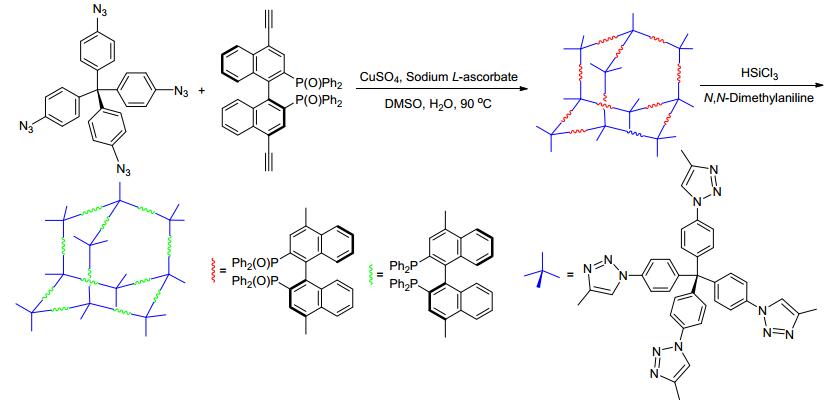
Scheme 1.
Synthesis and the deoxidation of the porous organic polymer, POP-BINAPO
Asymmetric Hydrogenation of β-Keto Esters Catalyzed by Ruthenium Species Supported on Porous Organic Polymer
Shengnan Kong , Abaid Ullah Malik , Xuefeng Qian , Mouhai Shu , Wende Xiao

The achievement of enantiomerically pure chiral compounds has attracted much attention because of its importance in the synthesis of pharmaceuticals and chemicals. [1] Although chiral compounds can be obtained by the separation from their racemic mixtures with excellent yields, this method is regarded as inefficient and expensive due to the production of undesired enantiomer. Inspired by the high enantioselectivity of enzymes in living organisms, asymmetric catalysis has been developed with great progress since the late 1960s. [2~4] Among the asymmetric catalysis, heteroge-neous asymmetric catalysis has attracted more and more attention due to the advantages such as the easiness of the separation of the product and the recycling of the expensive catalysts containing the noble metals and the ligands.
In most of the heterogeneous asymmetric catalysis systems, the catalytic units are normally immobilized onto the supporting materials. So far various materials have been employed as the supporting materials, such as silica, [5, 6] dendrimers, [7, 8] polymers, [9] nanoparticles [10] and metal-organic frameworks (MOFs). [11, 12] Recently, the emergence of porous organic polymers (POPs) provides a new platform for the heterogeneous asymmetric catalysis.
Porous organic polymers (POPs), [13~15] including covalent organic frameworks (COFs), [16~19] porous organic frameworks (POFs), [20] microporous organic networks (MONs) and conjugated microporous polymers (CMPs) [21] are constructed from well-designed organic building blocks by coupling or condensation reactions. POPs exhibit large surface area, low skeleton density, high thermal and chemical stability as well as the poor solubility in water and common organic solvents. The structures of POPs could be flexibly designed and the pore size and surface area could be availably controlled. Therefore POPs have not only been applied in gas storage and separate, [22~24] electronics, [25] light harvesting [26] and emitting, [27] but also been used as the supporting materials for heterogeneous catalysis. [16, 28~31] POPs can be functionalized by grafting the chiral ligands onto the skeletons of the porous structures, and serve as the heterogeneous asymmetric catalyst after the composition with suitable metal ions. Cui and coworkers have reported the heterogeneous asymmetric conjugation addition catalyzed by the metallized chiral robust diene-based porous organic frameworks (POFs), [32] and efficient and recyclable heterogeneous asymmetric addition catalyzed by α, α, α', α'-tetraaryl-1, 3-dioxolane-4, 5-dimethanol (TADDOL) functionalized two-dimensional COFs in the presence of [Ti(OiPr)4]. [33] Wang and coworkers have reported the asymmetric Michael addition of nitro-alkenes to aldehydes in good to excellent yield with high enantioselectivity, [34] and high diastereoselectivity catalyzed by the Jørgensen-Hayashi catalyst embedded into a nanoporous polymer. [35] Furthermore, Jiang and coworkers have reported the metal-free heterogeneous organocatalysis of chiral COFs materials which display excellent activity, enantio-selectivity and recyclability. [36, 37] Among chiral functional groups employed in asymmetric catalysts, so-called "privileged" chiral ligands such as 1-(2-hydroxynaphthalen-1-yl)naph-thalen-2-ol (BINOL), 2, 2'-bis(diphenylphosphino)-1, 1'-bi-naphthalene (BINAP), salen, bisoxazoline and TADDOLate, have been proven to be highly efficient to a wide range of different reactions. [38~40]
The asymmetric hydrogenation of β-keto esters catalyzed by ruthenium complexes with chiral phosphine ligands has attracted much attention in both academia and industry. The original [RuX2(BINAP)] catalysts reported by Noyori and co-workers have particularly been useful for the reduction of β-keto esters. [41] From then BINAP was frequently used to explore new ways of developing heterogeneous asymmetric catalysis. Lin et al. [42] reported novel chiral porous zirconium phosphonates for the highly enantioselective asymmetric hydrogenation of β-keto esters. Fan et al. [43] synthesized the first example of a thermomorphic system for asymmetric hydrogenation and liquid/solid separation of the chiral catalyst. Xiao et al. [44] presented a chiral cross-linked mesoporous polymer coordinated to a ruthenium species as an efficient heterogeneous catalyst. Li et al. [39] reported chiral CMPs could be utilized as the asymmetric hydrogenation of β-keto esters after coordination with ruthenium species. Ding et al. [45] synthesized one microporous polymers by Friedel-Crafts reaction, followed by loading a ruthenium species, the prepared catalysts could be applied for asymmetric hydrogenation.
In this work, we describe the synthesis of a porous organic polymer functionalized with chiral BINAPO via copper-catalyzed alkyne-azide click reaction (CuAAC), as illustrated in Scheme 1. After the deoxidization, the chiral BINAP functionalized polymer could be composited with [Ru(p-cymene)Cl2]2. The final material could be used as an efficient heterogeneous catalyst for the asymmetric hydrogenation of β-keto esters.
Enantiomerically pure chiral BINAP has widely been used as chiral ligand in heterogeneous asymmetric catalysis by immobilizing it onto various materials for the asymmetric catalysis. [39, 41, 44~50] Therefore it has been chosen as a functional building block for the synthesis of porous organic polymers (POPs) in this work. Inspired by the copper-catalyzed alkyne-azide click reaction between tetra-(4-azidophenyl)methane and 1, 4-dialkynylbenzene, [31] (S)-(4, 4'-diethynyl-2, 2'-bis(diphenylphosphino)-1, 1'-binaphthalene was chosen as the building block because of the similar linear structure to 1, 4-dialkynylbenzene. The desired material, POP-BINAPO was obtained with excellent yield by the click reaction between (S)-(4, 4'-diethynyl-2, 2'-bis(diphenylphosphino)-1, 1'-binaphthalene and tetra-(4-azidophenyl)methane. After the reduction with trichloro-silane in the presence of N, N-dimethylaniline, POP-BINAP was obtained with a very good yield.
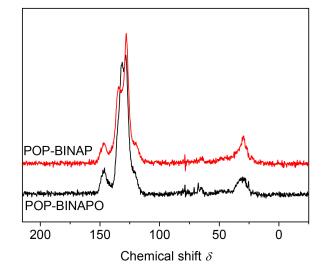
In the 13C CP/MAS NMR spectra of the polymers (Figure 1), signals in the range of δ 150~110 correspond to the aryl carbons of polymers. And there is nearly no signals at about δ 90, which indicates that the alkynyl group has been consumed during the preparation of the polymers.
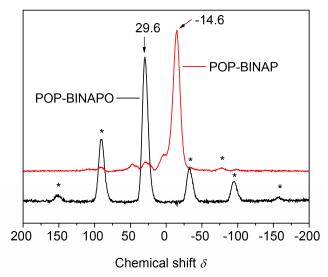
As show in Figure 2, POP-BINAPO exhibits a peak centered at about δ 29.6 ascribed to P=O, while PCP-BINAP gives the signal at δ -14.6 attributed to the reduced phosphine. The asterisks denote the spinning sidebands. The NMR results show the successful synthesis of POP-BINAPO and reduction of BINAPO to BINAP by the post-synthesis modification method.
In the Fourier transform infrared spectra of POP-BIN-APO, N=N stretch could be observed at 1600 cm-1, and C=C stretch could be observed at 1631 cm-1, which indicate the formation of the triazole ring. A strong peak could be observed at 1437 cm-1, which is ascribed to P=O. In the FT-IR spectra of POP-BINAP, it is clear that the P=O peak is very weak. The broad peak at ca. 3411 cm-1 is ascribed to water. These results further demonstrate the successful synthesis and reduction of the polymer, POP-BINAPO.
In the thermogravimetric analysis patterns, the synthesized polymers are stable up to 350 ℃. Their stabilities are very important for the supporting materials of the catalyst. The materials should be recycled without significant change of the structures and the decrease of the yields of the reactions. The low stability of the supporting materials may lead to the collapse of their structures under the experimental conditions, and this may decrease or lose the activity of the catalysts. The slight weight loss at lower temperature (less than 350 ℃) may result from the loss of the absorbed solvent. And the weight loss between 350 and 400 ℃ may come from the decomposition of the polymers with low molecular weight in materials at elevated temperatures under an N2 atmosphere. [51] The powder patterns X-ray diffraction of polymers indicates that the materials are amorphous.
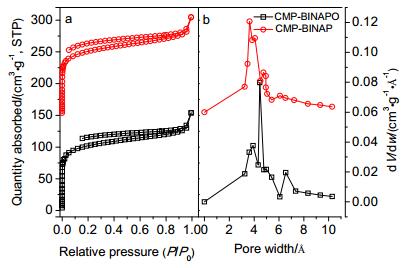
The N2 sorption isotherms were measured at 77 K, as shown in Figure 3a. The Brunauer-Emmett-Teller surface areas of POP-BINAPO and POP-BINAP are 326 and 316 m2•g-1, respectively. The pore volume and the micropore volume of POP-BINAPO are 0.2612 and 0.1465 cm3•g-1, respectively. The total pore volume and micropore volume of POP-BINAP are 0.2622 and 0.1423 cm3•g-1, respectively. The samples of POP-BINAPO and POP-BINAP gave only one sharp adsorption-desorption loop at low P/P0, indicating the existence of micropores in the materials. This could also be confirmed pore size distributions curves calculated by density functional theory (DFT) method (Figure 3b). The images of transmission electron microscopy (TEM) exhibit clear particles of the structures, although they stick together. And images of TEM also show that there are micropores inside the polymer, which is in accordance with the nitrogen adsorption isotherms.
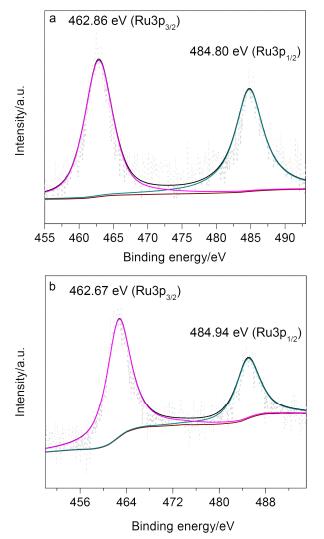
Ruthenium(Ⅱ) ion can be loaded onto the material POP-BINAP via metal-ligand coordination. The ruthe-nium precursor, [Ru(p-cymene)Cl2]2 was coordinated with the POP-BINAP after stirring in methanol at 50 ℃ for 5 h. The chemical state of ruthenium in Ru/BINAP was determined by X-ray photoelectron spectroscopy (Figure 4). The Ru3p signal was chosen as the Ru3d peak strongly overlaps with the C1s. The binding energies at 462.86 and 484.80 eV for Ru/POP-bipy in Figure 4a correspond to RuⅡ3p3/2 and RuⅡ3p1/2, respectively. [52~55] In Figure 4b, the peaks at 462.67 and 484.94 eV were also ascribed as RuⅡ3p3/2 and RuⅡ3p1/2, respectively. The little shift may resulted from the slight different chemical environments of Ru(Ⅱ) after the reactions.
The inductive coupled plasma (ICP) emission spectrometeranalyses show that ruthenium content of the catalyst. The ruthenium concentration in the composite Ru/POP-BINAP was 7.51 wt% before the catalyst reaction. While the content of Ru was 5.70 wt% after five times cycles. The decrease of catalytic activity may be resulted from the leaching of ruthenium during the reactions. The result of energy-dispersive X-ray spectroscopy (EDX) analysis for the Ru/POP-bipy catalyst was (wt%): C 77.08, N 2.10, O 9.24, P 3.20, Ru 6.41, Cl 1.96. The EDX on TEM confirms the presence of Ru metal in catalysts, and its content is generally consistent with ICP, which approve that the ruthenium species are highly dispersed in the catalysts
To evaluate the catalytic activity of the ruthenium modified POP, Ru/POP-BINAP, ethyl benzoylacetate was chosen as the model substrate for the asymmetric hydrogenation reaction of β-keto esters, and the reaction conditions such as pressure, temperature and reaction time were optimized (Table 1). Preliminary studies using the Ru/POP-BINAP catalytic system began with a solvent screen. Considering the most reported solvents for the hydrogenation reaction are alcohols, methanol and ethanol have been chosen as the solvents for the reaction. [12, 39, 42, 56]
 下载:
导出CSV
下载:
导出CSV
 |
|||||||
| Entry | p/MPa | T/℃ | t/h | Solvent | Yield b/% | Conv./% | eec/% |
| 1 | 2 | 85 | 20 | EtOH | 98 | 100 | 81 |
| 2 | 1 | 85 | 20 | EtOH | 64 | 73 | 61 |
| 3 | 3 | 85 | 20 | EtOH | 95 | 100 | 77 |
| 4 | 2 | 75 | 20 | EtOH | 83 | 91 | 71 |
| 5 | 2 | 95 | 20 | EtOH | 97 | 100 | 73 |
| 6 | 2 | 85 | 15 | EtOH | 76, 75 d | 85, 85 d | 80, 80 d |
| 7 | 2 | 85 | 25 | EtOH | 98 | 100 | 79 |
| 8 | 2 | 85 | 20 | MeOH | 82, 14 e | 100 | 83, 80 e |
| aReaction condition: substrate (1 mmol) in ethanol (1 mL), substrate/cata-lyst=200 (molar ratio). b Isolated yield. c Determined by HPLC using a Chiral OD-column. d The same reaction time was tried for another time. e Product of transterification: (S)-methyl 3-hydroxy-3-phenylpropanoate. | |||||||
Product of transterification was observed when methanol was used as the solvent, [56] while excellent yield and ee value were obtained when ethanol was used as the solvent, therefore ethanol was selected to perform the hydrogenation. We first performed the hydrogenation under different pressure (1, 2 and 3 MPa) at the temperature of 80 ℃ (Table 1, Entries 1~3). The results of lower ee values and yield illustrated that lower or higher pressure may have a negative influence on the enantioselectivity and conversation, especially for lower H2 pressure. We next selected the temperature of the reaction (Entries 4, 5). It was observed that the conversation of the reaction decreased to 91% when the reaction temperature changed to 75 ℃, the conversation reached 100% when the temperature increased to 95 ℃, but the ee values decreased when the reaction temperature was at 75 ℃ or 95 ℃, therefore the best reaction temperature was at 85 ℃. Taking into account that the reaction time may affect the outcome of the hydrogenation, and the reaction were performed under different reaction time. The effect of different reaction time was investigated at 85 ℃ (Entries 6 and 7). By shortening the reaction time to 15 h, the conversation of the reaction decreased to 85%, while prolonging the time to 25 h, the conversation of the reaction reached 100%. The ee values did not change significantly when the reaction time changed to 15 h or 25 h. It indicated that 20 h is the suitable time for the reaction.
The Ru/POP-BINAP catalytic system exhibited complete conversions and high enantioselectivities at 85 ℃ under H2 pressure of 2 MPa in ethanol. Various β-ketoesters could be converted into the corresponding chiral alcohol with excellent yields and the enantiomeric excess value is as high as 92% (Table 2). Ru/POP-BINAP is applicable to a wide range of substrates with good electronic and steric tolerance, the yields are excellent and the ee values are in the range of 76%~81% (Table 2, Entries 1~5). The results are close to the reported enantiomeric excess values for the reac-tions. [6, 43, 57] The enantiomeric excess values increased to 88%~92% when ethyl 3-oxo-4-phenylbutanoate and the derivatives were used as the substrates (Entry 6~13). The lower enantioselectivitiy of catalyst system for ethyl benzoylacetate and its derivatives maybe result from the weak π-π stack between the phenyl group of ethyl benzoylacetate and a phenyl group from BINAP. This week π-π stack could form and stabilize an unfavorable transition state, and leads to the decrease of the enantiomeric excess for ethyl benzoylacetate and its derivatives. [57] But for ethyl 3-oxo-4-phenylbutanoate and its derivatives, the π-π stack may not be easy to form, and it is propitious to increase the enantiomeric excess values.
下载:
导出CSV
 |
||||
| Entry | 1 | R | Yield b/% | ee c/% |
| 1 | 1a | Ph | 98 | 81 |
| 2 | 1b | 2-ClC6H4 | 92 | 76 |
| 3 | 1c | 3-CH3C6H4 | 99 | 80 |
| 4 | 1d | 2-CH3C6H4 | 98 | 81 |
| 5 | 1e | 4-CH3C6H4 | 98 | 76 |
| 6 | 1f | PhCH2 | 99 | 89 |
| 7 | 1g | 4-F-C6H4CH2 | 96 | 92 |
| 8 | 1h | 2-F-C6H4CH2 | 95 | 92 |
| 9 | 1i | 4-MeOC6H4CH2 | 95 | 88 |
| 10 | 1j | 3-MeOC6H4CH2 | 98 | 89 |
| 11 | 1k | 4-CH3C6H4CH2 | 98 | 89 |
| 12 | 1l | 4-ClC6H4CH2 | 94 | 89 |
| 13 | 1m | 4-CF3C6H4CH2 | 99 | 89 |
| 14 | 1n | CH3 | 99 | 96 |
| 15 | 1p | (CH3)2CH | 99 | 92 |
| a Reaction condition: substrate (1 mmol) in ethanol (1 mL), substrate/cata-lyst=200 (molar ratio), 2 MPa H2, 85 ℃, 20 h. b Isolated yield, conversion: 100%. c Determined by HPLC using a Chiral OD-column. | ||||
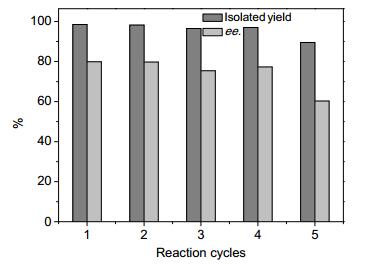
For the recycling ability (Figure 5), ethyl benzoylacetate was selected as the substrate. In the first four cycles, the yields and the enantiomeric excess values are nearly stable. But the yield and the enantiomeric excess value decreased in the fifth cycle. The decrease of the catalytic activity and enantiomeric selectivity of the Ru/POP-BINAP catalytic system may be due to the unknown change of the microenvironment of the activity center during the catalytic procedures. [39] During the recycling of the catalyst, the chiral phos-phorous ligands maybe partly oxidized and leading to the leaching of ruthenium, which could be a possible deactivation reason based on ICP analysis.
In summary, a porous organic polymer with BINAPO group has been synthesized by "click-based" reaction and reduced to BINAP using postsynthesis modification method. The materials have been charac-terized by several analytical techniques such as NMR, FT-IR, XRD, nitrogen absorption, thermogravimetric analysis, scanning electron microscopy, and trans-mission electron microscopy. After the modification with ruthenium(Ⅱ) species, the composite material Ru/POP-BINAP exhibited excellent catalytic activities and enantioselectivities for the asymmetric hydro-genation reaction of β-keto esters. Ru/POP-BINAP can be reused four cycles without significant loss of the catalytic activities and enantioselectivitiy.
(S)-(-)-2, 2'-Bis(diphenylphosphinyl)-1, 1'-binaphthale-ne ((S)-BINAPO), 2-methyl-3-butyn-2-ol, Pd(PPh3)4, trichlorosilane (HSiCl3), and [Ru(p-cymene)Cl2]2 were purchased from Sahn chemical technology (Shanghai) Co., Ltd (China). L-Sodium ascorbate was purchased from Alfa-Aesar (USA). Other reagents were obtained from Shanghai Ling Feng Chemical Reagent Co., Ltd (China). (S)-4, 4'-Dibromo-2, 2'-bis(diphenylphosphino)-1, 1'-binaph-thalene [58] and tetra(4-azidophenyl)methane [59] were prepared according to the literature methods. All solvents were dried by the standard methods before use.
Nitrogen isotherms were measured at 77 K using a Quantachrome Nova 4200 E Porosimeter. The samples were outgassed at 150 ℃ for 20 h. The specific surface area, pore size, pore volume and pore distribution were calculated by the Nonlocal Density Function Theory (NLDFT). FT-IR spectra were performed on a Perkin-Elmer Paragon 1000 spectrometer in the range 4000~400 cm-1. The 31P MAS NMR spectra were recorded at 161.97 MHz using 4-mm MAS probe with a spinning rate of 10 kHz, 256 scans, a recycle delay of 2 s and a contact time of 1 ms. The chemical shifts were referenced to the phosphoric acid solution at δ 0.13C CP/MAS NMR were recorded at 100.62 MHz using 4-mm MAS probe with a spinning rate of 10 kHz, 2048 scans, a recycle delay of 2 s and a contact time of 1 ms. The chemical shifts were referenced to the adamantine with the up-field methane peak at δ 29.5. Powder X-ray diffraction patterns were conducted on a Rigaku D/MAX-2200/PC. Scanning electron microscopy (SEM) was performed using JSM-7401F instrument. High-resolution transmission electron microscope (HR-TEM) images were taken on a JEOLJEM-3010 microscope operating at 200 kV. Thermogravimetric analysis (TGA) was recorded using a Pyris 1 TGA, and the samples were heated from 50 ℃ to 800 ℃ at the rate of 20 ℃/min in nitrogen atmosphere. The loading and the leaching of ruthenium(Ⅱ) ion were detected by inductively coupled plasma-atomic emission spectro-metry (ICP-AES, iCAP 6000 Radial).
(S)-4, 4'-Dibromo-2, 2'-bis(diphenylphosphinyl)-1, 1'-bi-naphthalene (4.855 g, 6.0 mmol), CuI (0.096 g, 0.50 mmol), Pd(PPh3)4 (0.361 g, 0.31 mmol) and 2-methyl-3-butyn-2-ol (1.75 mL, 18.06 mmol) were added in a 250 mL of Schlenk flask. The flask was degassed and purged with nitrogen three times before addition of freshly distilled and degassed triethylamine (Et3N) (100 mL). The resulting mixture was refluxed overnight under stirring. After cooling to room temperature, the resultant precipitate was filtered off, and then Et3N was removed under vacuum. The crude product was purified by column chromatography using ethyl acetate/ petroleum ether (V:V=2:1) as the eluent. The product was obtained as a light yellow solid, [60] yield 3.869 g (79%). m.p. 263 ℃ (dec.); 1H NMR (400 MHz, CDCl3) δ: 8.25 (d, J=8.4 Hz, 2H), 7.71~7.66 (m, 4H), 7.59~7.56 (d, J=12 Hz, 2H), 7.44~7.37(m, 10H), 7.30~7.22 (m, 8H), 6.82~6.68 (m, 4H), 1.71 (s, 12H); 13C NMR (101 MHz, CDCl3) δ: 143.2, 132.6, 132.5, 132.1, 131.3, 128.4, 128.3, 128.2, 128.0, 127.4, 126.4, 126.0, 99.9, 80.1, 65.9, 31.7;31P NMR (162 MHz, CDCl3) δ: 28.10.
In a 250 mL of round flask, (S)-(4, 4'-bis(3-hydroxy-3-methyl-but-1-yn-1-yl)-2, 2'-bis(diphenylphosphino)-1, 1'-binaphthalene) (5.129 g, 6.26 mmol) was dissolved in toluene (100 mL), powdered KOH (1.682 g, 30.0 mmol) was added to the flask and the mixture was refluxed under stirring for 1 h. After cooling to room temperature, the mixture was filtered and the solvent was removed under vacuum. The crude product was purified by column chromatography using ethyl acetate/petroleum ether/dichloromethane (V:V:V=2:5:1) as the eluent. The product was obtained as a light yellow solid, [60] yield 2.277 g (52%). m.p. 285 ℃ (dec.); 1H NMR (400 MHz, CDCl3, ) δ: 8.34 (d, J=8.4 Hz, 2H), 7.71~7.66 (m, 6H), 7.46~7.37 (m, 10H), 7.30~7.22 (m, 8H), 6.84~6.75 (m, 4H), 3.46 (s, 2H); 13C NMR (101 MHz, CDCl3) δ: 143.6, 134.3, 134.0, 133.2, 132.9, 132.6, 132.5, 132.1, 131.8, 131.5, 131.3, 129.7, 128.7, 128.3, 128.2, 128.1, 127.4, 126.6, 126.1, 119.8, 83.0, 81.7;31P NMR (162 MHz, CDCl3)δ: 28.94.
In a 100 mL of Schlenk flask, tetra(4-azidophenyl)me-thane (147 mg, 0.30 mmol), (S)-(4, 4'-diethynyl-2, 2'-bis-(diphenylphosphino)-1, 1'-binaphthalene) (422 mg, 0.60 mmol), CuSO4•5H2O (60 mg, 0.24 mmol) and L-sodium ascorbate (52 mg, 0.26 mmol) were dissolved in DMSO (50 mL) and water (3 mL). The mixture was degassed by three evacuation/nitrogen fill cycles and stirred at 90 ℃ for 3 d. After cooling to room temperature, the resulting yellow solid was collected by filtration and washed with water three times. The isolated solid was suspended in water (50 mL), to which EDTA-2Na (745 mg, 2 mmol) was added, the mixture was stirred at room temperature for 12 h, and the solid was collected by filtration. The solid was treated with EDTA-2Na for another two times in the same procedure. The solid was collected by filtration, washed sequentially with water, N, N-dimethylformamide (DMF) and methanol, dried under vacuum. The material was further purified by Soxhlet extraction in a mixture of dichloromethane/me-thanol/THF/acetone (V:V:V:V=1:1:1:1) for 2 d, and dried under vacuum. POP-BINAPO was obtained as a yellow solid, [61] yield 533 mg (94%).
POP-BINAP was synthesized by the deoxidization of POP-BINAPO, the detailed process can be described as follows. A 100 mL of three-necked round flask was equipped with a thermometer, a reflux condenser and a constant pressure dropping funnel. The flask was vacuumed and purged with nitrogen three times, and then POP-BINAPO (707 mg), N, N-dimethylaniline (2 mL) and freshly distilled and degassed toluene (35 mL) were added to the flask. Then trichlorosilane (1.5 mL, dissolved in 10 mL toluene) was added dropwise with care using dropping funnel. The mixture was stirred at room temperature for half an hour and refluxed under stirring for 20 h. The reaction mixture was cooled to 0 ℃ in an ice-water bath, to which 25 mL of degassed 10% NaOH (aq.) was added and stirred for half an hour. The solid was isolated by filtration, washed with degassed water for three times followed by degassed dichloromethane, and dried under vacuum. POP-BINAP was obtained as a yellow-brown powder, [12] yield 690 mg.
CAUTION: All the procedures during the preparation of POP-BINAP should be operated under nitrogen atmosphere. The tail gas was inflammable and should be handled with great care.
[Ru(p-cymene)Cl2]2 (6.1 mg, 0.01 mmol) and POP-BINAP (21.2 mg, 0.022 mmol) were added in a 25 mL of Schlenk tube. The tube was evacuated and purged with nitrogen three times before addition of freshly distilled and degassed methanol (4 mL). The mixture was heated at 50 ℃ for 5 h, cooled to room temperature. Then the solvent was removed under reduced pressure to get the catalyst Ru/POP-BINAP as a brownish solid. The resultant catalyst was equally put into four vials (6.5 mg each) containing substrate (1.0 mmol) in degassed ethanol (1 mL), then the vials were taken into an autoclave. The autoclave was purged with hydrogen and the finally pressure was maintained to 2 MPa and stirred the mixture at 85 ℃ for 20 h. After cooling to ambient temperature and release of the hydrogen with care, the autoclave was opened and the solvent was removed by evaporation. The products were purified by column chromatography of silica gel eluting with ether/ethyl acetate (V:V=50:1). The enantiomeric excess values (ee) were determined by HPLC.
(S)-Ethyl-3-hydroxy-3-phenylpropanoate (2a): [62] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=12.0 min, tminor=8.2 min; ee=80%.1H NMR (400 MHz, CDCl3) δ: 7.40~7.27 (m, 5H), 5.14 (dd, J=8.8, 4.0 Hz, 1H), 4.19 (q, J=7.2 Hz, 2H), 2.77~2.72 (m, 2H), 1.27 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.6, 142.6, 128.6, 127.9, 125.8, 70.4, 61.0, 43.4, 14.3.
(S)-Methyl-3-hydroxy-3-phenylpropanoate: Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=16.8 min, tminor=10.5 min; ee=80%.1H NMR (400 MHz, CDCl3) δ: 7.39~7.27 (m, 5H), 5.14 (dd, J=9.2, 4.0 Hz, 1H), 3.72 (s, 3H), 2.81~2.68 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 172.9, 142.6, 128.7, 128.0, 125.8, 70.4, 52.0, 43.3.
(S)-Ethyl-3-(2-chlorophenyl)-3-hydroxypropanoate (2b): [62] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=13.4 min, tminor=6.7 min; ee=76%.1H NMR (400 MHz, CDCl3) δ: 7.64~7.62 (dd, J=16, 7.6 Hz, 1H), 7.34~7.29 (m, 2H), 7.26~7.20 (m, 1H), 5.49 (dd, J=9.6, 2.4 Hz, 1H), 4.20 (q, J=7.2 Hz, 2H), 2.86 (m, 1H), 2.58 (dd, J=16.4, 9.6 Hz, 1H), 1.28 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.7, 139.9, 131.5, 129.5, 128.9, 127.3, 127.2, 67.1, 61.1, 41.5, 14.3.
(S)-Ethyl-3-hydroxy-3-(m-tolyl)propanoate (2c): [62] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=9.7 min, tminor=7.2 min; ee=80%.1H NMR (400 MHz, CDCl3) δ: 7.26~7.21 (t, J=7.6 Hz, 2H), 7.17~7.09 (dd, J=25.2, 7.6 Hz, 2H), 5.10 (dd, J=8.8, 4.0 Hz, 1H), 4.19 (q, J=7.2 Hz, 2H), 2.72 (m, 2H), 2.36 (s, 3H), 1.27 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.6, 142.5, 138.4, 128.7, 128.6, 126.5, 122.8, 70.5, 61.0, 46.4, 43.4, 21.6, 14.3.
(S)-Ethyl-3-hydroxy-3-(o-tolyl)propanoate (2d): [62] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=12.5 min, tminor=8.0 min; ee=81%.1H NMR (400 MHz, CDCl3)δ: 7.51 (d, 1H), 7.24~7.13 (m, 3H), 5.36 (dd, J=8.8, 3.6 Hz, 1H), 4.21 (q, J=7.2 Hz, 2H), 2.67 (m, 2H), 2.35 (s, 3H), 1.29 (t, J=7.2 Hz, 3H); 13C NMR (CDCl3) δ: 172.8, 140.6, 134.4, 130.6, 127.7, 126.5, 125.3, 67.1, 61.1, 42.2, 19.1, 14.3.
(S)-Ethyl-3-hydroxy-3-(p-tolyl)propanoate (2e): [62] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=98/2; flow rate=0.3 mL/min; 210 nm; tmajor=57.8 min, tminor=54.4 min; ee=76%.1H NMR (400 MHz, CDCl3) δ: 7.26 (d, J=8.0 Hz, 2H), 7.16 (d, J=8.0 Hz, 2H), 5.10 (dd, J=9.0, 3.6 Hz, 1H), 4.18 (q, J=7.2 Hz, 2H), 2.79~2.70 (m, 2H), 2.34 (s, 3H), 1.26 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.6, 139.7, 137.6, 129.3, 125.7, 70.3, 61.0, 43.4, 21.3, 14.3.
(S)-Ethyl-3-hydroxy-4-phenylbutanote (2f): [63] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=8.8 min, tminor=7.2 min; ee=89%.1H NMR (400 MHz, CDCl3) δ: 7.33~7.21 (m, 5H), 4.30~4.24 (m, 1H), 4.19~4.13 (q, J=7.2 Hz, 2H), 2.90~2.85 (dd, J=13.6, 7.2 Hz, 1H), 2.79~2.74 (dd, J=13.6, 6.4 Hz, 1H), 2.53~2.42 (m, 2H), 1.26 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.9, 137.8, 129.6, 128.7, 126.7, 69.2, 60.9, 43.0, 40.6, 14.3.
(S)-Ethyl-4-(4-fluorophenyl)-3-hydroxybutanoate (2g): Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=8.2 min, tminor=6.6 min; ee=92%.1H NMR (400 MHz, CDCl3) δ: 7.20~7.16 (m, 2H), 6.99 (m, 2H), 4.25~4.20 (m, 1H), 4.16 (q, J=7.2 Hz, 2H), 2.78 (m, 2H), 2.46 (m, 2H), 1.26 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.8, 161.9 (q, J=243.1 Hz, 1C), 133.5 (q, J=3.2 Hz, 1C), 131.0 (q, J=7.8 Hz, 1C), 115.4 (q, J=21 Hz, 1C), 69.1, 60.9, 42.1, 40.1, 14.3; MS (MALDI-TOF) calcd for C12H15FO3Na[M+Na] +: 249.0903, found 249.0915.
(S)-Ethyl-4-(2-fluorophenyl)-3-hydroxybutanoate (2h): Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=8.8 min, tminor=6.5 min; ee=92%.1H NMR (400 MHz, CDCl3) δ: 7.27~7.19 (m, 2H), 7.10~7.01 (m, 2H), 4.30 (m, 1H), 4.16 (q, J=7.2 Hz, 2H), 3.04 (s, 1H), 2.87 (m, 2H), 2.55~2.43 (m, 2H), 1.26 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.9, 161.4 (q, J=243.6 Hz, 1C), 132.0 (q, J=4.6 Hz, 1C), 128.5 (q, J=8.1 Hz, 1C), 124.8 (q, J=15.6 Hz, 1C), 124.2 (q, J=3.6 Hz, 1C), 115.5 (q, J=22.1 Hz, 1C), 68.1, 60.9, 40.5, 36.0, 14.3; MS (MALDI-TOF) calcd for C12H15FO3Na [M+Na] +: 249.0903, found 249.0908.
(S)-Ethyl-3-hydroxy-4-(4-methoxyphenyl)butanoate (2i): [63] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=10.9 min, tminor=8.5 min; ee=88%.1H NMR (400 MHz, CDCl3) δ: 7.14 (d, J=8.8 Hz, 2H), 6.85 (d, J=8.8 Hz, 2H), 4.24~4.20 (m, 1H), 4.18~4.13 (q, J=7.2 Hz, 2H), 3.79 (s, 3H), 2.83~2.68 (m, 2H), 2.54~2.38 (m, 2H), 1.26 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.9, 158.4, 130.5, 129.7, 114.1, 69.3, 60.9, 55.4, 42.1, 40.6, 14.3.
(S)-Ethyl-3-hydroxy-4-(3-methoxyphenyl)butanoate (2j): Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=10.9 min, tminor=9.2 min; ee=89%.1H NMR (400 MHz, CDCl3) δ: 7.25~7.21 (t, J=7.6 Hz, 1H), 6.82~6.77 (m, 3H), 4.30~4.23 (m, 1H), 4.18~4.13 (dd, J=14, 7.2 Hz, 2H), 3.80 (s, 3H), 2.88~2.82 (m, 1H), 2.76~2.72 (m, 1H), 2.50~2.44 (m, 2H), 1.26 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.9, 159.8, 139.3, 129.6, 121.9, 115.2, 112.1, 69.1, 60.9, 55.3, 43.1, 40.6, 14.3. MS (MALDI-TOF) calcd for C13H18O4Na[M+Na]+: 261.1103, found 261.1114.
(S)-Ethyl-3-hydroxy-4-(p-tolyl)butanoate (2k): Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=7.8 min, tminor=6.4 min; ee=89%.1H NMR (400 MHz, CDCl3) δ: 7.11 (s, 4H), 4.27~4.20 (m, 1H), 4.18~4.13 (q, J=7.2 Hz, 2H), 2.86~2.80 (dd, J=13.6, 7.2 Hz, 1H), 2.75~2.70 (dd, J=13.6, 6 Hz, 1H), 2.49~2.43 (m, 2H), 2.33 (s, 3H), 1.26 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 200.9, 167.3, 137.1, 130.2, 129.7, 129.5, 61.5, 49.8, 48.3, 21.2, 14.2. MS (MALDI-TOF) calcd for C13H18O3Na [M+Na]+: 245.1154, found 245.1160.
(S)-Ethyl-4-(4-chlorophenyl)-3-hydroxybutanoate (2l): [64] Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=9.8 min, tminor=6.9 min; ee=89%.1H NMR (400 MHz, CDCl3) δ: 7.29~7.26 (m, 2H), 7.16 (d, J=8.4 Hz, 2H), 4.26~4.21 (m, 1H), 4.19~4.13 (q, J=7.2 Hz, 2H), 2.85~2.80 (dd, J=13.6, 7.2 Hz, 1H), 2.76~2.71 (m, 1H), 2.48~2.40 (m, 2H), 1.26 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 127.8, 136.3, 132.6, 130.9, 128.7, 68.9, 61.0, 42.2, 40.5, 14.3.
(S)-Ethyl-3-hydroxy-4-(4-(trifluoromethyl)phenyl)buta-noate (2m): m.p. 32.1~34.3 ℃; Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=90/10; flow rate=1.0 mL/min; 210 nm; tmajor=9.7 min, tminor=6.1 min; ee=89%.1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J=8.4 Hz, 2H), 7.35 (d, J=8 Hz, 2H), 4.31~4.25 (m, 1H), 4.19~4.14 (q, J=7.2 Hz, 2H), 2.92~2.82 (m, 2H), 2.54~2.43 (m, 2H), 1.27 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 172.8, 142.1, 129.9, 129.1 (q, J=32.2 Hz, 1C), 125.5 (q, J=3.7 Hz, 1C), 124.2 (q, J=270.1 Hz, 1C), 68.7, 61.0, 42.7, 40.6, 14.3; MS (MALDI-TOF) calcd for C13H15F3O3Na[M+Na]+: 299.0871, found 299.0880.
(S)-Ethyl-3-hydroxybutanoate (2n): Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=95/5; flow rate=1.0 mL/min; 230 nm; tmajor=9.3 min, tminor=7.0 min; ee=96%.1H NMR (400 MHz, CDCl3) δ: 4.21~4.11 (m, 3H), 3.68 (s, 3H), 2.49~2.42 (m, 2H), 1.20 (d, J=6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 173.0, 64.3, 76.8, 64.3, 60.8, 42.9, 22.5, 14.3.
(S)-Methyl-3-hydroxy-4-methylpentanoate (2p): Optical purity is determined by HPLC with a ChiralCel OD-H column: 25 ℃; V(Hexane)/V(i-PrOH)=97/3; flow rate=0.8 mL/min; 210 nm; tmajor=12.2 min, tminor=7.6 min; ee=92%.1H NMR (400 MHz, CDCl3)δ: 4.16 (q, J=7.2 Hz, 2H), 3.79~3.74 (m, 1H), 2.64 (s, 1H), 2.51~2.35 (m, 2H), 1.74~1.65 (m, 1H), 1.26 (t, J=7.2 Hz, 3H), 0.92 (dd, J=11.2, 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ: 173.7, 72.8, 60.8, 38.6, 33.3, 18.2 (d, J=56.9 Hz, 1C), 14.3.
For recycling the catalyst, the catalyst was separated by centrifugation (performed in a glovebox), washed with ethanol for 3 times under N2 and dried under vacuum (using standard Schlenk-type techniques), the catalyst was collected and used for the next cycle.
Supporting Information Fourier transform infrared spectra, powder patterns of X-ray diffraction, images of Scanning Electron Microscopy and Transmission Electron Microscopy of the materials POP-BINAPO and POP-BINAP, 1H NMR and13C NMR spectra for all products associated with this article. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn
Collins, A. N.; Sheldrake, G.; Crosby, J. Chirality in Industry, Volume Ⅱ, New York, 1998.
Liu, X.; Han, Z. B.; Wang, Z.; Ding, K. L. Sci. China Chem. 2014, 57, 1073. doi: 10.1007/s11426-014-5134-7
Zhang, K. F.; Li, F.; Nie, J.; Ma, J. A. Sci. China Chem. 2014, 57, 265. doi: 10.1007/s11426-013-5017-3
van Leeuwen, P. W.; Kamer, P. C.; Claver, C.; Pàmies, O.; Diéguez, M. Chem. Rev. 2010, 111, 2077. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21087011
Yang, Q. H.; Liu, J.; Zhang, L. Sci. China Chem. 2010, 53, 351. doi: 10.1007/s11426-010-0046-7
Wang, P. Y.; Liu, X.; Yang, J.; Yang, Y.; Zhang, L.; Yang, Q. H.; Li, C. J. Mater. Chem. 2009, 19, 8009. doi: 10.1039/b913808k
Wang, Z. J.; Deng, G. J.; Li, Y.; He, Y. M.; Tang, W. J.; Fan, Q. H. Org. Lett. 2007, 9, 1243. doi: 10.1021/ol0631410
Deng, G. J.; Fan, Q. H.; Chen, X. M.; Liu, D. S.; Chan, A. S. Chem. Commun. 2002, 15, 1570.
Bleschke, C.; Schmidt, J.; Kundu, D. S.; Blechert, S.; Thomas, A. Adv. Synth. Catal. 2011, 353, 3101. doi: 10.1002/adsc.v353.17
Yasukawa, T.; Miyamura, H.; Kobayashi, S. Chem. Soc. Rev. 2014, 43, 1450. doi: 10.1039/C3CS60298B
Yoon, M.; Srirambalaji, R.; Kim, K. Chem. Rev. 2011, 112, 1196.
Falkowski, J. M.; Sawano, T.; Zhang, T.; Tsun, G.; Chen, Y.; Lockard, J. V.; Lin, W. J. Am. Chem. Soc. 2014, 136, 5213. doi: 10.1021/ja500090y
Farha, O. K.; Bae, Y. S. Hauser, B. G.; Spokoyny, A. M.; Snurr, R. Q.; Mirkin, C. A.; Hupp, J. T. Chem. Commun. 2010, 46, 1056. doi: 10.1039/b922554d
Lu, S. L.; Jin, Y. H.; Gu, H. W.; Zhang, W. Sci. China Chem. 2017, 60, 999. doi: 10.1007/s11426-017-9078-7
Lu, X. L.; Zhou, T. Y.; Wu, D. F.; Wen, Q.; Zhao, X.; Li, Q. W.; Xiang, Q.; Xu, J. Q.; Li, Z. T. Chin. J. Chem. 2015, 33, 539. doi: 10.1002/cjoc.201500181
Liu, G. F.; Sheng, J. H.; Zhao, Y. L. Sci. China Chem. 2017, 60, 1015.
万刚, 付宇昂, 郭佳宁, 向中华, 化学学报, 2015, 73, 557. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxxb201506008&dbname=CJFD&dbcode=CJFQZhou, B. L.; Chen, L. Acta Chim. Sinica 2015, 73, 487(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxxb201506008&dbname=CJFD&dbcode=CJFQ
Spitler, E. L.; Dichtel, W. R. Nat. Chem. 2010, 2, 672. doi: 10.1038/nchem.695
Wan, S.; Guo, J.; Kim, J.; Ihee, H.; Jiang, D. Angew. Chem., Int. Ed. 2008, 47, 8826. doi: 10.1002/anie.v47:46
Zou, X. Q.; Ren, H.; Zhu, G. S. Chem. Commun. 2013, 49, 3925. doi: 10.1039/c3cc00039g
Sun, C. J.; Zhao, X. Q. Wang, P. F.; Wang, H.; Han, B. H. Sci. China Chem. 2017, 60, 1067.
Jiang, J. X.; Su, F.; Niu, H.; Wood, C. D.; Campbell, N. L.; Khimyak, Y. Z.; Cooper, AI. Chem. Commun. 2008, 8, 486.
Huang, N.; Day, G.; Yang, X.; Drake, H.; Zhou, H. C. Sci. China Chem. 2017, 60, 1007. doi: 10.1007/s11426-017-9084-7
李艳强, 贲腾, 裘式纶, 化学学报, 2015, 73, 605. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxxb201506012&dbname=CJFD&dbcode=CJFQLi, Y.; Ben, T.; Qiu, S. l. Acta Chim. Sinica 2015, 73, 605(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxxb201506012&dbname=CJFD&dbcode=CJFQ
Feng, X. L.; Liang, Y. Y.; Zhi, L. J.; Thomas, A.; Wu, D. Q.; Lieberwirth, I.; Kolb, U.; Müllen, K. Adv. Funct. Mater. 2009, 19, 2125. doi: 10.1002/adfm.v19:13
Chen, L.; Honsho, H.; Seki, S.; Jiang, D. L. J. Am. Chem. Soc. 2010, 19, 6742.
Xu, Y.; Nagai, A.; Jiang, D. Chem. Commun. 2013, 49, 1591. doi: 10.1039/C2CC38211C
Chen, L.; Yang, Y.; Jiang, D. L. J. Am. Chem. Soc. 2010, 132, 9138. doi: 10.1021/ja1028556
王昌安, 王为, 化学学报, 2015, 73, 498. doi: 10.3866/PKU.WHXB201412162Wang, C. A.; Wang, W. Acta Chim. Sinica 2015, 73, 498(in Chinese). doi: 10.3866/PKU.WHXB201412162
Heitbaum, M.; Glorius, F.; Escher, I. Angew. Chem., Int. Ed. 2006, 45, 4732. doi: 10.1002/(ISSN)1521-3773
Ren, X. M.; Kong, S. N.; Shu, Q. D.; Shu, M. H. Chin. J. Chem. 2016, 34, 373. doi: 10.1002/cjoc.201500797
Dong, J.; Liu, Y.; Cui, Y. Chem. Commun. 2014, 50, 14949. doi: 10.1039/C4CC07648F
Wang, X. R.; Han, X.; Zhang, J.; Wu, X. W.; Liu, Y.; Cui, Y. J. Am. Chem. Soc. 2016, 138, 12332. doi: 10.1021/jacs.6b07714
Wang, C. A.; Zhang, Z. K.; Yue, T.; Sun, Y. L.; Wang, L.; Wang, W. D.; Zhang, Y.; Liu, C.; Wang, W. Chem. Eur. J. 2012, 18, 6718. doi: 10.1002/chem.201200753
An, W. K.; Han, M. Y.; Wang, C. A.; Yu, S. M.; Zhang, Y.; Bai, S.; Wang, W. Chem.-Eur. J. 2014, 20, 11019. doi: 10.1002/chem.201403002
Xu, H.; Gao, J.; Jiang, D. L. Nat. Chem. 2015, 7, 905. doi: 10.1038/nchem.2352
Xu, H.; Chen, X.; Gao, J.; Lin, J.; Addicoat, M.; Irle, S.; Jiang, D. Chem. Commun. 2014, 50, 1292. doi: 10.1039/C3CC48813F
Ma, L. Q.; Wanderley, M. M.; Lin, W. B. ACS Catal. 2011, 1, 691. doi: 10.1021/cs2001303
Wang, X.; Lu, S. M.; Li, J.; Liu, Y.; Li, C. Catal. Sci. Technol. 2015, 5, 2585. doi: 10.1039/C5CY00038F
Yoon, T. P.; Jacobsen, E. N. Science 2003, 299, 1691. doi: 10.1126/science.1083622
Noyori, R.; Ohkuma, T.; Kitamura, T.; Takaya, H.; Sayo, N.; Kumobayashi, H.; Akutagawa, S. ChemInform 1988, 19, 5856.
Hu, A. G.; Ngo, H. L.; Lin, W. B. Angew. Chem. 2003, 115, 6182. doi: 10.1002/(ISSN)1521-3757
Huang, Y. Y.; He, Y. M.; Zhou, H. F.; Wu, L.; Li, B. L.; Fan, Q. H. J. Org. Chem. 2006, 71, 2874. doi: 10.1021/jo052092m
Sun, Q.; Meng, X.; Liu, X.; Zhang, X.; Yang, Y.; Yang, Q.; Xiao, F. S. Chem. Commun. 2012, 48, 10505. doi: 10.1039/c2cc35192g
Wang, T.; Yuan, L.; Chen, X. K.; Li, C. Y.; Jiang, M.; Song, X.; Ding, Y. J. RSC Adv. 2016, 6, 28447. doi: 10.1039/C5RA23597A
Bayston, D. J.; Fraser, J. L.; Ashton, M. R.; Baxter, A. D.; Polywka, M. E. C.; Moses, E. J. Org. Chem. 1998, 63, 3137. doi: 10.1021/jo972330g
Kesanli, B.; Lin, W. B. Chem. Commun. 2004, 36, 2284.
Fan, Q. H.; Chen, Y. M.; Chen, X. M.; Jiang, D. Z.; Xi, F.; Chan, A. S. C. Chem. Commun. 2000, 31, 789.
Seki, T.; McEleney, K.; Crudden, C. M. Chem. Commu. 2012, 48, 6369. doi: 10.1039/c2cc31247f
Deng, G. J.; Yi, B.; Huang, Y. Y.; Tang, W. J.; He, Y. M.; Fan, Q. H. Adv. Synth. Catal. 2004, 346, 1440. doi: 10.1002/(ISSN)1615-4169
Patel, H. A.; Je, S. H.; Park, J.; Jung, Y.; Coskun, A.; Yavuz, C. T. Chem. Eur. J. 2014, 20, 772. doi: 10.1002/chem.v20.3
Qiu, D. F.; Zhao, Q.; Bao, X. Y.; Liu, K. C.; Wang, H. W.; Guo, Y. C.; Zhang, L. F.; Zeng, J. L.; Wang, H. Inorg. Chem. Commun. 2011, 14, 296. doi: 10.1016/j.inoche.2010.11.019
Geneste, F.; Moinet, C.; Ababou-Girard, S.; Solal, F. Inorg. Chem. 2005, 44, 4366. doi: 10.1021/ic048231k
Florea, M.; Sevinci, M.; Pârvulesscu, V. I.; Lemay, G.; Kaliaguine, S. Microporous Mesoporous Mater. 2011, 44, 483.
Marwan, J.; Addou, T.; Bélanger, D. Chem. Mater. 2005, 17, 2395. doi: 10.1021/cm047871i
Dai, L. X. Angew. Chem. Int. Ed. 2004, 43, 5726.; Padhi S. K.; Titu, D.; Pandian, N. G.; Chadha, A. Tetrahedron 2006, 62, 5133.
(a) Hu, A. ; Ngo, H. L. ; Lin, W. ChemInform 2004, 35, 2501.
(b) Chen, L. ; Ma, M. L. ; Peng, Z. H. ; Chen, H. Chin. J. Org. Chem. 2008, 28, 1724(in Chinese).
(陈丽, 马森林, 彭宗海, 陈华, 有机化学, 2008, 28, 1724.
Berthod, M.; Saluzzo, C.; Mignani, G.; Lemaire, M. Tetrahedron:Asymmetry 2004, 15, 639. doi: 10.1016/j.tetasy.2003.12.033
Pandey, P.; Farha, O. K.; Spokoyny, A. M.; Mirkin, C. A.; Kanatzidis, M. G.; Hupp, J. T.; Nguyen, S. B. T. J. Mater. Chem. 2011, 21, 1700. doi: 10.1039/c0jm03483e
Shu, W. F.; Guan, C. W.; Guo, W. H.; Wang, C. Y.; Shen, Y. J. J. Mater. Chem. 2012, 22, 3075. doi: 10.1039/c1jm15535k
Plietzsch, O.; Schilling, C. I.; Grab, T.; Grage, S. L.; Ulrich, A. S.; Comotti, A.; Sozzani, P.; Muller, T.; Bräse, S. New. J. Chem. 2011, 35, 1577. doi: 10.1039/c1nj20370c
Sun, Y.; Wan, X.; Guo, M.; Wang, D.; Dong, X.; Pan, Y.; Zhang, Z. G. Tetrahedron:Asymmetry 2004, 15, 2185. doi: 10.1016/j.tetasy.2004.04.034
Capozzi, G.; Roelens, S.; Talami, S. J. Org. Chem. 1993, 58, 7932. doi: 10.1021/jo00079a049
Hutton, J. A.; Goncalves, V.; Brannigan, J. A.; Paape, D.; Wright, M. H.; Waugh, T. M.; Roberts, S. M; Bell, A. S.; Wilkinson, A. J.; Smith, D. F.; Leatherbarrow, R. J.; Tate, E. W. J. Med. Chem. 2014, 57, 8664. doi: 10.1021/jm5011397

Figure 3 Nitrogen adsorption isotherms (a) measured at 77 K and pore size distribution (b) of POP-BINAP and POP-BINAPO

Figure 5 Effect of Ru/POP-BINAP recycling on the yield and ee of hydrogenation of ethyl benzoylacetate
Table 1. Optimization of reaction conditions for asymmetric hydrogenationa
|
|||||||
| Entry | p/MPa | T/℃ | t/h | Solvent | Yield b/% | Conv./% | eec/% |
| 1 | 2 | 85 | 20 | EtOH | 98 | 100 | 81 |
| 2 | 1 | 85 | 20 | EtOH | 64 | 73 | 61 |
| 3 | 3 | 85 | 20 | EtOH | 95 | 100 | 77 |
| 4 | 2 | 75 | 20 | EtOH | 83 | 91 | 71 |
| 5 | 2 | 95 | 20 | EtOH | 97 | 100 | 73 |
| 6 | 2 | 85 | 15 | EtOH | 76, 75 d | 85, 85 d | 80, 80 d |
| 7 | 2 | 85 | 25 | EtOH | 98 | 100 | 79 |
| 8 | 2 | 85 | 20 | MeOH | 82, 14 e | 100 | 83, 80 e |
| aReaction condition: substrate (1 mmol) in ethanol (1 mL), substrate/cata-lyst=200 (molar ratio). b Isolated yield. c Determined by HPLC using a Chiral OD-column. d The same reaction time was tried for another time. e Product of transterification: (S)-methyl 3-hydroxy-3-phenylpropanoate. | |||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Asymmetric hydrogenation of β-keto esters catalyzed by Ru/POP-BINAPa
|
||||
| Entry | 1 | R | Yield b/% | ee c/% |
| 1 | 1a | Ph | 98 | 81 |
| 2 | 1b | 2-ClC6H4 | 92 | 76 |
| 3 | 1c | 3-CH3C6H4 | 99 | 80 |
| 4 | 1d | 2-CH3C6H4 | 98 | 81 |
| 5 | 1e | 4-CH3C6H4 | 98 | 76 |
| 6 | 1f | PhCH2 | 99 | 89 |
| 7 | 1g | 4-F-C6H4CH2 | 96 | 92 |
| 8 | 1h | 2-F-C6H4CH2 | 95 | 92 |
| 9 | 1i | 4-MeOC6H4CH2 | 95 | 88 |
| 10 | 1j | 3-MeOC6H4CH2 | 98 | 89 |
| 11 | 1k | 4-CH3C6H4CH2 | 98 | 89 |
| 12 | 1l | 4-ClC6H4CH2 | 94 | 89 |
| 13 | 1m | 4-CF3C6H4CH2 | 99 | 89 |
| 14 | 1n | CH3 | 99 | 96 |
| 15 | 1p | (CH3)2CH | 99 | 92 |
| a Reaction condition: substrate (1 mmol) in ethanol (1 mL), substrate/cata-lyst=200 (molar ratio), 2 MPa H2, 85 ℃, 20 h. b Isolated yield, conversion: 100%. c Determined by HPLC using a Chiral OD-column. | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: