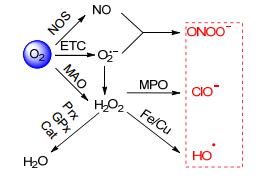
图式 1.
线粒体中ROS的转化示意图
Scheme 1.
Conversion of ROS in mitochondria

活性氧簇(reactive oxygen species, ROS)是生物体中普遍存在的一类活性物质.一般来说, 活性氧簇主要包含以下六种:单线态氧(1O2)、羟基自由基(•OH)、超氧根自由基(O2·-)、过氧化氢(H2O2)、过氧化亚硝酰(ONOO-)以及次氯酸(HOCl/ClO-).其中, ONOO-也可以被认为是活性氮簇(RNS)的一份子, 在这里为便于概述, 将其归于ROS一类.不正常的ROS水平会引起细胞中的氧化应力发生变化, 从而引起一系列的细胞功能紊乱, 最终导致各种疾病的产生, 如癌症和神经退行性疾病(帕金森症、阿尔茨海默症等)[1, 2].过去数十年的研究表明, ROS作为细胞中的第二信使, 对于调控生理过程中的氧化还原平衡具有十分重要的意义[3].因此, 研究这些活性物质的生理意义对于了解人类健康与疾病就显得很有必要.
线粒体是细胞“制造”ROS的主要场所[4].如Scheme 1所示, 细胞中的ROS一般都起源于分子氧.分子氧通过捕获电子传递链(ETC)上的电子形成O2·-. O2·-可以通过超氧歧化酶(SOD)催化或者自发歧化形成H2O2. H2O2也可以通过单胺氧化酶(MAO)催化氧化分子氧形成.一氧化氮(NO)是分子氧在一氧化氮合成酶(NOS)存在下形成的, 它可以和O2·-形成ONOO-. H2O2在微量的Fe2+或Cu+存在下, 经历Fenton反应或者Haber-Weiss反应形成•OH; 也可以在髓过氧化物酶(MPO)催化下与Cl-作用形成HOCl/ClO-.在这几种ROS中, •OH、ONOO-以及HOCl/ClO-的氧化性最强, 被称为高活性氧簇(hROS).
鉴于ROS在生理过程中扮演着重要的角色, 目前很多研究小组发展了多种方法来检测细胞或组织中的ROS水平[5, 6].由于其高灵敏性、高选择性、无损检测以及高时空分辨率, 荧光分析法越来越受到科学家们的青睐[7~10].近10年来, 有很多类型的荧光传感器被报道用于检测ROS, 有基于纳米材料的, 有基于大分子的, 也有基于小分子的[11~13].本综述主要就各种活性氧的小分子荧光探针的研究进展进行小结, 而侧重点则是各种荧光探针与ROS的反应类型、作用机理以及设计思路.
HOCl的pKa为7.6, 在生理条件下, 细胞中的次氯酸是以HOCl/ClO-的平衡形式存在的.在生命过程中, HOCl主要是和蛋白质的侧链或者肽键反应, 如甲硫氨酸残基等.每106个嗜中性粒细胞产生HOCl的速率是0.47 nmol•min-1[14], 同时HOCl的寿命也非常短, 因此, 细胞中的HOCl基础水平是很低的.目前所有的次氯酸小分子荧光探针都是基于其强氧化性设计而成.
2007年Nagano课题组[15]报道了第一例高选择性HOCl/ClO-小分子荧光探针的化合物1.如Eq. 1所示, 作者利用HOCl对硫醚的氧化作用, 使得无荧光的螺环状态的探针1在与HOCl作用后变成强荧光的开环产物.该反应位点与HOCl以外的其它活性氧基本不反应.最后, 作者利用该探针监测了猪的中性细胞吞噬酵母过程的完成, 因为该过程会伴随HOCl的产生.
|
|
(1) |
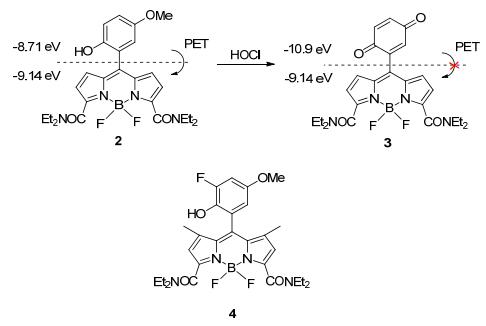
Yang课题组[16]在2008年报道了基于氟硼二吡咯的荧光探针2.该化合物本身由于苯酚部分向荧光团发生光诱导电子转移(PET)而不发射荧光; 在加入HOCl后, 苯酚被氧化成醌, PET过程被抑制, 荧光大大增强.探针2对次氯酸的选择性比较高, 但是其氧化产物3会被HOCl进一步氧化导致荧光变弱.为了克服这个缺点, 该课题组在2的结构上进行修饰, 得到化合物4 (Scheme 2)[17].化合物4具备和2一样的选择性及高灵敏度, 但是不易被氧化, 稳定性更高.这两个探针都被用于细胞中内源性HOCl的检测.
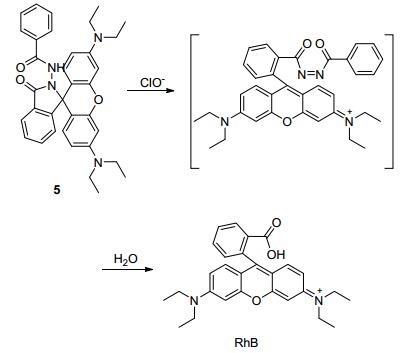
2008年, Ma课题组[18]巧妙地设计了一个基于罗丹明骨架并含有二苯甲酰肼反应位点的ClO-荧光探针5.如Scheme 3所示, 该探针本身呈螺环结构, 无荧光; 与次氯酸根作用后, 会发生氧化水解反应, 得到发射橙色荧光的开环产物罗丹明B (RhB), 并且伴随有肉眼可见的颜色变化——无色到紫色.通过pH滴定实验, 作者发现探针只在碱性条件下对ClO-有响应, 说明化合物是与ClO-而不是HOCl作用.该探针合成简单, 选择性优异, 灵敏度高, 其检测限为27 nmol•L-1, 但是水溶性不好, 反应时间也略长(30 min).
2009年, Lin课题组[19]报道了一例基于肟氧化的次氯酸根探针6 (Eq. 2).在次氯酸根作用下, 探针的荧光从439 nm红移至509 nm.通过1H NMR表征, 作者认为氧化产物是醛7.该探针第一次实现了对次氯酸根的比率识别.
|
|
(2) |
利用2, 4-二硝基苯腙可以被氧化为醛的特性, Lin课题组[20]基于分子内电荷转移(ICT)机制设计了探针8.他们原以为探针8在与ClO-作用前后会发射出不同颜色的荧光, 从而实现对ClO-的比率识别(Eq. 3).但是, 实验结果表明, 探针8本身荧光非常弱, 只能表现出“turn-on”的荧光响应.作者推测可能是因为探针8的荧光被硝基淬灭了, 所以他们又设计了其类似物9.化合物9本身具有强荧光, 其发射峰在与ClO-反应后从585 nm蓝移至505 nm, 实现了对ClO-的比率识别.最后, 探针9被成功应用于人乳腺癌细胞(MCF-7细胞)中ClO-的比率荧光成像.
|
|
(3) |
2013年, Han课题组[21]报道了一例可以可逆识别HOCl/H2S氧化还原对的探针10.探针本身荧光较弱, 作者推测是甲氧基苯基硒和氟硼二吡咯荧光团产生了PET过程; 在HOCl的氧化作用下, Se被氧化成Se=O, PET过程得到抑制, 从而荧光荧光增强(Eq. 4).该氧化过程只在HOCl作用下发生, 其它ROS并不能诱导该过程.而探针10的氧化产物在H2S作用下又可以被还原生成10.其它还原剂, 如谷胱甘肽(GSH)、二硫苏糖醇(DTT)等, 对该还原反应的诱导作用十分有限.最后, 作者利用探针10实现了小鼠巨噬细胞(RAW264.7)中HOCl和H2S的可逆成像, 为研究该氧化还原对在细胞中的生理作用提供了潜在的监测手段.
|
|
(4) |
2014年, Peng课题组[22]报道了一例可以对肿瘤细胞中的基础HOCl水平进行成像的超灵敏荧光探针11.如Eq. 5所示, 该探针分子内存在一个吡咯环和氟硼二吡咯荧光团之间的PET过程, 导致该化合物没有荧光.而作者认为相较于其它常见的一些可以产生PET过程的电子给体(如N原子), 吡咯环的电荷密度更高, 因此其引起的PET更有效, 这大大提高了探针11对HOCl的灵敏度, 其对HOCl的检测限可低达0.56 nmol•L-1.作者利用探针11的超灵敏性, 检测出子宫颈癌细胞(HeLa细胞)和MCF-7细胞中的基础HOCl浓度分别为9.45和8.23 nmol•L-1.
|
|
(5) |
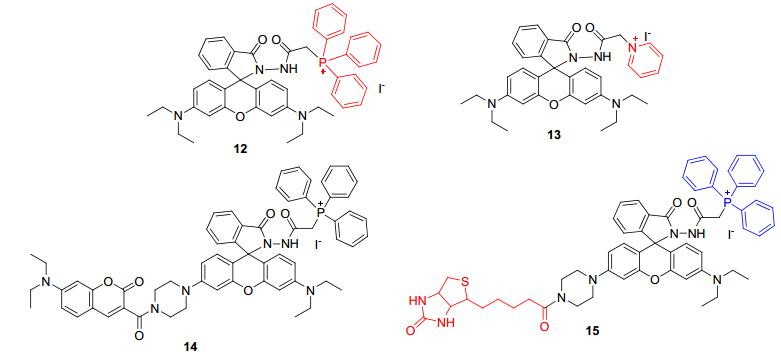
Yu课题组在Ma课题组[18]工作基础上, 对探针5结构进行改造, 将二苯甲酰肼结构中的苯环换成吡啶盐和三苯基膦盐, 得到探针12和13(图 1)[23].这两个基团的引入大大改善了探针的水溶性以及生物相容性, 同时可以使得探针具备检测细胞线粒体中ClO-的能力.实验表明这两个探针都能够对ClO-的高选择性、高灵敏性识别, 并且可以对细胞线粒体和小鼠中的外源性ClO-进行成像.随后, 该课题组[24]又设计报道了基于同一反应机理的探针14和15.探针14能够通过荧光共振能量转移(FRET)机理对ClO-实现比率识别, 被成功用于细胞线粒体中ClO-的定量检测.探针15具有肿瘤细胞靶向的生物素结构, 被证明可以实现肿瘤细胞和线粒体双定位[25].
最近, Yang课题组[26]报道了一例高选择性ClO−荧光探针16.在对二苯酚结构上引入两个氯原子, 可以提高探针对ClO-的选择性.通过ESI-MS表征发现, 探针16在ClO-作用下, 得到强荧光性的荧光素或卤代荧光素产物(Eq. 6).利用该探针的高灵敏性, 作者对噬菌体中的内源性ClO-进行了荧光成像, 并成功地对不同生长时期的斑马鱼体内的ClO-进行了造影.
作为一种内源性的强氧化物质, ONOO−在1990年才首次被发现.人们发现, ONOO−可以作为生物分子的硝化试剂, 如酪氨酸残基等. NO和O2·-形成ONOO−的速率大约为1010 L•mol-1•s-1, 形成方式为扩散控制.免疫细胞中的ONOO-正常浓度一般是纳摩尔级别; 当浓度不正常时, 可以高达100 μmol•L-1, 这样就会引起免疫紊乱等相关疾病.但是, 想要确切了解ONOO-在病理中扮演的角色并不简单, 因为这种活性物质的生理半衰期非常短(<10 ms).因此, 设计开发快速灵敏检测ONOO-的荧光探针具有十分重要的临床意义.
|
|
(6) |
利用苯甲醚衍生的高活性酮可以在ONOO-氧化下生成二烯酮双环氧乙烷结构这一特点, 2006年, Yang课题组[27]报道了第一例高选择性的ONOO-荧光探针17 (Eq. 7).在15 equiv.的ONOO-存在下, 探针17的荧光可以有7~8倍的增强.作者在与探针17共培养的神经元细胞中加入5-氨基-3-(4-吗啉基)-1, 2, 3-噁二唑鎓盐酸盐(SIN-1, 一种ONOO-释放剂), 发现有明显的荧光增强.
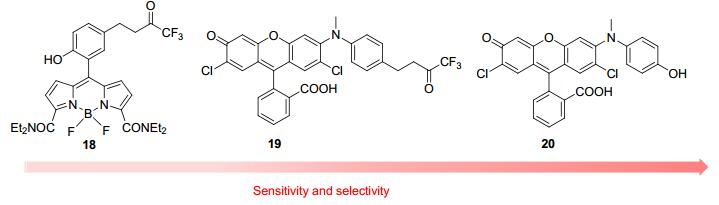
探针17的不足之处在于它和ONOO-的反应转化率太低, 导致灵敏度不够, 反应时间也较长(15 min).作者[28]猜测可能是芳基醚键断裂较为困难, 所以, 该课题组对反应位点进行改良, 设计了探针18 (图 2).化合物18的苯环部分和氟硼二吡咯部分存在PET过程, 所以无荧光; 和ONOO-反应后, PET被抑制, 18荧光得到恢复.其荧光强度在7 equiv. ONOO-存在下增强69倍, 而且反应速度可以减少至2 s, 但是•OH依然可以产生很大干扰.进一步的, 该课题组[29]将苯甲醚部分中的C—O键变换成C—N键, 得到探针19.相对于17和18, 19对ONOO-的选择性更高, 灵敏度也得到提升.最近, 该课题组[30]还报道了化合物20.和化合物18和19相比, 该探针的反应位点变为氨基苯酚, 其选择性和灵敏度都大大增加, 检测限可以达到10 nmol•L-1.作者利用探针20可以监测活细胞中内源性和外源性的ONOO-, 并且还实现了对小鼠心脏平滑肌中内源性ONOO-的双光子荧光成像.
ONOO−是很强的硝化试剂, 而一般情况下, 硝基的引入会淬灭荧光, 所以ONOO-荧光探针的设计就要避免荧光团被硝化.而Nagano课题组[31]却反其道而行之, 在2006年报道了一例利用ONOO-硝化能力的高选择性ONOO-探针21.如Eq. 8所示, 探针21的苯环部分由于电荷密度很高, 可以和氟硼二吡咯荧光团之间发生a-PET过程, 导致其荧光非常弱(量子产率Φ≈0);在CH3CN溶液中, ONOO-可以对21的苯环结构进行硝化, 使得其电荷密度大大降低, 从而抑制了a-PET过程并且d-PET过程也不会发生, 因此21的荧光得到大幅增强(Φ=0.687).但是由于21的水溶性很差, 反应性也不够高, 因此不能在水溶液中检测ONOO-.于是, 作者就将苯环结构中的一个醚键替换成了活性更高、水溶性更好的羟基, 得到化合物22和23.这两个化合物在水溶液中也可以对ONOO-产生很明显的荧光信号增强, 而其它活性氧与该探针基本不反应.
作为很多酶的活性位点的重要组成部分, Se是一种很重要的抗氧化剂并参与了生物体中的氧化还原循环. 2011年, Han课题组[32]首次将Se引入有机小分子荧光探针24中, 实现了对ONOO-的选择性识别.探针24以花青素作为近红外荧光团骨架, 4-苯基硒基苯胺作为反应位点, 通过PET过程淬灭24的荧光.在ONOO-将硒氧化后, 花青素荧光得到恢复.该反应对ONOO-的选择性比较好, 反应时间需要大约10 min.而氧化产物25在生物硫醇(如GSH和Cys)还原下可转化为24, 恢复率可以达到97%, 循环次数在5次以上(Eq. 9).作者将这种循环应用到了RAW264.7细胞中的氧化还原监测, 并利用探针24监测了RAW264.7细胞在脂多糖(LPS)、抗干扰素-γ (IFN-γ)和佛波酯(PMA)刺激下产生的ONOO-.而几乎是同一时间, Tang课题组[33]也报道了一例氧化还原可逆的有机硒近红外荧光探针26 (Eq. 10).只是探针25在被ONOO-氧化后发生的是荧光淬灭现象, 而且还原剂是还原性抗坏血酸(ASCH2).作者将这种循环应用到了RAW264.7细胞中的氧化还原监测, 并且发现26可以靶向进入细胞的线粒体中.
|
|
(7) |
|
|
(8) |
|
|
(9) |
在前面工作基础上, Han课题组[34]将2-苯基碲基-苯甲酰肼引入到花青素中, 构建了新型的ONOO-/GSH氧化还原荧光探针27 (Eq. 11).作者证明探针27具有线粒体靶向性, 并且可以对RAW264.7细胞中的内源性ONOO−进行监测.最后, 作者将探针27应用到了细胞和小鼠中的氧化还原平衡的荧光成像.
|
|
(10) |
|
|
(11) |
Nagano课题组[35]曾经报道过花青素Cy7的双键可以被ROS氧化断裂.但是该反应首先是缺乏选择性, 其次就是这是一个荧光淬灭过程, 不适合用于荧光成像.于是, Yu课题组[36]对这个反应加以利用, 设计合成了基于香豆素吡啶盐共轭结构的探针28 (Eq. 12).该探针在水溶液中荧光很弱, 与ONOO-作用后, 在490 nm处出现一个强的荧光发射峰.通过ESI-MS和1H NMR等表征手段, 作者证明了香豆素醛产物的生成.其它活性氧都不能引起此种荧光变化.最后, 该探针被用于RAW264.7细胞中内源性ONOO-的荧光成像.
|
|
(12) |
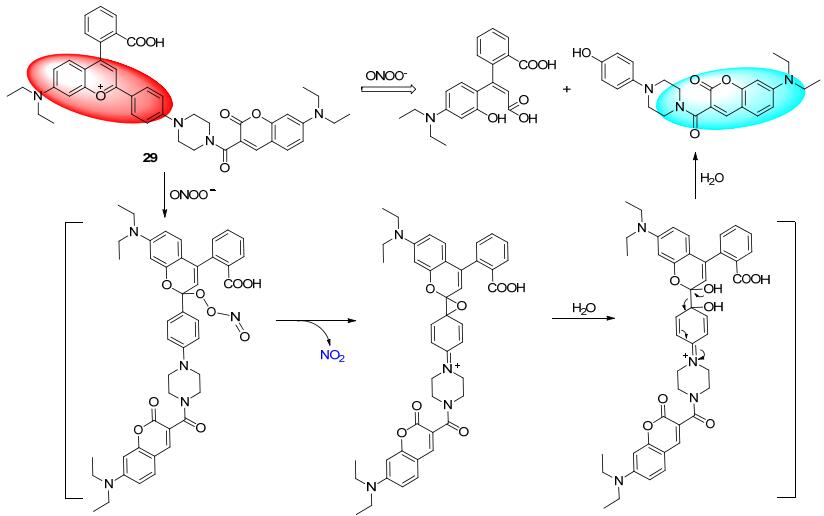
最近, Yuan课题组[37]报道了一例基于荧光共振能量转移(FRET)机制的ONOO-比率荧光探针29.作者提供了一种新的比率探针设计思路.对近20种荧光团与各种ROS的反应活性进行考察, 筛选出分别对ONOO-能够选择性作用以及表现惰性的两种荧光团, 然后通过桥联基组合为探针.探针29由于FRET机制, 发射红色荧光; 与ONOO-作用后, 经历氧化水解过程, 长波荧光团结构被破坏, 短波荧光团香豆素得到释放, 体系荧光表现为蓝绿色.该探针设计思路新颖, 对ONOO-的选择性和灵敏度都很高, 最终被成功地用于小鼠炎症模型中ONOO-的过度表达成像.
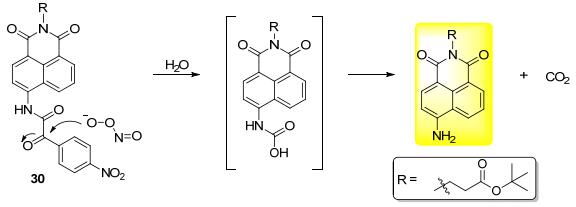
利用α-羰基酰胺对ONOO-的选择性反应, Tang课题组[38]报道了一例双光子荧光探针30 (Scheme 5).由于PET机制, 探针荧光非常弱; 在ONOO-作用下经过氧化水解, 得到4-氨基萘二酰亚胺荧光团.在这篇文章中, 作者对两种常用药物乙酰氨基酚和托卡朋引起的肝损伤进行了考察, 发现服用药物后的小鼠肝组织切片中有明显的荧光增强, 说明该探针可以对肝损伤引起的氧化应激产生灵敏的荧光响应, 因此探针具有检查药物引起的肝损伤的临床应用潜力.
细胞中的H2O2是由O2·-通过超氧岐化酶催化或者自发歧化形成的, 转化速率分别为≈105和≈109 L•mol-1• s-1.而体内的抗氧化性酶, 如过氧化物酶(Prx)和谷胱甘肽过氧化物酶(GPx), 则将体内的H2O2浓度保持在比较稳定的范围(10-9~10-6 mol•L-1).和其它ROS相比, H2O2是一种较温和的氧化剂, 其半衰期比较短(≈1 ms).
2004年, Chang课题组[39]将频哪醇硼酸酯引入荧光素, 设计了H2O2荧光探针31 (Eq. 13).在与H2O2作用后, 探针31脱去硼酸酯, 水解得到产物荧光素, 荧光增强.作者较为详细地研究了其它活性氧, 如O2·-, tBuOO•, ClO-, •OH和过氧化叔丁醇(TBHP)对该探针的影响.研究发现, tBuOO•和•OH都能引起31的荧光增强, 但都比H2O2引起的增强(>500倍)小.作者认为31对氧化性并不是最强的H2O2的选择性较好, 可能是因为这个反应只是一个脱保护的过程, 而不是氧化过程.作者将该探针用于活的人源胚胎肾细胞(HEK293T细胞)中外源性H2O2的荧光成像, 得到了比较好的结果.这是第一例用于细胞中H2O2荧光成像的探针.
|
|
(13) |
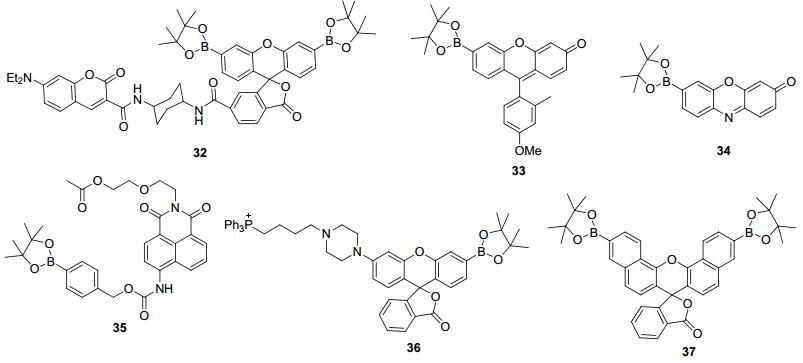
随后, 在2006~2008年, Chang课题组[40~44]又相继开发了一系列基于硼酸酯脱保护的H2O2荧光探针32~37(图 3).其中, 为了能够实现对H2O2的定量检测, 作者[40]首先合成了基于FRET机制的比率荧光探针32.利用探针32, 作者研究了在各种药物刺激下酵母线粒体中内源性H2O2浓度的变化.但是, 不管前面报道的探针31, 还是32, 它们的绝对荧光强度都比较低, 因此在生物样品中的成像应用会受到很大限制.于是, 该课题组合成了单一硼酸酯保护的探针33和34[41].与前面双保护的探针相比, 33和34自身的荧光要更强一点, 同时, 在与H2O2作用后, 其绝对荧光强度也是明显的增强, 但是, 在荧光增强倍数上, 要比双保护的探针弱.利用探针33, 作者首先考察了人皮肤鳞癌细胞(A431细胞, 过度表达生长因子受体)在各种生长因子诱导下的H2O2生成; 然后作者研究了A431细胞和产后老鼠神经元细胞产生H2O2的分子途径, 发现这两种细胞产生H2O2的途径是类似的, 都与表皮生长因子和Nox酶相关, 并证明了在细胞信号转达的过程中会有H2O2产生.
探针35是Chang课题组[42]设计合成的第二例H2O2比率荧光探针.探针35本身在475 nm处发射蓝色荧光; 在与H2O2作用后, 其发射波长红移至540 nm, 产生的比率(F540/F475)变化有12倍.作者利用35的比率特性研究了在PMA刺激下的RAW264.7细胞中的H2O2变化.发现在受PMA诱导后, 吞噬小泡处的荧光变化要比细胞其它部位明显, 说明吞噬过程会产生H2O2.和前面的探针相比, 36中额外引入了具有线粒体靶向性的三苯基膦盐.作者验证了36可以在多种细胞的线粒体中富集并且对线粒体中H2O2浓度的变化产生荧光信号响应[43].百草枯作为一种能够引起细胞氧化应力的小分子, 可以诱导产生类帕金森的表现型, 因此作者利用探针36研究了HeLa细胞在百草枯刺激下的氧化应力, 发现百草枯可以诱导细胞线粒体产生H2O2, 说明H2O2可能与帕金森疾病的产生相关.在实现了细胞中H2O2的比率检测、靶向性成像后, Chang课题组[44]设计合成了探针37以实现对细胞中H2O2的近红外成像.荧光成像实验表明, 37具备很好的细胞穿透能力并且可以对RAW264.7细胞中的外源性H2O2进行成像, 但是由于萘并荧光素的绝对荧光亮度比前面所用的荧光素等荧光团要弱, 所以利用该探针研究细胞中的氧化应激的尝试均告失败.
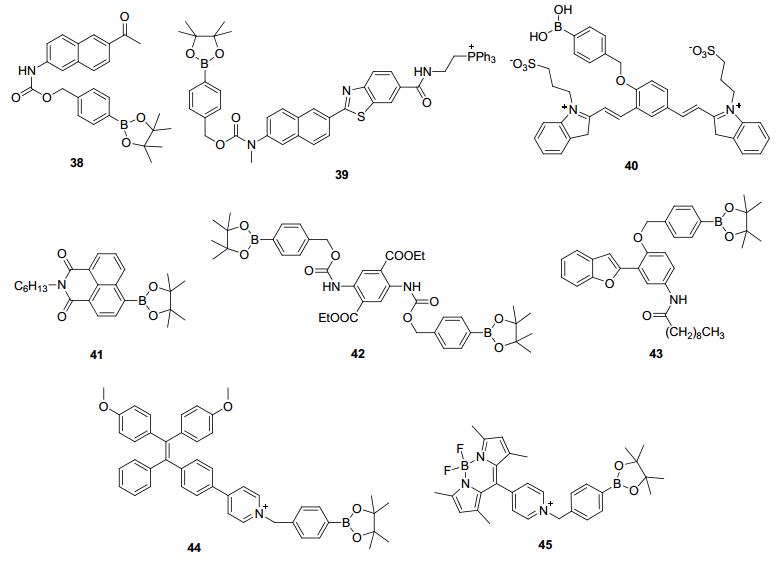
除了Chang课题组, 其他课题组也以各种荧光团为骨架设计合成了基于硼酸或硼酸酯的H2O2荧光探针(图 4). Cho等课题组[45, 46]报道了H2O2的双光子荧光探针38和39, 并利用它们研究了大鼠海马脑片中外源性H2O2的分布. Shabat和Satchi-Fainaro课题组[47]则在他们改良的花青素骨架上设计合成了H2O2近红外荧光探针40, 并成功地对小鼠中外源性和LPS刺激下的内源性H2O2进行了荧光成像. Zang课题组[48, 49]另辟蹊径, 设计了探针41和42并将它们制备成材料, 从而对H2O2蒸汽进行了快速灵敏的荧光显影. Jiang和Xue课题组[50]制备了基于激发态分子内质子转移(ESIPT)机制的探针43, 对H2O2实现了比率识别. Zhang课题组[51]则以四苯基乙烯为骨架设计了基于聚合诱导发光(AIE)机制的探针44, 并通过对H2O2的荧光响应实现了在葡萄糖氧化酶存在下对葡萄糖的选择性识别. Shao课题组[52]则设计了水溶性的荧光探针45, 并成功地将其应用于天使鱼在外源性H2O2刺激下的活体成像.
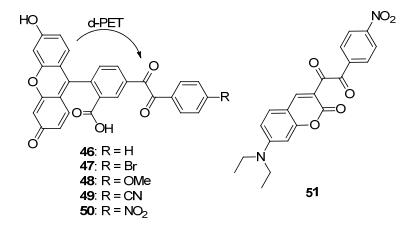
Sawaki等[53]曾经报道过二苯乙二酮可以在H2O2作用下发生Baeyer-Villiger反应生成苯甲酸酐, 进一步水解即可得到苯甲酸(Scheme 6). 2011年, Nagano课题组[54]利用该结构对H2O2的特异性响应设计合成了化合物46~50(图 5).由于荧光素的激发态和二苯乙二酮会发生d-PET过程, 所以这几个化合物的荧光都很弱, 尤其是探针50的量子产率只有0.004.研究发现, 取代基R吸电性越强, 探针与H2O2反应效果越好.因此, 作者引入吸电性更强的氰基(49)和硝基(50)以改善探针的性质.探针49和50的荧光在H2O2作用下可以增强到150倍, 但是反应速率还是只能和硼酸类的探针相当.由于探针50的信噪比更高, 因此作者利用50研究了RAW264.7和A431细胞在各种药物诱导下的H2O2生成. 2015年, Liu课题组[55]也利用该反应设计了探针51, 并利用其对肿瘤细胞中的基础H2O2水平进行了单光子或双光子荧光成像.
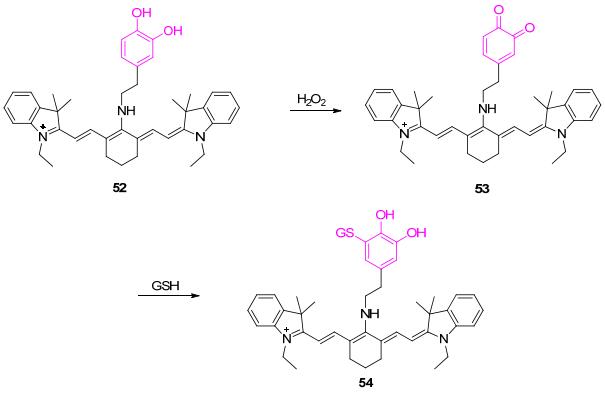
2012年, Han课题组[56]设计报道了一例基于多巴胺氧化的近红外荧光探针52 (Scheme 7).该化合物在755 nm处具有较强荧光; 在H2O2作用下, 多巴胺的邻苯二酚结构被氧化成醌式, 得到氧化产物53, 并发生分子内d-PET过程, 造成荧光淬灭; 该过程同时伴有蓝色至紫色的比色变化.其它ROS对该反应无影响.有意思的是, 化合物53在GSH和Cys的作用下, 可以得到加成产物54, 消除PET过程, 促进体系荧光恢复, 恢复程度可达95%.需要注意的是, 该系列氧化-还原反应并不是可逆进行的.最后, 作者利用探针52对几种细胞中的氧化还原变化进行了监测, 并实现了小鼠海马体组织中的H2O2氧化及硫醇修复过程的成像.
2014年, Yu课题组[57]报道了含硒的化合物55.在二甲亚砜(DMSO)/HEPES (V:V=1:99)的溶液中, 该化合物由于分子内存在PET过程而几乎没有荧光; 在H2O2的氧化下, PET被抑制, 55的荧光得到110倍的增强.氧化产物56的结构通过NMR和HRMS得到确认.其它活性氧, 如1O2, •OH, O2·-, ClO−, ONOO−, tBuOO•以及TBHP, 都不能引起55的荧光发生变化.有意思的是, 不同于探针55, 氧化产物56具有较强的固体荧光(Φ=0.114) (Eq. 14).作者猜测可能是由于探针中十二烷基链的引入阻止了分子间的作用, 避免了聚集诱导的荧光淬灭.
2016年, Lin课题组[58]利用H2O2对氧鎓的亲核性, 设计合成了探针57.该探针具有近红外荧光, 在H2O2的氧化作用下, 水解得到发射蓝绿色荧光的香豆素羧酸产物, 从而实现了H2O2的比率荧光识别(Eq. 15).利用探针57的高选择性和灵敏性, 作者对斑马鱼中的H2O2进行了比率荧光成像.
|
|
(14) |
|
|
(15) |
近期, Stains课题组[59]对罗丹明结构进行改造, 将荧光团中的氧原子换成硼酸, 得到探针58.该探针可以对H2O2进行高选择性和高灵敏性的识别, 并且在与H2O2作用前后产生比率荧光响应(Eq. 16).细胞实验表明探针58具有良好的细胞膜渗透能力, 并且可以对细胞中内源性H2O2的浓度变化进行灵敏的荧光成像.在此基础上, 作者还合成了硅取代探针59.与H2O2作用后, 该探针表现出更大的荧光发射波长变化, 同样也具备良好的细胞内H2O2成像应用潜力.
|
|
(16) |
O2·-在细胞的信号传达中具有非常重要的意义.细胞中的分子氧大概有0.15%~2%会通过ETC被转化成O2·-.虽然细胞产生O2·-的速率比较快, 但由于其在各种情况下可被转化成其它ROS, 所以细胞中O2·-稳态浓度是比较低的.和其它ROS相比, O2·-的活性相对来说是比较低的, 所以, 用于检测其它ROS的一些反应可能就不适用于检测O2·-.因此, 虽然被发现较早, 但相对于其它ROS, 针对O2·-的荧光识别研究进展缓慢.
2004年, Tang课题组[60]报道了苯并噻唑啉衍生物60 (Eq. 17).该化合物本身无荧光, 但是在碱性的NaS2O4(可产生O2·-)溶液中, 可以被氧化成苯并噻唑衍生物并发射绿色荧光.该探针对O2·-的选择性较好, •OH以及H2O2都不会产生干扰.通过流动注射荧光法, 作者测定了洋葱中SOD酶的活性.在这个工作基础上, 该课题组[61]又报道了化合物61.探针61对O2·-的检测限可以低达1.68 nmol•L-1.利用61, 作者实现了对RAW264.7细胞中O2·-浓度变化的成像.最近, 该课题组[62]又利用O2·-对苯并噻唑啉的氧化作用设计合成了具有线粒体靶向功能的荧光探针62.作者选用二苯并噻唑啉基富勒烯作为荧光团.该化合物在O2·-作用下可以在512 nm处发射荧光, 而且选择性保持优秀.作者发现化合物62具有良好的双光子荧光性能, 并利用该特性实现了对小鼠在LPS刺激下免疫响应产生的O2·-的成像.
|
|
(17) |
2005年, Maeda课题组[63]报道了一个专一性较高的O2·-荧光探针63 (Eq. 18).探针63与O2·-的作用并非基于常见的氧化还原机制, 而是利用了O2·-的亲核性.在黄嘌呤氧化酶/黄嘌呤体系中或者KO2作用下, 探针63的2, 4-二硝基苯磺酰基会被移除, 从而得到荧光大大增强的荧光素类似物产物.该反应可以在10 min内完成.作者利用63研究了PMA诱导的人嗜中性细胞中O2·-的产生, 可以得到比较好的结果.在这个工作的基础上, 该课题组对化合物63的结构进行了优化.对苯磺酰苯环上的取代基进行各种替换, 作者[64]最终得到了对O2·-性能最优的探针64.在pH 7.4的水溶液中, 探针64对O2·-的检测限是探针63的10倍.其它方面, 如选择性、生物应用性, 探针64都要比63更加的优秀.
|
|
(18) |
基于邻二苯酚可以被选择性O2·-氧化成邻二苯醌, Tang课题组[65]以三聚氰胺为基本骨架设计合成了探针65.化合物65的一个特点就是既可以在单光子激发下发光, 也可以在双光子激发下发光, 激发波长分别是491和800 nm.这是第一例O2·-的双光子荧光探针.探针与O2·-的反应产物在GSH的还原作用下可以变回探针65, 因此该探针又可以被用来对细胞中O2·-引起的氧化还原应激进行成像(Eq. 19).作者利用该探针对多种药物刺激下的肝细胞中O2·-水平变化进行了成像检测, 并实现了药物刺激下斑马鱼和小鼠中O2·-浓度变化的体内成像.
|
|
(19) |
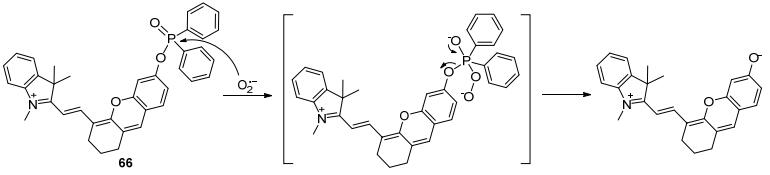
Zhang课题组[66]在以半碳菁染料为骨架设计合成了近红外荧光探针66.该探针以二苯基亚磷酸酯为反应位点, 可以高选择性地对O2·-产生“turn-on”的荧光响应.作者认为, 二苯基亚磷酸酯与O2·-的作用是亲核取代-消除, 并非通过氧化机理.探针66被证明具有良好的对细胞内源性O2·-进行荧光成像的能力, 并被用于斑马鱼和小鼠中O2·-的活体成像(Scheme 8).
近期, Tang课题组[67]以萘二酰亚胺为荧光团骨架设计合成了内质网靶向的O2·-荧光探针67.该探针以苯并噻唑林为反应位点, 以对甲基苯磺酰胺结构为内质网靶向基, 以PET为荧光变化机理, 同时具备单光子与双光子成像功能.利用67的高灵敏性, 作者尝试了一系列生物荧光分析, 发现和正常小鼠相比, 糖尿病型的小鼠腹部和肝组织中的O2·-水平更高, 并表明这与内质网紊乱相关.而利用同一反应类型, Zhang和Liu课题组[68]报道了喹啉-苯并噻唑林荧光探针68.该探针与O2·-作用后的产物具有双光子荧光特性, 激发波长为820 nm.作者利用该探针检测了LPS诱导的肺部炎症模型中O2·-的过度生成.
|
|
(20) |
•OH参与了体内生物大分子如DNA和蛋白质的降解过程.因此, 在癌症治疗的放射疗法中•OH扮演了重要角色.因为其在人类健康与疾病中的重要意义, 对•OH检测也得到了很多关注.但是, 大部分实验方法都面临着灵敏度低、难定量等困难, 如电子自旋共振光谱法(ESR).而关于•OH的荧光检测法在过去约20年间虽然也得到了一定的发展, 但离真正的临床应用依然还有很长一段距离.检测方法的发展比较缓慢和•OH的半衰期极短(≈1 ns)不无关系; 另一个检测难题就是•OH在体内的浓度比较低, 这对检测方法的灵敏性要求很高.
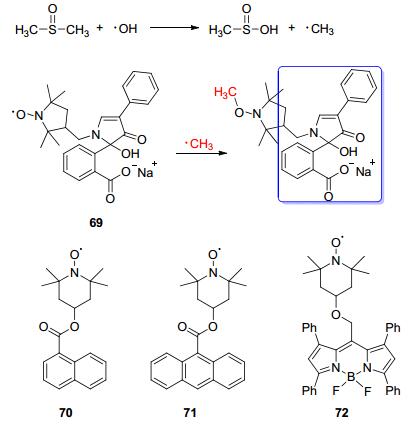
1993年, Pou课题组和合作者[69]报道了一例含氮氧结构的探针69.该探针在DMSO存在下可以对•OH产生荧光响应.作用机理大致如下:首先是•OH和DMSO作用产生甲基自由基•CH3; 然后•CH3与探针中的氮氧作用, 抑制了氮氧自由基对探针分子的荧光淬灭作用, 从而使得探针荧光增强(Scheme 9).但是该探针同样对O2·-也会有荧光增强响应.探针69在•OH作用下发生荧光增强的同时会出现ESR信号的减弱, 这也说明了荧光检测法和ESR相比的一个优势.作者随后将PMA处理过的嗜中性细胞与探针69培养, 然后加入硫酸亚铁继续培养, 可以看到细胞中出现明显的荧光增强.在这个探索性工作之后, 其他课题组也相继报道了一些基于该机制的•OH荧光探针(Scheme 9).
Guo课题组[70, 71]在先后报道了以萘和蒽作为荧光团的探针70和71.他们将69中的五元环的氮氧自由基换成了六元环的四甲基哌啶氮氧化物TEMPO.这两个探针本身也都由于氮氧自由基的淬灭作用只显示很微弱的荧光.在•OH作用下, 这两个化合物的荧光就会出现明显增强.但由于这两个探针的发射波长很短, 其生物应用就受到了很大限制.
Tang课题组[72]在2010年报道了以氟硼二吡咯作为荧光骨架的探针72.在pH=7.4的缓冲溶液中, 探针本身荧光非常微弱.但是在DMSO存在下, •OH的作用可以使得其在601 nm处产生明显的荧光增强.动力学实验表明72和•OH的反应可以在5 min内完成.作者将72分别和肝癌细胞与正常肝细胞共同培养, 发现前者中出现的荧光要比后者中的强, 说明了癌细胞中的•OH浓度要比正常细胞中更高.
氮氧自由基对•OH的选择性较高, 反应也较为快速, 但是这两种物质的反应并非直接进行的, 而是需要中间介质的存在, 如DMSO.这种局限性要求研究者开发新的能够直接检测•OH的反应型探针. 2010年, Lin课题组[73]报道了可以直接对•OH进行比率识别的荧光探针73 (Eq. 21).与•OH作用后, 探针73的荧光从495 nm红移至651 nm, 得到半花菁类染料74.其它ROS不会引起73发生类似变化.相比于氮氧自由基, 探针73和•OH的反应进行较慢, 需要1 h以上, 其准一级方程的表观速率常数是5.14×10−4 s−1.利用73, 作者实现了对HeLa细胞在PMA诱导下产生的•OH的比率荧光成像.利用同样的反应位点, 该课题组[74]于近期报道了•OH荧光探针75.该探针与•OH的作用速度非常快, 可以在0.5 min内完成, 但是延长反应时间会有副反应发生, 导致体系荧光淬灭.作者推测这是由体系中过量的氧化物所导致.探针75同样可被用于细胞中内源性•OH的成像.
|
|
(21) |
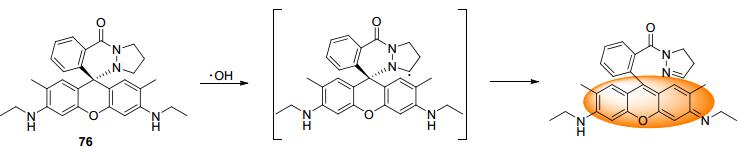
2013年, Tae和Shin课题组[75]报道了基于羟基自由基夺氢作用的荧光探针76.探针76可以在•OH作用下发生开环反应得到在550 nm处发射强烈荧光的产物.该反应可以在2 min内快速完成, 专一性也很高.作者随后将探针76成功应用于A549和RAW264.7细胞在Fenton试剂(FeSO4/H2O2=1/10)作用下•OH浓度升高的荧光成像.随后, 作者还成功地尝试了对两种细胞在PMA诱导下产生•OH的荧光成像, 证明了76具有良好的成像应用前景.
2014年, Yi课题组[76]报道了一例可以对•OH进行比率识别的荧光探针77.在H2O/N, N-二甲基甲酰胺(DMF) (V:V=98:2)溶液中, 探针77在552 nm处可以发射萘二酰亚胺的特征荧光, 并且在426 nm处有很微弱的萘啶特征荧光峰.在•OH作用下, 可以得到在418 nm发射强荧光的产物, 反应可以在2 min内完成.该探针对•OH的选择性比较高, 其它ROS引起的荧光变化相对来说都很微弱.通过对反应产物的核磁和质谱分析, 作者认为探针77在•OH作用下生成了一个羟基化的萘啶衍生物, 该衍生物可以在418 nm处发射荧光; 而萘二酰亚胺部分则变成了无荧光的物质.该探针还具有良好的生物相容性以及低毒性, 可被用于对RAW264.7细胞中外源性和内源性的•OH进行比率荧光成像.
最近, Xu课题组[77]报道了一例噻吩嗪-碳菁共轭化合物78.由于其高密度电子云, S原子可以被•OH氧化成亚砜结构, 因此, 探针78与•OH作用后出现明显的荧光增强响应.而探针自带的正电荷使得化合物主要富集在细胞线粒体中.探针78可以被用于对细胞中内源性的•OH进行荧光成像, 也可以被用于多种细菌受到药物污染后•OH水平升高的定性检测.作者开创性地利用探针78检测了不同尺寸的TiO2NPs (5 nm和40 nm)在光照条件下对细胞的氧化应激作用, 发现小尺寸的TiO2NPs产生•OH能力更强, 并因此产生的对斑马鱼的毒性也更强.
1O2作为氧气分子电子激发态最低的单线态氧, 被人们所知已有约80年历史.从大气科学到生物医学, 1O2都扮演者非常重要的角色.在细胞中, 1O2是一把双刃剑, 它既可以氧化生物大分子如DNA、蛋白质等引起细胞功能紊乱, 也可以杀死癌细胞治疗疾病.例如, 现在研究比较多的光动力疗法就必须有1O2的参与.在体内, 1O2会不断生成与猝灭, 其半衰期到现在并没有一个明确的共识. 1O2由于是一种激发态形式, 所以在体内极不稳定, 也给人们对细胞中1O2的了解带来了很大的麻烦.
大部分的1O2探针的设计都是基于蒽环和1O2的反应.在1992年, 曾有报道称9, 10-二苯基蒽(DPA)在1O2的作用下可以形成内环氧化合物, 从而实现对1O2的检测[78].但是该检测法是基于紫外吸收的变化, 而不是荧光的变化, 所以其灵敏度较低. 1999年, Nagano课题组[79]对该方法进行了改进.他们将DPA结构引入荧光素的9位, 构建了一系列1O2荧光探针79~81 (Eq. 24).由于DPA和荧光素之间发生PET过程, 导致这三个化合物的荧光都很弱; 当DPA和1O2作用生成内环氧化合物后, PET被抑制, 荧光素荧光恢复.作者对这三个化合物的氧杂蒽环上进行了不同的卤代, 希望通过这种方式稳定荧光素的羟基负离子, 使得探针能够在pH=7左右的生理条件下也能够有很好的荧光变化.这三个化合物的pKa分别是6.6、5.7和5.3, 证明了卤素原子的引入确实使得荧光素的羟基去质子化形式更加稳定.其它物质如O2·-、H2O2以及NO不会和这三个探针作用; 随后, Nagano课题组[80]利用PET机制进一步地优化了上述三个化合物的结构, 设计合成了82.和79相比, 82与1O2的反应速率增加了31倍, 灵敏性增加了53倍.
|
|
(22) |
|
|
(23) |
|
|
(24) |
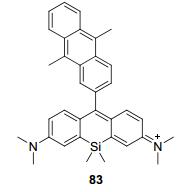
2014年, Majima等[81]利用硅取代的罗丹明制备了近红外的1O2荧光探针83 (图 6).该探针对1O2的选择性非常好, 其它ROS基本不会产生干扰.光照实验表明83在光照下不会自生成1O2.细胞定位实验表明使用低浓度的83和HeLa细胞培养后, 可以发现83具有很好的线粒体靶向性; 但是83浓度过高时, 就会失去靶向能力并开始表现出细胞毒性.最后, 作者利用83监测了细胞在光敏剂作用下产生的1O2, 这对光动力疗法的可视化提供了一条新的思路.
DPA结构和1O2的作用虽然具有比较高的专一性, 但是作用时间很长, 需要1 h以上. 2011年, Tang课题组[82]在花青素结构上引入组氨酸, 制备得到了1O2近红外荧光探针84 (Eq. 25).在Tris-HCl缓冲液(pH=7.4, 20 mmol•L-1)中, 由于组氨酸的咪唑基和花青素结构之间发生PET, 探针84处于无荧光状态; 在H2O2-NaOCl(能够定量产生1O2)作用下, 组氨酸咪唑基被氧化, PET过程被抑制, 于是花青素794 nm处的荧光得到恢复.该反应只需要10 min就可以完成.细胞定位实验表明84具有线粒体靶向性; 而且该探针也可以被用于检测PMA诱导下RAW264.7细胞中的内源性1O2.
|
|
(25) |
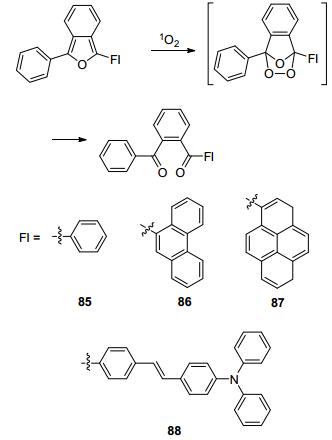
1, 3-二苯基异苯并呋喃(85)在水中可以很快地与1O2反应得到一个二酮产物, 但是该反应过程发生的荧光变化是一个“on-off”过程(Scheme 11)[83]. 2013年, Nam和You课题组[84]利用该反应设计合成了一系列新型的1O2荧光探针.他们为了得到比率型的荧光探针, 在85的结构基础上进行改良, 通过引入各种荧光团, 得到86~88.经过荧光实验, 化合物86~88都能在1O2作用下发生比率型荧光变化, 比率信号变化分别可以达到80倍、352倍和14倍.对比四个探针与1O2的反应速率, 88最快, 85最慢, 作者推测共轭体系的扩大可以使反应速率得到提高.选择性实验表明化合物86~88与1O2的反应具有很强的专一性.由于化合物87具有最佳的膜渗透性, 作者利用该探针实现了对RAW264.7细胞在PMA诱导下产生1O2的荧光成像.
有机小分子荧光探针发展到今天, 已经较为成熟.人们可以根据不同的机理设计不同结构的小分子探针, 但对于细胞或者活体中物质的荧光成像仍然面临着一些困难, 比如探针的毒性、在细胞或器官中的保留能力以及生物相容性等.为了设计具有生物应用前景的荧光探针, 人们要进行不断的尝试, 把化学与生物医学进行有效的结合, 才能得到令人满意的结果.
随着荧光分析仪器的发展, 荧光成像越来越精细, 单细胞和细胞器的成像变得越来越容易, 因此利用荧光探针对微小范围内的底物进行实时成像检测也变得更为容易.为了满足仪器所需, 设计光稳定性强、荧光亮度高的染料也是研究者所需面临的问题.鉴于ROS在生命进程中独特的生理意义, 开发性能越来越优良的ROS荧光探针将会成为长期的热点研究领域.
Suh, Y. A.; Arnold, R. S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A. B.; Griendling, K. K.; Lambeth, J. D. Nature 1999, 401, 79. doi: 10.1038/43459
Koh, C. H.; Whiteman, M.; Li, Q. X.; Halliwell, B.; Jenner, A. M.; Wong, B. S.; Laughton, K. M.; Wenk, M.; Masters, C. L.; Beart, P. M.; Bernard, O.; Cheung, N. S. J. Neurochem. 2006, 98, 1278. doi: 10.1111/jnc.2006.98.issue-4
Rhee, S. G. Science 2006, 312, 1882. doi: 10.1126/science.1130481
Dickinson, B. C.; Srikun, D.; Chang, C. J. Curr. Opin. Chem. Biol. 2010, 14, 50. doi: 10.1016/j.cbpa.2009.10.014
Peteu, S. F.; Boukherroub, R.; Szunerits S. Biosens. Bioelectron. 2014, 58, 359. doi: 10.1016/j.bios.2014.02.025
Yamato, M.; Egashira, T.; Utsumi, H. Free Radical Biol. Med. 2003, 35, 1619. doi: 10.1016/j.freeradbiomed.2003.09.013
de Silva, A. P.; Gunaratne, H. Q. N.; Gunnlaugsson, T.; Huxley, A. J. M.; McCoy, C. P.; Rademacher, J. T.; Rice, T. E. Chem. Rev. 1997, 97, 1515. doi: 10.1021/cr960386p
Berezin, M. Y.; Achilefu, S. Chem. Rev. 2010, 110, 2641. doi: 10.1021/cr900343z
Que, E. L.; Domaille, D. W.; Chang, C. J. Chem. Rev. 2008, 108, 1517. doi: 10.1021/cr078203u
Pradhan, T.; Jung, H. S.; Jang, J. H.; Kim, T. W.; Kang, C.; Kim, J. S. Chem. Soc. Rev. 2014, 43, 4684. doi: 10.1039/C3CS60477B
Chen, X.; Tian, X.; Shin, I.; Yoon, J. Chem. Soc. Rev. 2011, 40, 4783. doi: 10.1039/c1cs15037e
Chen, X.; Wang, F.; Hyun, J. Y.; Wei, T.; Qiang, J.; Ren, X.; Shin, I.; Yoon, J. Chem. Soc. Rev. 2016, 45, 2976. doi: 10.1039/C6CS00192K
Nagano, T. J. Clin. Biochem. Nutr. 2009, 45, 111. doi: 10.3164/jcbn.R09-66
Aratani, Y.; Koyama, H.; Nyui, S.-I.; Suzuki, K.; Kura, F.; Maeda, N. Infect. Immun. 1999, 67, 1828. http://www.ncbi.nlm.nih.gov/pubmed/10085024
Kenmoku, S.; Urano, Y.; Kojima, H.; Nagano, T. J. Am. Chem. Soc. 2007, 129, 7313. doi: 10.1021/ja068740g
Sun, Z.-N.; Liu, F.-Q.; Chen, Y.; Tam, P. K. H.; Yang, D. Org. Lett. 2008, 10, 2171. doi: 10.1021/ol800507m
Hu, J. J.; Wong N.-K.; Gu, Q.; Bai, X.; Ye, S.; Yang, D. Org. Lett. 2014, 16, 3544. doi: 10.1021/ol501496n
Chen, X.; Wang, X.; Wang, S.; Shi, W.; Wang, K.; Ma, H. Chem. Eur. J. 2008, 14, 4719. doi: 10.1002/(ISSN)1521-3765
Lin, W.; Long, L.; Chen, B.; Tan, W. Chem.-Eur. J. 2009, 15, 2305. doi: 10.1002/chem.v15:10
Yuan, L.; Lin, W.; Song, J.; Yang, Y. Chem. Commun. 2011, 47, 12691. doi: 10.1039/c1cc15762k
Wang, B.; Li, P.; Yu, F.; Song, P.; Sun, X.; Yang, S.; Lou, Z.; Han, K. Chem. Commun. 2013, 49, 1014. doi: 10.1039/C2CC37803E
Zhu, H.; Fan, J.; Wang, J.; Mu, H.; Peng, X. J. Am. Chem. Soc. 2014, 136, 12820. doi: 10.1021/ja505988g
Hou, J.-T.; Wu, M.-Y.; Li, K.; Yang, J.; Yu, K.-K.; Xie, Y.-M.; Yu, X.-Q. Chem. Commun. 2014, 50, 8640. doi: 10.1039/C4CC02673J
Hou, J.-T.; Li, K.; Yang, J.; Yu, K.-K.; Liao, Y.-X.; Ran, Y.-Z.; Liu, Y.-H.; Zhou, X.-D.; Yu, X.-Q. Chem. Commun. 2015, 51, 6781. doi: 10.1039/C5CC01217A
Li, K.; Hou, J.-T.; Yang, J.; Yu, X.-Q. Chem. Commun. 2017, 53, 5539. doi: 10.1039/C7CC01679D
Hu, J. J.; Wong, N.-K.; Lu, M.-Y.; Chen, X.; Ye, S.; Zhao, A. Q.; Gao, P.; Kao, R. Y.-T.; Shen, J.; Yang, D. Chem. Sci. 2016, 7, 2094. doi: 10.1039/C5SC03855C
Yang, D.; Wang, H.-L.; Sun, Z.-N.; Chung, N.-W.; She, J.-G. J. Am. Chem. Soc. 2006, 128, 6004. doi: 10.1021/ja0603756
Sun, Z.-N.; Wang, H.-L.; Liu, F.-Q.; Chen, Y.; Tam, P. K. H.; Yang, D. Org. Lett. 2009, 11, 1887. doi: 10.1021/ol900279z
Peng, T.; Yang, D. Org. Lett. 2010, 12, 4932. doi: 10.1021/ol102182j
Peng, T.; Wong, N.-K.; Chen, X.; Chan, Y.-K.; Ho, D. H.-H.; Sun, Z.; Hu, J. J.; Shen, J.; El-Nezami, H.; Yang, D. J. Am. Chem. Soc. 2014, 136, 11728. doi: 10.1021/ja504624q
Ueno, T.; Urano, Y.; Kojima, H.; Nagano, T. J. Am. Chem. Soc. 2006, 128, 10640. doi: 10.1021/ja061972v
Yu, F.; Li, P.; Li, G.; Zhao, G.; Chu, T.; Han, K. J. Am. Chem. Soc. 2011, 133, 11030. doi: 10.1021/ja202582x
Xu, K.; Chen, H.; Tian, J.; Ding, B.; Xie, Y.; Qiang, M.; Tang, B. Chem. Commun. 2011, 47, 9468. doi: 10.1039/c1cc12994e
Yu, F.; Li, P.; Wang, B.; Han, K. J. Am. Chem. Soc. 2013, 135, 7674. doi: 10.1021/ja401360a
Oushiki, D.; Kojima, H.; Terai, T.; Arita, M.; Hanaoka, K.; Urano, Y.; Nagano, T. J. Am. Chem. Soc. 2010, 132, 2795. doi: 10.1021/ja910090v
Hou, J.-T.; Yang, J.; Li, K.; Liao, Y.-X.; Yu, K.-K.; Xie, Y.-M.; Yu, X.-Q. Chem. Commun. 2014, 50, 9947. doi: 10.1039/C4CC04192E
Cheng, D.; Pan, Y.; Wang, L.; Zeng, Z.; Yuan, L.; Zhang, X.; Chang, Y. T. J. Am. Chem. Soc. 2017, 139, 285. doi: 10.1021/jacs.6b10508
Li, Y.; Xie, X.; Yang, X.; Li, M.; Jiao, X.; Sun, Y.; Wang, X.; Tang, B. Chem. Sci. 2017, 8, 4006. doi: 10.1039/C7SC00303J
Chang, M. C. Y.; Pralle, A.; Isacoff, E. Y.; Chang, C. J. J. Am. Chem. Soc. 2004, 126, 15392. doi: 10.1021/ja0441716
Albers, A. E.; Okreglak, V. S.; Chang, C. J. J. Am. Chem. Soc. 2006, 128, 9640. doi: 10.1021/ja063308k
Miller, E. W.; Tulyathan, O.; Isacoff, E. Y.; Chang, C. J. Nat. Chem. Biol. 2007, 3, 263. doi: 10.1038/nchembio871
Srikun, D.; Miller, E. W.; Domaille, D. W.; Chang, C. J. J. Am. Chem. Soc. 2008, 130, 4596. doi: 10.1021/ja711480f
Dickinson, B. C.; Chang, C. J. J. Am. Chem. Soc. 2008, 130, 9638. doi: 10.1021/ja802355u
Albers, A. E.; Dickinson, B. C.; Miller, E. W.; Chang, C. J. Bioorg. Med. Chem. Lett. 2008, 18, 5948. doi: 10.1016/j.bmcl.2008.08.035
Chung, C.; Srikun, D.; Lim, C. S.; Chang, C. J.; Cho, B. R. Chem. Commun. 2011, 47, 9618. doi: 10.1039/c1cc13583j
Masanta, G.; Heo, C. H.; Lim, C. S.; Bae, S. K.; Cho, B. R.; Kim, H. M. Chem. Commun. 2012, 48, 3518. doi: 10.1039/c2cc00034b
Karton-Lifshin, N.; Segal, E.; Omer, L.; Portnoy, M.; Satchi-Fainaro, R.; Shabat, D. J. Am. Chem. Soc. 2011, 133, 10960. doi: 10.1021/ja203145v
Xu, M.; Han, J.-M.; Zhang, Y.; Yang, X.; Zang, L. Chem. Commun. 2013, 49, 11779. doi: 10.1039/c3cc47631f
Xu, M.; Han, J.-M.; Wang, C.; Yang, X.; Pei, J.; Zang, L. ACS Appl. Mater. Interfaces 2014, 6, 8708. doi: 10.1021/am501502v
Li, G.; Zhu, D.; Liu, Q.; Xue, L.; Jiang, H. Org. Lett. 2013, 15, 924. doi: 10.1021/ol4000845
Hu, F.; Huang, Y.; Zhang, G.; Zhao, R.; Zhang, D. Tetrahedron Lett. 2014, 55, 1471. doi: 10.1016/j.tetlet.2014.01.056
Xu, J.; Li, Q.; Yue, Y.; Guo, Y.; Shao, S. Biosens. Bioelectron. 2014, 56, 58. doi: 10.1016/j.bios.2013.12.065
Sawaki, Y.; Foote, C. S. J. Am. Chem. Soc. 1979, 101, 6292. doi: 10.1021/ja00515a023
Abo, M.; Urano, Y.; Hanaoka, K.; Terai, T.; Komatsu, T.; Nagano, T. J. Am. Chem. Soc. 2011, 133, 10629. doi: 10.1021/ja203521e
Zhang, K.-M.; Dou, W.; Li, P.-X.; Shen, R.; Ru, J.-X.; Liu, W.; Cui, Y.-M.; Chen, C.-Y.; Liu, W.-S.; Bai, D.-C. Biosens. Bioelectron. 2015, 64, 542. doi: 10.1016/j.bios.2014.09.073
Yu, F.; Li, P.; Song, P.; Wang, B.; Zhao, J.; Han, K. Chem. Commun. 2012, 48, 4980. doi: 10.1039/c2cc30985h
Liao, Y.-X.; Li, K.; Wu, M.-Y.; Wu, T.; Yu, X.-Q. Org. Biomol. Chem. 2014, 12, 3004. doi: 10.1039/c4ob00206g
Dong, B.; Song, X.; Kong, X.; Wang, C.; Tang, Y.; Liu, Y.; Lin, W. Adv. Mater. 2016, 28, 8755. doi: 10.1002/adma.201602939
Zhou, X.; Lesiak, L.; Lai, R.; Beck, J. R.; Zhao, J.; Elowsky, C. G.; Li, H.; Stains, C. I. Angew. Chem., Int. Ed. 2017, 56, 4197. doi: 10.1002/anie.201612628
Tang, B.; Zhang, L.; Zhang, L. L. Anal. Biochem. 2004, 326, 176. doi: 10.1016/j.ab.2003.11.023
Gao, J. J.; Xu, K. H.; Tang, B.; Yin, L. L.; Yang, G. W.; An, L. G. FEBS J. 2007, 274, 1725. doi: 10.1111/j.1742-4658.2007.05720.x
Li, P.; Zhang, W.; Li, K.; Liu, X.; Xiao, H.; Zhang, W.; Tang, B. Anal. Chem. 2013, 85, 9877. doi: 10.1021/ac402409m
Maeda, H.; Yamamoto, K.; Nomura, Y.; Kohno, I.; Hafsi, L.; Ueda, N.; Yoshida, S.; Fukuda, M.; Fukuyasu, Y.; Yamauchi, Y.; Itoh, N. J. Am. Chem. Soc. 2005, 127, 68. doi: 10.1021/ja047018k
Maeda, H.; Yamamoto, K.; Kohno, I.; Hafsi, L.; Itoh, N.; Nakagawa, S.; Kanagawa, N.; Suzuki, K.; Uno, T. Chem.-Eur. J. 2007, 13, 1946. doi: 10.1002/(ISSN)1521-3765
Zhang, W.; Li, P.; Yang, F.; Hu, X.; Sun, C.; Zhang, W.; Chen, D.; Tang, B. J. Am. Chem. Soc. 2013, 135, 14956. doi: 10.1021/ja408524j
Zhang, J. J.; Li, C. W.; Zhang, R.; Zhang, F. Y.; Liu, W.; Liu, X. Y.; Lee, S. M. Y.; Zhang, H. X. Chem. Commun. 2016, 52, 2679. doi: 10.1039/C5CC09976E
Xiao, H. B.; Liu, X.; Wu, C. C.; Wu, Y. H.; Li, P.; Guo, X. M.; Tang, B. Biosens. Bioelectron. 2017, 91, 449. doi: 10.1016/j.bios.2016.12.068
Li, R. Q.; Mao, Z. Q.; Rong, L.; Wu, N.; Lei, Q.; Zhu, J. Y.; Zhuang, L.; Zhang, X. Z.; Liu, Z. H. Biosens. Bioelectron. 2017, 87, 73. doi: 10.1016/j.bios.2016.08.008
Pou, S.; Huang, Y. I.; Bhan, A.; Bhadti, V. S.; Hosmane, R. S.; Wu, S. Y.; Cao, G. L.; Rosen, G. M. Anal. Biochem. 1993, 212, 85. doi: 10.1006/abio.1993.1295
Yang, X.-F.; Guo, X.-Q. Anal. Chim. Acta 2001, 434, 169. doi: 10.1016/S0003-2670(01)00821-2
Yang, X. F.; Guo, X. Q. Analyst 2001, 126, 1800. doi: 10.1039/b103208a
Li, P.; Xie, T.; Duan, X.; Yu, F.; Wang, X.; Tang, B. Chem.-Eur. J. 2010, 16, 1834. doi: 10.1002/chem.v16:6
Yuan, L.; Lin, W.; Song, J. Chem. Commun. 2010, 46, 7930. doi: 10.1039/c0cc02390f
Wang, J. Y.; Liu, Z. R.; Ren, M. G.; Kong, X. Q.; Liu, K. Y.; Deng, B. B.; Lin, W. Y. Sens. Actuators, B 2016, 236, 60. doi: 10.1016/j.snb.2016.04.163
Kim, M.; Ko, S.-K.; Kim, H.; Shin, I.; Tae, J. Chem. Commun. 2013, 49, 7959. doi: 10.1039/c3cc44627a
Meng, L.; Wu, Y.; Yi, T. Chem. Commun. 2014, 50, 4843. doi: 10.1039/C4CC00975D
Liu, F.; Du, J.; Song, D.; Xu, M. Y.; Sun, G. P. Chem. Commun. 2016, 52, 4636. doi: 10.1039/C5CC10658C
Steinbeck, M. J.; Khan, A. U.; Karnovsky, M. J. J. Biol. Chem. 1992, 267, 13425.
Umezawa, N.; Tanaka, K.; Urano, Y.; Kikuchi, K.; Higuchi, T.; Nagano, T. Angew. Chem., Int. Ed. 1999, 38, 2899. doi: 10.1002/(ISSN)1521-3773
Tanaka, K.; Miura, T.; Umezawa, N.; Urano, Y.; Kikuchi, K.; Higuchi, T.; Nagano, T. J. Am. Chem. Soc. 2001, 123, 2530. doi: 10.1021/ja0035708
Kim, S.; Tachikawa, T.; Fujitsuka, M.; Majima, T. J. Am. Chem. Soc. 2014, 136, 11707. doi: 10.1021/ja504279r
Xu, K.; Wang, L.; Qiang, M.; Wang, L.; Li, P.; Tang, B. Chem. Commun. 2011, 47, 7386. doi: 10.1039/c1cc12473k
Aubry, J.-M.; Pierlot, C.; Rigaudy, J.; Schmidt, R. Acc. Chem. Res. 2003, 36, 668. doi: 10.1021/ar010086g
Song, D.; Cho, S.; Han, Y.; You, Y.; Nam, W. Org. Lett. 2013, 15, 3582. doi: 10.1021/ol401421r

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


























 下载:
下载:
















 下载:
下载: