

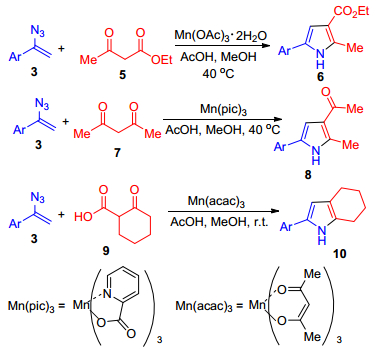
图式 1.
烯基叠氮与1, 3-二羰基化合物反应合成吡咯衍生物
Scheme 1.
Synthesis of pyrroles from vinyl azides and 1, 3-dicarbonyl compounds

含氮杂环化合物普遍存在于天然产物、药物化学以及材料化学中[1], 发展制备多种含氮杂环化合物的合成方法在有机合成化学中一直是热门的研究方向[1f, 1g].虽然已经发展了多种多样的方法制备含氮杂环化合物[2], 但反应较为复杂, 所以以易制备的合成子为原料, 可选择性控制取代基的通用方法仍然需要发展.
α-芳基烯基叠氮是一类重要的有机合成中间体, 通过市售底物即可制备得到.在过去的合成方法中, 烯基叠氮主要是通过烯烃与IN3反应制得[3], 反应经过两步完成, 首先, NaN3与ICl原位反应得到的IN3与烯烃发生加成反应, 反应完成后, 混合液中加入水和乙醚, 分离有机相并干燥; 第二步, 通过叔丁醇钾消除碘化氢, 制得α-芳基烯基叠氮.由于反应过程中需要用到爆炸性的试剂, 从而限制了其在有机合成中的应用.近年来, 随着合成方法学的发展, 化学家们使用非爆炸性的试剂, 如叠氮基三甲基硅烷等, 实现了高区域、高立体、高收率的选择性地制备烯基叠氮类化合物[4].
1864年, 德国化学家Griess[5]制得苯基叠氮类化合物, 此后涌现出很多叠氮衍生物的合成方法, 并以此发展了多种快速简单地构建含氮杂环化合物的新合成方法[6].然而, 由于其爆炸性、毒性、不稳定性、低选择性等因素阻碍了其在合成领域中的应用.在过去的一个世纪中, 此类化合物并未引起人们的注意, 但是随着实验安全技术的提高、新的表征手段的出现及新颖的合成方法的发展, 人们对α-芳基烯基叠氮类化合物的认识更加全面, 利用α-芳基烯基叠氮作为关键的合成子构建结构复杂的N-杂环化合物的工作也越来越多, 受到国内外研究学者的广泛关注, 大量有关构建N-杂环化合物的研究成果已经被报道[7].
α-芳基烯基叠氮可以参与多种反应, 广泛用于构建各种含氮杂环化合物, 并且此类化合物结构中的离去基团氮气具有很好的离去能力, 使其具有很高的反应活性, 在有机合成中具有广泛的应用.此外, 烯烃与叠氮相连, 使α-芳基烯基叠氮具有独特的性质, 能够充当亲电试剂、亲核试剂和自由基接受体. α-芳基烯基叠氮多样的反应途径, 能生成高反应活性的中间体, 为发展新颖的反应提供了可能.本综述重点阐述近年来α-芳基烯基叠氮参与合成含氮杂环化合物的研究进展, 以及对其应用前景进行展望.
2H-氮丙啶是一类重要的有机化合物, 可用于制备多种含多官能团的非环状和环状含氮化合物[8].氮丙啶类化合物具有高反应活性的三元不饱和含氮杂环结构, 在反应中, 可作为亲电试剂、亲核试剂、亲二烯体等, 在有机合成中具有广泛的应用[9].由于许多2H-氮丙啶化合物结构高度扭曲而不稳定, 很难制备和处理[10], 所以由烯基叠氮转化为2H-氮丙啶是目前最为常用的方法.通常, 烯基叠氮1通过光解或热解的方式生成相应的2H-氮丙啶类化合物2 (Eq. 1)[11].
|
|
(1) |
在热裂解的过程中, 通常需要高温, 其常用溶剂有甲苯、1, 4-二氧六环、庚烷等, 因这些溶剂沸点在98~110 ℃之间, 所以产物氮丙啶衍生物与溶剂难以分离. 2003年, Somfai等[12]以二氯甲烷作溶剂、150 ℃条件下, 将α-芳基烯基叠氮转化为相应的氮丙啶衍生物, 该方法的优点是产率高, 反应时间短, 且产物易于分离.
2003年, Gudmundsdóttir课题组[13]报道了在微波、不添加溶剂的条件下, α-芳基烯基叠氮3转化为2H-氮丙啶4反应(Scheme 2), 该反应与传统的光分解和热活化相比:无污染、反应时间短、转化率高.
|
|
(2) |
吡咯及其衍生物是一类重要的含氮杂环化合物, 在许多合成药物试剂[14]、天然产物[15]及材料化学[16]等领域具有广泛的应用, 如亚铁血红素、叶绿素、胆色素、维生素B12等.正是由于其广泛的应用性及生物活性, 吡咯环的合成方法一直受到广泛关注[17].
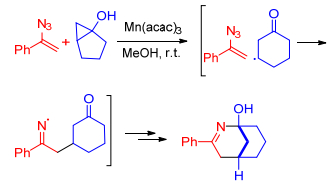
Chiba课题组[18]首次报道了利用三价锰络合物作为催化剂, 催化α-芳基烯基叠氮3与1, 3-二羰基化合物的反应, 从而制备多取代的吡咯类化合物(Scheme 1), 该反应可能是一个自由基反应历程:在醋酸锰催化下, 1, 3-二羰基化合物生成α-羰基自由基, 然后加成到α-芳基烯基叠氮化合物中得到亚胺基自由基, 随后与分子内的羰基发生环化反应, 最终得到吡咯类化合物. 1, 3-二羰基化合物为β-酮酯5、1, 3-二元酮7、β-酮酸9, 每一种1, 3-二羰基化合物对应的锰的催化剂不同, 这主要取决于1, 3-二羰基化合物的性质和Mn(Ⅲ)催化剂的氧化还原性[19].因此, 当α-芳基烯基叠氮与β-酮酯反应时, Mn(OAc)3·2H2O是有效催化剂; 当α-芳基烯基叠氮与1, 3-二元酮反应时, 则需要氧化性更强的催化剂Mn(pic)3; 而α-芳基烯基叠氮与β-酮酸反应时, Mn(acac)3更适合.结果表明:这些[3+2]环加成反应具有较好的底物普适性和较高的产率.
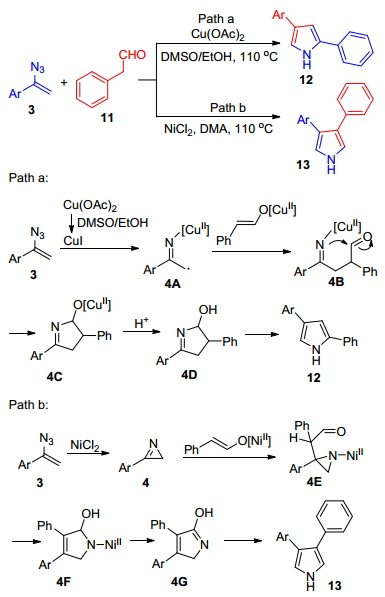
2012年, Jiao课题组[20]报道了一个新颖高效的环化反应:在铜络合物[Cu(OAc)2]或者镍络合物(NiCl2)催化下, α-芳基烯基叠氮3与苯乙醛11发生环化反应, 分别合成2, 4-和3, 4-二芳基取代的吡咯衍生物12和13 (Scheme 2).该方法通过改变过渡金属催化剂, 从而实现高度的区域选择性, 与已经报道的酸性或碱性的条件下制备多取代的吡咯衍生物的方法相比, 该方法的反应条件较为温和、中性、简单且不需要添加剂.当以Cu(OAc)2为催化剂时, 在二甲基亚砜(DMSO)/EtOH作用下, Cu(Ⅱ)还原为Cu(Ⅰ), 在其催化下, α-芳基烯基叠氮3分解得到自由基中间体4A, 然后, 4A与苯乙醛11的烯醇互变异构体发生偶联反应, 得到中间体4B, 随后发生分子内的亲核反应得到中间体4C, 之后中间体4C再质子化得到中间体4D, 最后脱水反应得到最终产物12 (Scheme 2, Path a).当以NiCl2为催化剂时, 烯基叠氮3在NiCl2作用下得到2H-氮丙啶4, 中间体4受到苯乙醛的烯醇互变异构体的亲核进攻得到中间体4E, 随后, 中间体4E发生开环反应, 得到五元环状化合物4F, 再发生β-羟基消除反应得到中间体4G, 最后互变异构得到最终产物13.
2015年, Liu课题组[21]研究发现:在金络合物催化下, 炔胺14与α-芳基烯基叠氮3可发生[3+2]环加成反应来制备吡咯衍生物15 (Eq. 3).在该环加成反应中, α-芳基烯基叠氮作为前体化合物, 在高温下先转化为2H-氮丙啶类化合物, 2H-氮丙啶再作为三原子合成子参与到[3+2]环加成反应中, 该反应的区域选择性依赖于α-芳基烯基叠氮的取代基.
|
|
(3) |
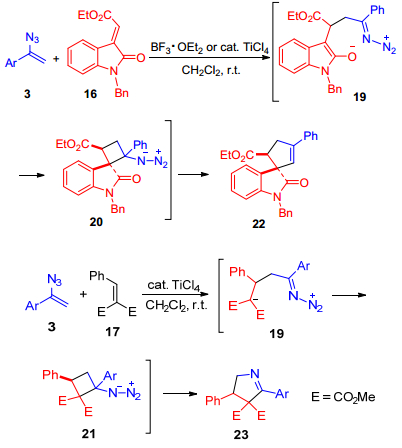
2016年, Chiba课题组[22]报道了在Lewis酸催化下, α-芳基烯基叠氮3与缺电子的烯烃发生共轭加成反应, 有效地构建了以吡咯啉为化合物骨架的方法.在该反应中, α-芳基烯基叠氮作为烯胺类型的亲核试剂与α, β-不饱和亲电试剂发生共轭加成反应.控制实验表明, α-芳基烯基叠氮与α, β-不饱和亲电试剂16和17发生的反应过程主要包含共轭加成反应, 并分别得到中间体18和19, 随后分别发生环化反应得到含氮环丁烷中间体20和21, 最后发生扩环反应得到对应的目标产物22和23 (Scheme 3).
噻唑是一类独特的杂环化合物, 其结构中含有一个硫原子和一个氮原子, 广泛存在于天然产物和生物活性分子中, 是许多天然和合成药物中重要分子的核心骨架[23].例如, 噻唑环是青霉素及其衍生物(该类化合物通常具有杀菌、抗逆转录病毒、抗真菌、抗组胺剂以及抗甲状腺的活性)的核心部分.
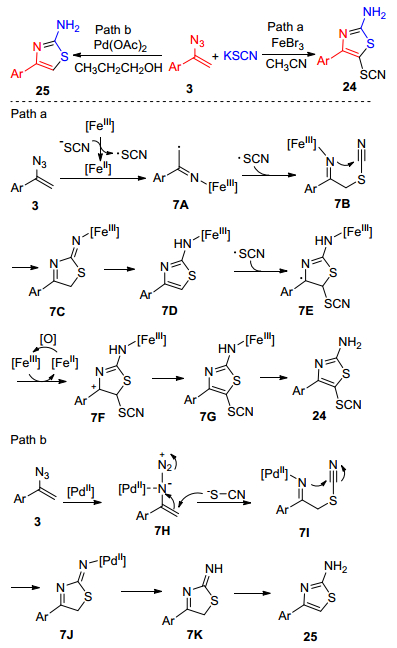
2015年, Zhang等[24]报道了在醋酸钯或溴化铁催化下, α-芳基烯基叠氮(3)与硫氰化钾的反应, 分别得到4-取代-2-氨基噻唑(24)和4-取代-5-硫氰基-2-氨基噻唑(25) (Scheme 4). 2-氨基噻唑及其衍生物是最为重要的含氮杂环化合物之一, 广泛存在于天然产物和药物中, 具有生物活性[25].研究表明, 在溴化铁催化下, 该反应可能通过自由基反应过程, 而在醋酸钯催化下该反应则是离子反应模式, 这两种反应都具有温和的反应条件及较高的选择性.当FeBr3为催化剂时(Path a, Scheme 4), 溴化铁单电子氧化硫氰酸根, 得到硫氰酸根自由基和亚铁离子, α-芳基烯基叠氮(3)在亚铁离子催化下得到自由基中间体7A, 硫氰酸根自由基与7A结合得到中间体7B, 随后发生分子内的亲核反应得到环状中间体7C, 7C通过1, 5-氢迁移反应生成中间体7D, 硫氰酸根自由基进攻中间体7D生成自由基7E, 再进一步氧化得到中间体7F, 7F再通过去质子化和质子化得到最终产物24.当Pd(OAc)2为催化剂时(Path b, Scheme 4), Pd(Ⅱ)与烯基叠氮配位得到中间体7H, 硫氰酸根亲核进攻7H生成中间体7I, 随后再发生分子内的亲核反应得到中间体7J, 7J通过质子化、芳香化后生成最终产物25.
咪唑及其衍生物广泛存在于天然产物[26]和药物[27]中, 是一类重要的氮杂环化合物, 有关构建咪唑骨架化合物的研究已经取得了较大的进展[28].然而, 新颖、高效地构建多取代的咪唑及其衍生物的研究仍然是热门的话题.
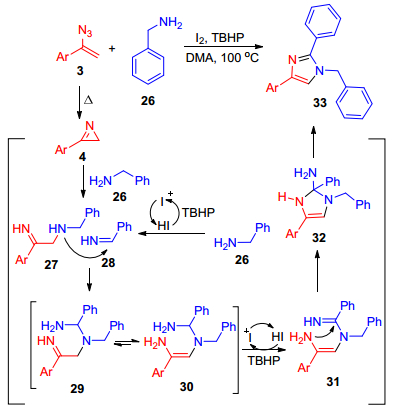
2015年, Yan课题组[29]报道了一种合成方法:在碘单质的催化下, α-芳基烯基叠氮化合物3与苄胺发生氧化串联环化反应, 从而合成出取代的咪唑及其咪唑衍生物.该反应采用I2/过氧化叔丁醇(TBHP)为催化剂/氧化剂体系, 以N, N-二甲基乙酰胺(DMA)作为溶剂, 在100 ℃条件下进行反应(Scheme 5), 该反应具有很好的普适性.可能的反应过程如下:首先, 底物3热分解为2H-氮丙啶4, 之后与26发生亲核加成反应得到中间体27[30], 随后, 27进攻由26氧化得到的亚胺28, 生成中间体29, 转化为中间体30, 得到化合物31, 发生分子内的环化反应得到中间体32, 最后, 再消除一分子伯胺得到最终产物33 (Scheme 5).
吡唑及其衍生物是最为重要的含氮杂环化合物之一, 在有机合成和药物合成中有广泛的应用[31].吡唑拥有多种生物活性, 包括止痛、抗炎、退烧、抗心律失常、镇静、松弛肌肉、精神兴奋、抗痉挛、一元胺氧化酶抑制剂、抗糖尿病和抗菌等.因其作用谱广、药效强烈等特点, 使得吡唑类化合物受到越来越多的关注.
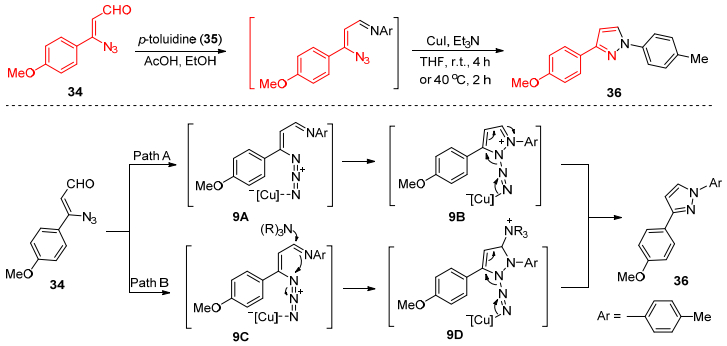
2011年, Rao课题组[32]报道了一个利用铜络合物催化形成氮氮键的合成方法, 该反应是以碘化亚铜为催化剂, 将α-芳基烯基叠氮醛34与芳胺35发生串联反应生成吡唑类化合物36 (Scheme 6), 该串联反应过程包括铜催化的分子内的环化反应和氮气的脱去过程, 具有较高的反应活性, 较好的底物普适性和较高的产率.反应可能通过两种路径完成.路径A:叔胺可能单纯地作为配体增加碘化亚铜的催化能力.在得到络合物9A后, 发生分子内的环化反应, 生成中间体9B, 然后, 脱去一分子的氮气, 在铜催化剂解离下生成最终产物36.路径B:首先, 叔胺亲核进攻亚胺9C, 得到中间体9D, 随后亲核性的氮进攻亲电性的叠氮生成中间体9E, 最后消除叔胺和氮气得到最终产物36.
喹啉及其衍生物普遍存在于药物化学、化学工业中[33], 是许多重要化合物的核心骨架, 如金鸡纳碱、奎宁丁、沙奎那韦、环丙沙星、加替沙星、氯碘羟喹、奥沙尼喹等.此外, 喹啉还用作有机合成中的起始原料[34]和导电材料[35].因此, 对喹啉及其衍生物的合成研究受到广泛的关注.常规的合成方法具有反应时间长、污染严重、底物不易获得、需要贵金属参与等缺点, 所以仍然需要发展操作简单、原子经济性高、对环境无伤害的方法来制备这类化合物.
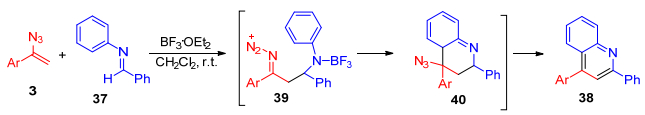
2014年, Chiba等[36]报道了在BF3·OEt2催化下, α-芳基烯基叠氮3与N-芳基醛亚胺37发生[4+2]环化反应, 合成出多取代的喹啉类化合物38 (Scheme 7).反应过程可能包含α-芳基烯基叠氮亲核进攻被BF3·OEt2活化了的N-不饱和醛亚胺, 得到中间体39, 然后发生分子内的环化反应得到四氢喹啉40, 随后消除一分子叠氮酸、芳香化得到喹啉类化合物38.该方法提供了一种简明的途径来制备在药物和材料领域都有广泛应用的喹啉类化合物.
异喹啉在生物活性分子、天然产物以及合成的药物试剂中是非常重要的结构单元[37].在过渡金属催化的反应中异喹啉类化合物还可以作为手性配体[38], 在材料科学中还可用于有机发光二极管中[39].由于异喹啉的应用广泛, 其合成受到很大的关注.
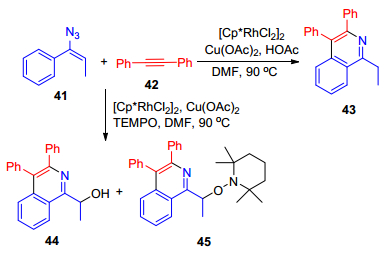
2011年, Chiba课题组[40]报道了在铑/铜双金属催化体系下, α-芳基烯基叠氮41与内炔烃42反应合成多取代的异喹啉衍生物的研究(Scheme 8).研究表明, 铑和铜在这个多步反应的催化循环中扮演着重要的协同催化作用.实验发现, 当试验中加入2, 2, 6, 6-四甲基哌啶氧化物(TEMPO)代替HOAc时, 苄基自由基中间体被TEMPO所捕获, 得到不同的产物.作者推测该反应经过下列步骤: (1)一价铜参与α-芳基烯基叠氮的去氮还原形成亚胺中间体, 该过程可能通过2H-氮丙啶中C—N键的断裂制得. (2)形成亚胺铑(Ⅲ)中间体, 然后邻位碳氢活化, 炔烃插入及C—N键还原消除, 从而得到异喹啉衍生物.
2015年, Liu课题组[41]报道了α-芳基烯基叠氮46和内炔烃47的串联反应, 提供了一个简单的途径来制备三氟乙基取代的异喹啉衍生物48.该反应是以Rh(Ⅲ)-Cu(Ⅱ)为双金属催化体系, 并以Togni’s试剂49作为三氟甲基源, 使此反应具有很好的普适性和较高的化学选择性, 适合多种α-芳基烯基叠氮和内炔烃(Eq. 4).该反应首次将传统的铑催化的C—H活化与Togni’s试剂相结合, 实现了一步构建复杂的三氟乙基取代的化合物, 该方法对构建具有生物活性的含氟化合物提供了新的思路.
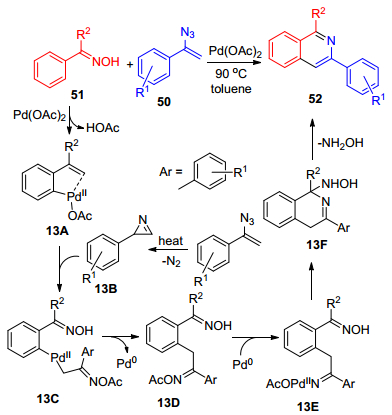
2016年, Deng等[42]报道了合成异喹啉类化合物52的研究, 该方法是以甲苯作为溶剂, 在90 ℃条件下, 以醋酸钯(Ⅱ)为催化剂, α-芳基烯基叠氮50与肟51发生环化反应(Scheme 9).在此反应中, 肟充当定位基和氧化剂, 从而具有非常好的官能团兼容性, 提供了一种在温和条件下合成不同类型的异喹啉化合物的制备方法.此外, 15N同位素标记实验表明, 异喹啉中的氮元素来自于烯基叠氮化合物.反应过程中, 肟51在钯(Ⅱ)催化下得到关键中间体13A, 烯基叠氮热分解为中间13B, 13B迁移插入中间体13A中得到中间体13C, 13C通过还原消除反应得到中间体13D, 进一步通过氧化加成N—O键得到中间体13E, 随后13E发生分子内的缩合反应得到13F, 最后释放一分子的羟胺得到最终产物52.
|
|
(4) |
菲啶及其衍生物是许多天然产物、生物碱以及其它合成的具有生物活性和应用价值的分子的核心部分(Figure 1).因其生物活性和光电性质引起了广泛的关注[43].因此, 已经发展出很多方法来构建这类多环化合物[44].
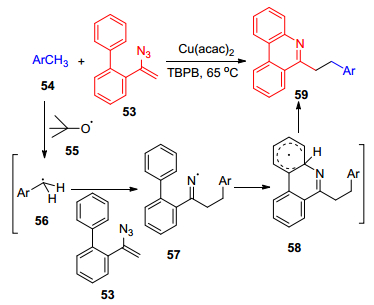
2016年, Guo等[45]报道了一个在铜催化下, α-联芳基烯基叠氮53与化合物54苄基的Csp3—H的氧化环化反应, 制备多种官能团取代的菲啶类化合物的研究成果(Scheme 10).除了苄基的碳氢键外, 该催化体系还适用于其它未被活化的Csp3—H键, 例如醚类、烷烃等, 使其反应具有很好的底物普适性.机理研究表明, 该反应是个自由基反应过程:首先, 金属铜通过催化单电子转移氧化或者过苯甲酸特丁酯的热力学均裂, 得到叔丁氧基自由基55, 叔丁氧基自由基再夺取甲苯上的氢得到苄基自由基56, 再加到α-联芳基烯基叠氮53的双键上, 得到亚氨基自由基57并释放出一分子的氮气, 最后, 自由基57发生分子内的环化反应得到自由基58, 自由基58通过单电子氧化并失去H+得到目标产物59.
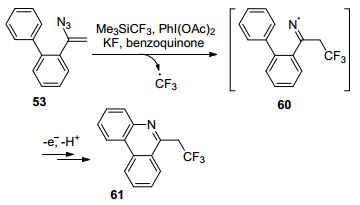
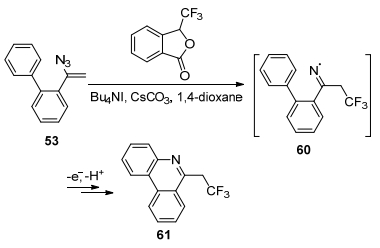
2014年, Chiba课题组[46]发展了α-联芳基烯基叠氮53的分子内自由基环化反应, 提供了一种简洁的方法来制备多氟烷基取代的含氮多环芳香烃化合物61 (Scheme 11).将易处理且易获得的Me3SiCF3作为反应的多氟烷基自由基源, 在PhI(OAc)2氧化下得到CF3自由基, 将其加到烯基叠氮碳上形成CF3亚胺基自由基60, 然后被α-芳基烯基叠氮邻位的芳基捕获, 形成碳氮键, 经环化、芳香化后得到三氟乙基取代的菲啶类化合物61.该反应中引入多氟烷基官能团可能会增加这些化合物在药物发现和材料基础应用中的价值.
2016年, Studer课题组[47]报道了以Togni试剂作为CF3源、Bu4NI作为引发剂, 用电子催化α-联芳基烯基叠氮53的氟烷基化反应, 制备三氟乙基菲啶类化合物61 (Scheme 12).该反应无需加入过渡金属, 反应过程是通过引发剂Bu4NI释放一个电子, 使Togni试剂释放出三氟甲基自由基, 作用于烯基叠氮, 再发生分子内的环化反应, 进而得到目标产物.
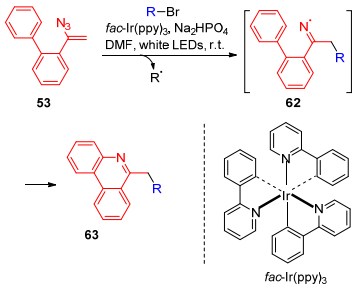
2016年, Yu课题组[48]报道了以可见光促进α-联芳基烯基叠氮53亚胺基自由基的形成, 制备菲啶衍生物63 (Scheme 13).在这反应中, 将烷基、三氟甲基自由基加入到烯基叠氮化合物中得到亚胺基自由基62, 然后发生分子内的芳香取代反应, 生成菲啶衍生物63.该反应条件温和, 反应普适性良好, 且产率较高, 与传统反应相比, 无需添加有毒锡试剂、紫外线照射、微波高温等.
吡啶是含有一个氮杂原子的六元杂环化合物, 在药物试剂、农用化学及材料化学中广泛存在(图 2)[49].在工业上, 吡啶可用作变性剂、助染剂, 及作为合成一系列产品(包括药品、消毒剂、染料等)的原料; 在有机合成中, 吡啶可用于制备手性的二氢和四氢吡啶以及哌啶, 这些化合物是重要生物活性分子的关键中间体.
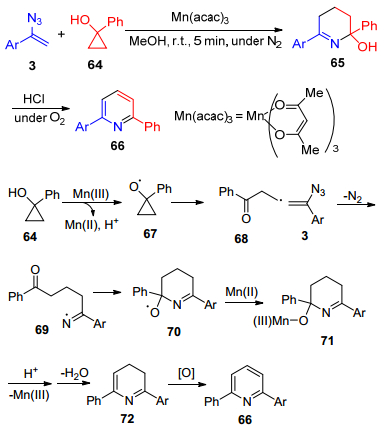
2009年, Chiba课题组[50]发展了以Mn(Ⅲ)催化α-芳基烯基叠氮3与被各种取代基取代的环丙醇化合物64的[3+3]环加成反应, 合成了吡啶类化合物66 (Scheme 14).该反应是采用环丙醇64作为β-羰基自由基68前体化合物.反应过程中单电子氧化剂Mn(Ⅲ)氧化环丙醇64, 得到β-羰基自由基68, 然后加成到烯基叠氮的双键上形成一个新的碳碳键和一个亚胺自由基69, 将亚胺自由基69与分子内的双键发生环化反应、还原反应、质子化及脱水, 再进一步氧化得到吡啶产物66.
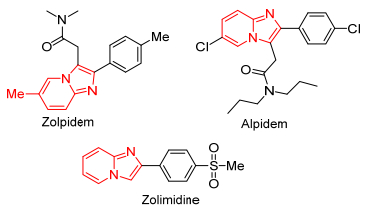
Imidazo[1, 2-a]pyridine是最为重要的双环5-6杂环化合物之一, 其在药物化学中具有广泛的应用, 如抗癌、抗分枝杆菌、抗利什曼虫、抗痉挛、抗病毒、抗糖尿病、质子泵抑制剂、杀虫活性等.其骨架也出现在市售药物中, 如佐利米定、唑吡坦、阿吡坦(图 3).因此, 科学家进行很多尝试对这种骨架进行结构修饰, 以求发现和发展新的治疗性药物.
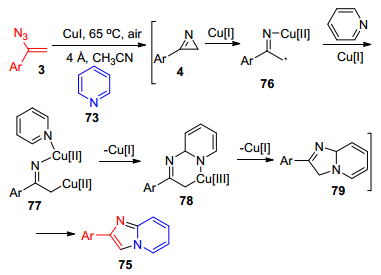
2014年, Adimurthy课题组[51]报道了在碘化亚铜催化、有氧条件下, 吡啶73、异喹啉74与α-芳基烯基叠氮3的Csp2—H官能团化反应, 合成Imidazo[1, 2-a]-pyridine杂环化合物75 (Scheme 15).该反应是直接以吡啶类化合物来构建imidazo[1, 2-a]pyridines (IPs), 具有重要的生物活性[52].该反应采用氧气作为氧化剂, 反应条件温和, 且唯一副产物是对空气无污染的氮气.类似地, α-芳基烯基叠氮3加热生成2H-氮丙啶4, 在Cu(Ⅰ)作用下4中的C—N发生断裂, 生成亚胺铜(Ⅱ)自由基76, 在Cu(Ⅰ)帮助下76与吡啶衍生物反应, 生成六元环状铜络合物78, 随后发生C—N键的还原消除反应、氧化反应得到最终产物75.
2-azabicyclo[3.3.1]nonane骨架结构存在于许多天然生物碱以及生物活性分子中[53].在多种生物的天然产物中, 发现超过300多种产物具有该骨架结构, 包括生物碱吗啡和士的宁.所以, 寻求简单易操作的方法制备含有2-azabicyclo[3.3.1]nonane骨架结构的化合物仍然备受化学家们的关注. 2009年, Chiba课题组报道了用Mn(Ⅲ)催化α-芳基烯基叠氮化合物与取代的双环环丙醇化合物(如bicyclo[3.1.0]-hexan-1-ol)反应, 合成了2-azabicyclo[3.3.1]non-2-en-1-ol类化合物(Scheme 16)[50].该反应是用bicyclo[3.1.0]hexan-1-ol作为β-羰基自由基源, 且只用了适量的催化剂Mn(acac)3 (5 mol%)就得到产率为89%的2-azabicyclo[3.3.1]non-2-en-1-ol.该反应具有很好的普适性, 合成了一系列的2-azabi-cyclo[3.3.1]non-2-en-1-ol衍生物.
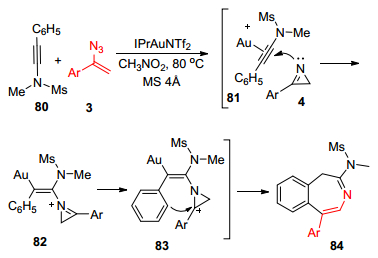
2015年, Liu课题组[21]报道了在金络合物催化下, 富电子芳基取代的炔胺80与α-芳基烯基叠氮3发生新颖的[4+3]环加成反应, 来制备1H-benzo[d]azepine衍生物84的研究(Scheme 17).作者推测该环加成反应过程中α-芳基烯基叠氮3先转化为氮丙啶类化合物4, 并以氮丙啶为该催化循环的活性中间体, 2H-氮丙啶随后加成到被金络合物活化了的叁键上81, 得到常见中间体82, 随后化合物炔胺上含富电子的苯基进攻氮丙啶环, 最终得到目标产物80.
本文总结了近年来α-芳基烯基叠氮类化合物在构建各种含氮杂环化合物反应机理、反应特点及应用研究.随着实验安全技术的提高以及对α-芳基烯基叠氮类化合物的认识更加全面, 越来越多的研究者利用α-芳基烯基叠氮作为关键的合成子来构建结构复杂的N-杂环化合物.含氮杂环化合物广泛存在于各种生物活性小分子和材料中, 促使研究者发展了各种合成方法. α-芳基烯基叠氮独特的理化性质为科学家发展新颖简洁地构建含氮杂环化合物方法打开了大门.尽管利用烯基叠氮构建含氮杂环化合物已经取得了很大的进步, 但基于烯基叠氮作为起始原料来构建含氮化合物, 以此来发展新颖的合成方法的研究将会结出更多果实, 相信这个领域的研究必将取得更大的发展.
For selected reviews, see: (a) Carey, J. S. ; Laffan, D. ; Thomson, C. ; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337.
(b) Welsch, M. E. ; Snyder, S. A. ; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347.
(c) Dandapani, S. ; Marcaurelle, L. A. Curr. Opin. Chem. Biol. 2010, 14, 362.
(d) Tohme, R. ; Darwiche, N. ; Gali-Muhtasib, H. Molecules 2011, 16, 9665.
(e) Thomas, G. L. ; Johannes, C. W. Curr. Opin. Chem. Biol. 2011, 15, 516.
(f) Zhang, Z. ; Zheng, X. ; Guo, C. Chin. J. Org. Chem. 2016, 36, 1241.
(g) Zhang, J. ; Liu, J. ; Ma, Y. ; Cheng, P. Chin. J. Org. Chem. 2017, 37, 555.
For recent reviews, see: (a) Gribble, G. W. ; Joule, J. A. Progress in Heterocyclic Chemistry, Vol. 20, Elsevier, Oxford, 2008, and others in this series.
(b) Katritzky, A. R. ; Ramsden, C. A. ; Scriven, E. F. V. ; Taylor, R. J. K. Comprehensive Heterocyclic Chemistry Ⅲ, Pergamon, Oxford, 2008.
(c) Katritzky, A. R. ; Rees, C. W. ; Scriven, E. F. V. ; McKillop, A. Comprehensive Heterocyclic Chemistry Ⅱ, Pergamon, Oxford, 1996and references therein.
(a) Hassner, A. ; Fowler, F. W. J. Org. Chem. 1968, 33, 2686.
(b) Brä se, S. ; Gil, C. ; Knepper, K. ; Zimmermann, V. Angew. Chem. , Int. Ed. 2005, 44, 5188.
(c) Gu, P. ; Su, Y. ; Wu, X. P. ; Sun, J. ; Liu, W. ; Xue, P. ; Li, R. Org. Lett. 2012, 14, 2246.
(a) Nair, V. ; Tesmol, G. G. Tetrahedron Lett. 2000, 41, 3199.
(b) Zhu, W. ; Ma, D. Chem. Commun. 2004, 888.
(c) Telvekar, V. N. ; Takale, B. S. ; Bachhav, H. M. Tetrahedron Lett. 2009, 50, 5056.
(d) Kupracz, L. ; Hartwig, J. ; Wegner, J. ; Ceylan, S. ; Kirschning, A. Beilstein J. Org. Chem. 2011, 7, 1441.
(e) Li, X. ; Liao, S. ; Wang, Z. ; Zhang, L. Org. Lett. 2017, 19, 3687.
Griess, P. Liebigs Ann. Chem. 1866, 137, 39.
For recent reviews on organic azides, see: (a) Brä se, S. ; Gil, C. ; Knepper, K. ; Zimmermann, V. Angew. Chem. , Int. Ed. 2005, 44, 5188.
(b) Moses, J. E. ; Moorhouse, A. D. Chem. Soc. Rev. 2007, 36, 1249.
(c) Driver, T. G. Org. Biomol. Chem. 2010, 8, 3831.
Select examples: (a) Chiba, S. ; Wang, Y. -F. ; Lapointe, G. ; Narasaka, K. Org. Lett. 2008, 10, 313.
(b) Wang, Y. -F. ; Toh, K. K. ; Chiba, S. ; Narasaka, K. Org. Lett. 2008, 10, 5019.
(c) Wang, Y. -F. ; Chiba, S. J. Am. Chem. Soc. 2009, 131, 12570.
(d) Wang, Y. -F. ; Toh, K. K. ; Ng, E. P. J. ; Chiba, S. J. Am. Chem. Soc. 2011, 133, 6411.
(e) Wang, Y. -F. ; Toh, K. K. ; Lee, J. -Y. ; Chiba, S. Angew. Chem. , Int. Ed. 2011, 50, 5927.
(f) Chen, F. ; Shen, T. ; Cui, Y. ; Jiao, N. Org. Lett. 2012, 14, 4926.
(g) Wang, Y. -F. ; Lonca, G. H. ; Runigo, M. L. ; Chiba, S. Org. Lett. 2014, 16, 4272.
For some recent reviews, see: (a) Murphee, S. S. ; Padwa, A. In Progress Heterocyclic Chemistry, Vol. 9, Eds. : Scriven, E. F. V. ; Suschitzky, H., Pergamon Press, Oxford, 1997.
(b) Pearson, W. H. ; Lian B. W. ; Bergmeier, S. C. In Comprehensive Heterocyclic Chemistry Ⅱ, Vol. 1A, Eds. : Katritzky, A. R. ; Rees, C. W. ; Scriven, E. F. V., Pergamon Press, Oxford, 1996, Chapter 1.
(c) Heimgartner, H. Angew. Chem. , Int. Ed. 1991, 30, 238.
(a) Ray, C. A. ; Risberg, E. ; Somfai, P. Tetrahedron. 2002, 58, 5983.
(b) Palacios, F. ; de Retana, A. M. O. ; de Marigorta, E. M. ; de los Santos, J. M. Org. Prep. Proced. Int. 2002, 34, 219.
(c) Timén, A. S. ; Fischer, A. ; Somfai, P. Chem. Commun. 2003, 34, 1150.
For recent reviews on 2H-azirines, see: (a) Palacios, F. ; Retana, A. M. D. ; Marigorta, E. M. D. ; Santos, J. D. L. Eur. J. Org. Chem. 2001, 2401.
(b) Palacios, F. ; Retana, A. M. O. D. ; Marigorta, E. M. D. ; Santos J. M. D. L. Org. Prep. Proced. Int. 2002, 34, 219.
(c) Khlebnikov, A. F. ; Novikov, M. S. Tetrahedron 2013, 69, 3363.
(a) Hortmann, A. G. ; Robertson, D. A. ; Gillard, B. K. J. Org. Chem. 1972, 37, 322.
(b) Hassner, A. ; Fowler, F. W. J. Am. Chem. Soc. 1968, 90, 2869.
(c) Pinho e Melo, T. M. V. D. ; Lopes, C. S. J. ; Cardoso, A. L. ; Rocha Gonsalves, A. M. d'A. Tetrahedron 2001, 57, 6203.
Åsa Sjöholm Timén, Risberg E, Somfai P. ChemInform 2003, 44, 5339.
Singh, P. N. D.; Carter, C. L.; Gudmundsdóttir, A. D. Tetrahedron Lett. 2003, 44, 6763. doi: 10.1016/S0040-4039(03)01558-2
(a) Muchowski, J. M. Adv. Med. Chem. 1992, 1, 109.
(b) Cozzi, P. ; Mongelli, N. Curr. Pharm. Des. 1998, 4, 181.
(c) Fürstner, A. Angew. Chem. , Int. Ed. 2003, 42, 3582.
(d) Balme, G. Angew. Chem. , Int. Ed. 2004, 43, 6238.
(e) Andreani, A. ; Cavalli, A. ; Granaiola, M. ; Guardigli, M. ; Leoni, A. ; Locatelli, A. ; Morigi, R. ; Rambaldi, M. ; Recanatini, M. ; Roda, A. J. Med. Chem. 2001, 44, 4011.
(f) Baraldi, P. G. ; Nunez, M. C. ; Tabrizi, M. A. ; De Clercq, E. ; Balzarini, J. ; Bermejo, J. ; Esterez, F. ; Romagnodi, R. J. Med. Chem. 2004, 47, 2877.
(g) Srivastava, S. K. ; Shefali Miller, C. N. ; Aceto, M. D. ; Traynor, J. R. ; Lewis, J. W. ; Husbands, S. M. ; J. Med. Chem. 2004, 47, 6645.
Walsh, C. T.; Garneau-Tsodikova, S.; Howard-Jones, A. R. Nat. Prod. Rep. 2006, 23, 517. doi: 10.1039/b605245m
(a) Novák, P. ; Müller, K. ; Santhanam, K. S. V. ; Haas, O. Chem. Rev. 1997, 97, 207.
(b) Gale, P. A. ; Acc. Chem. Res. 2006, 39, 465.
For selected examples in recent years, see: (a) Wan, X. ; Xing, D. ; Fang, Z. ; Li, B. ; Zhao, F. ; Zhang, K. ; Yang, L. ; Shi, Z. J. Am. Chem. Soc. 2006, 128, 12046.
(b) Seregin, I. V. ; Gevorgyan, V. J. Am. Chem. Soc. 2006, 128, 12050.
(c) Martín, R. ; Rivero, M. R. ; Buchwald, S. L. Angew. Chem. , Int. Ed. 2006, 45, 7079.
(d) Binder, J. T. ; Kirsch, S. F. Org. Lett. 2006, 8, 2151.
(e) Su, S. ; Porco, J. A., Jr. J. Am. Chem. Soc. 2007, 129, 7744.
(f) Shindo, M. ; Yoshimura, Y. ; Hayashi, M. ; Soejima, H. ; Yoshikawa, T. ; Matsumoto, K. ; Shishido, K. Org. Lett. 2007, 9, 1963.
(g) St. Cyr, D. J. ; Arndtsen, B. A. J. Am. Chem. Soc. 2007, 129, 12366.
(h) Lu, Y. ; Arndtsen, B. A. Angew. Chem. , Int. Ed. 2008, 47, 5430.
Wang Y. F.; Toh K. K.; Chiba S.; Narasaka, K. Org. Lett. 2008, 10, 5019. doi: 10.1021/ol802120u
Richert, S. A.; Tsang, P. K. S.; Sawyer, D. T. Inorg. Chem. 1988, 27, 1814. doi: 10.1021/ic00283a027
Chen, F.; Shen, T.; Cui, Y.; Jiao, N. Org. Lett. 2012, 14, 4926. doi: 10.1021/ol302270z
Pawar, S. K.; Sahani, R. L.; Liu, R. S. Chem.-Eur. J. 2015, 21, 10843. doi: 10.1002/chem.v21.30
Zhu, X.; Chiba, S. Chem. Commun. 2016, 52, 2473. doi: 10.1039/C5CC10299E
Wu, X. J.; Kassie, F.; Mersch-Sundermann, V. Mutat. Res. 2005, 589, 81. doi: 10.1016/j.mrrev.2004.11.001
Chen, B.; Guo, S.; Guo, X.; Zhang, G.; Yu, Y. Org. Lett. 2015, 17, 4698. doi: 10.1021/acs.orglett.5b02152
(a) Zablotskaya, A. ; Segal, I. ; Germane, S. ; Shestakova, I. ; Domracheva, I. ; Nesterova, A. ; Geronikaki, A. ; Lukevies, E. Chem. Heterocycl. Compd. 2002, 38, 859.
(b) Franklin, P. X. ; Pillai, A. D. ; Rathod, P. D. ; Yerande, S. G. ; Nivsarkar, M. ; Padh, H. ; Sudarsanam, V. ; Vasu, K. K. Eur. J. Med. Chem. 2008, 43, 129.
(c) Liu, R. ; Huang, Z. ; Murray, M. G. ; Guo, X. ; Liu, G. J. Med. Chem. 2011, 54, 5747.
(d) Annadurai, S. ; Martinez, R. ; Canney, D. J. ; Eidem, T. ; Dunman, P. M. ; Abou-Gharbia, M. Bioorg. Med. Chem. Lett. 2012, 22, 7719.
(e) Smith, B. ; Chang, H. -H. ; Medda, F. ; Gokhale, V. ; Dietrich, J. ; Davis, A. ; Meuillet, E. ; Hulme, C. Bioorg. Med. Chem. Lett. 2012, 22, 3567.
(a) Zhong, J. Nat. Prod. Rep. 2009, 26, 382.
(b) Forte, B. ; Malgesini, B. ; Piutti, C. ; Quartieri, F. ; Scolaro, A. ; Papeo, G. Mar. Drugs 2009, 7, 705.
(c) Midoux, P. ; Pichon, C. ; Yaouanc, J. -J. ; Jaffres, P. -A. J. Pharmacol. 2009, 157, 166.
(d) Xiong, F. ; Chen, X. -X. ; Chen, F. -E. ; Tetrahedron: Asymmetry 2010, 21, 665.
(a) Lee, R. J. C. ; Timmermans, P. C. ; Gallaghr, T. F. ; Kumar, S. ; McNully, D. ; Blumenthal, M. ; Heys, J. R. Nature 1994, 372, 739.
(b) Antolini, M. ; Bozzoli, A. ; Ghiron, C. ; Kennedy, G. ; Rossi, T. ; Ursini, A. Bioorg. Med. Chem. Lett. 1999, 9, 1023.
(c) Wang, L. ; Woods, K. W. ; Li, Q. ; Barr, K. J. ; McCroskey, R. W. ; Hannick, S. M. ; Gherke, L. ; Credo, R. B. ; Hui, Y. -H. ; Marsh, K. ; Warner, R. ; Lee, J. Y. ; Zielinsky-Mozng, N. ; Frost, D. ; Rosenberg, S. H. ; Sham, H. L. J. Med. Chem. 2002, 45, 1697.
(d) Dietrich, J. ; Gokhale, V. ; Wang, X. -D. ; Hurley, L. H. ; Flynn, G. A. Bioorg. Med. Chem. 2010, 18, 292.
(e) Cho, H. -J. ; Gee, H. -G. ; Baek, K. -H. ; Ko, S. -K. ; Park, J. -K. ; Lee, H. ; Kim, N. -D. ; Lee, M. -G. ; Shin, I. J. Am. Chem. Soc. 2011, 133, 20267.
(a) Du, H. ; He, Y. ; Rasapalli, S. ; Lovely, C. -J. Synlett 2006, 965.
(b) Bellina, F. ; Cauteruccio, S. ; Rossi, R. Tetrahedron 2007, 63, 4571.
(c) Kaniyo, S. ; Yamamoto, Y. Chem. -Asian J. 2007, 2, 568.
(d) Bellina, F. ; Rossi, R. Adv. Synth. Catal. , 2010, 352, 1223.
Xiang, L.; Niu, Y.; Pang, X.; Yang, X. D.; Yan, R. L. Chem. Commun. 2015, 6598.
(a) Chen, F. ; Shen, T. ; Cui, Y. ; Jiao, N. Org. Lett. 2012, 14, 4926.
(b) Donthiri, R. R. ; Pappula, V. ; Reddy, N. N. K. ; Bairagi, D. ; Adimurthy, S. J. Org. Chem. 2014, 79, 11277.
(c) Li, T. ; Xin, X. ; Wang, C. ; Wang, D. ; Wu, F. ; Li, X. ; Wan, B. Org. Lett. 2014, 16, 4806.
(a) Lan, R. ; Liu, Q. ; Fan, P. ; Lin, S. ; Fernando, S. R. ; McCallion, D. ; Pertwee, R. ; Makriyannis, A. J. Med. Chem. ; 1999; 42, 769.
(b) Ballesteros, P. ; Claramunt, R. M. ; Escolastico, C. ; Maria, M. D. S. ; Elguero, J. J. Org. Chem. 2002, 57, 1873.
(c) Barreiro, E. J. ; Camara, C. A. ; Verli, H. ; Brazil-Más, L. ; Castro, N. G. ; Cintra, W. M. ; Aracava, Y. ; Rodrigues, C. R. ; Fraga, C. A. M. J. Med. Chem. 2003, 46, 1144.
(d) Christodoulou, M. S. ; Liekens, S. ; Kasiotis, K. M. ; Haroutounian, S. A. Bioorg. Med. Chem. 2010, 18, 4338.
(e) Rashad, A. E. ; Hegab, M. I. ; Abdel-Megeid, R. E. ; Micky, J. A. ; Abdel-Megeid, F. M. E. Bioorg. Med. Chem. 2008, 16, 7102.
(f) Fustero, S. ; Sánchez-Roselló, M. ; Barrio, P. ; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984.
Hu, J.; Cheng, Y.; Yang, Y.; Rao, Y. Chem. Commun. 2011, 47, 10133. doi: 10.1039/c1cc13908h
(a) Michael, J. P. Nat. Prod. Rep. 1997, 14, 605.
(b) Xu, M. ; Wagerle, T. ; Long, J. K. ; Lahm, G. P. ; Barry, J. D. ; Smith, R. M. Bioorg. Med. Chem. Lett. 2014, 24, 4026.
(a) Franciò, G. ; Faraone, F. ; Leitner, W. Angew. Chem., Int. Ed. 2000, 39, 1428.
(b) Rueping, M. ; Antonchick, A. P. ; Theissmann, T. Angew. Chem. , Int. Ed. 2006, 45, 3683.
(a) Bakhshi, A. ; Bhalla, G. J. Sci. Ind. Res. 2004, 63, 715.
(b) Kim, J. I. ; Shin, I. -S. ; Kim, H. ; Lee, J. -K. J. Am. Chem. Soc. 2005, 127, 1614.
Zhu, X.; Wang, Y. F.; Zhang, F. L.; Chiba, S. Chem. Asian J. 2014, 9, 2458. doi: 10.1002/asia.201402421
(a) Kletsas, D. ; Li, W. ; Han, Z. ; Papadopoulos, V. Biochem. Pharmacol. 2004, 67, 1927.
(b) Muscarella, D. E. ; O'Brian, K. A. ; Lemley, A. T. ; Bloom, S. E. Toxicol. Sci. 2003, 74, 66.
(a) Sweetman, B. A. ; Müller-Bunz, H. ; Guiry, P. J. Tetrahedron Lett. 2005, 46, 4643.
(b) Durola, F. ; Sauvage, J. P. ; Wenger, O. S. Chem. Commun. 2006, 171.
(c) Lim, C. W. ; Tissot, O. ; Mattison, A. ; Hooper, M. W. ; Brown, J. M. ; Cowley, A. R. ; Hulmes, D. I. ; Blacker, A. J. Org. Process Res. Dev. 2003, 7, 379.
(a) Fang, K. H. ; Wu, L. L. ; Huang, Y. T. ; Yang, C. H. ; Sun, I. W. Inorg. Chim. Acta 2006, 359, 441.
(b) Zhao, Q. ; Liu, Q. S. ; Shi, M. ; Wang, C. ; Yu, M. ; Li, L. ; Li, F. ; Yi, T. ; Huang, C. Inorg. Chem. 2006, 45, 6152.
(c) Tsuboyama, A. ; Iwawaki, H. ; Furugori, M. ; Mukaide, T. ; Kamatani, J. ; Igawa, S. ; Moriyama, S. T. ; Miura, S. ; Takiguchi, T. ; Okada, S. ; Hoshino, M. ; Ueno, K. J. Am. Chem. Soc. 2003, 125, 12971.
Wang, Y.-F.; Toh, K. K.; Lee, J.-Y.; Chiba, S. Angew. Chem., Int. Ed. 2011, 50, 5927. doi: 10.1002/anie.v50.26
Liu, K.; Chen, S.; Li, X. G.; Liu, P. N. J. Org. Chem. 2015, 81, 265. https://www.sciencedirect.com/science/article/pii/S095656631630149X
Zhu, Z. Z.; Tang, X. D.; Li, X. W.; Wu, W. Q.; Deng, G. H.; Jiang, H. F. J. Org. Chem. 2016, 81, 1401. doi: 10.1021/acs.joc.5b02376
(a) Kock, I. ; Heber, D. ; Weide, M. ; Wolschendorf, U. ; Clement, B. J. Med. Chem. 2005, 48, 2772.
(b) Bernardo, P. H. ; Wan, K. -F. ; Sivaraman, T. ; Xu, J. ; Moore, F. K. ; Hung, A. W. ; Mok, H. Y. K. ; Yu, V. C. ; Chai, C. L. L. J. Med. Chem. 2008, 51, 6699.
(c) Cappoen, D. ; Jacobs, J. ; Van, T. N. ; Claessens, S. ; Diels, G. ; Anthonissen, R. ; Einarsdottir, T. ; Fauville, M. ; Verschaeve, L. ; Huygen, K. ; Kimpe, N. D. Eur. J. Med. Chem. 2012, 48, 57.
(d) Cappoen, D. ; Claes, P. ; Jacobs, J. ; Anthonissen, R. ; Mathys, V. ; Verschaeve, L. ; Huygen, K. ; Kimpe, N. D. J. Med. Chem. 2014, 57, 2895.
(e) Naidua, K. M. ; Nagesha, H. N. ; Singhb, M. ; Sriramc, D. ; Yogeeswaric, P. ; Sekhara, K. V. G. C. Eur. J. Med. Chem. 2015, 92, 415.
For selected examples, see: (a) Mehta, B. K. ; Yanagisawa, K. ; Shiro, M. ; Kotsuki, H. Org. Lett. 2003, 5, 1605.
(b) Gerfaud, T. ; Neuville, L. ; Zhu, J. Angew. Chem. , Int. Ed. 2009, 48, 572.
(c) Candito, D. A. ; Lautens, M. Angew. Chem. , Int. Ed. 2009, 48, 6713.
(d) Zhang, L. ; Ang, G. Y. ; Chiba, S. Org. Lett. 2010, 12, 3682.
(e) Deb, I. ; Yoshikai, N. Org. Lett. 2013, 15, 4254.
(f) Tummatorn, J. ; Krajangsri, S. ; Norseeda, K. ; Thongsornkleeb, C. ; Ruchirawat, S. Org. Biomol. Chem. 2014, 12, 5077.
(g) Li, J. ; Wang, H. ; Sun, J. ; Yang, Y. ; Liu, L. Org. Biomol. Chem. 2014, 12, 7904.
(h) Jiang, H. ; An, X. ; Tong, K. ; Zheng, T. ; Zhang, Y. ; Yu, S. Angew. Chem. , Int. Ed. 2015, 54, 4055.
Yang, J. C.; Zhang, J. J.; Guo, L. N. Org. Biomol. Chem. 2016, 14, 9806. doi: 10.1039/C6OB02012G
Wang, Y. F.; Lonca, G. H.; Le, R. M.; Chiba, S. Org. Lett. 2014, 16, 4272. doi: 10.1021/ol501997n
Mackay, E.; Studer, A. Chem.-Eur. J. 2016, 22, 13455. doi: 10.1002/chem.201602855
Sun, X.; Yu, S. Chem. Commun. 2016, 52, 10898. doi: 10.1039/C6CC05756J
Quesne, P. W. L. J. Nat. Prod. 1997, 60, 202. doi: 10.1007/s10482-017-0920-9
Wang, Y. F.; Chiba, S. J. Am. Chem. Soc. 2009, 131, 12570. doi: 10.1021/ja905110c
Donthiri, R. R.; Pappula, V.; Reddy, N. N. K.; Bairagi, D.; Adimurthy, S. J. Org. Chem. 2014, 79, 11277. doi: 10.1021/jo5021618
(a) Enguehard-Gueiffier, C. ; Gueiffier, A. Med. Chem. 2007, 7, 888.
(b) Lhassani, M. ; Chavignon, O. ; Chezal, J. -M. ; Teulade, J. -C. ; Chapat, J. -P. ; Snoeck, R. ; Andrei, G. ; Balzarini, J. ; De Clercq, E. ; Gueiffier, A. Eur. J. Med. Chem. 1999, 34, 271.
(c) Rupert, K. C. ; Henry, J. R. ; Dodd, J. H. ; Wadsworth, S. A. ; Cavender, D. E. ; Olini, G. C. ; Fahmy, B. ; Siekierka, J. Bioorg. Med. Chem. Lett. 2003, 13, 347.
For a review on synthesis of 2-azabicyclo[3. 3. 1] nonanes, see: Bonjoch, J. ; Diaba, F. ; Bradshaw, B. ; Farmàcia, F. Synthesis 2011, 993.

图式 1 烯基叠氮与1, 3-二羰基化合物反应合成吡咯衍生物
Scheme 1 Synthesis of pyrroles from vinyl azides and 1, 3-dicarbonyl compounds

图式 2 由NiCl2或Cu(OAc)2催化制备吡咯类化合物
Scheme 2 Pyrroles synthesis switched by copper and nickel catalysts

图式 4 钯、铁催化剂催化下制备2-氨基噻唑类化合物
Scheme 4 2-Aminothiazoles synthesis switched by palladium and iron catalysts

图式 7 烯基叠氮与N-不饱和亚胺的[4+2]环化反应
Scheme 7 Formal [4+2] annulation of vinyl azides with N-unsaturated aldimines

图式 9 醋酸钯催化烯基叠氮与肟的环化反应
Scheme 9 Pd(OAc)2 catalyzed cyclization between vinyl azide with oximes

图式 16 三氟乙基菲啶的合成
Scheme 16 Formation of trifluoroethyl phenanthridine via a radical cyclization.

图 2 吡啶和二氢吡啶衍生物
Figure 2 Examples of commercial pyridine or dihydropyridine derivatives

图式 14 烯基叠氮与环丙醇的[3+3]环化反应
Scheme 14 Mn(Ⅲ)-mediated formal [3+3]-annulation reactions of vinyl azides and cyclopropanols.

图式 15 铜(Ⅰ)催化吡啶与烯基叠氮的C—H官能团化反应
Scheme 15 Cu(Ⅰ)-catalyzed C—H functionalization of pyridines with vinyl azides

图式 16 烯基叠氮与环丙醇的环化反应
Scheme 16 Annulation reactions of vinyl azides and cyclopropanols.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:










 下载:
下载: