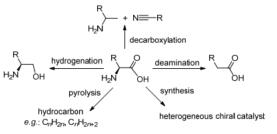
图式 1.
氨基酸的催化转化过程
Scheme 1.
Catalytic transformation process of amino acids

近些年来, 随着化石能源的消耗, 可再生的生物质[1](如草、树叶、纤维素等)、微生物(如蓝藻、蓝细菌)及富含蛋白质的废弃物[2]的转化利用越来越受到人们的关注.含有氨基和羧基两种官能团的氨基酸可以通过它们经发酵[3, 4]、酶催化法、化学转化法或萃取等方法得到.
氨基酸的催化转化利用有很多方面, 主要是利用氨基和羧基这两种官能团在不同条件下转化得到不同类型的产物(图 1).利用具有手性中心的氨基酸通过羧基加氢还原可以得到手性氨基醇, 利用氨基酸的脱羧反应可以得到胺类、腈类, 利用氨基酸的脱氨反应可以得到羧酸, 利用氨基酸的催化热解可以得到生物燃料, 利用氨基酸的手性还可以合成多相手性催化剂, 用于催化不对称aldol反应等.在图 1中, 当氨基酸的R不同时, 制备得到的产品也不同.同时R不同, 反应条件也会不同.如:当R中含羟基等杂原子的官能团时, 不仅要考虑不同官能团之间的相互影响, 同时还要考虑反应条件对反应的影响.而当R中含有不饱和的键(如苯环、双键等), 则要考虑反应条件如加氢还原、加氧氧化等的影响.对于具体的氨基酸, 要综合考虑所制备产品的应用价值和生产的经济性等方面, 选择合适的氨基酸转化类型.因此, 本文对氨基酸的不同反应类型的催化转化进行综述不仅具有理论意义, 还具有巨大的应用意义, 有助于实现氨基酸的综合高效转化利用.
氨基酸的催化转化主要涉及到催化剂等反应条件.均相催化是指催化剂与反应物同处于均匀物相中的催化反应, 通常具有催化效率高, 但是又具有催化剂难分离, 较难实现连续工作的缺点.而多相催化是指在两相界面上发生的催化反应, 通常具有催化剂易回收、可循环利用的优点, 这点对于贵金属催化的反应尤为明显.因此, 本文主要综述了近些年来氨基酸多相催化转化的研究进展.
手性氨基醇是一类具有手性的物质, 广泛用于农业、医药、化工、表面活性剂及手性助剂[5~7]等领域.其制备方法主要有拆分法和还原剂还原法.采用催化加氢还原法制备手性氨基醇不仅避免了手性拆分法和还原剂还原法生产成本高、污染问题等缺点, 而且催化加氢法具有原子利用率高、绿色环保等优点.
氨基酸的催化加氢主要涉及到催化剂和其它催化反应条件.催化剂的作用主要是降低反应的活化能, 提高反应的效率, 同时可以降低反应的温度、压力等.氨基酸加氢的实质是C=O双键的加成还原反应. C=O双键的加氢所涉及的催化剂活性组分主要有Ru、Pt等[8]贵金属和Fe、Ni、Cu等[9]非贵金属, 载体主要有活性炭[10]和含有Lewis酸性位的TiO2[11]、ZrO2[12].这种含酸性位的载体可以实现C=O双键的极化, 而活性组分可以活化氢气, 两者实现协同催化作用, 使反应高效进行.而氨基酸的加氢又与普通的羧酸加氢不同, 氨基酸同时含有的氨基和羧基官能团, 氨基的碱性和羧基的酸性, 使得氨基酸在pH不同的溶液中以不同的形式存在, 将会影响加氢反应的速率.同时氨基和羧基也会缩合生成肽类物质, 原料氨基酸与产物氨基醇也会酯化生成酯类物质.这些因素增加了氨基酸加氢的复杂性.而其它反应条件如温度、压力, 不仅会改变反应的速率, 而且会改变产物的选择性.因此, 选择合适的反应条件对反应尤为重要.
本节主要从氨基酸加氢的影响因素和加氢动力学模型及机理两方面来论述手性氨基酸加氢的研究进展.
氨基酸中的羰基与羟基氧原子上的未共用电子对共轭, 降低了羰基碳原子的亲电能力, 使得氨基酸加氢的难度较大, 反应条件较苛刻.
20世纪40年代, Adkins等[13]首次报道了Raney Ni催化剂催化氨基酸酯加氢到氨基醇.结果发现, 高比率的催化剂可以降低反应的温度, 但在反应过程中, 催化剂会部分溶解损失.
20世纪末, Antons等[14]在专利中报道了Ru基催化剂用于氨基酸的催化加氢.在考察载体对反应的影响时, 以Ru/Al2O3和Ru/C为催化剂时产物氨基醇的ee值分别为98.5%和98%;而以钌黑和RuO2为催化剂时, 其ee值分别为95%和93%.这表明负载型的催化剂可以得到略高光学纯度的氨基醇.同时RuO2催化剂重复循环利用5次, 其产物氨基醇的ee值没有变化.之后又报道了在单金属钌的基础上引入第二种金属Re[15], 以Ru/Re双金属催化剂催化一系列氨基酸加氢制手性氨基醇.其产物的收率均在31%~80%之间, ee值均高于98%.其中L-丙氨酸的加氢收率最高, 达到80% (Eq. 1).
|
|
(1) |
后来, Mägerlein等[16]也报道了以Ru/Re双金属催化剂催化L-丙氨酸加氢. Ru/Re双金属催化剂的催化活性约是钌黑催化剂的三倍, 反应仅需5 h原料就实现了完全转化, 同时产物的ee值基本保持不变.但作者并没有解释为什么Re加入后催化剂活性有明显提高.笔者认为, 可能是Re助剂的加入, 实现了Ru与Re之间的电子授受作用, 提高了催化剂的活性.在考察助剂量和前驱体对反应的影响时, 以Ru-Re/C为催化剂时, 其制备方法是将助剂Re负载在前驱体Ru/C上.结果显示, 当Re与Ru原子质量比为1时, L-脯氨酸加氢活性最高.同时不同前驱体制备的Ru-Re/C催化剂, 反应活性也相差很大. Ru/C前驱体还原温度越低, Ru分散度越高, 催化活性越高.
2014年, Metkar等[17]报道了过渡金属负载型催化剂催化赖氨酸加氢制赖氨醇的研究(Eq. 2).在考察催化剂活性组分对反应的影响时, 结果发现:其催化活性顺序是: Ru/C>Rh/C>Re/C≈Ir/C≈Ni/C≈Pd/C≈Pt/C≈0.接着又考察了载体对反应的影响, 发现Ru/SiO2和Ru/Al2O3的催化活性均低于Ru/C.而且, 这些氧化物载体的催化剂对赖氨醇的选择性也明显低于碳载体的催化剂.将反应后的Ru/C催化剂经水洗、干燥等过程, 重新用于反应.循环利用6次, 赖氨酸的转化率均维持在初始的95%左右, 赖氨醇的选择性均维持在初始的88%左右, 反应后液体中也没有检测到Ru粒子.因此该催化剂的稳定性较好, 商业应用前景较好.
|
|
(2) |
Gong等[18]以5% Ru-Rh/Al2O3为催化剂, 考察了L-丙氨酸的催化加氢.在383 K、4 MPa、磷酸过量的条件下, 反应4 h, 原料的转化率为96%, 产物L-丙氨醇的光学收率达到了99%, 该催化剂与Ru-Re/Al2O3相比降低了反应的压力, 催化效果优良.
在Ru基催化剂的基础上, 也有学者将Rh基催化剂用于氨基酸的加氢反应. Tamura等[19]报道了以Rh-MoOx/SiO2为催化剂, 可以实现高效的氨基酸选择性加氢.其制备方法是采用两步浸渍法, 第一步用Rh-Cl3·3H2O浸渍SiO2, 还原后得到Rh/SiO2催化剂, 第二步用(NH4)6Mo7O24·4H2O溶液浸渍Rh/SiO2催化剂, 还原后得到Rh-MoOx/SiO2催化剂.在反应中, 该催化剂与Rh/SiO2和MoOx/SiO2相比, 催化活性和选择性显著提高, 氨基醇的收率通常在90%~94%之间, 而且立体选择性不变.这说明, Rh与MoOx之间的协同作用是提高催化剂催化性能的关键.
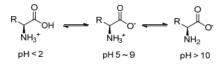
氨基酸同时含有的氨基和羧基两种官能团既可以与酸反应又能与碱反应.因此, 在不同酸碱性的溶液中氨基酸将以不同的形式存在[20], 如Scheme 2.
Jere[20]考察了相同过量的磷酸、硫酸和三氟甲磺酸对L-丙氨酸加氢的影响.反应6 h后, 原料转化率的顺序是:磷酸>三氟甲磺酸>硫酸.其中, 磷酸的反应速率约是硫酸的2倍.作者认为, 可能是这些过量的酸作为吸附物种占据了活性位, 而硫酸和三氟甲磺酸占据活性位的能力更强, 使得反应速率更慢.也可能是含硫元素的酸使催化剂中毒降低了反应速率.
在液体酸的基础上, 也有研究者将固体酸用于氨基酸的催化加氢. Tamura等[21]考察了一系列酸性添加物对L-丙氨酸催化加氢制L-丙氨醇的影响.结果发现, 催化反应活性顺序是: H3PO3>H-ZSM-5≈H-Beta≈H-USY≈H-Mordenite≈none>CeO2≈0, 其选择性均在75%~88%之间.这种固体酸的使用减少了产物的后处理, 有利于降低生产成本, 但反应效果不如添加液体酸好.
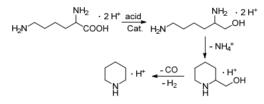
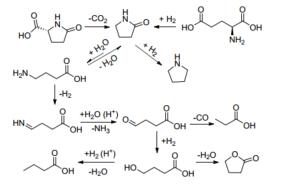
Metkar等[17]报道了酸量对赖氨酸催化加氢制赖氨醇的影响.当所用的酸量不足时, 原料最终不能完全转化.这是因为赖氨醇是一种碱性物质, 随着反应的进行, 溶液的pH值升高, 当酸量不能质子化赖氨酸时, 反应会停止.作者还发现赖氨醇的选择性与溶液的初始pH值有很大关系, 在较低的pH值(<1.3~1.5)下, 选择性会因为产物的环化形成哌啶而降低.因此稍高的pH有利于提高赖氨醇的选择性, 哌啶生成过程如Scheme 3.
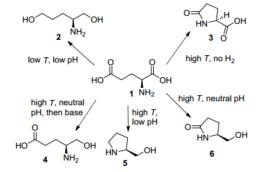
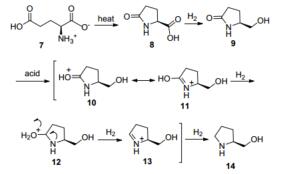
Holladay等[22]报道了以Ru/C为催化剂, 在不同条件下L-谷氨酸催化转化的研究.在氢气存在的条件下, L-谷氨酸1会发生羧基的还原, 其中在高温酸性或中性的条件下还会因为pH不同环化生成不同的产物L-脯氨醇5或L-焦谷氨醇6.而在无氢气的条件下, L-谷氨酸仅仅会发生环化生成L-焦谷氨酸3 (Scheme 4).
其催化加氢机理是:首先质子化的L-谷氨酸(7)在加热的条件下, 环化形成L-焦谷氨酸(8), 然后再经过催化加氢生成L-焦谷氨醇(9), 在酸催化的条件下, 焦谷氨醇会加速形成烯醇式11, 然后经过三步加氢生成最终产物L-脯氨醇14 (Scheme 5).
温度对氨基酸加氢的影响主要体现在改变反应的速率和产物的选择性.
Mägerlein等[16]以Ru-Re/C为催化剂考察了温度对L-缬氨酸加氢产物对映选择性的影响(Eq. 3), 当温度由353 K提高到423 K时, 产物的ee值由99.7%降至89.4%.
|
|
(3) |
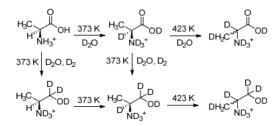
为了研究温度对反应过程中对映选择性的影响, Jere等[23]通过同位素标记法和对产物手性及反应速率的分析, 研究了温度对L-丙氨酸加氢制L-丙氨醇的机理和立体化学的影响.当反应温度为373 K, 反应6 h后, 原料的转化率为93%, ee值为99.2%, 产物收率为99.2%.当反应温度提高到423 K时, 原料45 min实现了完全转化, ee值为89%, L-丙氨醇的收率仅为78%.产物的收率与373 K时的相比有所降低, 主要是产物发生消旋化生成了D-丙氨醇, 同时C—C键氢解生成了乙胺.其消旋过程如Scheme 6.
一个多相催化的加氢反应要发生, 必须经历质量传递过程.其过程是:首先, 氢气从气相穿过气液界面进入液相; 然后, 氢气和反应底物从液相穿过液固表面到达催化剂粒子表面; 最后, 氢气和底物穿过多孔催化剂载体扩散到金属表面活性位上.而压力和搅拌速度均会通过影响氢气在反应液中的溶解率, 进而影响反应物的内外传质和反应的速率.
Wang[24]研究了压力对L-丙氨酸加氢转化速率的影响.所用丙氨酸与磷酸的摩尔比约为1:1.32, 在压力为2~7 MPa时, 原料的转化率由20.7%升高到45.3%.但是当压力升高到8 MPa时, 原料的转化率仅提高了1%左右.说明此时原料的转化速率与氢气的浓度无关, 催化剂表面吸附的氢已经饱和.
而He[25]同样研究了L-丙氨酸的加氢, 考察了一系列因素如丙氨酸与磷酸的摩尔比、反应温度、反应时间、反应压力对反应的影响.最终由Statistica软件分析了影响反应的最优组合和重要因素.结果发现, 压力是影响产物丙氨醇收率和对映选择性的首要因素, 其最优为8 MPa.
Zwietering[26]研究了催化剂颗粒完全悬浮时最小的搅拌速率.结果显示, 1 g催化剂溶解在100 g水中, 搅拌速率约为500 r·min-1时催化剂可以完全悬浮.因此在反应的过程中, 要消除外扩散对反应的影响, 就要保证反应在一定的搅拌速度下进行.
在动力学数据的基础上, 各学者又对各种类型的氨基酸加氢的动力学模型与机理进行了研究.当表面反应为速率控制步骤时, 动力学模型主要有Langmuir-Hin-shelwood (L-H)模型、Elet-Rideal (E-R)模型、Langmuir-Hinshelwood-Hougen-Watson (L-H-H-W)模型.动力学模型的建立, 使得可以直观地通过表达式来研究各参数对反应的影响情况, 为后续催化剂的设计及反应参数的调控提供了参考.
2004年, Jere等[27]对L-丙氨酸水相加氢制L-丙氨醇的动力学进行了研究.以磷酸为酸源, Ru/C为催化剂, 提出了L-H动力学模型, 认为质子化的丙氨酸和未解离的磷酸竞争催化剂表面的其中一个活性位, 氢气的解离吸附在另一个活性位.磷酸三元弱酸的性质一方面使得溶液的pH值保持稳定, 质子化的丙氨酸的数量保持不变, 维持反应的进行; 另一方面未解离的氨基酸会占据反应的活性位阻止反应.因此反应中丙氨酸的浓度与磷酸的浓度比是催化反应快慢的关键.而如果以强酸如硫酸作为质子化的酸, 依据L-H动力学模型, 则可以避免原料与酸的竞争吸附关系, 使反应高效进行, 但强酸的使用对设备的要求较高.
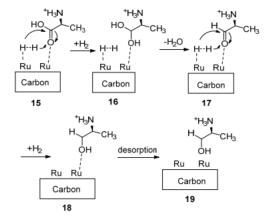
根据L-H动力学模型, 笔者认为Ru/C催化L-丙氨酸加氢的机理(Scheme 7)是:首先原料L-丙氨酸和氢气在碳载体上的Ru活性位上被活化, 分别发生碳氧双键的极化和氢气的解离15, 然后解离的氢气在极化的碳氧双键发生加成反应, 生成不稳定的二醇结构16, 接着脱去一分子水形成氨基醛17.该步加氢反应速率较慢, 是反应的速控步骤.然后再发生羰基的加氢反应, 生成产物丙氨醇19.
2017年, Bhandare等[28]考察了Ru/C催化的丝氨酸和谷氨酸水相加氢的动力学.作者提出了L-H-H-W动力学模型, 认为解离后化学吸附的H2和吸附的氨基酸在催化剂的表面活性位上竞争吸附一个位点, 形成一个慢的表面反应, 是反应的速控步骤.通常, 低温高压有利于H2在水中的溶解, 这就使得控制反应的温度与压力是控制催化反应快慢的关键.同时原料的浓度与催化剂的比值也会很大影响反应的快慢.
在单一氨基酸的基础上, 也有学者研究了混合氨基酸的加氢动力学. Pimparkar等[29]研究了丝氨酸、丙氨酸、缬氨酸三种混合氨基酸加氢制氨基醇的动力学研究.结果发现:多种底物的氨基酸存在将通过竞争吸附来影响加氢速率, 同时氨基酸不同, 竞争吸附的能力也不同, 三种氨基酸吸附能力的强弱是:缬氨酸>丝氨酸≈丙氨酸.缬氨酸强的吸附能力不仅使得自身加氢反应速率变慢, 也使得其它氨基酸加氢反应速率变慢.这种研究方法对混合氨基酸的加氢提供了思路.
综上, 在氨基酸进行加氢还原制备手性氨基醇时, 反应条件中温度主要影响了产物的ee值, 在373 K时产物氨基醇不会发生消旋化, 当温度逐渐升高时, 产物发生消旋化, ee值降低.因此在反应中不能单纯通过提高反应温度来加快反应速率; 酸量主要会影响原料的最终转化率, 采用弱酸(如磷酸)作为酸源时, 原料的最终转化率会随着酸量的增加呈现先增加而后降低的趋势, 因此存在最优的酸量值.而强酸(如硫酸)的使用会避免弱酸中转化率的降低, 但对设备提出了更高的要求.催化剂中Ru、Rh基催化剂展现了良好的催化效果, 文献表明:催化剂活性组分的分散度越高, 粒子粒径越小, 催化剂的活性越高.因此制备出更高活性的催化剂(如引入助剂、改性催化剂载体、改进优化催化剂制备方法等)是氨基酸加氢还原制备手性氨基醇的突破口.
胺类是一类重要的物质, 广泛用于染料、农药、药物[30]等方面.传统的有机合成方法[31]制取胺类不仅会用到有机试剂, 产生大量废液, 而且通常收率不高.由氨基酸脱羧反应一步法制胺类物质不仅绿色无污染, 而且收率也会比较高.
2015年, De Schouwer等[32]研究了一系列Pd和Pt负载型催化剂催化焦谷氨酸和谷氨酸脱羧反应制2-吡咯烷酮, 其中以Pd/Al2O3效果最好.在523 K, 0.6 MPa N2气氛下, 选择性为81%, 收率为70%, 副产物主要是丙酸.其转化路径如Scheme 8.
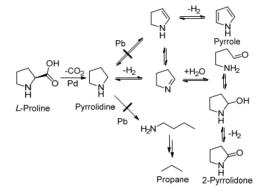
2016年, Verduyckt等[33]考察了脯氨酸脱羧生成吡咯烷的研究.在498 K, 0.6 MPa N2气氛下, 反应6 h, Pd/C和Pd/MOx催化剂均能使原料完全转化, 但产物的选择性最高仅为33%.当引入第二种金属Pb后, 产物的选择性有明显提高.产物的选择性最高为95%.作者认为, Pb的修饰使Pd有更多的电子, 使Pd与产物吡咯烷的N原子之间的作用减弱, 促进了仲胺的解吸, 避免了在Pd表面的开环水解等副反应的发生.同样的方式, 吡咯啉不仅很容易从Pd表面脱附, 避免了进一步脱氢生成吡咯.转化路径如Scheme 9.
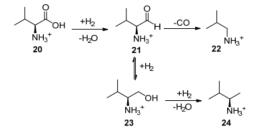
2017年, Verduyckt等[34]又考察了Ru/C催化的缬氨酸脱羧反应制初级胺.在423 K原料完全转化时, 异丁胺22的选择性为87%, 其副反应是缬氨酸在加氢还原的过程中生成的氨基醛21进一步加氢脱水, 生成2-氨基-3-甲基丁胺(24).转化过程如Scheme 10.
2015年, Claes等[35]研究了钌基负载型催化剂, 在氧气存在的条件下, 373 K反应24 h, 亮氨酸完全氧化脱羧为异戊腈, 选择性最高为85%. Claes之后又研究了一系列氨基酸的氧化脱羧(Eq. 4).
|
|
(4) |
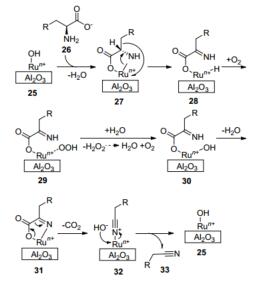
作者认为其反应机理是质子化的α-氨基酸26在Ru(OH)x/Al2O3 (25)上通过氨基的配体交换进入到催化循环中, 羧酸盐可能在Ru配位层上占据另一个配体.然后, Ru-氨化合物经过β-H消除进入到α-亚氨基羧酸盐和Ru-单氢物种28中. Run+(OH)催化剂的再生是通过H2O2的媒介使产生的O2嵌入到Ru-单氢物种29中.在这个反应条件下, H2O2很快被钌所分解.在第二阶段, Ru调控的α-亚氨基羧酸盐是一个脱羧反应, 释放出CO2, 生成腈类物质32.通过OH-或新鲜的反应物取代腈类产品33进入Run+实现催化剂的再生(Scheme 11).
同年, Claes等[36]又报道了以[Ni, Al]-LDH-WO4 (LDH为Layered Double Hydroxide, 层状双氢氧化物)为催化剂, 在NH4Br试剂的作用下, H2O2作为最终氧化剂, 将苯丙氨酸氧化脱羧为苯乙腈的反应(Eq. 5).产物的收率最高为89%.作者认为, 溴化物、羧酸盐与LDH表面的静电相互作用是实现高效催化的关键.
|
|
(5) |
催化机理如Scheme 12.首先[Ni, Al]-LDH-WO4催化剂上的离子WO42- (34)在H2O2的作用下被氧化为离子WO62- (35).然后35将Br-离子氧化为Br+离子, 自身被还原为34, 实现再生.而被氧化Br+离子在碱性条件下将苯丙氨酸36氧化为苯乙腈37, 自身被还原为Br-, 实现催化的循环.
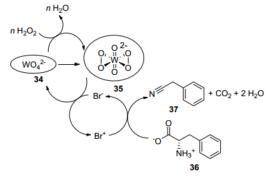
因此, 在氨基酸脱羧反应中, 不同的反应条件会生成不同类型的产物, 在隔绝空气、498 K以上温度的条件下, 会脱羧生成胺类物质; 而在O2气氛、低于373 K温度的条件下, 会氧化脱羧生成腈类物质.在众多的催化剂中, Pd基和Ru基催化剂效果较好.笔者认为, 主要原因是它们具有很好的断裂RCOOH键和活化C—N键的能力.
近年来, 随着人们生活质量的提高, 蛋白质、微生物污染物等废弃物的产生也越来越多.如果能将这些废弃物综合利用, 将会有巨大的应用前景.对于这些废弃物的水解产物氨基酸(如L-谷氨酸、L-天冬氨酸)由于其高度的官能团化致使其应用受限.因此将这些氨基酸去氨基化制备一元、二元或多元羧酸等化学品是多羧基官能团氨基酸利用的另一方面.
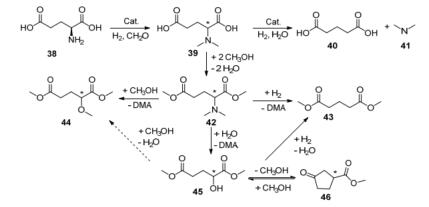
2017年, De Vos等[37]报道了L-谷氨酸的催化去氨基的反应.为了再充分利用含氮类物质, 作者采用了“两步法”反应(Scheme 13).首先将L-谷氨酸38转化为N, N-二甲基-L-谷氨酸(39), 该步中以Pd/C为催化剂, 甲醛作为烷基化试剂, 在温和的条件下, 39的收率为90%.然后再发生脱氨反应生成戊二酸40和二甲胺41, 该步以碱性的Pt/Al2O3为催化剂, 在473 K, 1.5 MPa H2的条件下反应24 h, 40的收率为49%.而副反应主要是39进一步水解脱氨、脱羧生成α-羟谷氨酸、γ-丁内酯.为了抑制水解反应, 作者用甲醇代替溶剂水, 39会酯化生成N, N-二甲基-L-谷氨酸二甲酯(42), 以Pt/TiO2为催化剂, 在498 K, 3 MPa H2的条件下反应24 h, 戊二酸二甲酯(43)的收率为76%, 与以水为溶剂的反应收率提高了约50%, 而副反应主要是42的水解脱氨和醇解脱氨.作者认为金属与中等强度酸(Lewis酸或Bronsted酸)的载体之间的相互作用是Pt/TiO2催化剂展现良好催化性能的关键.但该类反应也面临一些问题.比如, 虽然产物的收率较高, 但仍然有副产物的生成, 这将会增加产品提纯的成本.如何改性催化剂、优化工艺方法和路线、降低生产成本是研究的重点.
综上可以看出, 氨基酸催化还原脱氨的实质是氨基酸中C—N键的断裂, 其产物主要有羧酸及其衍生物和胺类物质.这种方法提供了制备羧酸及其衍生物的另一种思路.
氨基酸作为蛋白质的基本单位, 对一些碳氢含量较高的氨基酸进行催化热解研究一方面有助于研究裂解催化剂在氨基酸催化热解过程中的作用, 另一方面可以制备替代化石燃料的生物燃料. Liu等[38]以ZSM-5为催化剂, 考察了亮氨酸和脯氨酸在923 K下的热解情况.
其中, 亮氨酸产生的烃类化合物(29.6%的芳香烃类、34.9%的烯烃和8.1%的烷烃)比脯氨酸(25.3%的芳香烃类、14.0%的烯烃和5.5%的烷烃)多.作者认为主要是含N杂环的脯氨酸在催化热解的过程中产生的N杂环化合物很容易结焦, 不易进一步热解, 导致28.2%的含氮物质在剩余分解物中, 而亮氨酸的仅有4.3%.
在催化热解的过程中, 会产生NOx、HCN、NH3等含氮化合物, 这些高氮含量的物质会造成酸雨、光化学烟雾等污染.为了解决这些问题, Yi等[39]研究了在氨基酸催化热解中加入Fe/Ca混合添加剂对产生氮的影响.研究结果发现, Ca的加入可以将HCN转化为NH3, Fe的加入可以将NH3转化为N2.因此, Fe/Ca混合添加剂可以有效去除HCN和NH3等有害物质.
将Fe/Ca混合添加剂引入氨基酸的催化热解中巧妙地解决了副产物的污染问题, 降低了生产成本, 但在实际的应用中将氨基酸热解来制备生物燃料的成本较高, 应用难度较大, 但可以以此来研究生物质的催化热解.
不对称aldol反应是有机反应中一类重要的反应, 广泛用于构建碳-碳键、碳-杂原子键的形成反应.而氨基酸特殊的双官能团、手性及酸碱协同中心, 同时作为生物酶的组成单位, 因此在一些特定反应中应该具有酶的一些特性.据文献报道, 20世纪70年代, Eder等[40]报道了以L-脯氨酸为催化剂, 催化分子内的Robinson环化反应.之后几十年, 罕有报道以氨基酸为催化剂催化有机反应.到了本世纪初, 氨基酸作为手性催化剂可以催化不对称aldol反应被报道后[41].手性氨基酸催化剂的研究成为热点.但由于均相催化的缺点, 如易失活、低的结构和热力学稳定、催化剂的分离与回收利用困难, 这些缺点阻碍了手性氨基酸催化剂的广泛应用.而且催化剂的用量高达30 mol%[42]、产物ee值较低.但是如果能将均相催化剂多相化, 制备成负载或固载型催化剂, 将能很好地解决这些问题.手性氨基酸的多相化最常用的是利用氨基酸与载体之间的相互作用, 通过吸附、氢键作用、接枝等方法将氨基酸固(负)载在载体上.常用的载体有水滑石、介孔二氧化硅、MCM-41[43].
2009年, Yang等[44]采用共聚法合成了L-脯氨酰胺/ SiO2催化剂, 在催化对硝基苯甲醛和环己酮的反应(Eq. 6)中展现了良好的催化性能, 其ee值高达96%, TEM图表明, 所制备的催化剂呈现高度的有序, 有2D六边形介孔结构.高于其它无序介孔材料的催化性能(75%).这说明, 有序的介孔结构可以提高产物的对映选择性.
2006年, He等[45]将L-脯氨酸负载在阴离子黏土层间上, 合成了Mg/Al L-Pro LDHs催化剂, 将其用于苯甲醛和丙酮的不对称aldol反应(Eq. 7), 产物的收率最高为90%, ee值为94%.与均相L-脯氨酸相比, 其收率和ee值略有降低.但这种具有空间限域效应的催化剂一方面增强了脯氨酸的固载程度, 另一方面有效阻止了因热处理导致的L-脯氨酸的消旋化.
|
|
(6) |
|
|
(7) |
之后作者[46]又将L-脯氨酸嫁接到介孔SiO2上来催化直接不对称aldol反应和Knoevenagel-Michael串联反应(Eqs. 8, 9), 产物的收率均高于80%, ee值均高于90%.
|
|
(8) |
|
|
(9) |
催化剂循环利用4次后, 其催化效果基本不变.制备方法如Scheme 14.
将具有手性的氨基酸多相化后用于催化不对称aldol反应中, 解决了均相中的催化剂难分离、回收利用的问题, 但是与均相相比催化活性并没有明显提高.因此今后研究者要从催化剂的设计上(如催化剂的制备方法、载体的选择与修饰、手性氨基酸的选择与修饰等)考虑来提高催化的活性和产物的对映选择性.
以自然界中易获得的氨基酸为原料, 通过催化转化的方法实现氨基酸的综合高效利用, 是未来生物质化学研究的一个重要方向.但在利用的过程中(表 1), 也有一些技术难点.比如:氨基酸羧基还原制手性氨基醇, 虽然Ru基和Rh基催化剂展现了较好的催化性能, 但反应通常在高温高压下进行, 对设备和能耗的要求较高, 而通过加入助剂的方法又要考虑溶液的强酸性.氨基酸催化脱羧、催化脱氨、催化热解需要更高的温度, 能耗是考虑的重点.因此制备高性能的催化剂来降低反应的温度和压力是今后氨基酸催化转化利用研究的发展方向.而氨基酸作为多相手性催化剂实现的不对称aldol转化, 产物的收率和ee值有待进一步提高, 催化剂的设计与制备是研究的重点.
 下载:
导出CSV
下载:
导出CSV
| 产品 | 温度/K | 压力 | 催化剂 | 特殊添加剂 |
| 手性氨基醇 | 373 | 8 MPa H2 | Ru/C、Rh/SiO2 | 无机酸 |
| 胺类与腈类 | 525 | 0.6 MPa N2、O2 | Ru基、Pd基催化剂 | 无 |
| 羧酸及其衍生物 | 498 | 3 MPa H2 | Pd/C、Pt/TiO2 | 无 |
| 生物燃料(烃类) | 923 | 隔绝空气 | ZSM-5等裂解催化剂 | Fe/Ca混合物 |
| 多相手性催化剂 | 常用载体:水滑石、介孔二氧化硅、氧化铝、MCM-41 | |||
(a) Corma, A. ; Iborra, S. ; Velty, A. Chem. Rev. 2007, 107, 2411.
(b) Sheldon, R. A. Green Chem. 2014, 16, 950.
(c) Gilkey, M. J. ; Xu, B. ACS Catal. 2016, 6, 1420.
Tuck, C. O.; Pérez, E.; Horváth, I. T.; Sheldon, R. A.; Poliakoff, M. Science 2012, 337, 695. doi: 10.1126/science.1218930
Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Kesseler, M.; Stürmer, R.; Zelinski, T. Angew. Chem., Int. Ed. 2004, 43, 788. doi: 10.1002/(ISSN)1521-3773
Demain, A. L. Ind. Biotech. 2007, 3, 269. doi: 10.1089/ind.2007.3.269
Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc. 1987, 109, 5551. doi: 10.1021/ja00252a056
Rogers, G. A.; Parsons, S. M.; Anderson, D. C.; Nilsson, L. M.; Bahr, B. A.; Kornreich, W. D.; Kaufman, R.; Jacobs, R. S.; Kirtman, B. J. Med. Chem. 1989, 32, 1217. doi: 10.1021/jm00126a013
Corey, E. J.; Zhang, F. Y. Angew. Chem., Int. Ed. 1999, 38, 1931. doi: 10.1002/(ISSN)1521-3773
(a) Abdelrahman, O. A.; Heyden, A.; Bond, J. Q. ACS Catal. 2014, 4, 1171.
(b) Tan, J.; Cui, J.; Cui, X.; Deng, T.; Li, X.; Zhu, Y.; Li, Y. ACS Catal. 2015, 5, 7379.
(c) Zhou, M.; Zhang, H.; Ma, H.; Ying, W. Ind. Eng. Chem. Res. 2017, 56, 8833.
(a) Wang, F.; Zhang, Z. ACS Sustainable Chem. Eng. 2016, 5, 942.
(b) Adkins, H.; Pavlic, A. A. J. Am. Chem. Soc. 1947, 69, 3039.
(c) Zhu, Y.; Zhu, Y.; Ding, G.; Zhu, S.; Zheng, H.; Li, Y. Appl. Catal., A 2013, 468, 296.
(d) Zheng, X.; Lin, H.; Zheng, J.; Duan, X.; Yuan, Y. ACS Catal. 2013, 3, 2738.
Di, X.; Li, C.; Zhang, B.; Qi, J.; Li, W.; Su, D.; Liang, C. Ind. Eng. Chem. Res. 2017, 56, 4672. doi: 10.1021/acs.iecr.6b04875
Primo, A.; Concepción, P.; Corma, A. Chem. Commun. 2011, 47, 3613. doi: 10.1039/c0cc05206j
Fan, G.; Zhou, Y.; Fu, H.; Ye, X.; Li, R.; Chen, H.; Li, X. Chin. J. Chem. 2011, 29, 229. doi: 10.1002/cjoc.201190071
Adkins, H., Billica, H. R. J. Am. Chem. Soc. 1948, 70, 3121. doi: 10.1021/ja01189a085
(a) Antons, S.; Beitzke, B. DE 4428106, 1996 [Chem. Abstr. 1996, 124, 288759].
(b) Antons, S. DE 4444109, 1996 [Chem. Abstr. 1996, 125, 114175].
Antons, S.; Tilling, A. S.; Wolters, E. WO 9938838, 1999[Chem. Abstr. 1999, 131, 130283].
Mägerlein, W.; Dreisbach, C.; Hugl, H.; Tse, M. K.; Klawonn, M.; Bhor, S.; Beller, M. Catal. Today 2007, 121, 140. doi: 10.1016/j.cattod.2006.11.024
Metkar, P. S.; Scialdone, M. A.; Moloy, K. G. Green Chem. 2014, 16, 4575. doi: 10.1039/C4GC01167H
龚大春, 涂志英, 何红华, 韦萍, 欧阳平凯, 现代化工, 2007, 27, 151. doi: 10.3321/j.issn:0253-4320.2007.z1.035Gong, D.-C.; Tu, Z.-Y.; He, H.-H.; Wei, P.; Ou Yang, P.-K. Mod. Chem. Ind. 2007, 27, 151(in Chinese). doi: 10.3321/j.issn:0253-4320.2007.z1.035
Tamura, M.; Tamura, R.; Takeda, Y.; Nakagawa, Y.; Tomishige, K. Chem. Commun. 2014, 50, 6656. doi: 10.1039/c4cc02675f
Jere, F. T. Ph.D. Dissertation, Michigan State University, East Lansing, 2003. https://www.kbs.msu.edu/research/all-publications/these-and-dissertations/
Tamura, M.; Tamura, R.; Takeda, Y.; Nakagawa, Y.; Tomishige, K. Chem.-Eur. J. 2015, 21, 3097. doi: 10.1002/chem.201405769
Holladay, J. E.; Werpy, T. A.; Muzatko, D. S. In Proceedings of the Twenty-Fifth Symposium on Biotechnology for Fuels and Chemicals Held May 4~7, Breckenridge, Humana Press, Clifton, 2003, pp. 857~869. doi: 10.1007/978-1-59259-837-3_70
Jere, F. T.; Miller, D. J.; Jackson, J. E. Org. Lett. 2003, 5, 527. doi: 10.1021/ol0274211
王毅, 硕士论文, 天津大学, 天津, 2007.Wang, Y. M.S. Thesis, Tianjin University, Tianjin, 2007(in Chinese).
何洪华, 硕士论文, 南京工业大学, 南京, 2005.He, H.-H. M.S. Thesis, Nanjing Tech University, Nanjing, 2005(in Chinese).
Zwietering, T. N. Chem. Eng. Sci. 1958, 8, 244. doi: 10.1016/0009-2509(58)85031-9
Jere, F. T.; Jackson, J. E.; Miller, D. J. Ind. Eng. Chem. Res. 2004, 43, 3297. doi: 10.1021/ie034046n
Bhandare, S. G.; Vaidya, P. D. Ind. Eng. Chem. Res. 2017, 56, 3797. doi: 10.1021/acs.iecr.6b04406
Pimparkar, K. P.; Miller, D. J.; Jackson, J. E. Ind. Eng. Chem. Res. 2008, 47, 7648. doi: 10.1021/ie800351x
Roose, P.; Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. Amines, Aliphatic in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, Germany, 2015, pp. 1~55. doi: 10.1002/14356007.a02_001.pub2/abstract
Froidevaux, V.; Negrell, C.; Caillol, S.; Pascault, J. P.; Boutevin, B. Chem. Rev. 2016, 116, 14181. doi: 10.1021/acs.chemrev.6b00486
De Schouwer, F.; Claes, L.; Claes, N.; Bals, S.; Degrève, J.; De Vos, D. E. Green Chem. 2015, 17, 2263. doi: 10.1039/C4GC02194K
Verduyckt, J.; Van Hoof, M.; De Schouwer, F.; Wolberg, M.; Kurttepeli, M.; Eloy, P.; Gaigneaux, E. M.; Bals, S.; Kirschhock, C. E. A.; De Vos, D. E. ACS Catal. 2016, 6, 7303. doi: 10.1021/acscatal.6b02561
Verduyckt, J.; Coeck, R.; De Vos, D. E. ACS Sustainable Chem. Eng. 2017, 5, 3290. doi: 10.1021/acssuschemeng.6b03140
Claes, L.; Verduyckt, J.; Stassen, I.; Lagrain, B.; De Vos, D. E. Chem. Commun. 2015, 51, 6528. doi: 10.1039/C5CC00181A
Claes, L.; Matthessen, R.; Rombouts, I.; Stassen, I.; De Baerdemaeker, T.; Depla, D.; Delcour, J. A.; Lagrain, B.; DeVos, D. E. ChemSusChem 2015, 8, 345. doi: 10.1002/cssc.201402801
De Schouwer, F.; Cuypers, T.; Claes, L.; De Vos, D. E. Green Chem. 2017, 19, 1866. doi: 10.1039/C6GC03222B
Liu, G.; Wright, M. M.; Zhao, Q.; Brown, R. C.; Wang, K.; Xue, Y. Energy Convers. Manage. 2016, 112, 220. doi: 10.1016/j.enconman.2016.01.024
Yi, L.; Liu, H.; Lu, G.; Zhang, Q.; Wang, J.; Hu, H.; Yao, H. Energ. Fuel. 2017, 31, 9484. doi: 10.1021/acs.energyfuels.7b01413
Eder, U.; Sauer, G.; Wiechert, R. Angew. Chem., Int. Ed. 1971, 10, 496. doi: 10.1002/(ISSN)1521-3773
List, B.; Lerner, R. A.; Barbas, C. F. J. Am. Chem. Soc. 2000, 122, 2395. doi: 10.1021/ja994280y
List, B. Tetrahedron 2002, 58, 5573. doi: 10.1016/S0040-4020(02)00516-1
(a) Wang, J.-Z. M.S. Thesis, Beijing University of Chemical Technology, Beijing, 2011(in Chinese).
王玖钊, 硕士论文, 北京化工大学, 北京, 2011.
(b) Gruttadauria, M.; Giacalone, F.; Noto, R. Adv. Synth. Catal. 2009, 351, 33.
(c) Doyagüez, E. G.; Calderon, F.; Sanchez, F.; FernandezMayoralas, A. J. Org. Chem. 2007, 72, 9353.
Gao, J.; Liu, J.; Jiang, D.; Xiao, B.; Yang, Q. J. Mol. Catal. A:Chem. 2009, 313, 79. doi: 10.1016/j.molcata.2009.08.005
An, Z.; Zhang, W.; Shi, H.; He, J. J. Catal. 2006, 241, 319. doi: 10.1016/j.jcat.2006.04.035
An, Z.; Guo, Y.; Zhao, L.; Li, Z.; He, J. ACS Catal. 2014, 4, 2566. doi: 10.1021/cs500385s

图式 3 L-赖氨酸的催化加氢转化
Scheme 3 Transformation of L-lysine in the condition of catalytic hydrogenation

图式 4 L-谷氨酸的催化加氢转化
Scheme 4 Transformation of L-glutamic acid in the condition of catalytic hydrogenation

图式 6 温度对L-丙氨酸催化加氢对映选择性的影响
Scheme 6 Temperature on the effects of enantioselectivity in the catalytic hydrogenation of L-alanine

图式 8 L-焦谷氨酸和L-谷氨酸的催化转化
Scheme 8 Catalytic transformation of L-pyroglutamic acid and L-glutamic acid

图式 11 氨基酸催化氧化脱羧机理
Scheme 11 Mechanism of amino acid's catalytic oxidative de-carboxylation

图式 12 氨基酸氧化脱羧到腈类的机理
Scheme 12 Mechanism for oxidative decarboxylation of amino acids to nitriles
表 1 氨基酸转化制备不同化学产品所需的主要条件
Table 1. Main conditions of preparation of different chemical products from the transformation of amino acids
| 产品 | 温度/K | 压力 | 催化剂 | 特殊添加剂 |
| 手性氨基醇 | 373 | 8 MPa H2 | Ru/C、Rh/SiO2 | 无机酸 |
| 胺类与腈类 | 525 | 0.6 MPa N2、O2 | Ru基、Pd基催化剂 | 无 |
| 羧酸及其衍生物 | 498 | 3 MPa H2 | Pd/C、Pt/TiO2 | 无 |
| 生物燃料(烃类) | 923 | 隔绝空气 | ZSM-5等裂解催化剂 | Fe/Ca混合物 |
| 多相手性催化剂 | 常用载体:水滑石、介孔二氧化硅、氧化铝、MCM-41 | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

















 下载:
下载: