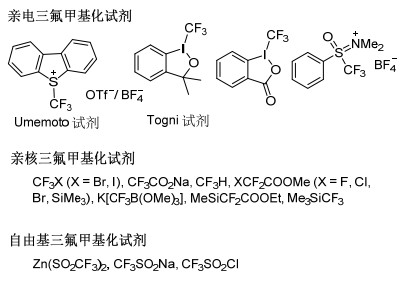
 图1
常见的三氟甲基化试剂
Figure1.
Common trifluoromethyl reagents
图1
常见的三氟甲基化试剂
Figure1.
Common trifluoromethyl reagents
引用本文:
惠人杰, 张士伟, 谭政, 吴小培, 冯柏年. 三氟甲基亚磺酸钠实现的三氟甲基化反应研究进展[J]. 有机化学,
2017, 37(12): 3060-3075.
doi:
10.6023/cjoc201709011

Citation: Hui Renjie, Zhang Shiwei, Tan Zheng, Wu Xiaopei, Feng Bainian. Research Progress of Trifluoromethylation with Sodium Trifluoromethanesulfinate[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3060-3075. doi: 10.6023/cjoc201709011
Citation: Hui Renjie, Zhang Shiwei, Tan Zheng, Wu Xiaopei, Feng Bainian. Research Progress of Trifluoromethylation with Sodium Trifluoromethanesulfinate[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3060-3075. doi: 10.6023/cjoc201709011
三氟甲基亚磺酸钠实现的三氟甲基化反应研究进展
English
Research Progress of Trifluoromethylation with Sodium Trifluoromethanesulfinate
Abstract:
Trifluoromethyl can increase the chemical and metabolic stability of drugs, improve its lipophilicity and bioavailability, and furthermore, enhance drug binding selectivities. Sodium trifluoromethanesulfinate (CF3SO2Na) is a stable inexpensive reagent, which has been widely used in the field of organic fluorine chemistry. The recent progress (2014~2017) in trifluoromethylation by employing CF3SO2Na as the trifluoromethyl source is summarized. In addition, the reactions of bifunctionalization, trifluoromethylation of aromatics, trifluoromethylthioization and other types of reactions are described respectively, with their applications and reaction mechanism. It is hoped that this review can be referred to in the studies of trifluoromethyl introduction.
-
Key words:
- sodium trifluoromethanesulfinate
- / trifluoromethylation
- / mechanism
-
自1957年抗癌药物5-氟尿嘧啶上市以来[1], 约15%~20%的现代药物(例如依法韦仑、甲氟喹和索拉非尼等)中均含有氟原子[2].氟的存在增加了有机药物的稳定性, 提高了化合物的脂溶性和渗透性, 也减少了药物耐药性的产生[3], 因此氟原子在现代药物设计和药物合成中具有举足轻重的地位[4].
氟原子本身具有高活性, 在反应中不易控制, 因此在药物合成和设计中多采用引入含氟基团的方法, 其中三氟甲基化是常用合成手段之一.三氟甲基的构建主要分为C(sp)—CF3、C(sp2)—CF3、C(sp3)—CF3[5], 常用的CF3的主要来源有CF3I、CF3SO2Cl、Langlois reagent (CF3SO2Na)[6]、Umemoto reagents[7]、Togni reagents[8]、Ruppert-Prakash reagent[9]、[(phen)CuCF3][10]、[(Ph3P)3Cu-(CF3)][11]、HCF2Cl[12]等(图 1).以三氟甲基为氟源的药物结构改良增加了候选药物的化学稳定性、代谢稳定性以及电负性, 改善了药物的亲脂性和生物利用度, 增强了药物的结合选择性[2], 目前在药物、农药、染料、有机材料等方面得到了广泛的运用[13].
图1
常见的三氟甲基化试剂
Figure1.
Common trifluoromethyl reagents
传统的CF3源多是气态(如CF3I, CF3Br)或需要多步合成(如Togni试剂, Umemoto试剂)得到, 这些性质严重限制了其在实际工艺中的应用.由于具有良好的反应性能和易于处理的特性, 固体试剂三氟甲磺酸钠(CF3SO2-Na, Langlois试剂)脱颖而出.该试剂最早开发于1989年, 作为CF3自由基的一种便利来源, 应用于单电子氧化反应[14].此后直到2011年, Baran等[6]报道了利用三氟甲基亚磺酸钠为三氟甲基源, 使用叔丁基过氧化氢(TBHP)作为氧化剂的杂环的三氟甲基化反应(Eq. 1).该方法具有可扩展性, 广泛适用于富电子和缺电子杂环, 操作简便.反应为自由基反应机理, CF3SO2Na在TBHP的作用下形成三氟甲基自由基(Scheme 1).该研究作为一个重要的突破点, 为后续的三氟甲基化反应提供了重要思路, 出现了大量关于CF3SO2Na作为三氟甲基试剂的文献报道. 2014年Zhang等[15]对早期Langlois试剂参与的相关反应进行了综合报道, 重点叙述了三氟甲基亚磺酸钠作为三氟甲基化试剂的四类主要反应(Scheme 2), 包括: (1)对硼酸类、三氟硼酸钾类化合物的取代反应; (2)对烯烃、炔烃等不饱和键的加成反应; (3)对(杂)芳烃的取代反应; (4)对α, β-不饱和烯烃和炔烃的环化和三氟甲基化反应.
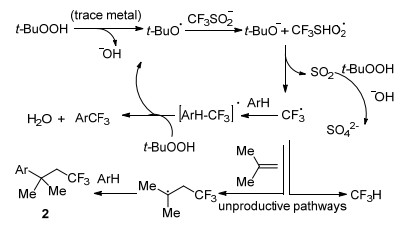 图式 1
TBHP-介导的三氟甲基化反应机理
Scheme1.
Mchanism of TBHP-mediated trifluoromethylation
图式 1
TBHP-介导的三氟甲基化反应机理
Scheme1.
Mchanism of TBHP-mediated trifluoromethylation
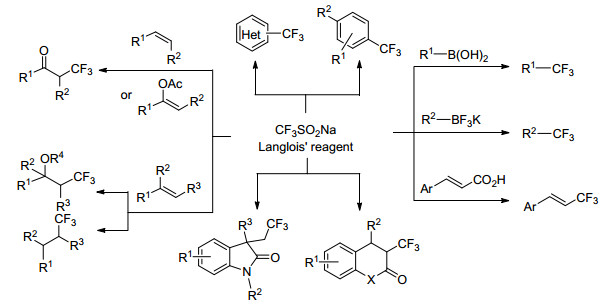 图式 2
CF3SO2Na对一些化合物的三氟甲基化
Scheme2.
Trifluoromethylation of organic compounds with CF3SO2Na
图式 2
CF3SO2Na对一些化合物的三氟甲基化
Scheme2.
Trifluoromethylation of organic compounds with CF3SO2Na
本文对2014年至2017年报道的以CF3SO2Na作为三氟甲基化试剂的反应进行了归纳.根据反应类型, 按照双官能团化、芳烃三氟甲基化、三氟甲硫基化及其他反应的分类介绍了CF3SO2Na在不同反应中的应用及相关的反应机理, 希望为今后三氟甲基的引入提供参考.
1 CF3SO2Na对各种不饱和键的双官能团化反应
Zhang小组[16]利用Cu(Ⅰ)作为催化剂, CF3SO2Na作为氟源, 实现了对芳基(杂芳基)烯醇乙酸酯的加成, 合成了一系列α-三氟甲基酮衍生物(Eq. 2).该方法反应条件温和, 反应迅速, 电子效应对该反应没有显著的影响.对反应机理进行了初步研究, 使用不同剂量的TEMPO作为自由基捕捉剂, 产物随着TEMPO的增加而减少并伴随着TEMPO-CF3的生成, 当加入4 equiv.的TEMPO时, 仅有痕量的产物生成, 由此判断该反应可能为自由基机理(Scheme 3).
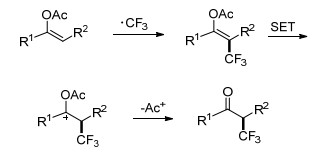 图式 3
α-CF3取代的酮可能的机理
Scheme3.
Possible mechanism of α-CF3-substituted ketones
图式 3
α-CF3取代的酮可能的机理
Scheme3.
Possible mechanism of α-CF3-substituted ketones
CF3取代的碘乙烯是一种有价值的合成结构单元, 在许多偶联反应中都有利用. Liu小组[17]在水性介质中使用三氟甲基亚磺酸钠和五氧化二碘这两种固体试剂, 对各种烯烃和炔烃进行选择性的碘三氟甲基化, 得到主要产物β-CF3-烯基碘及烷基碘(Scheme 4).该方法避免了传统方法中使用CF3I所带来的缺点, 各种芳基和烷基取代的烯烃在该反应条件下都有较高的产率.当利用炔烃取代烯烃时, 在NaHCO3的作用下, 获得一系列(E)-β-CF3-烯基碘.对反应机理进行了研究, 利用2-甲基-2-亚硝基丙烷(MNP)作为自由基自旋捕捉剂, 通过电子自旋共振(ESR)实验验证了该反应为单电子氧化的自由基反应(Scheme 5).
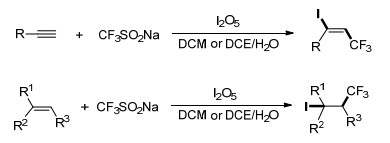 图式 4
烯烃和炔烃的碘三氟甲基化
Scheme4.
Strategies for iodotrifluoromethylation of alkenes and alkynes
图式 4
烯烃和炔烃的碘三氟甲基化
Scheme4.
Strategies for iodotrifluoromethylation of alkenes and alkynes
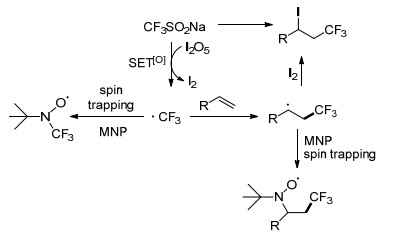 图式 5
可能的反应机理
Scheme5.
Possible reaction mechanism
图式 5
可能的反应机理
Scheme5.
Possible reaction mechanism
Li小组[18]报道了光照条件下, 用Ir作为催化剂, 进行末端烯烃和迈克尔受体的三氟甲基化反应(Eq. 3), 酯、酰胺、醚、醛、砜、酮和芳基硼酸酯等在该反应中都有很好的产率.末端烯烃的加成符合反马式规则, 而非末端烯烃则不适用于该反应; 环状迈克尔受体的双键虽能反应但产率不佳; 雌素酮类衍生物有着很好的反应效果.该小组通过使用CD3OD, 确认了CH3OH在整个反应过程中仅作为质子化试剂提供了烯烃β位的H, 而并未参与氧化还原过程.该小组提出反应可能为自由基机理.反应中亚磺酸根阴离子还原激发的*Ir(Ⅲ)催化剂以产生三氟甲基自由基, 三氟甲基自由基下一步与烯烃反应生成相对稳定的亚甲基自由基.亚甲基自由基氧化Ir(Ⅱ), 生成Ir(Ⅲ), 最终完成催化循环(Scheme 6).
 图式 6
可能的反应机理
Scheme6.
Possible reaction mechanism
图式 6
可能的反应机理
Scheme6.
Possible reaction mechanism
Qing小组[19]报道了铁介导的苯乙烯和脂肪族烯烃的氯三氟甲基化反应(Eq. 4), 其中FeCl3作为Cl源, CF3SO2Na作为CF3源.该方法为合成氯三氟甲基化合物的开辟了新的途径, 值得注意的是, 该方法对乙烯雌激素和乙烯基-N-苯甲酰基-L-酪氨酸乙酯的三氟甲基化也有很好的效果, 为其他天然药物的三氟甲基化提供了一条可能途径.关于机理, 该小组提出在FeCl3和K2S2O8处理后, 通过单电子转移(SET), CF3SO2Na失去一个电子生成三氟甲基自由基.然后, 烯烃1与三氟甲基自由基反应, 得到中间体A.中间体A可以被FeCl3和K2S2O8进一步氧化以产生阳离子中间体B, 其通过氯化物阴离子进行亲核攻击以形成氯三氟甲基化产物2 (Scheme 7).
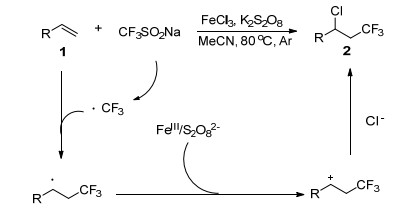 图式 7
可能的反应机理
Scheme7.
Possible reaction mechanism
图式 7
可能的反应机理
Scheme7.
Possible reaction mechanism
Vicic小组[20]使用MnCl2·4H2O/O2体系对苯乙烯衍生物进行三氟甲基化, 反应条件温和, 产率在40%~80%.该反应体系从CF3SO2Na中获得游离CF3自由基, 使其与苯乙烯衍生物反应, 选择性生成相应的β-三氟甲基化醇和α-三氟甲基化酮(Eq. 5), 产物比例为10:1左右.在反应结束后加入还原剂, 可获得单一产物β-三氟甲基化醇.
Maiti小组[13]报道了通过苄基自由基中间体合成α-三氟甲基酮的反应.该方法为一种用(杂)芳基乙炔直接合成α-三氟甲基酮的新方法(Eq. 6).含不同取代基的芳基炔烃在该反应条件下均表现出良好的活性, 直链烷烃、环状烷烃或杂环(如噻吩、吡啶、嘧啶、吲哚、苯并噻唑等)取代的炔烃都适用于该方法.机理表明, 三氟甲烷亚磺酸钠与银催化剂首先形成三氟甲基自由基, 随后三氟甲基自由基进攻炔烃, 产生α-苯乙烯自由基中间体, 该反应中间体与氧气发生反应后生成过氧化物, 最终进一步氧化后通过烯醇互变得到终产物(Scheme 8, Eq. 7).
 图式 8
可能的反应机理
Scheme8.
Possible reaction mechanism
图式 8
可能的反应机理
Scheme8.
Possible reaction mechanism
Qing小组[21]报道了在TBHP/CuCl反应体系中CF3SO2Na对烯烃的双三氟甲基化反应(Eq. 8).该反应是一条简便的双三氟甲基路线, 克服了之前该类反应产率低、适用范围小的缺点.该反应通过自由基机理进行.反应中, 通过调节CF3自由基的浓度实现该反应的化学选择性; 而CuCl的增加可以减少二聚体的形成, 对烯烃的双三氟甲基化有明显的促进作用.此外, 一些1, 5-二烯反应后得到了相应1, 4-双(三氟甲基化)化合物.
早在2013年, Wu[22], Zhang[23]和Sodeoka[24]的小组就分别报道过使用过渡金属和Togni试剂对烯丙醇类化合物进行三氟甲基化的反应.考虑到过渡金属的毒性和Togni试剂的高售价, Gao小组[25]探索出了一种在无金属催化下使用CF3SO2Na作为CF3源的新方法(Scheme 9).该方法不仅对反应底物有着良好的适用性, 还实现了对α, α-二芳基烯丙醇的三氟甲基化.反应机理为在(NH4)2S2O8存在下加热, 三氟甲基亚磺酸钠生成三氟甲基自由基后与芳基烯醇双键反应, 随后发生芳基迁移, 生成中间体, 中间体失去一个质子后氧化生成最终产物(Scheme 10).
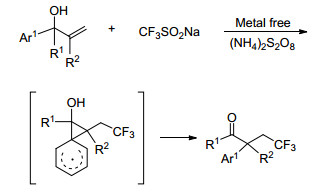 图式 9
烯丙醇的芳基三氟甲基化
Scheme9.
Aryltrifluoromethylation of allylic alcohols
图式 9
烯丙醇的芳基三氟甲基化
Scheme9.
Aryltrifluoromethylation of allylic alcohols
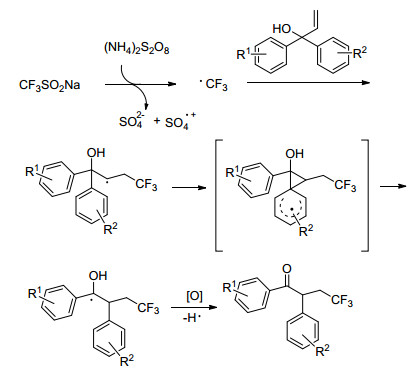 图式 10
可能的反应机理
Scheme10.
Possible reaction mechanism
图式 10
可能的反应机理
Scheme10.
Possible reaction mechanism
Huang小组[26]利用CF3SO2Na对α-溴代苯乙烯类化合物进行加成, 得到一系列2-(三氟甲基)苯乙酮衍生物(Eq. 9).该方法具有广泛的适用性, 可以在不使用任何额外的化学计量的有机氧化剂或过渡金属催化剂的情况下生成各种α-三氟甲基酮.控制实验表明, K2S2O8在自由基引发过程中扮演着重要角色, 并且O2的存在促进了自由基链式反应的持续进行, 使反应向着产物生成方向进行(Scheme 11).
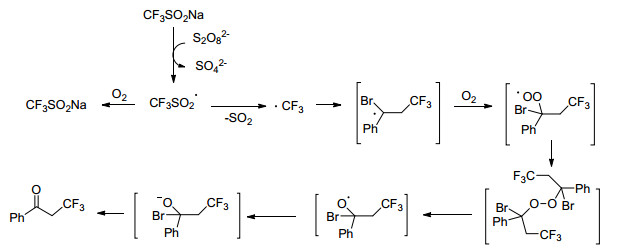 图式 11
可能的反应机理
Scheme11.
Possible reaction mechanism
图式 11
可能的反应机理
Scheme11.
Possible reaction mechanism
Huang小组[27]使用O2或空气在NMP中生成过氧化物, 并与CF3SO2Na反应得到三氟甲基自由基, 使之与苯乙烯类化合物形成β-CF3取代的叔醇, 且不需要额外的氧化剂或者过渡金属催化剂(Scheme 12).初步的机理实验证明O2的扩散速度可影响反应的产率(Scheme 12).反应过程中, 电子效应对α-甲基苯乙烯的衍生物并没有明显的影响, 即使是容易发生过氧化的苯硫基取代的α-甲基苯乙烯在该反应条件下也有较好的产率.结果显示, 杂芳基和非共轭烯均适用于该方法.
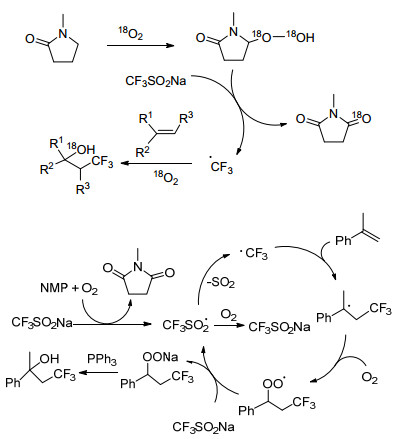 图式 12
α-取代的苯乙烯的三氟甲基化及其可能的反应机理
Scheme12.
Trifluoromethylation of α-substituted styrenes and their possible reaction mechanism
图式 12
α-取代的苯乙烯的三氟甲基化及其可能的反应机理
Scheme12.
Trifluoromethylation of α-substituted styrenes and their possible reaction mechanism
Xia小组[28]使用紫外光在感光剂(如苯甲酮、蒽醌等)存在下对烯烃进行双官能化(Scheme 13), 通过自由基环化过程直接构建C(sp3)—CF3键和C—C键, 得到多种菲类和蒽酮类化合物(Scheme 14).针对该反应的条件适用性研究表明, α, β-不饱和芳香酮选用苯甲酮作为感光剂反应效果最好; γ, δ-不饱和芳香酮选用蒽醌效果最佳.该反应操作简单、原子利用率高、有良好的官能团耐受性和广泛的适用范围, 是一条较为理想的合成路径.
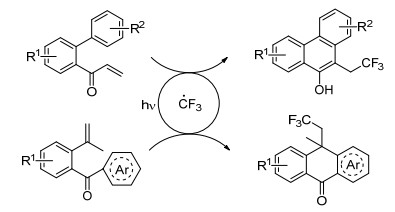 图式 13
在紫外光下用CF3SO2Na进行三氟甲基化
Scheme13.
Trifluoromethylation with CF3SO2Na via UV light irradiation
图式 13
在紫外光下用CF3SO2Na进行三氟甲基化
Scheme13.
Trifluoromethylation with CF3SO2Na via UV light irradiation
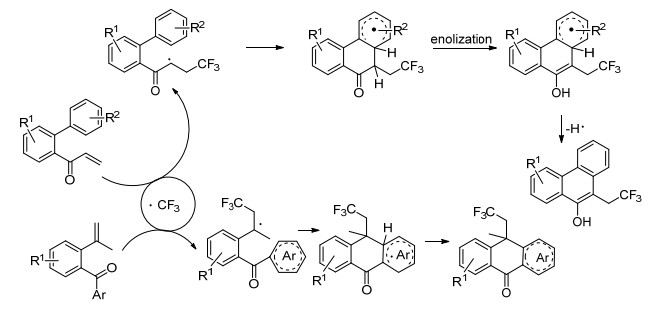 图式 14
可能的反应机理
Scheme14.
Possible reaction mechanism
图式 14
可能的反应机理
Scheme14.
Possible reaction mechanism
Fu小组[29]使用N-烯丙基酰胺在无金属催化下进行三氟甲基化, 并制备相应的噁唑啉(Eq. 10).该路线是第一例合成CF3取代的噁唑啉的实例, 为环状化合物的三氟甲基化提供了一条有效的途径.在合成不同取代基的产物后发现, N-烯丙基酰胺的Ar上含给电子基团比含吸电子基团的反应效果好.此外, 空间位阻对该反应也有一定的影响, 在尝试合成手性噁唑啉时, 使用(R)-N-(1-苯基烯丙基)苯甲酰胺作为底物时, 得到了一对非对应异构体.推测其反应机理为, PhI(OAc)2与CF3SO2Na反应生成Ⅰ和Ⅱ, 并在加热条件下Ⅰ和Ⅱ解离出Ⅳ或Ⅴ, 加入N-烯丙基酰胺后得到Ⅵ, 并在Ⅲ的作用下发生Ⅵ分子内环化生成目标产物(Scheme 15).
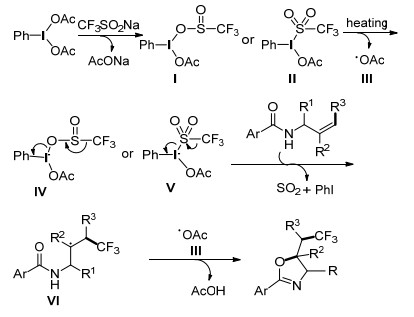 图式 15
含有CF3的噁唑啉的可能反应机理
Scheme15.
Possible mechanism of CF3-containing oxazolines
图式 15
含有CF3的噁唑啉的可能反应机理
Scheme15.
Possible mechanism of CF3-containing oxazolines
2012年, Liu研究小组[30]报道了使用TMSCF3/CsF作为CF3源, Pd/Yb催化的烯烃三氟甲基化.三氟甲基试剂和催化剂比较昂贵且反应条件苛刻, Liang小组[5]报道了一种使用铜催化的芳基丙烯酰胺内环化生成羟吲哚同时引入三氟甲基的方法(Scheme 16).该反应使用了相对廉价的CF3SO2Na, 在常温下水性介质中反应, 避免了有机溶剂的挥发, 水性介质的可循环利用也更环境友好.该反应能有效合成各种含CF3的羟基吲哚.初步机理研究表明, 该反应涉及到了C(sp3)-PdⅣ(CF3)中间体, 其经历了还原消除以生成C(sp3)—CF3键.
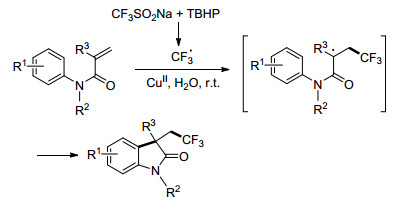 图式 16
铜催化的N-芳基丙烯酰胺的三氟甲基化
Scheme16.
Copper-catalyzed trifluoromethylation of N-aryl-acrylamides
图式 16
铜催化的N-芳基丙烯酰胺的三氟甲基化
Scheme16.
Copper-catalyzed trifluoromethylation of N-aryl-acrylamides
Yang小组[31]使用CF3SO2Na作为三氟甲基化试剂, 并以过硫酸钾作为氧化剂, 对N-芳基丙烯酰胺类化合物进行三氟甲基化, 得到CF3取代的羟吲哚衍生物(Eq. 11).苯胺部分的取代基差异对反应产率没有明显影响, 芳基对位含F、Cl、Br、CN取代基的N-芳基丙烯酰胺表现出良好的活性, 这为进一步对CF3取代的羟吲哚衍生物进行结构修饰提供了可能.当2 equiv.的TEMPO加入后, 反应完全抑制, 表明该反应通过自由基机理进行(Scheme 17).
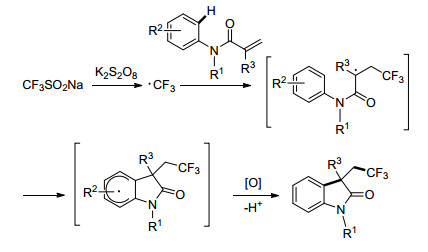 图式 17
可能的反应机理
Scheme17.
Possible reaction mechanism
图式 17
可能的反应机理
Scheme17.
Possible reaction mechanism
Qiu小组[32]发现了一种使用铜催化活泼炔烃进行双官能化三氟甲基化反应的方法, 并得到了相应的3-(三氟甲基)-螺[4,5]三烯酮类化合物(Eq. 12), 产物是许多天然产品和药物的常见结构.该反应实现了同时形成两个碳-碳单键和一个碳-氧双键的炔双官能化.当2.5 equiv. TEMPO加入反应体系中, 反应被有效抑制, 仅检测到痕量产物, 由此表明反应可能通过自由基机理反应(Scheme 18).
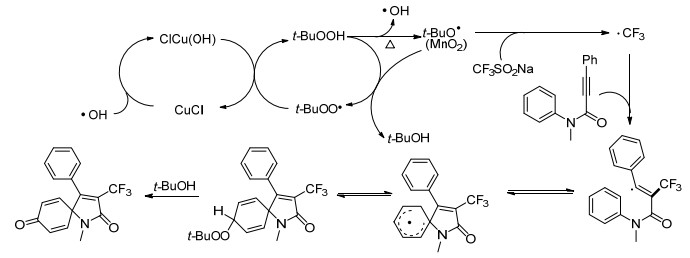 图式 18
可能的反应机理
Scheme18.
Possible reaction mechanism
图式 18
可能的反应机理
Scheme18.
Possible reaction mechanism
Liu小组[33]报道了N-芳基甲基丙烯酰胺在I2O5和CF3SO2Na的作用下发生自由基串联反应, 实现了环化和三氟甲基化, 得到相应的CF3取代的羟吲哚类化合物(Scheme 19).电子自旋共振(ESR)研究表明, 反应过程中涉及到自由基过程.该小组仍在对该反应进行进一步的研究.
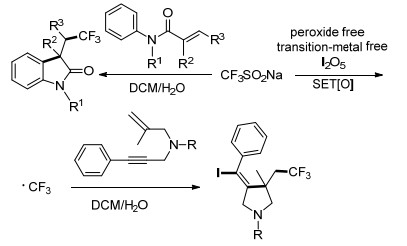 图式 19
I2O5介导的N-芳基甲基丙烯酰胺的环化和三氟甲基化
Scheme19.
I2O5-mediated trifluoromethylation/cyclization of N-arylmethacrylamides
图式 19
I2O5介导的N-芳基甲基丙烯酰胺的环化和三氟甲基化
Scheme19.
I2O5-mediated trifluoromethylation/cyclization of N-arylmethacrylamides
2017年, Kumar小组[34]在无金属、氧化剂以及可见光诱导的条件下, 使用三氟甲基亚磺酸钠, 实现了对不稳定烯烃的三氟甲基化(Eq. 13), 用于合成各种含CF3的结构, 诸如苯并呋喃、苯并噻吩和吲哚等.其反应机理是光诱导产生三氟甲基自由基, 与烯烃加成, 然后环化, 提供将电子转移到PQ并产生乙烯基阳离子的乙烯基.或者, 电子转移可以从CF3加成的烯烃部分发生, 形成碳正离子, 进行正离子环化以产生乙烯基碳正离子.随后向乙烯基正离子中加入水, 除去氢气形成三氟甲基化的C3-芳酰基/酰化杂环(Scheme 20).
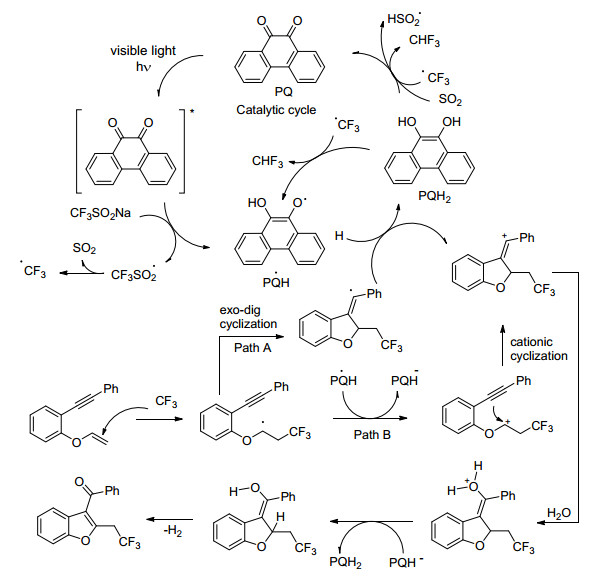 图式 20
可能的反应机理
Scheme20.
Possible reaction mechanism
图式 20
可能的反应机理
Scheme20.
Possible reaction mechanism
用于乙烯基三氟甲基化的常见底物有乙烯基硼酸、乙烯基磺酸盐、乙烯基卤化物、乙烯基硼酸盐、乙烯基羧酸或硝基烯烃等预官能化烯烃.该方法虽然具有良好的反应性和区域选择性, 但是制备预官能化底物需要进行多步合成. Loh小组[35]开发了在TBHP存在下使用CF3SO2Na作为三氟甲基源的苯乙烯衍生物的有效铜(Ⅰ)催化三氟甲基化(Eq. 14).机理研究表明,反应可能通过自由基途径进行, 该反应提供了一种直接获取三氟甲基苯乙烯衍生物的方法, 值得一提的是, 在该反应中苯乙烯衍生物获得优异的顺反选择性(Scheme 21).
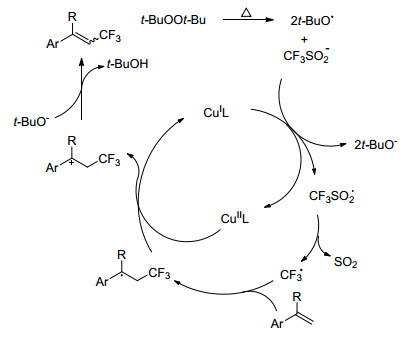 图式 21
可能的反应机理
Scheme21.
Possible reaction mechanism
图式 21
可能的反应机理
Scheme21.
Possible reaction mechanism
2017年, Shen小组[36]报道了一种在Blue LED照射下, 苯乙烯通过CF3SO2Na引入三氟甲基的方法(Eq. 15), 反应通过自由基机理进行, 是对经典催化羧化反应的补充.该方法实现了此前在羧化领域未能达到的设想.该小组对这一方面的研究还在进行进一步探索.
2 CF3SO2Na参与的三氟甲基自由基芳基化反应
Li课题组[37]使用Cu(Ⅰ)作为催化剂对香豆素及其衍生物进行三氟甲基化(Scheme 22), 并对苯环上不同取代基的电子效应进行了研究, 发现含强供电子基团的香豆素衍生物反应效果最佳, 其次是含弱供电子基团的香豆素衍生物, 含吸电子基团的香豆素衍生物反应效果最差.值得关注的是该小组将连续流微反应器引入到了该反应中并取得良好的效果, 放大反应也得到了相对理想的结果, 此方法为以后的大规模生产提供了一种新思路.该小组提出反应可能的机理, 首先Cu(Ⅰ)还原CF3SO2Na, 通过单电子转移生成三氟甲基自由基, 然后三氟甲基自由基进攻香豆素衍生物, 形成更为稳定的中间体, 该中间体被Cu(Ⅱ)氧化, 最后去质子化得到最终产物(Scheme 23).
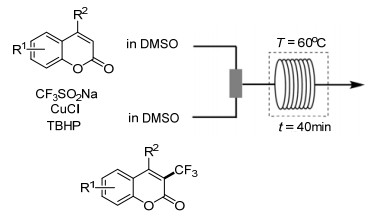 图式 22
不饱和α-三氟甲基酮和3-三氟甲基香豆素的合成
Scheme22.
Synthesis of unsaturated α-trifluoromethyl ketones and 3-trifluoromethyl coumarins
图式 22
不饱和α-三氟甲基酮和3-三氟甲基香豆素的合成
Scheme22.
Synthesis of unsaturated α-trifluoromethyl ketones and 3-trifluoromethyl coumarins
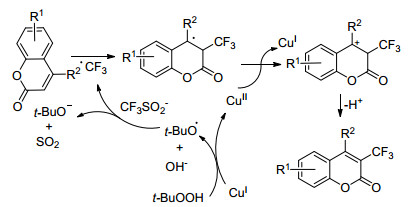 图式 23
可能的反应机理
Scheme23.
Possible reaction mechanism
图式 23
可能的反应机理
Scheme23.
Possible reaction mechanism
Qing小组[38]报道了用芳香四氟硼酸重氮盐在铜催化下合成三氟甲基化产物和三氟甲磺酰化产物的反应(Eqs. 16, 17).该反应是对Sandmeyer三氟甲基化反应的补充和延伸, 具有较高的应用价值.反应以二甲亚砜(DMSO)为溶剂, Cu2O催化时得到三氟甲磺酰化产物; 以TBHP为氧化剂, 在CuBF4(MeCN)4/Tpy作用下, 得到三氟甲基化产物.两种反应条件都比较温和, 且适用范围较广.实验证明CF3自由基参与反应, 该小组提出可能的机理为重氮盐脱去氮气生成芳基自由基, 同时, Cu(Ⅰ)将t-BuOOH转变成t-BuO自由基, 然后其将CF3SO2Na转化为CF3自由基, 在Cu盐的作用下, 最终生成三氟甲基化产物(Scheme 24).
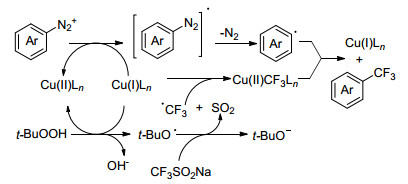 图式 25
可能的反应机理
Scheme24.
Possible reaction mechanism
图式 25
可能的反应机理
Scheme24.
Possible reaction mechanism
前期报道的关于乙酰苯胺结构的三氟甲基化反应, 存在反应底物单一、三氟甲基试剂价格高、反应条件苛刻等缺点, 难以广泛应用. Dai课题组[39]在前人的基础上对此类反应进行了优化, 在无金属催化下使用CF3SO2Na对乙酰苯胺类化合物进行邻位三氟甲基化(Eq. 18).据Xi[40a]和Shi[40b]小组此前的报道, 乙酰氨基与新戊酰胺应为邻位导向基团, 而Dai小组在试验过程中发现了不同结果.他们使用了对位取代的苯胺结构对其进行了三氟甲基化, 发现除对位为强吸电子基团的苯胺化合物, 其余化合物的反应效果均不理想.由此确认了乙酰胺基对于邻位三氟甲基化反应具有重要作用.在反应体系加入1或4 equiv.的TEMPO, 没有观察到预期的三氟甲基化产物, 在反应液中观察到TEMPO-CF3复合物, 这些结果表明反应是通过自由基途径进行的.
咪唑杂环具有多种生物活性, 是重要的药效基团, 虽然人们在其结构修饰方面做了大量的工作, 但对咪唑杂环的三氟甲基化却未引起人们的足够重视. 2015年, Chen的小组[41]提出了一条咪唑杂环三氟甲基化的有效合成途径(Eq. 19).他们以[Bmim]BF4/H2O (V:V=1:1)为溶剂, 使用Cu(Ⅱ)和TBHP将三氟甲基自由基从CF3SO2Na中游离出来, 使其与咪唑杂环形成中间产物, 再在TBHP的作用下氧化、脱氢得到最终产物(Scheme 25).
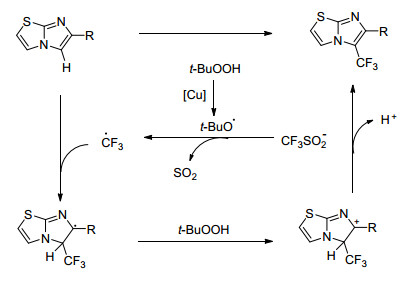 图式 25
可能的机理
Scheme25.
Possible mechanism
图式 25
可能的机理
Scheme25.
Possible mechanism
近年来报道的三氟甲基化主要为过渡金属和过氧化物体系如Cu(SO3CF3)2/t-BuOOH, LnPd/TESCF3或TMSCF3, RuCl2(PPh3)3/CF3SO2Cl, ReO3Me/Togni reagents, Ru(phen)3Cl2/CF3SO2Cl等[42], 不仅反应成本较高, 产物的后处理也相对复杂.考虑到以上缺点, Lipshutz小组[43]以水作为溶剂在室温下, 使用水溶性胶束作为反应体系(Eq. 20), 不仅避免了过渡金属的使用, 减少了CF3SO2Na和TBHP的一半用量, 而且反应溶液可循环使用4~5次, 表现出良好的绿色环保性能.
Zhou小组[44]开发了一种Mn(OAc)3催化的方法, 对香豆素的3位直接进行三氟甲基化得到相应的产物.该方法亦可应用于喹啉酮和嘧啶酮(Eq. 21).与大多数三氟甲基化反应相同, 该反应是一个自由基的反应.在底物拓展过程中发现, 当8位存在取代基时, 香豆素的3位和苯环上均可被三氟甲基取代, 对反应效果有一定影响.该小组提出反应机理为在Mn(OAc)3存在下, CF3SO2Na生成CF3自由基, 然后进攻香豆素的3位, 形成相对稳定的中间体, 在Mn(OAc)3作用下, 最终去质子化得到终产物(Scheme 26).
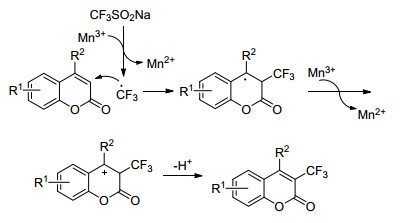 图式 26
可能的反应机理
Scheme26.
Possible reaction mechanism
图式 26
可能的反应机理
Scheme26.
Possible reaction mechanism
Zhang小组[45]实现了在相对温和条件下使用CuCl/ Ag2CO3作为催化剂对α, β-不饱和羧基脱羧, 并使用CF3SO2Na引入CF3基团, 得到具有优异E/Z选择性的末端三氟甲基取代的苯乙烯结构(Eq. 22).加入1或4 equiv.的TEMPO, 原反应明显被抑制, 证明该反应通过自由基途径进行(Scheme 27).
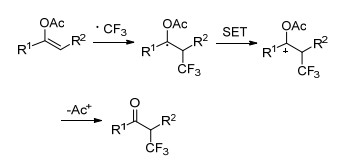 图式 27
可能的反应机理
Scheme27.
Possible reaction mechanism
图式 27
可能的反应机理
Scheme27.
Possible reaction mechanism
2013年, Baudoin小组[46]报道了一种氯化钴催化的使用Togni试剂实现N, N-二烷基腙的三氟甲基化的方法, 但该方法存在着底物范围窄、反应产率低、原料昂贵的缺点. 2017年, 我们发现了一种更高效的方法[47](Eq. 23), 即以三氟甲基亚磺酸钠作为三氟甲基源, 醋酸碘苯作为氧化剂, 实现N, N-二烷基腙的三氟甲基化.该反应条件温和, 底物范围广, 而且反应产率很高.反应首先由PhI(OAc)2与三氟甲磺酸钠反应产生Ⅰ, 进而产生Ⅱ和三氟甲基两个自由基, 然后CF3自由基被腙捕获产生三氟甲酰胺基氮原子, 随后在Ⅱ作用下去质子, 得到目标产物(Scheme 28).
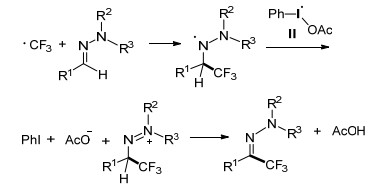 图式 28
腙三氟甲基化的反应机理
Scheme28.
mechanism for trifluoromethylation of hydrazones
图式 28
腙三氟甲基化的反应机理
Scheme28.
mechanism for trifluoromethylation of hydrazones
2017年, Wu小组[48]报道了无金属催化下在烯胺上引入三氟甲基的方法, 避免了成本高、有污染的缺点(Eq. 24).其反应机理如Scheme 29所示, 首先, TBHP作为氧化剂氧化CF3SO2Na产生CF3自由基.随后, 缺电子的CF3基团选择性地攻击电负性更强的β-C(路径a), 生成含CF3的自由基中间体A, 在氧化条件下生成B, 脱掉氢形成中间体C.最终产物是中间体C的互变异构体, 是热力学稳定的产物.但是不能排除在TBHP存在下可能产生的中间体D, 与CF3基团直接偶联形成亚胺C(路径b)的可能性.
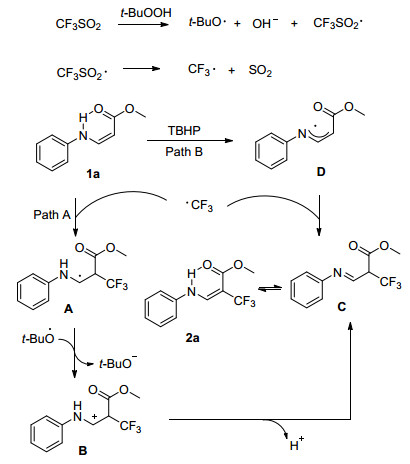 图式 29
可能的反应机理
Scheme29.
Possible reaction mechanism
图式 29
可能的反应机理
Scheme29.
Possible reaction mechanism
Mizuno的小组[49]使用CF3SO2Na(Langlois'试剂)作为CF3源对(异)芳烃的直接C—H三氟甲基化, 使用O2作为末端氧化剂.该小组首次报道, 在催化量的磷钼钒酸的存在下, 各种取代的苯和杂芳族化合物均可以转化为相应的三氟甲基化产物(Eq. 25).反应通过自由基途径进行(Scheme 30).反应的应用范围较广泛, 比如含取代的苯, 萘, 吡嗪, 吡啶, 喹啉, 噻吩等均可进行三氟甲基化.其反应机理如, 首先, 发生从CF3SO2Na到杂多酸催化剂(HPA)的单电子转移得到还原的HPA和CF3SO2中间体(步骤1), 然后立即歧化成CF3和SO2(步骤2).而后, CF3与芳烃反应生成自由基中间体A(步骤3), 随后HPA接受来自中间体A的电子和质子, 得到相应的三氟甲基化产物(步骤4).在步骤1和步骤4中形成的还原HPA被氧气再氧化.
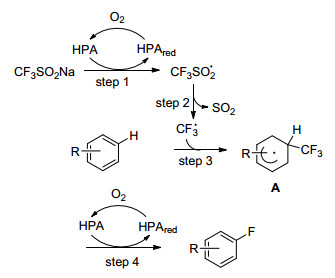 图式 30
HPA催化三氟甲基化的可能反应机理.
Scheme30.
A possible reaction mechanism for HPA-catalyzed trifluoromethylation
图式 30
HPA催化三氟甲基化的可能反应机理.
Scheme30.
A possible reaction mechanism for HPA-catalyzed trifluoromethylation
Kroutil小组[50]提出使用生物催化剂(漆酶), t-BuOOH以及CF3SO2Na或Baran’s亚硫酸锌的三氟甲基引入未保护的酚类的概念(Eq. 26).该方法依赖于两种自由基物质的重组, 即漆酶和CF3自由基直接产生的苯酚自由基阳离子.反应机理研究表明(Scheme 31), 该反应首先是漆酶通过单电子转移, 氧化苯酚形成酚自由基阳离子.后者再与CF3自由基反应, 得到阳离子中间体, 重新分解成最终产物.通过生物催化生成的两种自由基物质和化学反应中的另一种重组, 实现了基本的C—CF3键形成.此外, 该方法显示出高官能团耐受性, 允许醛, 酯和酮的转化而不分解, 这使得该方法适用于后期三氟甲基化.该方法在具有高区域选择性的温和反应条件下进行.
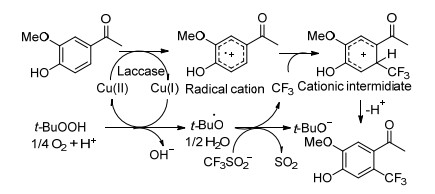 图式 31
可能的反应机理
Scheme31.
Possible reaction mechanism
图式 31
可能的反应机理
Scheme31.
Possible reaction mechanism
3 CF3SO2Na参与的三氟甲硫基化反应
由于三氟甲硫基(SCF3)的吸电子效应和极高的亲油性(π=1.44), 三氟甲基硫化物(CF3SR)在各种药物、农药和功能材料的合成中起着重要的作用, 具有很大的潜力[51].三氟甲磺酰(CF3SO2)单元作为SCF3来源为三氟甲基硫醇化开辟了新的领域.与其它SCF3试剂相比, 三氟甲磺酸钠(CF3SO2Na, Langlois试剂)容易获得、便宜且易于处理.
Yu小组[52]报道了在PPh3作用下, CuCl和CF3SO2Na在氮气保护下生成CuSCF3后, 在1, 3-二甲基-2-咪唑啉酮(DMI)溶剂中110 ℃氮气保护下对(杂)芳基碘进行三氟甲基硫醇化的反应(Eqs. 27, 28).电子效应对该反应影响不明显, (杂)芳环碘化物上被供电子和吸电子基团取代时, 都表现出良好的反应效果(74%~93%).
2017年, Lu小组[53]开发了一种新的无过渡金属催化的反应途径, 即在PCl3存在下, 用CF3SO2Na直接对富马酸芳烃进行三氟甲基硫醇化和三氟甲基亚砜化(Eqs. 29, 30).值得注意的是, PCl3在反应中同时用作还原和氯化试剂, 表现出良好的原子经济性.其反应机理为(Scheme 32), CF3SO2Na可以与PCl3反应形成中间体A, 其被吲哚攻击两次, 得到D和中间体B.中间体B与PCl3反应生成P(OH)Cl2 (C).通过PCl3和C还原D得到相应的吲哚三氟甲硫醚, 但是不能排除吲哚与其它物质的三氟甲基亚砜氧化反应.
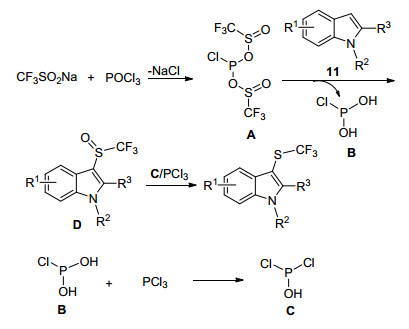 图式 32
可能的反应机理
Scheme32.
Possible reaction mechanism
图式 32
可能的反应机理
Scheme32.
Possible reaction mechanism
Zhang小组[54]用亚磷酸二乙酯作为还原剂和CuCl作为催化剂形成C—SCF3键; Vicic等[55]报道了用CF3SO2Na合成CuSCF3的方法, 并且所产生的CuSCF3可以直接用于(异)芳基碘化物的三氯甲基硫醇化; Shibata组[56]开发了吲哚衍生物与三氟甲磺酰氯(CF3SO2Cl)的三氟甲基硫醇.然而, 这三种系统在相对高(110 ℃)和低(78℃至室温)的温度下操作. Cai小组[51]开发了在温和条件下吲哚衍生物的过渡金属的直接三氟甲基硫醇化.该方案的底物范围也可扩展到吡咯和烯胺(Eq. 31).其反应机理如Scheme 33所示, 首先CF3SO2Na、三苯基膦和N-氯亚酰胺反应产生三氟甲基亚硫酰氯, 同时形成三苯基氧化膦和邻苯二甲酰亚胺钠.吲哚经CF3SCl亲电攻击, 得到三氟甲硫基化产物, 同时发生质子化产生邻苯二甲酰亚胺.
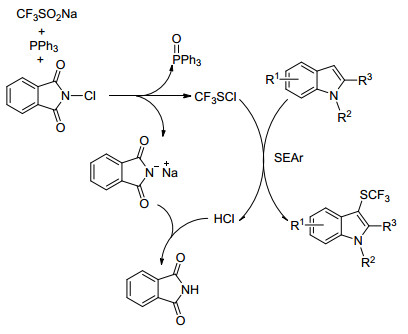 图式 33
可能的反应机理
Scheme33.
Possible reaction mechanism
图式 33
可能的反应机理
Scheme33.
Possible reaction mechanism
4 CF3SO2Na参与的一些其他反应
已有大量文献报道了在芳基上引入三氟磺酰基的合成方法, 但在乙烯基上引入三氟磺酰基的方法却寥寥无几.基于对烯烃和炔烃的三氟磺酰化的文献报道, Qing小组[57]首次提出了对烯烃C—H的直接三氟磺酰化(Eq. 32).该方法具有良好的反应选择性, 对于含不同取代基的苯乙烯化合物具有良好的耐受性, 但若邻位存在位阻较大的基团, 反应则难以进行.推测烯烃可能先与碘形成了一个环正离子的过渡态, 接着CF3SO2Na对其亲核进攻后脱去一分子HI, 得到最终产物(Scheme 34).
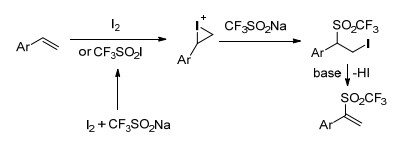 图式 34
乙烯基三氟甲磺酸酯可能的反应机理
Scheme34.
Proposed reaction mechanism of vinyl triflones
图式 34
乙烯基三氟甲磺酸酯可能的反应机理
Scheme34.
Proposed reaction mechanism of vinyl triflones
ArSO2CF3结构经常被用于治疗癌症和免疫疾病等药物的研究中, 因此SO2CF3的引入具有重要的意义[58]. Smyth小组[59]用Pd2(dba)3和Rockphos的组合体系, 有效地实现了芳香族和杂芳族化合物的三氟甲磺酸化(Eq. 33).
远端氟化酮适用于氟化杂环和生物活性分子片段的合成, 因此探索适用于获得这些化合物的方法和过程就显得尤为重要. 2017年, Kananovich小组[60]在乙酸铜(Ⅱ)催化剂和三氟甲磺酸钠存在下, 用叔丁基过氧化使得环丙烷环裂解以制备β-三氟甲基酮(Eq. 34).其反应机理(Scheme 35)可能是在Cu(Ⅰ)催化剂存在下, 叔丁基过氧化氢氧化三氟甲磺酸钠, 产生三氟甲基自由基.然后与铜(Ⅱ)或铜(Ⅰ)反应, 生成作为实际的三氟甲基化试剂的三氟甲基铜(Ⅲ)和铜(Ⅱ)络合物.两种物质可能通过预配体交换步骤导致环丙醇的环裂解, 并且随后发生的单电子转移转变成β-氧羰基或通过亲电子攻击生成β-金属酮中间体.后者经铜(Ⅰ)的还原性消除产生β-三氟甲基酮, 同样的产物也可以通过β-氧羰基自由基途径形成.
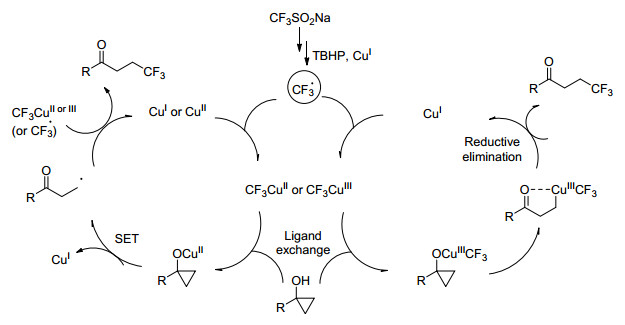 图式 35
可能的反应机理
Scheme35.
Possible reaction mechanism
图式 35
可能的反应机理
Scheme35.
Possible reaction mechanism
硝基亚砜是合成多种含氮化合物的通用结构单元的原料. Selander小组[61]使用三氟甲烷亚磺酸钠, 在氧化剂存在下对硝基亚砜实现了三氟甲基的取代, 合成了一种全新的化合物, 实现了N-三氟甲基化的突破(Eq. 35).机理探索实验说明(Scheme 36), 首先由Cu和TBHP共同作用产生三氟甲基自由基, 与硝基化合物反应生成的中间体再通过氢供体氢醌提供的氢, 生成产物.同时, 氢醌脱氢产生的半醌自由基进一步被氧化成苯醌.
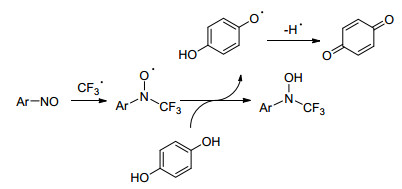 图式 36
可能的反应机理
Scheme36.
Possible reaction mechanism
图式 36
可能的反应机理
Scheme36.
Possible reaction mechanism
近年来, 含有N—O键的底物在过渡金属催化的氧化偶联反应中起了很大的应用.由于极化的N—O键可被过渡金属还原裂解, 所以N—O键可用作合成有价值含氮产物的催化剂内部氧化剂.通过避免过量添加氧化剂, 产生较少的废物, 实现改善官能团的相容性. Selander小组[62]使用肟衍生物作为内部氧化物, 直接合成β-三氟甲基化的酰胺(Eq. 36).其反应机理如Scheme 37所示, 在CuF2, CF3SO2Na和Ac2O的存在下, 将乙酸肟还原成酰胺, 然后通过铜(Ⅱ)的还原产生三氟甲基自由基, 同时伴随N—O键的切割, 随后通过向烯酰胺中添加自由基然后氧化中间体以再生Cu(Ⅱ), 最终形成三氟甲基化的酰胺.与使用Togni试剂的烯酰胺的三氟甲基化相比, 该方法更加经济.
 图式 37
可能的反应机理
Scheme37.
Possible reaction mechanism
图式 37
可能的反应机理
Scheme37.
Possible reaction mechanism
全氟烷基磺酰基基团由于其具有强吸电能力和高亲脂性, 已经成为新型有机支架构建中最重要的功能之一. Zhang小组[63]探索了CF3SO2Na在氮气氛下在室温下在二氯甲烷中与炔基(苯基)碘鎓盐反应, 短时间内得到各种炔三氟甲磺酸酯和炔全氟烷基砜, 具有良好的收率(Eq. 37).碘鎓盐中芳基乙炔基部分的苯环上取代基的位置对反应有很大的影响.机理探索实验表明是自由基反应, 反应通过亚烷基卡宾中间体进行.该方法的优点是反应时间短、条件温和.
Zheng小组[64]报道了一种利用原位形成的硫代碳酰氟完成异硫氰酸酯合成的方法(Eq. 38).其可能的反应机理如Scheme 38所示, CF3SO2Na分别与HPO(OEt)2和CuI反应生成中间体A和B. A和B可以进一步与HPO(OEt)2反应生成中间体C, 经历过渡态D脱氟形成硫代羰基氟化物E.或在加热条件下, 由中间体B直接脱氟, 原位形成硫代碳酰氟E.所得硫代碳酰氟E与伯胺反应形成相应的异硫氰酸酯产物.该方法的优点在于, 避免了异硫氰酸酯传统合成方法中所需的CS2、光气或二氯硫化碳等有毒气体的使用, 使得反应更加安全和环境友好.
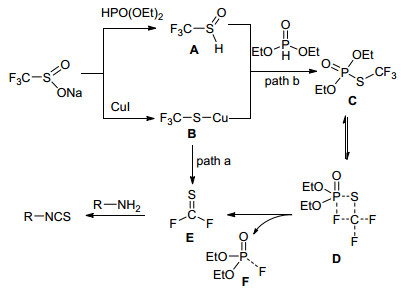 图式 38
可能的反应机理
Scheme38.
Possible reaction mechanism
图式 38
可能的反应机理
Scheme38.
Possible reaction mechanism
5 展望
CF3的引入能够有效地改变药物的理化性质及生物活性, 具有重大的研究意义. CF3SO2Na具有稳定、廉价的特点, 在有机氟化学领域有广泛的应用.但目前三氟甲基的引入方面依旧存在着底物范围窄、产率低、造价昂贵、反应不能放大等一系列缺点, 这些都将是今后氟化学中有意义的研究内容.
-
-
[1]
Grem J. L. Invest. New Drugs 2000, 18, 299 doi: 10.1023/A:1006416410198
-
[2]
Krik, K. L. Org. Process Res. Dev. 2008, 12, 305 doi: 10.1021/op700134j
-
[3]
Yang, B.; Xu, X. H.; Qing, F. L. Org. Lett. 2015, 17, 1906. doi: 10.1021/acs.orglett.5b00601
-
[4]
Liu, X.; Xu, C.; Wang, M. Chem. Rev. 2015, 115, 683. doi: 10.1021/cr400473a
-
[5]
Yang, F.; Klumphu, P.; Liang, Y. M. Chem. Commun. 2014, 50, 936. doi: 10.1039/C3CC48131J
-
[6]
Ji, Y.; Brueckl, T.; Baxter, R. D.; Fujiwara, Y.; Seiple, I. B.; Su, S.; Blackmond, D. G.; Baran, P. S. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14411. doi: 10.1073/pnas.1109059108
-
[7]
Umemoto, T. Chem. Rev. 1996, 96, 1757. doi: 10.1021/cr941149u
-
[8]
Eisenberger, P.; Gischig, S.; Togni, A. Chemistry 2006, 12, 2579. doi: 10.1002/(ISSN)1521-3765
-
[9]
Ryota, H.; Toshiaki, I.; Kohsuke, A. Chemistry 2014, 20, 2750. doi: 10.1002/chem.v20.10
-
[10]
Morimoto, H.; Tsubogo, T.; Litvinas, N. D. Angew Chem., Int. Ed. 2011, 50, 3793. doi: 10.1002/anie.v50.16
-
[11]
Tomashenko, O. A.; Escudero, A. E. C.; Martínez, B. M. Angew Chem., Int. Ed. 2011, 50, 7655. doi: 10.1002/anie.201101577
-
[12]
Zheng, J.; Lin, J. H.; Deng, X. Y. Org. Lett. 2015, 17, 532. doi: 10.1021/ol503548s
-
[13]
Maji, A.; Hazra, A.; Maiti, D. Org. Lett. 2014, 16, 4524. doi: 10.1021/ol502071g
-
[14]
(a) Lefebvre, Q. Synlett 2016, 28, 19.
(b) Tordeux, M.; Langlois, B. R.; Wakselman, C. J. Org. Chem. 1989, 54, 2452. -
[15]
Zhang, C. Adv. Synth. Catal. 2014, 356, 2895. doi: 10.1002/adsc.201400370
-
[16]
Lu, Y.; Li, Y.; Zhang, R. J. Fluorine Chem. 2014, 161, 128. doi: 10.1016/j.jfluchem.2014.01.020
-
[17]
Hang, Z.; Li, Z.; Liu, Z. Q. Org. Lett. 2014, 16, 3648. doi: 10.1021/ol501380e
-
[18]
Zhu, L.; Wang, L. S.; Li, B. Chem. Commun. 2016, 52, 6371. doi: 10.1039/C6CC01944G
-
[19]
Yang, B.; Xu, X. H.; Qing, F. L. Chin. J. Chem. 2016, 34, 465. doi: 10.1002/cjoc.201500641
-
[20]
Yang, Y.; Liu, Y.; Jiang, Y.; Zhang, Y.; Vicic, D. A. J. Org. Chem. 2015, 80, 6639. doi: 10.1021/acs.joc.5b00781
-
[21]
Yang, B.; Xu, X. H.; Qing, F. L. Org. Lett. 2015, 17, 1906. doi: 10.1021/acs.orglett.5b00601
-
[22]
Liu, X.; Xiong, F.; Huang, X.; Xu, L.; Li, P.; Wu, X. Angew. Chem., Int. Ed. 2013, 52, 6962. doi: 10.1002/anie.201302673
-
[23]
Chen, Z. M.; Bai, W.; Wang, S. H.; Yang, B. M.; Tu, Y. Q.; Zhang, F. M. Angew. Chem., Int. Ed. 2013, 52, 9781. doi: 10.1002/anie.201304557
-
[24]
Egami, X.; Shimizu, R.; Usuiac, Y.; Sodeoka, M. Chem. Commun. 2013, 49, 7346. doi: 10.1039/c3cc43936d
-
[25]
Huang, H. L.; Yan, H.; Gao, G. L. Asian J. Org. Chem. 2015, 4, 674. doi: 10.1002/ajoc.201500096
-
[26]
Lu, Q.; Liu, C.; Huang, Z. Chem. Commun. 2014, 50, 14101. doi: 10.1039/C4CC06328G
-
[27]
Liu, C.; Lu, Q.; Huang, Z. Org. Lett. 2015, 17, 6034. doi: 10.1021/acs.orglett.5b03035
-
[28]
Li, B.; Fan, D.; Yang C; Xia W. Org. Biomol. Chem. 2016, 14, 5293. doi: 10.1039/C6OB00912C
-
[29]
Yu, J.; Yang, H.; Fu, H. Adv. Synth. Catal. 2014, 356, 3669. doi: 10.1002/adsc.v356.17
-
[30]
Mu, X.; Wu, T.; Wang, H. Y.; Guo, Y. L.; Liu, G. S. J. Am. Chem. Soc. 2012, 134, 878. doi: 10.1021/ja210614y
-
[31]
Wei, W.; Wen, J.; Yang, D. J. Org. Chem. 2014, 79, 4225. doi: 10.1021/jo500515x
-
[32]
Hua, H. L.; He, Y. T.; Qiu, Y. F. Chem.-Eur. J. 2015, 21, 1468. doi: 10.1002/chem.201405672
-
[33]
Zhang, L. Z.; Li, Z. J.; Liu, Z. Q. Cheminform 2014, 46, 3648.
-
[34]
Jana, S.; Verma, A.; Kadu, R.; Kumar, S. Chem. Sci. 2017, 8, 6633. doi: 10.1039/C7SC02556D
-
[35]
Wu, L. H.; Zhao, K.; Shen, Z. L.; Loh, T. P. Org. Chem. Front. 2017, 4, 1872. doi: 10.1039/C7QO00416H
-
[36]
Martin, R.; Reddy, Y. V.; Shen, Y. Angew. Chem., Int. Ed. 2017, 56, 10915. doi: 10.1002/anie.201706263
-
[37]
Zhang, X.; Huang, P.; Li, Y. Org. Biomol. Chem. 2015, 13, 10917. doi: 10.1039/C5OB01516B
-
[38]
Zhang, K.; Xu, X. H.; Qing, F. L. J. Org. Chem. 2015, 80, 7658. doi: 10.1021/acs.joc.5b01295
-
[39]
Wu, M.; Ji, X.; Dai, W. J. Org. Chem. 2014, 79, 8984. doi: 10.1021/jo501221h
-
[40]
(a) Cai, S. J.; Chen, C.; Sun, Z. L.; Xi, C. J. Chem. Commun. 2013, 49, 4552.
(b) Zhang, L. S.; Chen, K.; Chen, G. H.; Li, B. J.; Luo, S.; Guo, Q. Y.; Wei, J. B.; Shi, Z. J. Org. Lett. 2013, 15, 10. -
[41]
Ji, X. M.; Wei, L.; Chen, F. RSC Adv. 2015, 46, 29766.
-
[42]
(a) Cho, E. J.; Senecal, T. D.; Kinzel, T. Y.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679.
(b) Oishi, M.; Kondo H.; Amii, H. Chem. Commun. 2009, 1909.
(c) Chu, L.; Qing, F. L. Org. Lett. 2010, 12, 5060.
(d) Senecal, T. D.; Parsons, A. T.; Buchwald, S. L. J. Org. Chem. 2011, 76, 1174.
(e) Jiang, X.; Chu, L.; Qing, F. L. J. Org. Chem. 2012, 77, 1251.
(f) Herrmann, A. T.; Smith, L. L.; Zakarian, A. J. Am. Chem. Soc. 2012, 134, 6976.
(g) Sato, K.; Omote, M.; Ando, A.; Kmadaki, I. Org. Lett. 2004, 6, 4359.
(h) Shimizu, R.; Egami, H.; Nagi, T.; Chae, J.; Hamashima, Y.; Sodeoka, M. Tetrahedron Lett. 2010, 51, 5947.
(i) Liu, T.; Shen, Q. Org. Lett. 2011, 13, 2342.
(j) Nagib, D. A. M.; MacMillan, D. W. C. Nature 2011, 480, 224. -
[43]
Fennewald, J. C.; Lipshutz, B. H. Green Chem. 2014, 16, 1097. doi: 10.1039/C3GC42119H
-
[44]
Cao, X.; Pan, X.; Zhou, P. Chem. Commun. 2014, 50, 3359. doi: 10.1039/c3cc49689a
-
[45]
Lu, Y.; Li, Y.; Zhang, R. J. Fluorine Chem. 2014, 161, 128. doi: 10.1016/j.jfluchem.2014.01.020
-
[46]
Pair, E.; Monteiro, N.; Bouyssi, D.; Baudoin, O. Angew. Chem., Int. Ed. 2013, 52, 5346. doi: 10.1002/anie.201300782
-
[47]
Tan, Z.; Zhang, S. W.; Zhang, Y. J. Org. Chem. 2017, 82, 9384. doi: 10.1021/acs.joc.7b01359
-
[48]
Jiang, H. F.; Huang, W.; Yu, Y.; Yi, S. J.; Li, J. W.; Wu, W. Q. Chem. Commun. 2017, 53, 7473. doi: 10.1039/C7CC03125D
-
[49]
Li, C. F.; Suzuki, K.; Yamaguchi, K.; Mizuno, N. New J. Chem. 2017, 41, 1471. http://pubs.rsc.org/en/content/articlepdf/2017/nj/c7nj03057f
-
[50]
Simon, R. C.; Busto, E.; Richter, N.; Resch, V.; Houk, K. N.; Kroutil W. Nat. Commun. 2016, 7, 13323. doi: 10.1038/ncomms13323
-
[51]
Bu, M.; Lu, G.; Cai, C. T. Org. Chem. Front. 2017, 4, 266. doi: 10.1039/C6QO00622A
-
[52]
Yang, Y.; Xu, L.; Yu, S. Chem.-Eur. J. 2016, 22, 858. doi: 10.1002/chem.201504790
-
[53]
Zhao, X.; Wei, A. Q.; Yang, B.; Li, T. J.; Li, Q.; Qiu, D.; Lu, K. J. Org. Chem. 2017, 82, 9175. doi: 10.1021/acs.joc.7b01226
-
[54]
Jiang, L.; Qian, J.; Yi, W.; Lu, G.; Cai, C.; Zhang, W. Angew. Chem., Int. Ed. 2015, 54, 14965. doi: 10.1002/anie.201508495
-
[55]
Yang, Y.; Xu, L.; Yu, S.; Liu, X.; Zhang, Y.; Vicic, D. A. Chem.-Eur. J. 2016, 22, 858. doi: 10.1002/chem.201504790
-
[56]
Chachignon, H.; Maeno, M.; Kondo, H.; Shibata, N.; Cahard, D. Org. Lett. 2016, 18, 2467. doi: 10.1021/acs.orglett.6b01026
-
[57]
Hua, L. N.; Li, H.; Qing, F. L. Org. Biomol. Chem. 2016, 14, 8443. doi: 10.1039/C6OB01567K
-
[58]
(a) Shangary, S.; Johnson, D. E. Leukemia 2003, 17, 1470.
(b) Oltersdorf, T.; Elmore, S. W.; Shoemaker, A. R.; Armstrong, R. C.; Augeri, D. J.; Belli, B. A.; Bruncko, M.; Deckwerth, T. L.; Dinges, J.; Hajduk, P. J.; Joseph, M. K.; Kitada, S.; Korsmeyer, S. J.; Kunzer, A. R.; Letai, A.; Li, C.; Mitten, M. J.; Nettesheim, D. G.; Ng, S.; Nimmer, P. M.; O'Connor, J. M.; Oleksijew, A.; Petros, A. M.; Reed, J. C.; Shen, W.; Tahir, S. K.; Thompson, C. B.; Tomaselli, K. J.; Wang, B.; Wendt, M. D.; Zhang, H.; Fesik, S. W.; Rosenberg, S. H. Nature 2005, 435, 677;
(c) Morizawa, Y.; Okazoe, T.; Wang, S. Z.; Sasaki, J.; Ebisu, H.; Nishikawa, M.; Shinyama, H. J. J. Fluorine Chem. 2001, 109, 83. -
[59]
Smyth, L. A. J. Org. Chem. 2016, 81, 1285. doi: 10.1021/acs.joc.5b02523
-
[60]
Konik, Y. A.; Kudrjashova, M.; Konrad N.; Kaabel, S.; Järving, I.; Lopp, M.; Kananovich, D. G. Org. Biomol. Chem. 2017, 15, 4635. doi: 10.1039/C7OB00680B
-
[61]
van der Werf, A.; Hribersek, M.; Selander, N. Org. Lett. 2017, 19, 2374. doi: 10.1021/acs.orglett.7b00908
-
[62]
Yang, H. B.; Selander, N. Org. Biomol. Chem. 2017, 15, 1771. doi: 10.1039/C7OB00203C
-
[63]
Han, J. B.; Yang, L.; Chen, X.; Zha, G. F.; Zhang, C. P. Adv. Synth. Catal. 2016, 358, 4119. doi: 10.1002/adsc.201600717
-
[64]
Liao, Y. Y.; Deng, J. C.; Ke, Y. P.; Zhong, X. L.; Xu, L.; Tang, R. Y.; Zheng, W. Chem. Commun. 2017, 53, 6073. doi: 10.1039/C7CC02373A
-
[1]
-

图式 2 CF3SO2Na对一些化合物的三氟甲基化
Scheme 2 Trifluoromethylation of organic compounds with CF3SO2Na

图式 4 烯烃和炔烃的碘三氟甲基化
Scheme 4 Strategies for iodotrifluoromethylation of alkenes and alkynes

图式 12 α-取代的苯乙烯的三氟甲基化及其可能的反应机理
Scheme 12 Trifluoromethylation of α-substituted styrenes and their possible reaction mechanism

图式 13 在紫外光下用CF3SO2Na进行三氟甲基化
Scheme 13 Trifluoromethylation with CF3SO2Na via UV light irradiation

图式 16 铜催化的N-芳基丙烯酰胺的三氟甲基化
Scheme 16 Copper-catalyzed trifluoromethylation of N-aryl-acrylamides

图式 19 I2O5介导的N-芳基甲基丙烯酰胺的环化和三氟甲基化
Scheme 19 I2O5-mediated trifluoromethylation/cyclization of N-arylmethacrylamides

图式 22 不饱和α-三氟甲基酮和3-三氟甲基香豆素的合成
Scheme 22 Synthesis of unsaturated α-trifluoromethyl ketones and 3-trifluoromethyl coumarins

图式 30 HPA催化三氟甲基化的可能反应机理.
Scheme 30 A possible reaction mechanism for HPA-catalyzed trifluoromethylation
-


 下载:
下载:






































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 320
- 文章访问数: 13945
- HTML全文浏览量: 4839
 下载:
下载:
