, 6-二氰基-1, 4-苯醌(DDQ)就会进行脱氢芳构化的作用.
|
|
(4) |
2010年Kozlov等[25]报道了以萘胺、芳醛衍生物、1, 3-环己二酮衍生物为原料, 通过Knoevenagel缩合反应合成了含吖啶环骨架的具有荧光特性的化合物6a和6b (Eq. 5).用大功率振荡器时发现电子可以从Sn (n=1~3)到S0而发光, 可用于医学和生物学荧光标记.通过荧光猝灭法研究不同的吸电子和供电子基团的影响, 发现荧光猝灭效应是由激发态的电子转移引起的, 这可能是因为吸电子或供电子基团使电子发生转移, 另一个可能是与共轭结构形成π键使电子离域.
|
|
(5) |
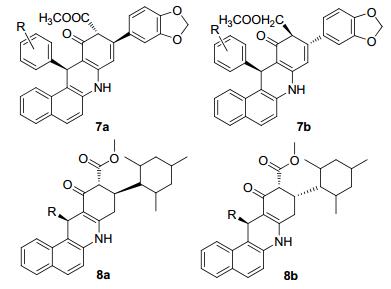
同年Kozlov等[26]用同样的合成方法改变活性亚甲基为手性双甲酮, 也合成了含吖啶环骨架的具有荧光特性的化合物7a和7b(图 1).并对该化合物的发光性能进行了分析, 但在生物活性方面没有去做相应的测试, 可以看到吖啶衍生物7a和7b中有亚甲二氧基的结构片段, 与木质素的结构相似.木质素是从五味子果仁中提取, 五味子为常用的中药, 对迁延性和慢性病毒性肝炎血清谷丙转氨酶(SGPT)升高患者有较好的疗效, 也可用于艾滋病的治疗, 因此吖啶环与木质素结构相似的亚甲二氧基片段的组合可能会有潜在的药用价值.而在2011年该课题组[27]报道了通过在β-双酮上引入均三甲基苯与芳香醛和2-氨基萘反应获得了苯并吖啶的两种异构体化合物8a和8b(图 1), 该类化合物在治疗老年痴呆症上也有一定的药理活性[28~30], 因老年性痴呆症具有遗传性, 遗传性的疾病与DNA有着密切的关系, 而吖啶通过嵌入到DNA链中, 可能会在某种方面带来治疗的作用.
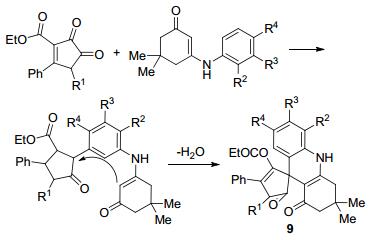
2010年Maslivets等[31]报道了一种合成螺杂环吖啶的方法(Scheme 2), 以二酮吡咯衍生物和烯胺酮衍生物为原料在无水间二甲苯中回流5 h, 得到了螺环吖啶化合物9, 收率为70%~81%, 并且通过单晶衍射确定了其结构, 这种方法为螺杂环吖啶类化合物的合成提供了简便渠道.是通过分子内的合环来实现螺环的方法, 除了这种方法, 还可以通过在准备螺环化的碳原子上设计合成有一个羟基和一个长链的羟基, 再进行分子内的脱水缩合得到螺环化合物.
2015年Poriel等[32]合成了含吖啶环结构的具有物理活性的有机半导体化合物10 (Eq. 6), 可作为天蓝色磷光有机发光二极管.发光二极管大多都是由金属制造的, 而近年来有机发光二极管是个比较热门的课题, 有机发光二极管的日益成熟会使我们减轻对重金属的依赖以及对环境的污染.磷光是由激发三重态的最低振动能级到各基态振动能级间跃迁产生的, 发光基团往往带有大的共轭平面, 吖啶正好具有这样的结构, 吖啶环上的电子通过共轭键可离域到其他环上, 再由激发态到基态以发光的方式辐射能量, 另一个原因可能是吸电子基团或原子导致共轭环上电子云密度降低从而使其荧光强度有所减弱, 而磷光强度则会相应的增强, 正好该化合物就满足了这两种要求.
|
|
(6) |
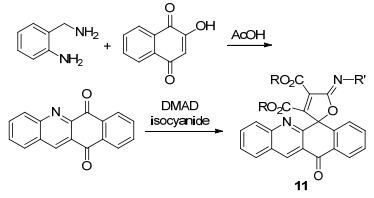
2012年Shaabani等[33]在合成中意外得到了螺杂环吖啶化合物11 (Scheme 3), 经过表征确定了该化合物的结构, 收率为67%~88%.螺杂环吖啶化合物的合成利用三组分一锅法, 室温搅拌24 h所得, 原料中异腈碳氮叁键的极化的电子在合环反应中起到了偶极加成的重要作用.
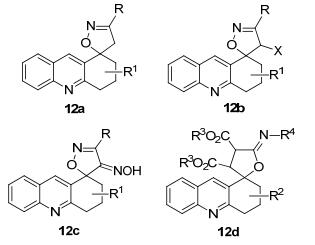
本课题组也正在进行螺杂环化合物12a~12d的合成和应用方面的研究(图 2).螺环化合物是在螺碳原子由两个共轭的平面十字相交而形成, 在结构上与其他吖啶衍生物通过单键连接有较大的不同, 是否会像其他螺环化合物一样具有发光特性, 有待进一步的证明, 可能会有意想不到的特性.
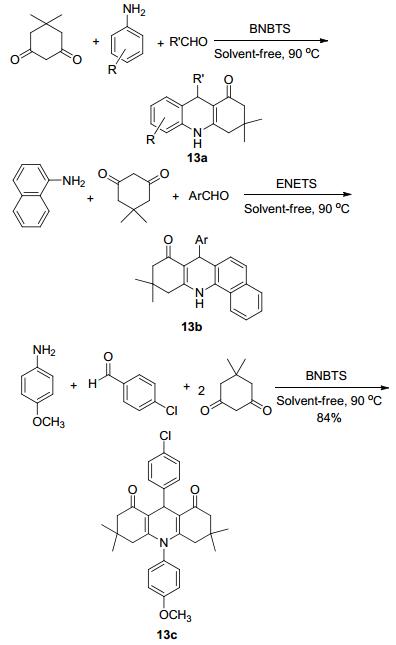
2010年Vaghei等[34]报道了三组分一锅法直接合成吖啶酮衍生物的简便渠道, 以苯胺、醛、达米酮为原料, 利用一种高效率的催化剂对甲苯磺酰甲基亚硝酰胺(BNBTS), 在反应温度为90 ℃和无溶剂条件下, 反应25~50 min, 合成了吖啶酮衍生物13a~13c (Scheme 4), 收率为75%~93%.该反应中通过改变5, 5-二甲基-1, 3-环己二酮的比例与不同的胺与醛反应, 获得了各种取代基的1, 8-二酮吖啶和苯并吖啶衍生物.这种方法具有操作简单、后处理方便、成本低、产率高等优点.在大多数这一类反应中可以得出的一点是苯胺可以是硝基苯, 在一锅法中还原硝基苯得到苯胺后继续后面的反应, 而不用还原成苯胺后提纯再进行后面的反应.可在反应中进行多米诺式一锅法的反应, 省去了中间多余的步骤.
2014年Kaya等[35]报道了一种绿色的一锅法合成磺胺吖啶衍生物14, 以达米酮、N-(4-二氨基甲烷)苯磺酰胺、芳香醛作为原料, 水作溶剂, 在室温下反应获得(Eq. 7), 收率为74%~80%.并对人类碳酸酐酶同功酶的抑制作用进行了研究, 具有较好的效果.前面讲过吖啶环是可以嵌入到DNA链中的, 而在吖啶环上引入有活性的基团或活性结构片段, 可能会有不同的药理活性, 这都是我们所期待的结果.
2014年Singh等[36]报道了通过三组分一锅法合成一种新的苯并吖啶二酮15, 以2-羟基-1, 4-萘醌、苯甲醛、苯胺为原料, 在前人研究的基础上, 通过改变加入原料的顺序和延长反应时间得到了意想不到的芳基苯并吖啶二酮产物(Eq. 8).再经过条件优化, 收率高达92%.在传统的方法中多数用的是1, 3-二酮, 而该方法使用了一种1, 4-二酮, 会使产物吖啶分子具有多环的结构, 目标产物也具有较多的活性反应片段.
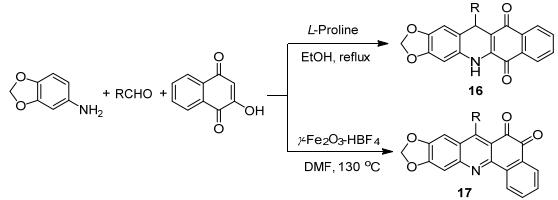
2015年Wu等[37]报道了以3, 4-亚甲二氧基苯胺、醛、2-羟基-1, 4-萘醌为原料, L-脯氨酸作催化, 在乙醇中回流得到18个含吖啶环骨架的化合物16等(Scheme 5), 收率为72%~99%, 而其中12-(3, 4, 5-三甲氧基苯基)-5, 10-二氢-苯并[i][1, 3]二氧戊环[4, 5-b]-吖啶-6, 11-二酮具有抗癌活性.同年该研究组[38]继续报道了以同样的原料, 用可磁力回收的氟硼酸催化剂, 合成了含吖啶环骨架的化合物17, 共合成19种新的化合物, 收率达72%~92%, 其中12-(2-氟苯基)-苯并[h][1, 3]-二氧戊环[4, 5-b]吖啶-10, 11-二酮具有抗癌活性.该反应的催化剂可磁力回收, 能够重复使用6次且不失活性.磁力回收催化剂是比较新异的手段, 对催化剂的分离和循环利用带来了很大的便利.
|
|
(7) |
|
|
(8) |
2015年Shivashankar等[39]报道了用1, 2-二醇替代传统的芳香醛, 四乙酸铅作催化剂, 在乙醇中进行回流反应一锅法合成吖啶衍生物18 (Eq. 9), 该反应中1, 2-二醇均裂形成苯甲醛再进行反应, 底物扩展收率为82%~95%, 该反应具有反应时间短、条件温和、后处理方便等优点.这也可以说是多米诺式的反应, 在反应中1, 2-二醇通过被Pb(OAc)4氧化均裂后形成了苯甲醛, 随后进行常见的芳醛、β-双酮和胺三组分反应合成了吖啶酮, 在这里需要控制β-双酮的用量, 如果是2 equiv.就可以形成1, 8-二酮吖啶衍生物, 而1 equiv.就会形成1-氧代吖啶衍生物.
|
|
(9) |
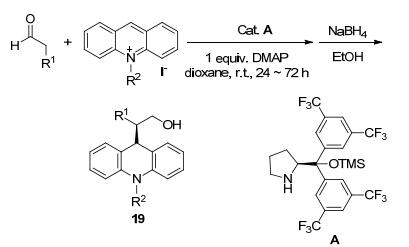
2012年Xiao等[40]报道了利用有机手性催化剂二芳基脯氨醇硅醚的仲胺(A)催化进行醛和吖啶盐的不对称1, 4-加成, 获得了9-H被取代的手性吖啶衍生物19 (Scheme 6).而9-C为手性碳原子的吖啶化合物在大多数吖啶合成中是较少见的.
2013年Guillena等[41]报道了在无溶剂条件下用邻氨基苯甲醛的衍生物和对位取代的环己酮为原料, 以脯氨酰胺衍生物作为手性催化剂经过Friedländer反应合成了具有手性的克林活性药物的类似物20 (Eq. 10), 克林类化合物具有与吖啶和喹啉相似的结构, 众所周知吖啶和喹啉类衍生物都具有很重要的药理活性.
|
|
(10) |
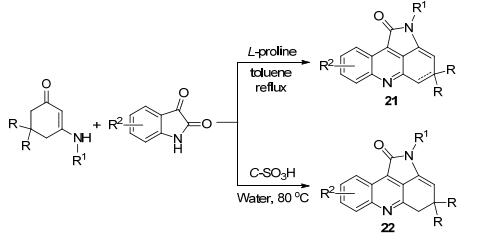
2012年Shi等[42]报道了L-脯氨酸作催化剂, 以靛红和烯胺酮为原料经过多米诺反应直接形成新颖的吡咯并吖啶化合物21, 收率为60%~93%.该反应由吲哚醌的C—N键直接断裂形成.而在2016年Zhang等[43]以同样的原料, 用可重复使用的磺化含碳材料作为异相催化剂合成了吡咯并吖啶衍生物22, 进行底物扩展收率达到78%~89% (Scheme 7).
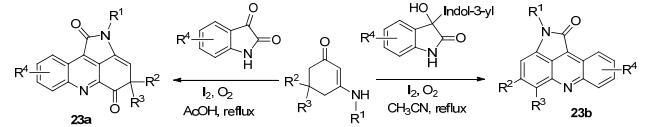
2013年Ji等[44]报道了一种用靛红和烯胺酮为原料, I2/O2促进的多米诺反应一锅法合成了含吖啶环骨架的吡咯并吖啶化合物23a和23b, 收率分别为21%~71%和45%~82% (Scheme 8).该反应中C—H键的活化是没有经过过渡金属参与的反应, 多米诺反应的机制也省略了多步复杂的反应.该文献中阐述了化合物23b的R3基团的迁移的原因可能是碘自由基夺取了邻碳原子的氢导致R3基团迁移至邻碳原子.
从以上的几个反应中可以看出, 具有烯胺酮结构的环己酮和靛红反应形成的都是稠环的吖啶衍生物, 如果把烯胺酮结构上的氮原子换成氧原子, 可能会形成相对应的稠环氧杂蒽衍生物并有着不同的应用.
2010年Larock等[45]报道了在温和的条件下合成9-甲基吖啶的一种方法(Eq. 11), 乙腈作为溶剂, CsF作为苯环中形成炔键的引发剂, 用2-氨基乙酰苯和邻三甲基硅苯三氟甲磺酸脂发生[4+2]的环加成反应, 在65 ℃下反应24 h得到9-取代的吖啶24.这也是吖啶合成中典型的合环反应, 通过离去基团的离去, 再进行亲核反应完成合环.在这个反应中CsF起到让TMS基团离去作用, 再由OTf基团的离去, 形成一个炔键, 最后进行亲核加成, 完成合环.
2012年Liu等[46]报道了以四元环酮和活性亚甲基酮经过[4+2]环加成制备了高度取代的氨基苯酚, 在经过环化后得到了吖啶的衍生物25a和25b (Scheme 9).这种方法的优点是在氨基苯酚环上提前引入活性基团去合成更多具有活性取代基的吖啶衍生物, 避免了在模型分子中再引入其他官能团的反应步骤, 为相应化合物的合成中提供了方便, 但要注意的一点是吸电子和供电子基团对反应的产率具有一定的影响.
|
|
(11) |
2013年Verma等[47]报道了以喹啉并吡喃和炔烃作原料, 通过钯催化的[3+2]合环、开环、再进行分子内的羟醛缩合一锅法合成了含吖啶环结构的吡咯[3, 2, 1-de]吖啶类化合物26 (Eq. 12), 收率为60%~85%.而含吡咯并吖啶类化合物本身就具有广泛的抗肿瘤[48]、抗真菌[49]等生物活性, 在结构上的改变可能在某种程度上提高了其活性.
2015年Mukhopadhyay等[50]报道了羧酸化合物与杯状芳烃化合物反应得到了一种有机催化剂, 以苯并吡咯、1, 3-环己二酮、胺、丁炔二酸二酯为原料, 通过该催化剂催化反应得到具有吖啶环骨架的多环化合物27 (Eq. 13), 收率为67%~77%.我们都知道催化剂在化学反应中有着举足轻重的地位, 在众多催化剂中, 金属催化剂会在产物中有残留, 会有金属污染, 不是首选的绿色催化剂, 而有机催化剂是一种理想的选择.
|
|
(12) |
|
|
(13) |
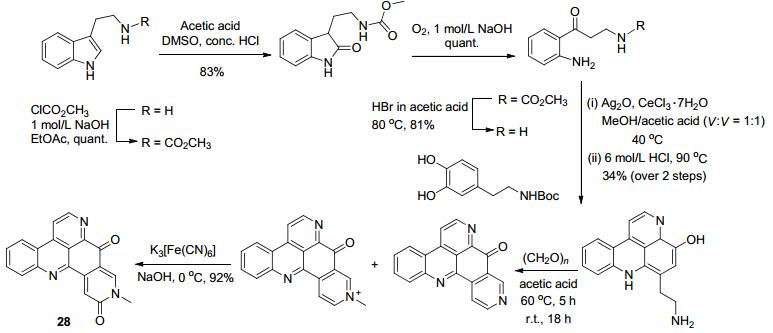
2016年Copp等[51]报道合成了具有生物活性的海洋生物碱仿生化合物, 通过非天然的原料合成了含有吡啶并吖啶环的具有细胞毒性[52]的天然产物28 (Scheme 10), 总收率达到14%.生物碱是一类生物体内一种含氮化合物, 它不仅存在于植物中, 也存在于动物、微生物和海洋生物中, 人们已经发现了很多有活性的生物碱且将其用于抗肿瘤、抗菌、抗病毒等方面的研究.在许多疾病治疗中, 生物碱类药物价值已经受到了人们的普遍关注.
2016年Ji等[53]用易得的苯胺通过调整溶剂和磷配体, 进行1, 4-钯迁移的分子内的Heck反应制备了吲哚并吖啶29 (Eq. 14), 并进行底物扩展, 收率达到60%~88%.此报道称该化合物具有较高的三重态能量, 发出的蓝色磷光比普通的磷光强, 可作为较理想的三重态蓝色磷光发射体.如果该化合物作为发射蓝色磷光的发光体, 其稳定性和寿命方面的性能有待进一步的研究.
|
|
(14) |
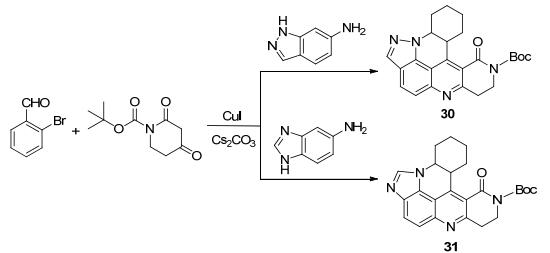
2016年Wang等[54]报道了邻卤代苯甲醛、叔丁基2, 4-氧代哌啶-1-羧酸酯、1H-吲唑-6-胺或3H-苯并[d]咪唑-5-胺在无配体条件下碘化铜催化三组分一锅法获得稠杂环化合物30和31, 收率分别为79%~92%, 76%~89% (Scheme 11).形成双吖啶环结构的过程中, 醛、1, 3-二酮、胺可以形成双吖啶的一部分, 另一部分是由碘化亚铜催化生成的.像这种有双吖啶环的结构比起单吖啶环结构的化合物有哪些特殊的作用, 需进一步深入地研究.
多数文献中, 稠环的吖啶衍生物偏向药用活性的报道较多, 而在发光性质方面的研究较少, 吖啶本身就具有荧光性质, 在稠环吖啶衍生物中, 都是多环大共轭的刚性平面结构, 那么该类化合物在荧光和磷光方面有什么样的性质和特性?是否和其他具有发光性质的吖啶衍生物有着相似或着更独特的性质?这些都需要我们在将来的研究中去探索和发现.
在2010年Buchwald等[55]利用钯配体控制分子内的环化来制备了5H-二苯氮
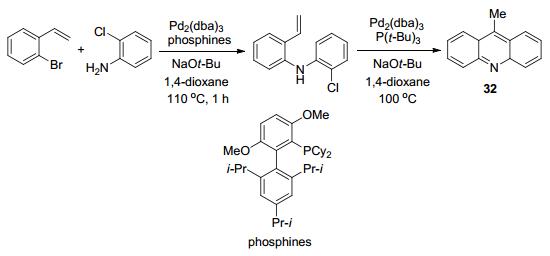
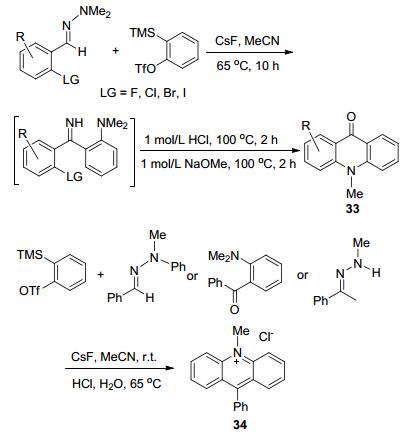
2012年Larock等[56]报道了一种N-甲基吖啶衍生物合成方法和吖啶盐的新型的合成途径, 以腙和苯炔为反应原料获得了收率为75%~94%的N-甲基吖啶酮的衍生物33和31%~88%的吖啶盐34 (Scheme 13).通过腙和苯炔的反应来提前设计好吖啶酮羰基的位置也是一种很好的反应路线, 前面已提到过, 吖啶环是可以嵌入DNA链中的, 而吖啶酮上的羰基作为反应位点, 可以通过亲核等反应获得更多的吖啶衍生物, 结构上的差异可能会带来未知的潜在活性.
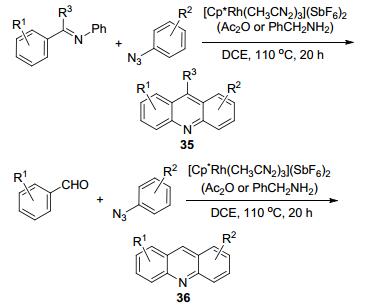
2013年Ellman等[57]报道了通过胺化、分子内亲电取代和串联等反应得到了9-H被取代的吖啶衍生物35和9-C被没有取代基的吖啶36的合成(Scheme 14), 该方法以N-苯基亚胺和叠氮苯为原料, Rh(Ⅲ)作为催化剂, 2 equiv.的乙酸酐作为溶剂而合成, 收率为32%~91%.这是一种典型的分子间合环的反应, 该方法的巧妙之处是叠氮化合物通过热分解除去了一分子的氮气, 从而留下一个氮原子正好用于合成吖啶中的氮原子.
2013年Guo等[58]首次报道了一种钯催化、胺化、环化、芳构化一锅法合成不对称吖啶衍生物37的方法(Eq. 15).该方法省去了多步操作, 收率为36%~99%.能够环化形成吖啶衍生物的亮点在于苯甲醛邻位的离去基团起到一个关键的作用.
|
|
(15) |
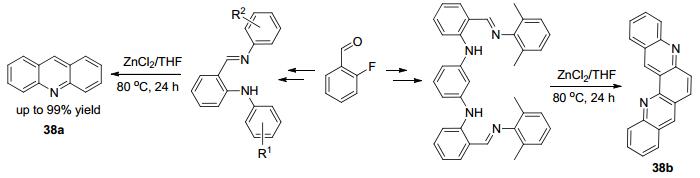
2014年Mu等[59]报道了他们在设计合成邻芳基氨基苯的席夫碱的锌配体时得到并不是所想要的产物, 而是形成了吖啶衍生物38a和38b, 从而发现了一种氯化锌促进的简洁高效的吖啶衍生物的合成方法(Scheme 15).在这个反应中氯化锌起到一个形成碳正离子的作用, 使碳氮双键上的电子发生极化, 最后通过亲核反应完成合环.
2014年Du等[60]报道了一种无金属催化, 以PhI(OAc)2促进反应, 邻位甲醛取代的二苯胺为原料进行分子内交叉偶联合成吖啶衍生物39的方法(Eq. 16), 总共合成了17种化合物, 收率为40%~82%.因为是无金属催化的反应, 所以药物开发中避免了金属的残留.又是交叉偶联反应, 是一种绿色、原子经济型的合成方法.这个反应原料类似于前面介绍的Guo等的Pd催化的反应的中间体的模型.
|
|
(16) |
2014年Chen等[61]报道了以邻氰基苯胺和二芳基碘鎓盐为原料通过串联反应合成喹唑啉时, 发现当二者比例为1:2时, 生成了9-取代的吖啶衍生物40 (Eq. 17).并通过单晶衍射确定了其结构, 进一步做了底物扩展, 收率为62%~92%.在该反应中一份子的二芳基碘鎓盐作为吖啶的一部分, 另一份是作为9-C取代基进行反应.
|
|
(17) |
2015年Xu等[62]报道了通过优化传统的反应条件合成多种吖啶衍生物41 (Eq. 18), 收率为59%~99%.这个合成方法和前面介绍的Guo等的合成方法在原料方面比较相似, 主要区别在于反应条件上的不同, 但在产物的模型分子上有很大的相似之处, 几乎是一样的.苯甲醛的邻位上一个是离去基团, 另一个是卤素, 通过钯作为催化剂形成C—N键, 而有离去基团就不用钯作催化剂, 比起贵金属还是离去基团较好一些.
|
|
(18) |
2016年Mukhopadhyay等[63]报道了溴化铜存在下对甲苯磺酸作催化剂, 氰化甲烷作溶剂, 在空气中氧化2 equiv.的烯胺酮, 82 ℃下反应18 h, 经过交叉偶联和重排获得1, 8-二酮吖啶衍生物42 (Scheme 16), 底物扩展收率达到42%~78%.这个合成方法的主要特点在于第二步的重排反应使交叉偶联反应的产物很巧妙地重排, 分子内合环形成了含有吖啶环结构的化合物.
2016年Zhikharko等[64]首次报道了吖啶并吖啶的吖啶衍生物43, 以N1, N5-双(芳基亚甲基)萘-1, 5-二胺和1, 3-环己二酮为原料经过Mannich反应、Hofmann-Martius重排、分子内脱水缩合, 再进行Mannich反应, 最后进行Hofmann-Martius重排等反应过程获得该化合物, 收率为21%~85% (Eq. 19).该合成路线的想法较独特, 亮点在于萘分子上, 1-C和5-C上有相同的取代基, 所以这两个位置都能起同样的反应, 只要提供2 equiv.的β-双酮就能得到一种两个吖啶衍生物错位相并的吖啶衍生物.
|
|
(19) |
2011年Natrajan等[65]报道了一种较为简便的合成N-烷基化的吖啶衍生物44的方法, N-烷基化的吖啶在诸多领域有着重要的应用, 传统的反应是以吖啶内酯与1, 3-丙磺酸内酯在常用的溶剂中进行反应, 1, 3-丙磺酸内酯是一个致癌物, 并且转化率很低, 得到的收率也低, 须用大量的1, 3-丙磺酸内酯进行反应, 对环境也造成了相当大的污染.为了开发一种高转化率、高收率的反应, 进而该研究组对溶剂进行探索, 发现使用离子液体[BMIM][PF6]和[BMIM][BF4]时的转化率和收率高, 收率为81%~93%, 不会有大量的聚合物产生, 后处理方式简单(Eq. 20).有离子液体参与的反应使N-烷基化进行得更加容易.反应中应用离子液体替代普通溶剂优点是反应速率比在普通溶剂中快几倍, 所用的离子液体和催化剂的混合液可以重复利用.研究表明, 在反应过程中离子液体起到溶剂和催化剂的双重作用.
|
|
(20) |
2012年Chatani等[66]报道了使用有机锌试剂选择性地活化吖啶的C—H键, 二芳基锌作芳基化试剂, 用各种金属催化剂去尝试反应时, 在用到镍作催化剂时发现主要产物为4-C被芳基化的吖啶衍生物45 (Eq. 21), 进行底物扩展收率达到52%~68%.在化学反应中, 有选择性地去连接基团是很有前景的, 我们在很多合成中进行底物扩展时, 需要研究在不同位置上的各基团对目标化合物在各种性能上带来的影响, 如果没有在相应位置上取代的原料, 那么选择性地取代在这里显得尤为重要.在这里有必要提一下, 9-C取代的吖啶的左右两边苯环在空间位阻是一样的. 4-C和5-C是具有一样的化学环境, 为什么只会在一个位置进行反应, 可能是因为金属催化剂和芳基化试剂的使用量以及取代基对空间位阻的影响而产生的结果.
|
|
(21) |
2015年Wang等[67]报道了容易制备且可回收的无金属的有机催化剂可将9-C无取代的吖啶氧化为吖啶酮46, 催化剂使用量的不同也使9-C无取代的吖啶进行了不同程度的氧化(Eq. 22), 收率可达63%.该反应具有条件温和、助催化剂廉价易得等优点.在9-C氧化可能是因为9-H活泼的原因.这也像前面提到过的选择性的取代, 使我们得到所期望的目标产物, 所以这种合成方法具有更广阔的前景.
|
|
(22) |
2016年Gill等[68]报道了一种绿色的方法合成N-取代的1, 8-二酮吖啶, 原料也是双甲酮、苯甲醛和胺的衍生物, 通过β-糊精作超分子催化, 水作为溶剂, 经过超声反应得到了N-取代的1, 8-二酮吖啶47 (Eq. 23), 收率为56.35%~94.56%.因为β-糊精是可生物降解和回收使用的超分子化合物, 而且该反应是用水作反应介质, 得到的产物收率也较高, 所以是个绿色的反应方法.在吖啶衍生物的合成中, 很多典型的反应路线都是以β-双酮和醛以及胺为原料合成吖啶衍生物, 不同处在于反应条件上的差异, 那我们就可以根据反应体系中的利弊等各种情况来选择最佳的合成路线.在反应过程中用到了超声辅助, 可能的原因是β-糊精是超分子, 超分子在水中是较难溶的物质, 超声就起到了溶解, 充分混合的作用.
|
|
(23) |
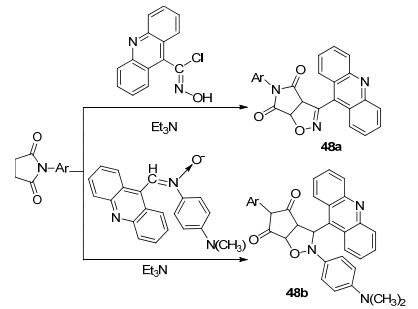
2010年我们课题组[69]报道了具有生物活性的吖啶衍生物48a和48b, 在三乙胺的作用下N-芳基-马来酸亚胺分别与α-氯代9-吖啶甲醛肟和N-(N, N-二甲氨基苯基)-C-(9-吖啶基)取代硝酮发生1, 3-偶极环加成反应得到13个化合物, 化合物48a和48b的收率分别为19%~48.1%和53.9%~82.7% (Scheme 17).当样品浓度为20 μg/mL时化合物48a (Ar=p-BrC6H4)和48b (Ar=p-HO2CC6H4)对抗癌药物靶点细胞分裂周期控制蛋白磷酸酯酶Cdc25A的抑制率分别为50.90%和51.22%.在该吖啶衍生物中可以看到有异噁唑结构片段, 而异噁唑化合物又具有很多的生物活性, 还具有很好的药理学特性, 它在降低人类血糖、消除疼痛、抵抗炎症、杀死有害细菌、控制以及减小艾滋病毒的危害等方面有较大的作用.另外, 它在农药和杀虫剂领域也有广泛的应用, 如一些异噁唑衍生物表现出农业化学效用, 具有抑制杂草和土壤细菌生长的效能, 吖啶本身也具有药理特性, 活性片段的组合具有更多的潜在可能.
在2010年, Sondhi等[70]报道了9-氯-2, 4-取代吖啶和9-硫氰酸-2, 4-取代吖啶分别与糠胺反应得到了22种吖啶衍生物49a和49b (Scheme 18), 收率为40%~96%.并对其进行了体外生物活性测试, 其中2-甲氧基-N-(2-(噻吩-2-基)乙基)吖啶-9-胺展现出41.17%的抗炎活性, 高于常用的标准药物布洛芬39%的抗炎活性.
2010年Patel等[71]报道了酸催化一锅法合成具有生物活性的吖啶衍生物50 (Scheme 19), 通过底物扩展, 收率为68%~72%.将该化合物进行了体外实验活性测试, 并与市售的药物进行了对比, 具有良好的抗菌效果.

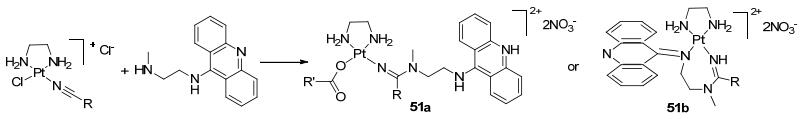
2011年Bierbach等[72]报道合成含有铂金属螯合吖啶混合的抗癌试剂51a和51b (Scheme 20), 这是通过铂腈与二元胺吖啶合成的抗癌活性试剂.虽合成的成本较高, 但具有一定的生物活性.
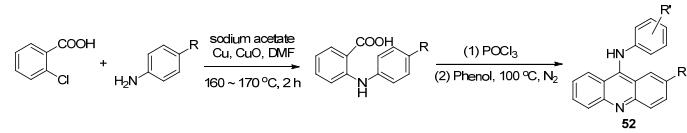
2013年Kumar等[73]报道9-氨基吖啶衍生物52 (Scheme 21)对人的宫颈癌细胞株和肺癌细胞株具有良好抗癌活性.该化合物通过改良Ullmann-Goldberg反应, 用乙酸钠替代碳酸钾, 铜作催化剂, 氧化铜作助催化剂进行反应, 将15~18 h的过夜反应的时间缩短到了2 h, 收率为73%~90%.比起金属钯, 较廉价的铜作催化剂可以降低合成的成本, 该反应的不同点在于是后来在9-C上修饰取代的基团, 这也是一个亮点所在.
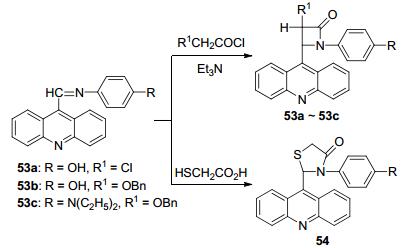
2014年我们课题组[74]合成了9个含吖啶基的单环β-内酰胺及噻唑啉酮衍生物, 以吖啶基甲醛芳亚胺席夫碱分别与氯乙酰氯、苄氧乙酰氯在三乙胺作用下产生的烯酮和巯基乙酸发生[2+2]环加成和合环反应, 收率为29.9%~42.1% (Scheme 22).并对所合成的化合物进行了体外抗癌活性测试和白细胞共同抗原活性筛选.结果表明53a, 53b, 53c和54等四个化合物具有生物活性.化合物53c对肿瘤细胞HL-60 (Leucocythemia人白血病细胞)生长的抑制率为79.4%.化合物53a, 53b和54对细胞分裂周期控制蛋白磷酸酯酶Cdc25B (Cell division cycle 25B phosphatase)的抑制率分别为80.64%、99.75%和99.34%.而化合物53a和54对CD45蛋白酪氨酸磷酸酶A的抑制率分别为86.12%和91.03%, 其余化合物的抑制率(IC50值)都小于50%.
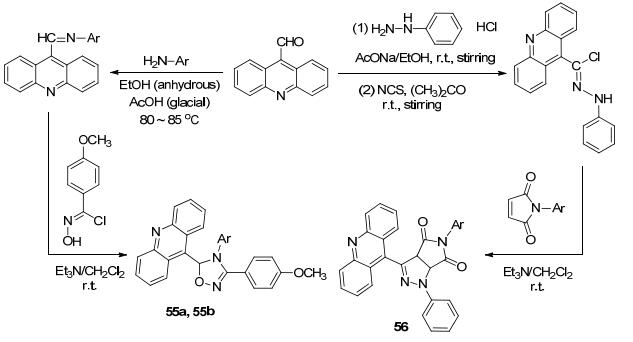
2016年我们课题组[75]报道合成了8种含吖啶环的天然产物类似物1, 2, 4-噁二唑啉55a和55b以及吡唑啉56, 作为潜在与DNA非共价结合抗肿瘤剂, 并对其进行了体外抗肿瘤活性筛选(Scheme 23), 以9-吖啶基甲醛为原料分别与芳胺和苯肼反应形成席夫碱和亚硝胺,再与N-羟基-4甲氧基酰亚胺和N-芳基马来酰亚胺进行1, 3-偶极环加成形成化合物55和56.结果表明, 其中化合物55a和55b对Cdc25B磷酸酯酶的抑制率达到57.10%和50.98%, 其余化合物的抑制率(IC50值)都小于50%, 为1, 2, 4-噁二唑作为抗癌化合物的研究带来了一定的工作基础.
就吖啶衍生物的生物活性这方面来说, 前人做过很多的研究, 在吖啶环不同的位置引入多种活性的基团, 通过活性测试等方法发现了具有重大生物学意义的吖啶衍生物.
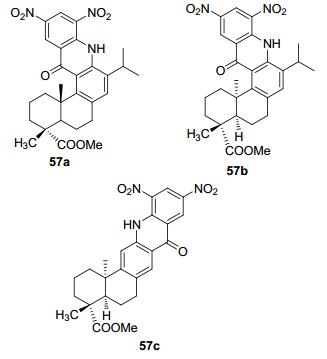
2011年, 王恒山等[76]合成了含有脱氢松香酸骨架的吖啶化合物, 并对其理化性质进行相应的研究.利用脱氢松香酸为原料, 经甲酯化、硝化、还原、关环和水解等多步操作合成了三种松香基吖啶酮化合物57a、57b、57c三种化合物(图 3).
并对这三种化合物进行了荧光和色价的研究, 发现其具有典型的吖啶酮化合物荧光性质, 而在色价方面57a、57b、57c三种化合物分别与天然红色素如梭子蟹壳红色素、赤豆红色素、市售紫甘薯红色素等接近, 色价略高, 具有色素实际应用的性能.松香的一些衍生化合物可作为油漆使用, 吖啶又是典型的发光基团.组合这两个结构, 可能会产生多用途的效果, 可以想象一下, 在交通要道涂抹了荧光油漆是不是可以大大增加夜间交通安全系数?未发掘的潜在应用有待化学工作者们更加深入地研究, 可以通过在这两种结构组合的基础上引入其他的基团来改变该化合物的发光特性.
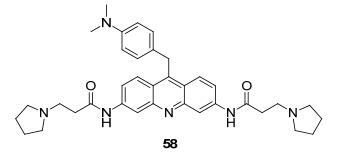
2013年Ladame等[77]报道了一种三取代的吖啶衍生物58(图 4), 具有作为染料和pH传感器等双重用途, 从酸性到碱性是由蓝色到黄褐色的变化过程中, 较小的质子浓度会展现出较明显的色差.电子的激发经常触发质子化和去质子化过程, 这导致了酸碱化合物在激发态下的分子性能可能与基态时存在很大差异, 不同的激发态到基态通过发光的形式辐射出不同波长的光, 观察到的颜色也就不一样了.
2012年Vilar等[78]报道了含吖啶结构的具有荧光活性的大环化合物59 (Eq. 24), 收率为85%.该化合物在不同的酸中表现出了不同强度的荧光活性, 尤其在H3PO4中荧光强度最好, 依据这样的荧光可以检测是否含有H3PO4, 可作为检测剂使用.
2013年Singh等[79]报道了光分解产生的吖啶醇60和其相应的酯偶联物可用于细胞的荧光成像研究(Eq. 25).还研究了在不同的质子浓度下对荧光强度的影响, 从结构上可以看出, 酯通过光照射进行水解, 产生羧酸和9-吖啶基甲醇, 可能不同的质子浓度会对9-吖啶基甲醛上的醇羟基产生影响, 从而导致荧光在发光强度上有所变化.这样一来可作为环境敏感剂.吖啶可以嵌入到DNA链中, 在细胞中可被内膜化和均匀化, 且可利用吖啶的发光特性来对细胞进行荧光成像, 因此可用吖啶作为荧光标记物.
|
|
(24) |
|
|
(25) |
2013年Quintavalla等[80]报道了含吖啶的1, 2-二氧杂环丁烷通过Ullmann反应生成吖啶酮, 再与2-金刚烷酮进行McMurry反应, 最后经过光氧化得到最终吖啶的1, 2-二氧杂环丁烷61, 并在吖啶环上不同位置上进行了甲基取代来观察荧光强度和产生荧光的难易程度.原理是该化合物通过加热分解产生单线激发态的吖啶而产生荧光(Scheme 24), 该化合物在加热温度小于100 ℃时就有荧光活性, 灵敏度更高, 可作为高灵敏度的荧光探针.鉴于不同位置取代的甲基对吖啶环上的电子密度产生不同影响, 通常邻对位使荧光增强, 间位使荧光减弱方法, 可控制发光强弱.在较低的温度就能使该化合物分解, 可适用于较接近常温的检测.
2015年张晶莹等[81]设计合成了具有光电效应的吖啶衍生物62 (Eq. 26), 并研究了其电致发光性能, 以吖啶二胺和碘苯加热回流24 h, 得到3, 6-二苯胺吖啶衍生物, 收率为60%.研究结果表明其具有良好的电致发光性能, 可发出绿色的光, 发射峰不随电压的变化而变化.如果对有机电致发光材料的研究有了突破就不再依赖稀缺的贵金属了, 在结构上可以看到吖啶环与两个苯环是通过氮原子连接, 总体上形成p-π大共轭的状态.可能大的共轭环容纳电子的能力较强, 大量的电子从激发态到基态所辐射的能量也越多, 在辐射强度上也有所增强, 发光强度也就随之增强.
|
|
(26) |
2015年Lee等[82]报道了用3-碘基-9-苯基咔唑和9, 9-二甲基吖啶通过钯催化的偶联反应获得了咔唑3-C与吖啶10-C偶联的吖啶化合物63 (Eq. 27).该化合物作为有机发光二极管的发光材料, 发出蓝色的磷光.通常空穴传输材料的给电子性较好, 多为芳香多胺类化合物, 这类化合物电子云分布广, 容易失去电子而形成带正电的空位, 当外部注入电子时, 通过空穴不断流动, 注入阳极与阴极的电荷在发光层结合时, 即可激发有机材料而发光.
|
|
(27) |
2015年赵燕苹等[83]报道了以吖啶酮为原料经过磺化和硝化得到5, 7-二硝基-2-磺基吖啶酮(64) (Eq. 28), 以修饰后的吖啶酮衍生物小分子为杂交指示剂构建了非标记型DNA电化学生物传感器, 利用指示剂在DNA杂交前后电化学信号的变化来反映杂交或错配过程, 以实现对目标DNA序列的测定.比起其他的电化学生物传感器, 5, 7-二硝基-2-磺基吖啶酮的制备过程较为方便.
|
|
(28) |
电致发光是两电极的电压产生电场激发的电子碰击发光中心, 导致电子在能级间的跃迁以辐射能量的方式表现出发光现象.像夹层式的发光结构在电极不断注入电子时, 共轭大环电子密度增高, 而且在电场的作用下电子被加速, 高速运动状态下发生碰撞, 在激发态以辐射的方式释放能量回到基态.这样一来, 共轭的大环结构化合物有作为电致发光材料的潜在可能性, 但是辐射的同时也伴随着放热, 所以电致发光的材料要有热稳定性, 寿命要很长, 并且发光的颜色不能随着电压的变化而变化, 这样才会有更好的应用前景.
吖啶因具有多种性能, 已在多种领域得到应用, 也日益受到科学家们的关注.吖啶因具有与生物体DNA结合的性能, 被科学家视为各种抗病毒物质的良好载体, 从而合成出了多种带有抗病毒活性的官能团的吖啶衍生物.通过研究其药理活性, 研制出了不少很好的抗病毒药物, 在药学尤其在抗癌药物研究领域有着独特的发展潜力.吖啶的荧光性能也被科学家用来研究新型高灵敏度的探针以及细胞的荧光成像, 在染料方面也具有实际应用的性能.除此之外, 在电致发光的有机半导体材料方面也显现出了潜在的应用价值, 是个较年轻的技术领域, 在电致发光方面有了突破就可以不再依赖稀缺的贵金属, 从而减少重金属对环境的污染, 所以对于吖啶在各种应用方面的研究也显得很必要.我们实验室也正在进行吖啶酮和吖啶螺异噁唑类的合成研究, 已经有了很好的进展, 后期会对合成出来的最终产物进行多方面的测试.
Martins, M. A. P.; Frizzo, C. P.; Moreira, D. N.; Buriol, L.; Machado, P. Chem. Rev. 2009, 109, 4140. doi: 10.1021/cr9001098
Kalirajan, R.; Muralidharan, V.; Jubie, S.; Gowramma, B.; Gomathy, S.; Sankar, S.; Elango, K. J. Heterocycl. Chem. 2012, 49, 748. doi: 10.1002/jhet.848
Bonse, S.; Santelli, R. C.; Barbe, J.; Krauth-Siegel, R. L. J. Med. Chem. 1999, 42, 5448. doi: 10.1021/jm990386s
Nadaraj, V.; Selvi, S. T.; Mohan, S. Eur. J. Med. Chem. 2009, 44, 976. doi: 10.1016/j.ejmech.2008.07.004
Wilson, W. R.; Thompson, L. H.; Anderson, R. F.; Denny, W. A. J. Med. Chem. 1989, 32, 31. doi: 10.1021/jm00121a007
Bastianelli, C.; Caia, V.; Cum, G.; Gallo, R.; Mancini, V. J. Chem. Soc., Perkin Trans. 21991, 679.
Jaycox, D.; Gribble, G. W.; Hacker, J. Heterocycl. Chem. 1987, 24, 1405. doi: 10.1002/jhet.v24:5
张秀平, 时惠麟, 颜闵, 医药工业, 1984, 3, 8. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=zhou198403003&dbname=CJFD&dbcode=CJFQZhang, X.-P.; Shi, H.-L.; Yan, M. Med. Ind. 1984, 3, 8(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=zhou198403003&dbname=CJFD&dbcode=CJFQ
Velingkar, V. S.; Dandekar, V. D. Chin. J. Chem. 2011, 29, 504. doi: 10.1002/cjoc.201190113
Ferreira, R.; Avinó, A.; Mazzini, S.; Eritja, R. Molecules 2012, 17, 7067. doi: 10.3390/molecules17067067
Geddes, C. D. Dyes Pigm. 2000, 45, 243. doi: 10.1016/S0143-7208(00)00025-5
Goel, A.; Kumar, V.; Singh, S. P.; Sharma, A.; Prakash, S.; Singh, C.; Anand, R. S. J. Mater. Chem. 2012, 22, 14880. doi: 10.1039/c2jm31052j
陈鹏, 王宇洋, 张宇模, 张晓安, 化学学报, 2016, 74, 669. doi: 10.11862/CJIC.2016.069Chen, P.; Wang, Y.-Y.; Zhang, Y. M.; Zhang, X. A. Acta Chim. Sinica 2016, 74, 669(in Chinese). doi: 10.11862/CJIC.2016.069
Bernthsen, A. Ann. 1878, 192, 1. http://www.drugfuture.com/OrganicNameReactions/ONR39.htm
Bernthsen, A. Ann. 1884, 224, 1. doi: 10.1002/jlac.18842240102/abstract
Popp, F. D. J. Org. Chem. 1962, 27, 2658. doi: 10.1021/jo01054a518
Roopan, S. M.; Nawaz Khan, F. R. Med. Chem. Res. 2011, 20, 732. doi: 10.1007/s00044-010-9381-7
Das, S.; Thakur, A. J. Green Chem. Lett. Rev. 2011, 4, 131. doi: 10.1080/17518253.2010.521775
Avila, J. M.; Vargas, F. D.; Camacho, S. P. D.; Rivero, I. A. RSC Adv. 2012, 2, 1827. doi: 10.1039/c1ra01135a
Kaur, B.; Kumar, H. J. Chem. Sci. 2013, 125, 989. doi: 10.1007/s12039-013-0431-9
Kaur, B.; Parmar, A.; Kumar, H. Heterocycl. Lett. 2011, 1, 59. doi: 10.6023/cjoc201710007
Kaur, B.; Parmar, A.; Kumar, H. Synth. Commun. 2012, 42, 453.
Puri, S.; Kaur, B.; Parmar, A.; Kumar, H. Heterocycl. Commun. 2009, 15, 57. doi: 10.1515/HC.2009.15.1.51
Gómez, A. H.; Herd, E.; Uzelac, M.; Cadenbach, T.; Kennedy, A. R.; Borilovic, I.; Aromí, G.; Hevia, E. Organometallics 2015, 34, 2614. doi: 10.1021/om501251q
Kozlov, N. G.; Bondarev, S. L.; Kadutskii, A. P.; Basalaeva, L. I. Zh. Org. Khim. 2010, 46, 209. doi: 10.1134/S1070428010110059
Kozlov, N. G.; Bondarev, S. L.; Knyukshto, V. N.; Odnoburtsev, B. A.; Basalaeva, L. I. Zh. Org. Khim. 2010, 46, 1639. doi: 10.1134/S1070428012110061
Kozlov, N. G. Zh. Org. Khim. 2011, 47, 1675.
Kozlov, N. G.; Gusak, K. N. Zh. Org. Khim. 2006, 42, 1680. doi: 10.1134/S107042800611011X
Kozlov, N. G.; Tereshko, A. B.; Gusak, K. N. Zh. Org. Khim. 2006, 42, 281. doi: 10.1134/S1070428011110091
Kozlov, N. G.; Gusak, K. N. Zh. Obshch. Khim. 2006, 76, 294. doi: 10.1134/S1070428011110091
Silaichev, P. S.; Dmitriev, M. V.; Aliev, Z. G.; Maslivets, A. N. Zh. Org. Khim. 2010, 46, 1173. doi: 10.1134/S107042801111011X
Romain, M.; Tondelier, D.; Geffroy, B.; Shirinskaya, A.; Jeannin, O.; Berthelot, J. R.; Poriel, C. Chem. Commun. 2015, 51, 1313. doi: 10.1039/C4CC08028A
Ghadari, R.; Hajishaabanha, F.; Mahyari, M.; Shaabani, A.; Khavasi, H. R. Tetrahedron Lett. 2012, 53, 4018. doi: 10.1016/j.tetlet.2012.05.107
Yeşildağ, I.; Ulus, R.; Başar, E.; Aslan, M.; Kaya, M.; Buülbül, M. Monatsh Chem. 2014, 145, 1027. doi: 10.1007/BF03246091
Ghorbani-Vaghei, R.; Malaekehpoor, S. M. J. Iran. Chem. Soc. 2010, 7, 957. doi: 10.1007/s00706-013-1145-x
Mahajan, S.; Khullar, S.; Mandal, S. K.; Singh, I. P. Chem. Commun. 2014, 50, 10078. doi: 10.1039/C4CC03079F
Yang, X. J.; Zhang, C.; Wu, L. Q. RSC Adv. 2015, 5, 18945. doi: 10.1039/C4RA16372A
Yang, X. J.; Zhang, C.; Wu, L. Q. RSC Adv. 2015, 5, 25115. doi: 10.1039/C5RA00887E
Jagadishbabu, N.; Shivashankar, K. RSC Adv. 2015, 5, 95240. doi: 10.1039/C5RA19595K
Caballero, A. B.; Guillena, G. G.; Nájera, C. J. Org. Chem. 2013, 78, 5349. doi: 10.1021/jo2025114
Liang, T.; Xiao, J.; Xiong, Z. Y.; Li, X. W. J. Org. Chem. 2012, 77, 3583. doi: 10.1021/jo400522m
Wang, H. Y.; Li, L. L.; Lin, W.; Xu, P.; Huang, Z. B.; Shi, D. Q. Org. Lett. 2012, 14, 4598. doi: 10.1021/ol302058g
Li, C. M.; Zhang, F. R. RSC Adv. 2016, 6, 75359. doi: 10.1039/C6RA18048E
Hao, W. J.; Wang, J. Q.; Xu, X. P.; Zhang, S. L.; Wang, S. Y.; Ji, S. J. J. Org. Chem. 2013, 78, 12362. doi: 10.1021/jo401773j
Rogness, D. C.; Larock, R. C. J. Org. Chem. 2010, 75, 2289. doi: 10.1021/jo1000687
Han, X. D.; Zhao, Y. L.; Meng, J.; Ren, C. Q.; Liu, Q. J. Org. Chem. 2012, 77, 5173. doi: 10.1021/jo300615t
Verma, A. K.; Reddy Kotla, S. K.; Aggarwal, T.; Kumar, S.; Nimesh, H.; Tiwari, R. K. J. Org. Chem. 2013, 78, 5372. doi: 10.1021/jo400539x
Gimenez-Arnau, E.; Missailidis, S.; Stevens, M. F. G. Anti-Cancer Drug Des. 1998, 13, 431. doi: 10.1007/s11164-014-1576-y
McCarthy, P. J.; Pitts, T. P.; Gunawardana, G. P.; Kelly-Borges, M.; Pomponi, S. A. J. Nat. Prod. 1992, 55, 1664. doi: 10.1021/np50089a016
Sarkar, P.; Mukhopadhyay, C. Green Chem. 2015, 17, 3452. doi: 10.1039/C5GC00156K
Khalil, I. M.; Barker, D.; Copp, B. R. J. Org. Chem. 2016, 81, 282. doi: 10.1021/acs.joc.5b02312
Marshall, K. M.; Barrows, L. R. Nat. Prod. Rep. 2004, 21, 731. doi: 10.1039/b401662a
Gu, Z. Y.; Liu, C. G.; Wang, S. Y.; Ji, S. J. Org. Lett. 2016, 18, 2379. doi: 10.1021/acs.orglett.6b00843
Ma, Y. G.; Qiang, W. W.; Li, C.; Zhang, M. M.; Wang, X. S. Monatsh. Chem. 2016, 147, 1233. doi: 10.1007/s00706-015-1625-2
Tsvelikhovsky, D.; Buchwald, S. L. J. Am. Chem Soc. 2010, 132, 14048. doi: 10.1021/ja107511g
Dubrovskiy, A. V.; Larock, R. C. J. Org. Chem. 2012, 77, 11232. doi: 10.1021/jo302378w
Lian, Y. J.; Hummel, J. R.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2013, 135, 12548. doi: 10.1021/ja406131a
Guo, H. M.; Mao, R. Z.; Wang, Q. T.; Niu, H. Y.; Xie, M. S.; Qu, G. R. Org. Lett. 2013, 15, 5460. doi: 10.1021/ol402596g
Su, Q.; Li, P.; He, M. N.; Wu, Q. L.; Ye, L.; Mu, Y. Org. Lett. 2014, 16, 18. doi: 10.1021/ol402732n
Zheng, Z. S.; Dian, L. Y.; Yuan, Y. C.; Negrerie, D. Z.; Du, Y. F.; Zhao, K. J. Org. Chem. 2014, 79, 7451. doi: 10.1021/jo5011697
Pang, X. L.; Chen, C.; Su, X.; Li, M.; Wen, L. R. Org. Lett. 2014, 16, 6228. doi: 10.1021/ol503156g
Wang, T. J.; Chen, W. W.; Li, Y. Xu, M. H. Org. Biomol. Chem. 2015, 13, 6580. doi: 10.1039/C5OB00755K
Sarkar, R.; Mukhopadhyay, C. Org. Biomol. Chem. 2016, 14, 2706. doi: 10.1039/C5OB02655E
Zhikharko, Y. D.; Kozlov, N. G.; Basalaeva, L. I. Zh. Org. Khim. 2016, 52, 383. doi: 10.1134/S1070428016030155
Natrajan, A.; Wen, D. Green Chem. 2011, 13, 913. doi: 10.1039/c0gc00758g
Hyodo, I.; Tobisu, M.; Chatani, N. Chem. Commun. 2012, 48, 308. doi: 10.1039/C1CC16582H
Zhang, Z. G.; Gao, Y.; Liu, Y.; Li, J. J.; Xie, H. X.; Li, H.; Wang, W. Org. Lett. 2015, 17, 5492. doi: 10.1021/acs.orglett.5b02877
Chate, A. V.; Rathod, U. B.; Kshirsagar, J. S.; Gaikwad, P. A.; Mane, K. D.; Mahajan, P. S.; Nikam, M. D.; Gill, C. H. Chin. J. Catal. 2016, 37, 146. doi: 10.1016/S1872-2067(15)61005-1
穆赫塔尔·伊米尔艾山, 王婷, 萨提瓦力迪·海力力, 库尔班·吾斯曼, 吐尔洪·买买提, 有机化学, 2010, 30, 1884. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract339487.shtmlImerhasan, M.; Wang, T.; Helil, S.; Osman, K.; Muhammad, T. Chin. J. Org. Chem. 2010, 30, 1884(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract339487.shtml
Sondhi, S. M.; Singh, J.; Rani, R.; Gupta, P. P.; Agrawal, S. K.; Saxena, A. K. Eur. J. Med. Chem. 2010, 45, 555. doi: 10.1016/j.ejmech.2009.10.042
Patel, M. M.; Mali, M. D.; Patel, S. K. Bioorg. Med. Chem. Lett. 2010, 20, 6324. doi: 10.1016/j.bmcl.2010.06.001
Graham, L. A.; Wilson, G. M.; West, T. K.; Day, C. S.; Kucera, G. L.; Bierbach, U. ACS Med. Chem. Lett. 2011, 2, 687. doi: 10.1021/ml200104h
Kumar, P.; Kumar, R.; Prasad, D. N. P. Arabian J. Chem. 2013, 6, 79. doi: 10.1016/j.arabjc.2012.04.039
力瓦依丁·买合苏提, 穆赫塔尔·伊米尔艾山, 麦麦提依明·马合木提, 萨特瓦尔迪·赫力力, 刘华君, 有机化学, 2014, 34, 1235. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract344261.shtmlMahsud, L.; Imerhasan, M.; Mahmud, M. A.; Helil, S.; Liu, H. J. Chin. J. Org. Chem. 2014, 34, 1235(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract344261.shtml
Haydar, G.; Imerhasan, M.; Eshbakova, K. A.; Kurbanbaeva, A. E. J. Pharm. Biol. Sci. 2016, 4, 41. doi: 10.6023/cjoc201710007
田小艳, 杨达, 潘英明, 童碧海, 苏桂发, 王恒山, 有机化学, 2011, 31, 346. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract339817.shtmlTian, X. Y.; Yang, D.; Pan, Y. M.; Tong, B. H.; Su, G. F.; Wang, H. S. Chin. J. Org. Chem. 2011, 31, 346(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract339817.shtml
Percivalle, C.; Mahmood, T.; Ladame, S. Med. Chem. Commun. 2013, 4, 211. doi: 10.1039/C2MD20173A
Centelles, V. M.; Burguete, M. I.; Galindo, F.; Izquierdo, M. A.; Kumar, D. K.; White, A. J. P.; Luis, S. V.; Vilar, R. J. Org. Chem. 2012, 77, 490. doi: 10.1021/jo202077v
Jana, A.; Saha, B.; Karthik, S.; Barman, S.; Ikbal, M.; Ghosh, S. K.; Singh, N. D. P. Photochem. Photobiol. Sci. 2013, 12, 1041. doi: 10.1039/c3pp25362g
Fusco, M. D.; Quintavalla, A.; Trombini, C.; Lombardo, M.; Roda, A.; Guardigli, M.; Mirasoli, M. J. Org. Chem. 2013, 78, 11238. doi: 10.1021/jo401683r
陈栋, 张学强, 张晶莹, 王悦, 高等学校化学学报, 2015, 36, 484. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=gdxxhxxb201503012Chen, D.; Zhang, X. Q.; Zhang, J. Y.; Wang, Y. Chem. J. Chin. Univ. 2015, 36, 484(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=gdxxhxxb201503012
Seo, J. A.; Jeon, S. K.; Gong, M. S.; Lee, J. Y.; Noh, C. H.; Kim, S. H. J. Mater. Chem. C 2015, 3, 4640. doi: 10.1039/C5TC00640F
赵燕苹, 硕士论文, 福建医科大学, 福州, 2015.Zhao, Y. -P. M. S. Thesis, Fujian Medical University, Fuzhou, 2015(in Chinese).

图式 2 螺环[吖啶-9, 3'-吡咯]-4'-羧酸酯的合成
Scheme 2 Synthesis of spiro[acridine-9, 3'-pyrrole]-4'-carboxy-lates

图式 3 螺-苯并[b]吖啶-6, 2'-呋喃衍生物的合成
Scheme 3 Synthesis of spiro-benzo[b]acridine-6, 2'-furan derivatives

图式 6 有机催化不对称吖啶衍生物的合成
Scheme 6 Synthesis of organic catalytic asymmetric acridine derivatives

图式 7 吡咯并[2, 3, 4-kl]吖啶-1-酮衍生物的合成
Scheme 7 Synthesis of pyrrolo[2, 3, 4-kl]acridin-1-one derivatives

图式 12 Pd-配体控制吖啶衍生物的合成
Scheme 12 Pd-ligand controlled synthesis of acridine derivatives

图式 15 ZnCl2促进的分子内环化合成吖啶衍生物
Scheme 15 Synthesis of acridine derivatives by ZnCl2-promoted intramolecular cyclization

图式 17 9-(吡咯并[3', 4'-d]异噁唑)吖啶衍生物的合成
Scheme 17 Synthesis of 9-(pyrrolino[3', 4'-d]isoxazole) acridines derivatives

图式 19 Bernthsen反应合成吖啶衍生物
Scheme 19 Bernthsen reaction synthesis of acridine derivatives

图式 21 Ullmann-Goldberg反应合成吖啶衍生物
Scheme 21 Synthesis of acridine derivatives by optimized Ullmann-Goldberg reactions

图 4 具有染料和荧光双重特性的吖啶衍生物
Figure 4 Acridine derivatives with dual properties of dyes and fluorescent

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:






























 下载:
下载:

















 下载:
下载: