 图式 1
偶氮苯基团导向的镍催化的邻位氘代反应
Scheme1.
Azo-induced and Ni catalyzed o-deuteration
图式 1
偶氮苯基团导向的镍催化的邻位氘代反应
Scheme1.
Azo-induced and Ni catalyzed o-deuteration
引用本文:
汪珊, 严沣, 汪连生, 朱磊. 基于导向策略的C—H键活化反应的研究进展[J]. 有机化学,
2018, 38(2): 291-303.
doi:
10.6023/cjoc201708055

Citation: Wang Shan, Yan Feng, Wang Liansheng, Zhu Lei. Recent Advances in Directing Group-Induced C-H Activation Reactions[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 291-303. doi: 10.6023/cjoc201708055
Citation: Wang Shan, Yan Feng, Wang Liansheng, Zhu Lei. Recent Advances in Directing Group-Induced C-H Activation Reactions[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 291-303. doi: 10.6023/cjoc201708055
基于导向策略的C—H键活化反应的研究进展
摘要:
C—H键是有机化合物中存在最广泛的化学键之一.过渡金属催化的C—H键活化反应具有反应效率高、原子经济性高、产生废物少等优点,因此发展新的C—H键的转化类型和方法十分重要.由于分子内经常存在高度稳定且活性相似的C—H键,所以导致传统的C—H键活化反应的区域选择性很差,难以真正被应用于天然产物或者药物分子等的合成中.向反应体系中引入导向基团后,不仅可以大大提高C—H键官能化反应的活性,更可以提高区域选择性,实现高效合成单一目标产物的目的.因此,探索导向基导向的C—H键直接官能化反应具有重要意义.将从不同导向原子的角度介绍近十年导向基辅助的C—H键活化反应,并对其作用机理进行相关阐述.
English
Recent Advances in Directing Group-Induced C-H Activation Reactions
Abstract:
C-H bonds are widely existed in almost all the organic compounds. Transition-metal-catalyzed C-H functionalizations usually have high reaction efficiency and high atom-economy. However, traditional strategies based on such transition-metal catalyzed C-H activations generally result in poor selectivities, because C-H bonds in one molecule facilely display similar reactivity. It is difficult to be utilized in preparation of natural products, pharmaceuticals and biomolecules. However, directing group can induce the metal to activate proximal C-H bonds via cyclometallated intermediates, improve the regioselectivity of the transformations. Therefore, it is extremely significant to deveplop auxiliary-induced C-H bonds activations. The research progress of directing group-induced C-H activation reactions and mechanisms for recent ten years are sumarized.
-
Key words:
- C-H activation
- / regioselectivity
- / directing group
- / transition-metal
-
C—H键是有机化合物中存在最广泛的化学键之一, 从最简单的甲烷到复杂的药物分子、生物大分子都含有大量的C—H键. C—H活化是通过断裂未活化的C—H键, 然后再在某个特殊的位点形成新的C—C键或者C—杂原子键(狭义上未活化C—H键是指pKa>25的C—H键).这种方法可以避免传统合成中的预活化过程, 比如制备卤化物、三氟甲磺酸酯等, 具有反应效率高、原子经济性高、产生废物少等优点, 是一种理想的合成方法[1~3].
C—H键活化反应固然有诸多优点, 有利于绿色化学的发展, 然而传统的C—H键活化反应也存在着不可避免的缺点和不足. 1967年, Fujiwara课题组[4]报道了第一例经历氯化苯乙烯-钯复合物中间体发生的苯环烯基化反应(Eq. 1).但是当氯苯这样的单取代的芳烃参与反应时, 反应体系变得十分复杂, 得到的是三种异构体的混合产物1~3.之所以会出现这种结果, 是因为氯苯中邻间对三种位置的C—H键的反应活性差别很小, 所以反应的区域选择性很差.对于一个单纯的化学反应而言, 如果没有区域、化学、立体等选择性是没有什么实用价值的. 1968年, 先驱有机金属化学家Halpern提出: “成功发展直接C—H活化的方法的研究仍需继续, 而这应该是全世界面临的最重要的问题和挑战之一”[5].
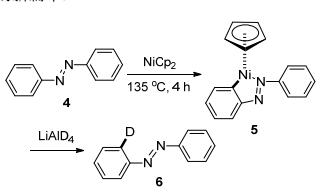
1963年, Kleiman和Dudeck课题组[6]报道了第一例Ni催化的邻位C—H键氘代反应(Scheme 1).在该反应中, 偶氮苯(4)中的偶氮基团作为导向基与金属Ni配位后, 选择性的活化邻位C—H键, 生成相应的Ni复合物5.复合物5与LiAlD4作用后只得到了邻位氘代的产物6.该反应证明, 如果向反应底物中引入某些可与金属配位的官能团, 就可以通过金属环化作用特定地活化与金属相邻的C—H键, 从而实现具有区域选择性的C—H官能化反应.这些导向基的引入解决了传统C—H键活化反应中选择性差的问题, 自此揭开了C—H键活化反应的新篇章.
图式 1
偶氮苯基团导向的镍催化的邻位氘代反应
Scheme1.
Azo-induced and Ni catalyzed o-deuteration
导向基团可分为弱配位基团和强配位基团.其中, 最经典导向基导向的C—H键活化反应中, 导向基团通常都是含N、S、P的强配位基团, 比如吡啶、噁唑啉、硫化物和磷酸酯等, 因为这些物质都是强的σ供体和π受体.以2-苯基吡啶(7)为例(Scheme 2), 通常这些物质经过与金属配位, 形成一个五元或者六元的环金属过渡态9.环金属过渡态9与偶联试剂继续反应得到产物10.
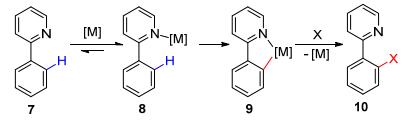 图式 2
经典的导向基作用机理
Scheme2.
Classic process of directing group
图式 2
经典的导向基作用机理
Scheme2.
Classic process of directing group
其中, “金属环化作用”是在20世纪60年代首次被提出, 这个过程包含两步: (1)后过渡金属催化C—H键断裂; (2)形成[M—R].形成的机理根据金属种类、底物、反应条件的不同而不同, 包括氧化加成、亲电活化、金属化/去质子化等作用.化合物11~16已经被公认的通过配位作用形成金属环化中间体(图 1), 包括Ni、Pd、Pt、Ir、Rh、Ru等多种金属都能进行这一过程.
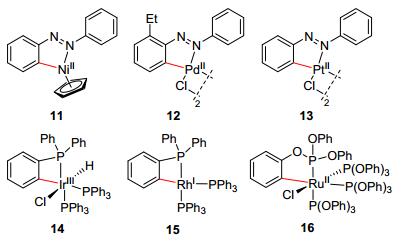 图1
公认的金属环化中间体
Figure1.
Well-defined metallacycles
图1
公认的金属环化中间体
Figure1.
Well-defined metallacycles
近几十年, 科学家们根据以上反应机理发展出各种类型的导向基团, 逐步丰富了导向基团所能适用的反应类型, 并且着手研究不同于强配位机理的弱配位反应历程.本文将从不同导向原子的角度介绍近10年导向基辅助的C—H键活化反应.
1 含O导向基
1.1 羟基导向的C—H活化反应
1997年, Miura课题组[7]首次报道了钯催化酚羟基导向的C—H键活化反应.该课题组成功地在碱性条件下实现了酚羟基导向的邻位芳基化反应.最近, Fan课题组[8]也以Pd(Ⅱ)作为催化剂, 叔丁基过氧化氢(t-BuOOH)作为氧化剂成功实现了酚羟基导向的邻位羟基化反应, 合成了2, 2'-联苯酚及其衍生物18 (Eq. 2).
根据实验结果提出相应的反应机理(Scheme 3).首先, 反应物17在碱的作用下生成阴离子19, 接着Pd(Ⅱ)与化合物配位生成化合物20.在导向基和配体的辅助下, Pd(Ⅱ)通过金属插入作用活化邻位C—H键, 并释放出LH.与此同时, t-BuOOH均裂生成自由基HO·和t-BuO·. t-BuO·与LH作用生成t-BuOH, 并释放自由基L·. HO·以及L·和化合物21进行氧化加成得到Pd(Ⅳ)中间体22, 继续经历还原消除生成Pd(Ⅱ)中间体23.中间体23通过质子化作用生成目标产物18, 同时Pd(Ⅱ)催化剂重生.
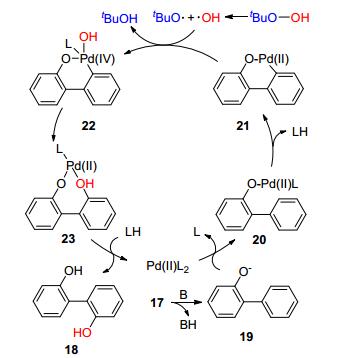 图式 3
化合物18的形成机理
Scheme3.
Proposed reaction mechanism for the formation of 18
图式 3
化合物18的形成机理
Scheme3.
Proposed reaction mechanism for the formation of 18
酚羟基导向的C—H键活化反应早已被报道并且得到了一定的发展.但是可能是由于酚羟基的配位能力比醇羟基强, 并且一级醇和二级醇相比于酚更容易发生β-H消除, 所以醇羟基导向的C—H活化反应直到2010年才被Yu课题组[9, 10]报道(Scheme 4).这类反应存在两个问题: (1) Pd与O的σ-螯合很弱; (2) Pd(Ⅱ)会氧化伯醇、仲醇, 分解三级醇.实验发现, 这类反应在弱碱和非极性芳烃溶剂中进行时可以解决上述两个问题.但是一般的非极性芳烃溶剂如甲苯等会和丙烯酸以及PhI(OAc)2发生反应, 并且由于它们是溶剂量的, 会极大地抑制目标反应.因此, 在不得以的情况下, 上述两个反应只有选择全氟取代的六氟苯作为溶剂.
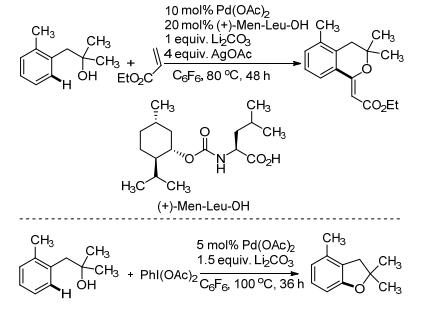 图式 4
Pd催化的醇羟基导向的邻位C—H活化反应
Scheme4.
Pd-catalyzed alcoholic hydroxyl-induced o-C—H activations
图式 4
Pd催化的醇羟基导向的邻位C—H活化反应
Scheme4.
Pd-catalyzed alcoholic hydroxyl-induced o-C—H activations
2011年, Gevorgyan课题组[11]报道了一种无痕的硅烷醇导向的C—H键烯基化反应(Eq. 3).由于导向基中硅原子上连接的叔丁基空间位阻较大, 所以该反应选择性地在位阻小的一侧生成单取代产物, 展现出很好的区域选择性.该反应在经历第一步钯盐催化偶联之后不用分离, 直接加入四丁基氟化铵即可将导向基去除, 实现了一锅法制备目标产物24.
1.2 羧酸和羧酸盐导向的C—H活化反应
如果羧酸作为反应物参与反应时, 羧基本身即可作为导向基, 无需再添加额外的导向基团, 那么这无疑是一个巨大的优势. Yu课题组[9, 12]在羧酸及其衍生物导向的C—H键官能化方面做出了杰出的贡献(Scheme 5, Eq. 4).经过大量实验发现, 向反应中引入配对阳离子, 如Na+, K+, NH4+甚至被质子化的N, N-二甲基甲酰胺(DMF), 都能有效地促进羧酸作为底物时Pd催化的C—H断裂过程.作者推测Pd可以和羧基产生多种多样的配位构象, 但是如果在K1构象26和K2构象25中平衡强烈的向K2构象25倾斜, 那么期望中的C—H键活化断裂将变得非常迟钝(Eq. 5).而配对阳离子与羧基形成K2配位25后, 会导致Pd与无位阻的氧原子发生K1模式26的单电子对配位, C—H活化反应才能进行, 他们也通过单晶衍射证实了这一推测[13].
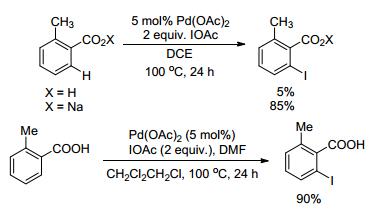 图式 5
Pd催化的羧基导向的邻位C—H键碘代反应
Scheme5.
Pd-catalyzed carboxyl-induced o-C—H iodination
图式 5
Pd催化的羧基导向的邻位C—H键碘代反应
Scheme5.
Pd-catalyzed carboxyl-induced o-C—H iodination
之后, 该课题组[13]继续将反应体系拓展到C—H/ R—BX2交叉偶联(Scheme 6).傅克烷基化反应是最常见的烷基化反应, 但是由于底物适用性差、区域选择性低、容易过度烷基化等缺陷, 使得其运用受到较多限制.该课题组报道的Pd催化的C—H/R—BX2交叉烷基化反应可以提供具有良好区域和化学选择性的单取代产物.更进一步地, 该课题组还将该反应拓展到C(sp3)—H的烷基化反应中, 成功实现了偶联, 只是C(sp3)—H烷基化反应产率较低.
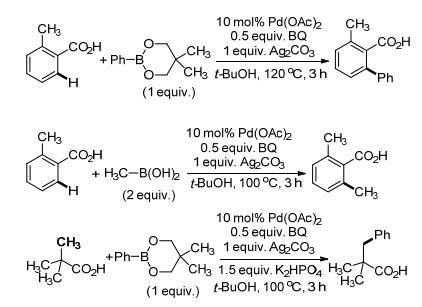 图式 6
Pd催化的C—H/R—BX2交叉偶联
Scheme6.
Pd-catalyzed C—H/R—BX2 cross-coupling
图式 6
Pd催化的C—H/R—BX2交叉偶联
Scheme6.
Pd-catalyzed C—H/R—BX2 cross-coupling
同时, 该课题组[14]还报道了Pd催化的羧基导向的氧化Heck反应(Eq. 6).值得注意的是, 该反应具有很高的区域选择性.他们认为关于如何控制反应位点, 可以从两方面出发: (1)底物控制, 通过添加不同的保护基, 调整适合的反应空间; (2)配体控制, 通过不同的配体改变中心金属的立体性质和电子效应.该反应选择了配体控制, 当有两个不同间位取代基的苯乙酸27参与反应时, 即存在不同的邻位反应位点时, N-单取代保护的氨基酸可以通过微妙的立体差异或者电子效应差异来区分不同的两个位点, 选择性地生成产物28或者29.
1.3 羰基导向的C—H活化反应
2010年, Yu课题组[15]报道了以CO作为碳源, 以羰基作为导向基团的苯胺衍生物30邻位羧基化反应(Scheme 7).他们在研究中发现, 对甲苯磺酸和醋酸与1, 4-二氧六环的存在对反应十分重要.而羧基化产物31在PCl3的作用下可以进一步转化成喹唑啉酮32.
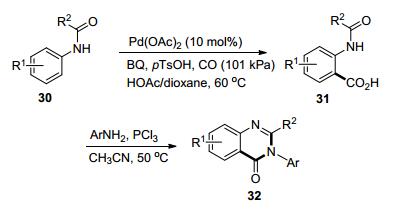 图式 7
Pd催化的CO作为碳源的羰基导向的邻位羧基化反应
Scheme7.
Pd-catalyzed carbonyl-induced o-carboxylation with CO as carbon source
图式 7
Pd催化的CO作为碳源的羰基导向的邻位羧基化反应
Scheme7.
Pd-catalyzed carbonyl-induced o-carboxylation with CO as carbon source
同年, Liu课题组[16]报道了Pd催化的苯酚酯的邻位C—H键的芳基化反应(Eq. 7).这个反应对水和空气都不敏感, 可以在室温下直接反应.并且他们首次用单晶衍射证实了苯酚酯的环钯化中间体33的存在.
2011年, Gaunt课题组[17]报道了Cu催化的α-芳基羰基化合物的间位芳基化反应(Scheme 8).该反应具有良好的官能团容忍性和优秀的区域选择性.即使在邻对位定位基存在时, 依然选择性地生成间位取代产物34~38.
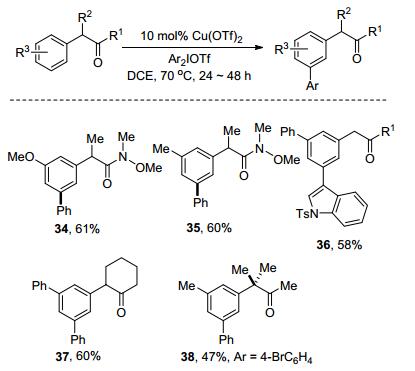 图式 8
Cu催化的α-芳基羰基化合物的间位芳基化反应
Scheme8.
Cu-catalyzed m-arylation of α-aryl carbonyl compound
图式 8
Cu催化的α-芳基羰基化合物的间位芳基化反应
Scheme8.
Cu-catalyzed m-arylation of α-aryl carbonyl compound
同年, Liu课题组[18]报道了Pd催化的芳基酮与磺酰胺发生的邻位酰胺化反应(Eq. 8). Pd(OTf)2的存在对该反应十分重要, 反应中间体酮的环钯化复合物通过单晶衍射得到了验证.另外, 该反应可以用于合成2-烷基吲哚、3-烷基吲哚以及2-氨基苯基酮等化合物.
除了酮羰基以外, 醛羰基也可以作为导向基团促进C—H活化反应的进行.最近, Cheng课题组[19]报道了Co催化1, 6-烯炔39与苯甲醛合成吡咯烷或二氢呋喃40的环化反应.当例如烯基或苯基二膦配体(dppen或dppben)等相对缺电的配体参与反应时, 醛基即作为导向基团辅助金属Co的邻位C—H键金属化作用, 进行邻位烷基化环化反应(Eq. 9).另外, 当相对富电的配体参与反应时, 金属Co不再活化苯甲醛邻位C—H键, 而是对醛基进行插入作用, 此时醛基不再作为导向基团.
2 含N导向基
2.1 含氮杂环导向的C—H活化反应
相较于含O导向基而言, 含N导向基种类更为繁多, 应用更为广泛.继Sanford和Yu等课题组[20, 21]在吡啶基辅助的C—H活化领域做出了先驱性工作之后, 许多科学家都对这一领域进行了探索. 2013年, Shi课题组[22]报道了吡啶基作为导向基团, 苯甲酸作为芳源的直接脱羧芳基化反应.无论是缺电或富电的基团取代的2-苯基吡啶都可以与缺电或富电的苯甲酸直接发生脱羧芳基化反应(Scheme 9), 并且这个反应避免了使用昂贵和高毒性的氧化剂.
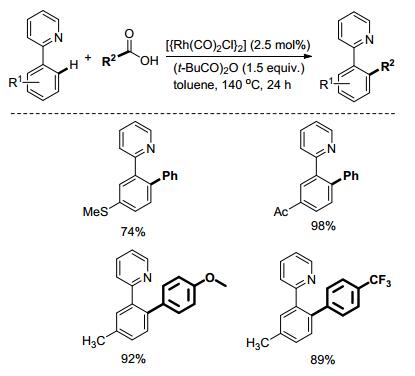 图式 9
吡啶导向的C—H键脱羧芳基化反应
Scheme9.
Pyridyl-induced C—H arylation by decarboxylation
图式 9
吡啶导向的C—H键脱羧芳基化反应
Scheme9.
Pyridyl-induced C—H arylation by decarboxylation
2016年, Yu课题组[23]也报道了2-苯基吡啶7, 以Cp*CoI2(CO)作为催化剂, 与端基炔烃41的邻位C—H键烯基化反应(Scheme 10).该反应的反应条件十分温和, 具有良好的官能团容忍性.甚至6-芳基嘌呤43也能顺利地进行反应, 并且高产率得到产物44.更进一步地, 该反应可以扩展反应物的π电子体系, 利用这一特性设计合成了具有线粒体靶向的荧光染料.
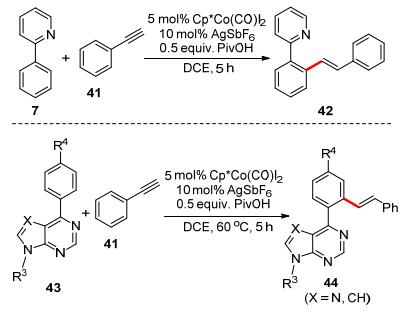 图式 10
2-苯基吡啶和6-芳基嘌呤的烯基化反应
Scheme10.
Alkenylation of 2-phenylpridine and 6-arylpurine
图式 10
2-苯基吡啶和6-芳基嘌呤的烯基化反应
Scheme10.
Alkenylation of 2-phenylpridine and 6-arylpurine
2013年, Kuang课题组[24]报道了Pd催化的1, 2, 3-三唑导向的邻位C—H键酰化反应(Scheme 11).该反应从醛出发合成了相应的芳香酮, 具有很高的原子经济性.氧化剂为过氧化叔丁醇(70%水溶液), 廉价易得、环境友好.另外, 该反应具有很好的官能团容忍性, 反应条件温和, 为应用于合成天然产物和其他有用化合物提供了可能性.
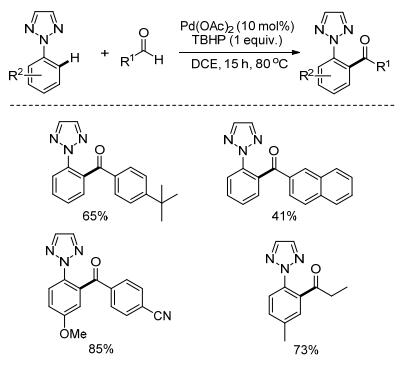 图式 11
Pd催化的1, 2, 3-三唑导向的邻位C—H键酰化反应
Scheme11.
Pd-catalyzed 1, 2, 3-azo-induced o-C—H acylation
图式 11
Pd催化的1, 2, 3-三唑导向的邻位C—H键酰化反应
Scheme11.
Pd-catalyzed 1, 2, 3-azo-induced o-C—H acylation
2014年, Yi课题组[25]报道了一个反应条件温和的Rh(Ⅲ)催化的吲哚C-2位烯基化反应(Scheme 12).该反应使用嘧啶基作为导向基团, 反应产率良好, 可以容忍多种官能团, 并且具有优秀的区域选择性和原子经济性.因此该合成方法可以用于构建具有C-2-烯基吲哚结构单元的活性生物分子.更进一步地, 产物45的导向基——嘧啶基可以很容易地被移除, 得到相应的C-2丙烯酸取代的吲哚46.而化合物46是一个非常重要的合成中间体, 可以用于合成很多重要的天然产物和药效团, 如Trp-P-2和MK-7246.
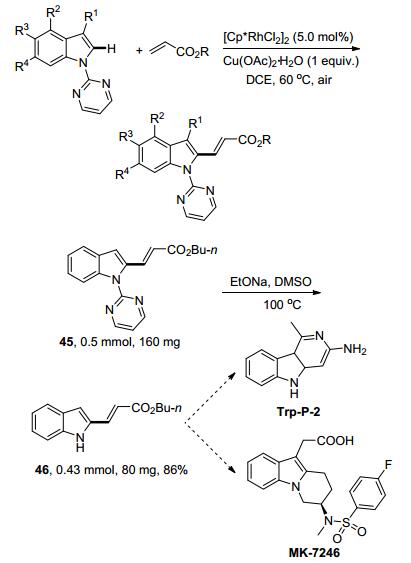 图式 12
Cp*Rh催化的嘧啶基诱导的吲哚C-2位烯基化反应
Scheme12.
Cp*Rh-catalyzed pyrimidyl-induced C-2 alkenylation of indole
图式 12
Cp*Rh催化的嘧啶基诱导的吲哚C-2位烯基化反应
Scheme12.
Cp*Rh-catalyzed pyrimidyl-induced C-2 alkenylation of indole
除了单齿导向基外, 二齿配体导向基也可以用于促进C—H键活化反应. 2005年Daugulis课题组[26]报道了Pd(OAc)2催化的以8-氨基喹啉片段作为导向基的C—H键的芳基化反应(Eq. 10).更加有意义的是, 导向基本身可以很容易地被移除, 生成相应的羧酸, 具有很好的实际应用价值.例如, Chen课题组[27]在强效抗有丝分裂剂Celogentin C的全合成中, 其中一步就是利用上述Daugulis报道的8-氨基喹啉酰胺与卤代烃的芳基化反应.该步骤采用亮氨酸的衍生物与碘代吲哚反应, 成功地得到区域和立体选择性的产物(Eq. 11).
无独有偶, Baran课题组[28]也利用该反应设计合成了他们的目标化合物Pipercyclobutanamide A.其中关键一步, 通过直接活化的C(sp3)—H, 依次反应得到了芳基化和烯基化的环烷烃(Scheme 13).
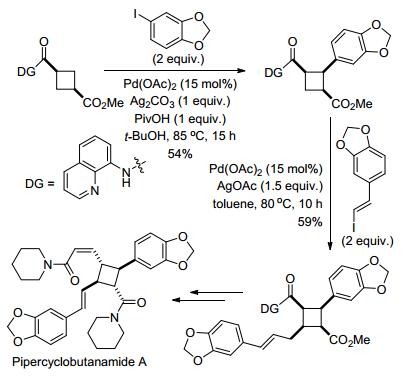 图式 13
Pipercyclobutanamide A全合成过程中的C—H活化步骤
Scheme13.
C—H activation steps in preparation of pipercyclobutanamide A
图式 13
Pipercyclobutanamide A全合成过程中的C—H活化步骤
Scheme13.
C—H activation steps in preparation of pipercyclobutanamide A
继Daugulis及其合作者在8-氨基喹啉片段导向的C—H活化反应上画下浓墨重彩的一笔之后, 许多科学家都参与了这一领域的研究.而这一片段也表现出其优秀的促进C—H活化的能力, 从昂贵的Pd[26], Ru[29, 30]等金属, 到廉价的Cu[31, 32], Fe[33, 34], Ni[35], Co[36, 37]等金属都能与其发生配位并促进反应的发生.
由于膦试剂具有很强的配位能力, 使得通过过渡金属催化的C—H键膦化反应构建C—P十分困难.而Yu课题组[32]发展了8-氨基喹啉片段导向的C—H键膦化反应(Eq. 12), 克服了这一难题.该反应利用廉价的铜盐作为催化剂, 并且反应条件十分温和, 并且选择性地生成邻位单取代的膦化产物, 具有很好的官能团容忍性.当取代基在间位时, 反应选择性地在位阻小的一侧进行.
另外, 这一片段不仅能导向C(sp2)—H键的反应, 也可以促进sp3 C—H键的反应[37, 38]. Ge课题组[37]报道了Co(acac)3催化的8-氨基喹啉基导向的C(sp3)—H胺化反应(Scheme 14).该反应可以用于直接合成单环和螺环β以及γ内酰胺.并且首次实现了Co催化的分子间C(sp3)—H脱氢胺化反应.
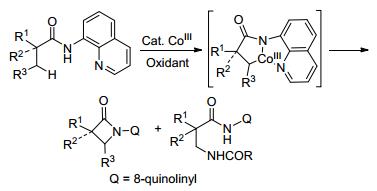 图式 14
8-氨基喹啉片段导向的C(sp3)—H键胺化反应
Scheme14.
8-Aminoquinolinyl-induced C(sp3)—H amination
图式 14
8-氨基喹啉片段导向的C(sp3)—H键胺化反应
Scheme14.
8-Aminoquinolinyl-induced C(sp3)—H amination
Yu课题组[39]报道了二齿导向基氨基取代的噁唑啉导向的C—H酰胺化和胺化反应(Scheme 15).磺酰胺、酰胺和苯胺类化合物都可以参与反应, 即使两种反应物中都含有多个杂原子反应依然可以顺利进行, 并以相对较高的产率得到相应的产物49~54.而这一特性对于修饰含有羧基的药物分子十分重要.值得注意的是, 该反应条件温和, 并且不需要加入Ag盐作为氧化剂或共催化剂.另外, 导向基可以很容易地被移除, 在以86%的产率得到羧酸产物47的同时, 可以回收93%的导向基团的制备原料48.
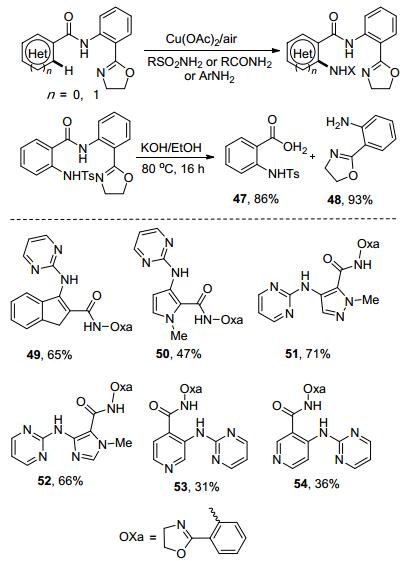 图式 15
Cu催化的C—H酰胺化和胺化反应
Scheme15.
Copper-catalyzed C—H amidation and amination
图式 15
Cu催化的C—H酰胺化和胺化反应
Scheme15.
Copper-catalyzed C—H amidation and amination
2.2 氮氧类官能团导向的C—H活化反应
2013年, You课题组[40]报道了Rh催化的N, N-二烷基苯胺氮氧化物55的邻位烯基化反应(Eq. 13).该反应不需要添加额外的氧化剂, 这是由于N+—O-结构在反应中除了起到导向作用以外, 还提供了氧化作用.在Rh催化下, N, N-二烷基苯胺氮氧化物55与苯乙烯56室温条件下就可以转化成N, N-二烷基的产物57.通过培养出的中间体的单晶[40], 证明该反应过程中形成了一个五元的铑复合物中间体58.
2015年, Song课题组[41]报道了Co催化的氮氧化物59的邻位醚化反应(Eq. 14), 其中以2-氨基吡啶氮氧化物作为二齿配位导向基团.这也是第一例烯烃的直接C—H键醚化反应.该反应证明了除Pd或Cu, Co也可以参与这一类型的C—H键活化反应, 并且拓展了Co催化的C—H键官能化反应类型.值得一提的是, 氮氧二齿配体可以很容易地被移除.
2.3 其他含氮化官能团诱导的C—H活化反应
除了上述提到的含N杂环和氮氧类导向基团外, 还有很多种其他类型的含N导向基团. 2011年, Yoshikai课题组[42]报道了亚胺邻位烯基化反应, 这个反应需要Co盐、三芳基膦、格氏试剂以及吡啶同时存在才能顺利进行(Scheme 16).该反应不仅具有良好的官能团容忍性, 还展现出了独特的区域选择性(60~70).当甲氧基、卤素和氰基等取代基在亚氨基的间位的时候, 反应选择性地在位阻更大的邻位C—H处进行.此外, 在酸性条件下, C—H活化产物可以以中等至良好的产率水解生成相应的酮或环化产物.
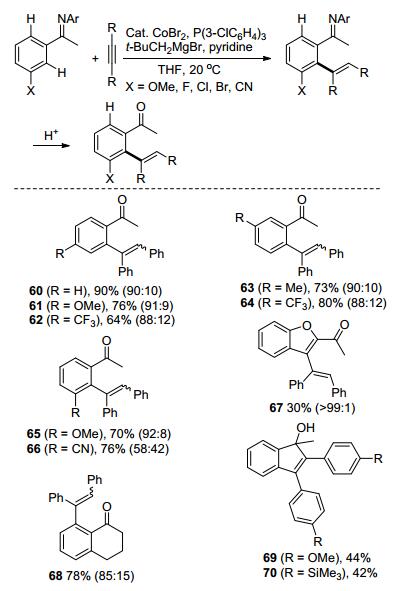 图式 16
Co催化的亚胺邻位烯基化反应
Scheme16.
Co-catalyzed o-alkenylation of imine
图式 16
Co催化的亚胺邻位烯基化反应
Scheme16.
Co-catalyzed o-alkenylation of imine
2007年, Hiroya课题组[43]报道了Pd催化的苯腙作为导向基团的分子内胺化反应, 该反应可以用于合成多取代的吲唑环(Eq. 15).该反应的条件温和, 在50 ℃下即可进行, 并具有良好的官能团容忍性, 如酯基、腈基、卤原子等多种官能团都能在反应后得以保留.
2012年, Bräse课题组[44]报道了三氮烯导向的邻位三氟甲基化反应(Scheme 17).值得注意的是, 通常三氟甲基金属中间体会在C—X键位点发生反应, 将卤素取代下来.但是该反应体系则很好地容忍了这些官能团的存在.另外有趣的是, 为了研究机理, 向反应中加入了5 equiv.的自由基抑制剂——2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO), 结果发现该反应完全被抑制.这样的结果更倾向于说明该反应经历了自由基历程.但是通常自由基反应的可控性差, 很难得到区域选择性的偶联产物, 因此该反应十分引人注目.除此之外, 三氮烯导向基也可以在温和的条件下被修饰成多种不同的官能团, 拓展了该反应的实际应用价值.
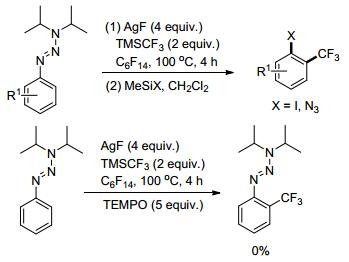 图式 17
Ag催化的三氮烯诱导的邻位三氟甲基反应
Scheme17.
Ag-catalyzed triazenyl-induced o-trifluoromethyla-tion
图式 17
Ag催化的三氮烯诱导的邻位三氟甲基反应
Scheme17.
Ag-catalyzed triazenyl-induced o-trifluoromethyla-tion
苄胺类化合物的重要性使得它们的C—H键官能化反应十分有意义. 2007年Shi课题组[45]报道了Pd催化的N, N-二甲基氨基导向的邻位C—H键烯基化反应(Scheme 18).值得注意的是, N, N-二甲基苄胺是一个很强的σ电子供体, 这使得它可以与过渡金属发生强配位.这种结合方式不需要占据配位点, 但增加了金属中心的电荷密度, 从而削弱了过渡金属的亲电能力, 不利于反应进行.理论上, 酸的存在则可以提高过渡金属的亲电能力使其进攻芳香环, 但是N, N-二甲基苄胺又是一个很强的布朗斯特碱.虽然其可以被质子化形成化合物71, 但是当其被质子化之后, 配位能力和导向能力将大大被减弱, 使得官能化反应只能通过傅克反应来进行, 可是这又不是具有区域选择性的C—H活化反应.这种矛盾使得N, N-二甲基苄胺作为导向基时反应十分困难.因此, 选择合适pKa的添加剂尤为重要, 最终选择了乙酸作为添加剂, 使得反应可以顺利进行.
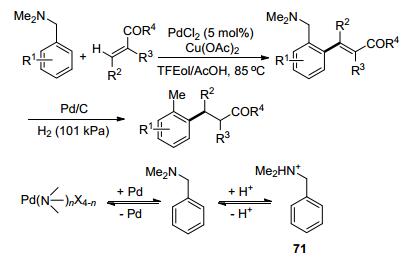 图式 18
Pd催化的N, N-二甲基氨基导向的邻位烯基化反应
Scheme18.
Pd-catalyzed N, N-dimethyl amino group-induced o-alkenylation
图式 18
Pd催化的N, N-二甲基氨基导向的邻位烯基化反应
Scheme18.
Pd-catalyzed N, N-dimethyl amino group-induced o-alkenylation
2012年, Yu课题组[46]报道了Pd催化的胍基导向的邻位C—H键活化反应(Scheme 19).通过适当改变反应条件, 可以选择性地实现邻位芳基化或烯基化反应.作者提出由于反应条件不同, 烯基化反应经历了Pd0/PdⅡ的反应机理, 而芳基化反应则应该是PdⅡ/PdⅣ反应历程.
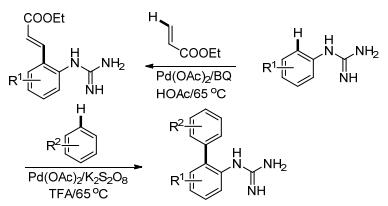 图式 19
Pd催化的胍基导向的邻位芳基化和烯基化反应
Scheme19.
Pd-catalyzed guanidyl-induced o-arylation and alkenylation
图式 19
Pd催化的胍基导向的邻位芳基化和烯基化反应
Scheme19.
Pd-catalyzed guanidyl-induced o-arylation and alkenylation
2012年, Yu课题组[47]设计合成了化合物72, 包含一种新型的远程导向基团(Scheme 20).这种导向基与经典的导向基不同, 是一种U形导向基.反应时, U形末端的氰基可以和Pd形成弱配位.之所以要设计这样一个基团是因为在间位定位或者对位定位的反应中, 在预过渡态中需要形成一个大于7元的环.而这种末端活化的导向基可以形成一个类似大环状烯烃的预过渡态, 从而解决了传统C—H键活化上的问题.末端的氰基与Pd发生弱配位也许可以缓解类大环状烯烃的环张力, 或者可能在间位C—H附近释放Pd, 提供一个很高的反应浓度.同时, 氰基的电子云密度很高, 从而弥补了它在配位能力上的不足, 也能促进反应进程.
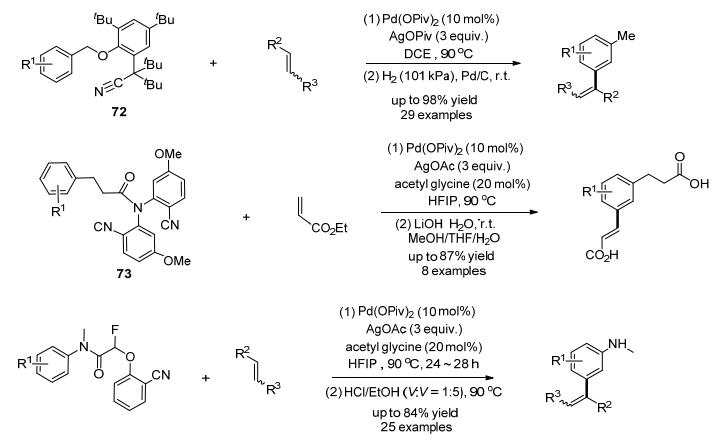 图式 20
Pd催化的氰基导向的间位烯基化反应
Scheme20.
Pd-catalyzed cyano group-induced m-alkenylation
图式 20
Pd催化的氰基导向的间位烯基化反应
Scheme20.
Pd-catalyzed cyano group-induced m-alkenylation
在进行了这个反应之后, 作者逐渐将反应体系扩展到可以移除的酰胺键上, 从而合成了化合物73, 74, 发展了另外两个反应[48], 使得导向基团可以很容易地被移除.并且, 该课题组发现这种类型的导向基还可以促进间位C—H键的芳基化反应[49](Eq. 16).在该反应中, 乙酰甘氨酸(MPAA)的存在对偶联反应的进行十分重要.另外, 四丁基铵盐的加入可以阻止钯盐在反应中生成没有催化活性的钯黑, 同时, 负离子的存在可以稳定反应中出现的钯正离子中间体.
3 其他导向基
3.1 含S化合物导向的C—H活化反应
2010年, Daugulis课题组[50]首次报道了烷基硫醚导向的β-C(sp3)—H键的芳基化反应(Eq. 17).该反应结果证明了硫原子也可以与Pd发生配位, 并促进C—H键活化过程.随后该课题组[51]对该类反应进行了进一步的研究, 并报道了甲硫醚导向的非天然氨基酸的合成(Scheme 21).研究发现当手性反应物75参与反应时, 可以得到手性保持的产物76, 经过简单的酰胺的酯解反应即可制备得到非天然氨基酸77.
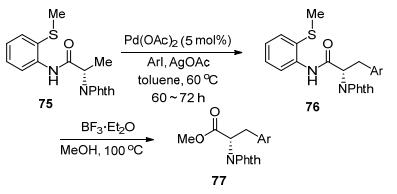 图式 21
硫醚诱导的非天然氨基酸的合成
Scheme21.
Thioether group-induced preparation of non-natural amino acid
图式 21
硫醚诱导的非天然氨基酸的合成
Scheme21.
Thioether group-induced preparation of non-natural amino acid
2011年, Yu课题组[52]设计了一个弱配位的导向基磺酰胺基, 他们发现这个基团可以被用于药物化学领域.例如, 化合物78是一个医药中间体, 经过一步简单的Pd催化的C—H键官能化, 可以分别和丙烯酸酯、甲基硼酸、苯基硼酸酯、IOAc和CO反应, 转化为6种不同的修饰体, 极大地促进了磺胺类药物的发展(Scheme 22).
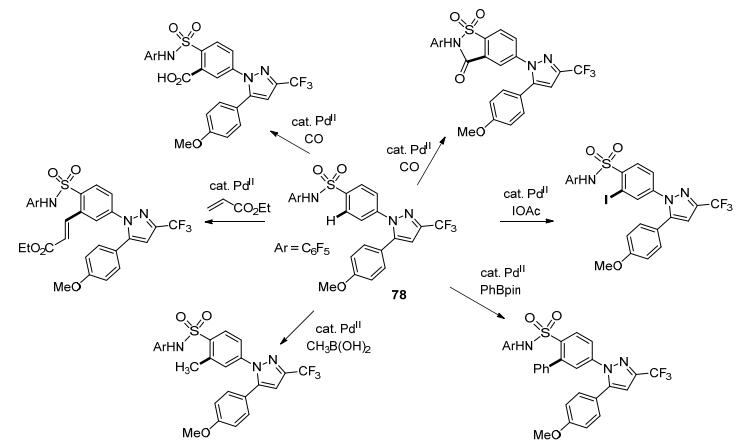 图式 22
Pd催化的磺酰氨基诱导的C—H活化反应
Scheme22.
Pd-catalyzed sulfonamino group-induced C—H activations
图式 22
Pd催化的磺酰氨基诱导的C—H活化反应
Scheme22.
Pd-catalyzed sulfonamino group-induced C—H activations
2014年, Zhang课题组[53]报道了Pd催化的亚砜基导向的C(sp2)—H键的烯基化反应(Scheme 23).各种各样的烯烃都可以选择性的在芳基亚砜苯环的邻位发生反应, 高效且原子经济性地生成目标产物.亚砜和苯环之间的碳原子个数在1~3之间时, 邻位的C—H键都可以被活化.另外, 亚砜可以轻易地被转换成其他官能团, 这是该反应的另一个优势.
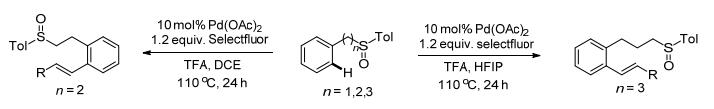 图式 23
亚砜基诱导的C—H键的烯基化反应
Scheme23.
Sulfoxide group-induced C—H alkenylations
图式 23
亚砜基诱导的C—H键的烯基化反应
Scheme23.
Sulfoxide group-induced C—H alkenylations
3.2 含P化合物导向的C—H活化反应
2013年, Chatani课题组[54]报道了Pd催化三芳基膦79合成磷杂茂80的反应(Scheme 24).在该反应中, 磷原子直接参与配位, 并且需要脱除一分子苯.这个反应操作简单, 有着很好的官能团容忍性.
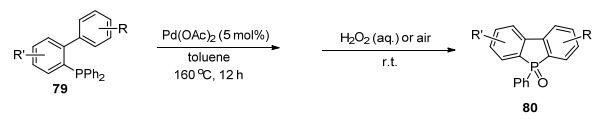 图式 24
Pd催化合成磷杂茂的反应
Scheme24.
Pd-catalyzed preparation of H-phosphole
图式 24
Pd催化合成磷杂茂的反应
Scheme24.
Pd-catalyzed preparation of H-phosphole
2014年, Yang课题组[55]报道了Pd催化R2(O)P-基团导向的邻位乙酰化反应(Scheme 25).该反应具有较高的实际应用价值, 可以用于合成生物活性化合物.例如, 由化合物(R)-81经(R)-82、(R)-83最终合成了手性配体(R)-MeO-MOP.
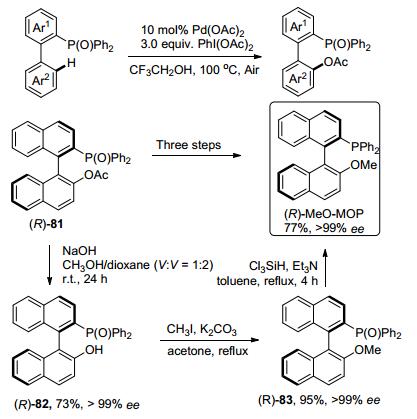 图式 25
R2(O)P基团诱导的邻位乙酰化反应
Scheme25.
R2(O)P group-induced o-acetylation
图式 25
R2(O)P基团诱导的邻位乙酰化反应
Scheme25.
R2(O)P group-induced o-acetylation
2013年, Lee课题组[56]报道了Pd催化的膦酰胺导向的邻位C—H键芳基化反应(Eq. 18).该反应的条件十分温和, 在室温下即可进行, 芳基来源为二芳基碘盐. Cu盐的存在至关重要, 当没有Cu盐时, 该反应几乎不能发生; 当加入3 equiv. CuO, 在室温下搅拌3 h后即可以84%的收率得到目标产物.
Kim课题组[57]报道了Pd催化的烷基磷酸酯基和单磷酸酯基导向的C—H键芳基化反应(Scheme 26).该反应有良好的官能团容忍性, 化合物84和85两类磷酸酯均能参与反应.其中85在温度高于110 ℃时会发生分解, 所以反应需要控制在合适温度. 85反应的产物86在TMSCHN2/MeOH存在的条件下, 室温搅拌30 min即可转化成相应的二甲基芳基磷酸酯.
 图式 26
磷酸酯诱导的C—H芳基化反应
Scheme26.
Phosphate-induced C—H arylations
图式 26
磷酸酯诱导的C—H芳基化反应
Scheme26.
Phosphate-induced C—H arylations
4 结论与展望
过渡金属催化的C—H键活化反应具有反应效率高、原子经济性高、产生废物少等优点, 这些优点都是有利于绿色化学发展的.但是由于分子内经常存在高度稳定且活性相似的C—H键, 所以传统的C—H键活化反应的区域选择性很差, 难以真正被应用于天然产物或者药物分子等的合成中.当向反应体系中引入导向基团后, 不仅可以大大提高C—H键官能化反应的活性, 更加可以提高区域选择性, 因此, 探索导向基导向的C—H键直接官能化反应具有重要意义.
目前科学家们对发展具有区域选择性的C—H活化反应已经做出了卓越的贡献, 但是该方向仍有较大的发展空间.首先, 虽然非贵金属催化的导向基诱导的C—H键活化已经有了一定发展, 但是目前绝大多数的这类反应依然依赖于贵金属催化, 所以, 进一步发展非贵金属催化的具有区域选择性的C—H键活化反应十分必要, 尤其是C(sp3)—H活化反应.其次, 剧烈、苛刻的反应条件, 不但为有机合成工作者们带来了不便, 而且会破坏药物分子、生物活性分子中存在的一些敏感官能团.因此, 应继续研发反应条件温和、官能团容忍性好这类反应[58].第三, 目前这类反应主要提高的是反应的区域选择性, 但是大多数的天然产物和药物分子都是手性化合物.因此, 提高反应的立体选择性也同样重要.第四, 目前导向反应主要局限于邻位反应, 只有极少量的间位和对位反应被报道.设计与开发新型的间位、对位导向基团应成为未来的发展方向.最后, 除发展新的反应外, 更加深入地研究反应历程、导向基配位方式等也尤为重要.
有机合成方法学通常会发展出新型的简单高效的合成方法, 这些方法可以运用于各类化合物的合成, 将有机合成方法学与实际应用结合起来.
-
-
[1]
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147. doi: 10.1021/cr900184e
-
[2]
Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. Chem. Soc. Rev. 2011, 40, 4740. doi: 10.1039/c1cs15083a
-
[3]
Ackermann, L. Acc. Chem. Res. 2014, 47, 281. doi: 10.1021/ar3002798
-
[4]
Moritanl, I.; Fujiwara, Y. Tetrahedron Lett. 1967, 8, 1119. doi: 10.1016/S0040-4039(00)90648-8
-
[5]
Bergman, R. G. Nature 2007, 446, 391. doi: 10.1038/446391a
-
[6]
Kleiman, J. P.; Dubeck, M. J. Am. Chem. Soc. 1963, 85, 1544. doi: 10.1021/ja00893a040
-
[7]
Satoh, T.; Kawamura, Y.; Miura, M.; Nomura, M. Angew. Chem., Int. Ed. 1997, 36, 1740. doi: 10.1002/(ISSN)1521-3773
-
[8]
Duan, S.; Xu, Y.; Zhang, X.; Fan, X. Chem. Commun. 2016, 52, 10529. doi: 10.1039/C6CC04756D
-
[9]
Lu, Y.; Wang, D. -H.; Engle, K. M.; Yu, J. -Q. J. Am. Chem. Soc. 2010, 132, 5916. doi: 10.1021/ja101909t
-
[10]
Wang, X.; Lu, Y.; Dai, H. -X.; Yu, J. -Q. J. Am. Chem. Soc. 2010, 132, 12203. doi: 10.1021/ja105366u
-
[11]
Huang, C.; Chattopadhyay, B.; Gevorgyan, V. J. Am. Chem. Soc. 2011, 133, 12406. doi: 10.1021/ja204924j
-
[12]
Mei, T. -S.; Giri, R.; Maugel, N.; Yu, J. -Q. Angew. Chem., Int. Ed. 2008, 47, 5215. doi: 10.1002/anie.v47:28
-
[13]
Giri, R.; Maugel, N.; Li, J. -J.; Wang, D. -H.; Breazzano, S. P.; Saunders, L. B.; Yu, J. -Q. J. Am. Chem. Soc. 2007, 129, 3510. doi: 10.1021/ja0701614
-
[14]
Wang, D. -H.; Engle, K. M.; Yu, B. -F.; Yu, J. -Q. Science 2010, 327, 315. doi: 10.1126/science.1182512
-
[15]
Giri, R.; Lam, J. K.; Yu, J. -Q. J. Am. Chem. Soc. 2010, 132, 686. doi: 10.1021/ja9077705
-
[16]
Xiao, B.; Fu, Y.; Xu, J.; Gong, T. -J.; Dai, J. -J.; Yi, J.; Liu, L. J. Am. Chem. Soc. 2010, 132, 468. doi: 10.1021/ja909818n
-
[17]
Duong, H. A.; Gilligan, R. E.; Cooke, M. L.; Phipps, R. J.; Gaunt, M. J. Angew. Chem., Int. Ed. 2011, 50, 463. doi: 10.1002/anie.201004704
-
[18]
Xiao, B.; Gong, T. -J.; Xu, J.; Liu, Z. -J.; Liu, L. J. Am. Chem. Soc. 2011, 133, 1466. doi: 10.1021/ja108450m
-
[19]
Santhoshkumar, R.; Mannathan, S.; Cheng, C. -H. J. Am. Chem. Soc. 2015, 137, 16116. doi: 10.1021/jacs.5b10447
-
[20]
Dick, A. R.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2004, 126, 2300. doi: 10.1021/ja031543m
-
[21]
Chen, X.; Hao, X. -S.; Goodhue, C. E.; Yu, J. -Q. J. Am. Chem. Soc. 2006, 128, 6790. doi: 10.1021/ja061715q
-
[22]
Pan, F.; Lei, Z.; Wang, H.; Li, H.; Sun, J.; Shi, Z. -J. Angew. Chem., Int. Ed. 2013, 52, 2063. doi: 10.1002/anie.v52.7
-
[23]
Wang, S.; Hou, J. -T.; Feng, M. -L.; Zhang, X. -Z.; Chen, S. -Y.; Yu, X. -Q. Chem. Commun. 2016, 52, 2709. doi: 10.1039/C5CC09707J
-
[24]
Wang, Z.; Tian, Q.; Yu, X.; Kuang, C. Adv. Synth. Catal. 2014, 5, 961.
-
[25]
Gong, B.; Shi, J.; Wang, X.; Yan, Y.; Li, Q.; Meng, Y.; Xu, H. E.; Yi, W. Adv. Synth. Catal. 2014, 356, 137. doi: 10.1002/adsc.201300700
-
[26]
Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154. doi: 10.1021/ja054549f
-
[27]
Feng, Y.; Chen, G. Angew. Chem., Int. Ed. 2010, 49, 958. doi: 10.1002/anie.200905134
-
[28]
Gutekunst, W. R.; Gianatassio, R.; Baran, P. S. Angew. Chem., Int. Ed. 2012, 51, 7507. doi: 10.1002/anie.201203897
-
[29]
Aihara, Y.; Chatani, N. Chem. Sci. 2013, 4, 664. doi: 10.1039/C2SC21506C
-
[30]
Rouqueta, G.; Chatani, N. Chem. Sci. 2013, 4, 2201. doi: 10.1039/c3sc50310k
-
[31]
Suess, A. M.; Ertem, M. Z.; Cramer, C. J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 9797. doi: 10.1021/ja4026424
-
[32]
Wang, S.; Guo, R.; Wang, G.; Chen, S. -Y.; Yu, X. -Q. Chem. Commun. 2014, 50, 12718. doi: 10.1039/C4CC06246A
-
[33]
Matsubara, T.; Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2014, 136, 646. doi: 10.1021/ja412521k
-
[34]
Ilies, L.; Ichikawa, S.; Asako, S.; Matsubara, T.; Nakamura. E. Adv. Synth. Catal. 2015, 357, 2175. doi: 10.1002/adsc.v357.10
-
[35]
Aihara, Y.; Chatani, N. J. Am. Chem. Soc. 2013, 135, 5308. doi: 10.1021/ja401344e
-
[36]
Grigorjeva, L.; Daugulis, O. Angew. Chem., Int. Ed. 2014, 53, 10209. doi: 10.1002/anie.201404579
-
[37]
Wu, X.; Yang, K.; Zhao, Y.; Sun, H.; Li, G.; Ge, H. Nat. Commun. 2015, 6, 6462. doi: 10.1038/ncomms7462
-
[38]
Tran, L. D.; Daugulis, O. Angew. Chem., Int. Ed. 2012, 51, 5278.
-
[39]
Shang, M.; Sun, S. -Z.; Dai, H. -X.; Yu, J. -Q. J. Am. Chem. Soc. 2014, 136, 3354. doi: 10.1021/ja412880r
-
[40]
Huang, X.; Huang, J.; Du, C.; Zhang, X.; Song, F.; You, J. Angew. Chem., Int. Ed. 2013, 52, 12970. doi: 10.1002/anie.201307174
-
[41]
Zhang, L. -B.; Hao, X. -Q.; Zhang, S. -K.; Liu, Z. -J.; Zheng, X. -X.; Gong, J. -F.; Niu, J. -L.; Song, M. F. Angew. Chem., Int. Ed. 2015, 54, 272. doi: 10.1002/anie.201409751
-
[42]
Lee, P. -S.; Fujita, T.; Yoshikai, N. J. Am. Chem. Soc. 2011, 133, 17283. doi: 10.1021/ja2047073
-
[43]
Inamoto, K.; Saito, T.; Katsuno, M.; Sakamoto, T.; Hiroya, K. Org. Lett. 2007, 15, 2931. http://www.ncbi.nlm.nih.gov/pubmed/17595097
-
[44]
Hafner, D. -C. A.; Bräse, S. Angew. Chem., Int. Ed. 2012, 51, 3713. doi: 10.1002/anie.v51.15
-
[45]
Cai, G.; Fu, Y.; Li, Y.; Wan, X.; Shi, Z. J. Am. Chem. Soc. 2007, 129, 7666. doi: 10.1021/ja070588a
-
[46]
Shao, J.; Chen, W.; Giulianotti, M. A.; Houghten, R. A.; Yu, Y. Org. Lett. 2012, 14, 5452. doi: 10.1021/ol302533w
-
[47]
Leow, D.; Li, G.; Mei, T. -S.; Yu, J. -Q. Nature 2012, 486, 518. doi: 10.1038/nature11158
-
[48]
Tang, R. -Y.; Li, G.; Yu, J. -Q. Nature 2014, 507, 215. doi: 10.1038/nature12963
-
[49]
Wan, L.; Dastbaravardeh, N.; Li, G.; Yu, J. -Q. J. Am. Chem. Soc. 2013, 135, 18056. doi: 10.1021/ja410760f
-
[50]
Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2010, 132, 3965. doi: 10.1021/ja910900p
-
[51]
Tran, L. D.; Daugulis, O. Angew. Chem., Int. Ed. 2012, 51, 5188. doi: 10.1002/anie.201200731
-
[52]
Dai, H. -X.; Stepan, A. F.; Plummer, M. S.; Zhang, Y. H.; Yu, J. -Q. J. Am. Chem. Soc. 2011, 133, 7222. doi: 10.1021/ja201708f
-
[53]
Wang, B.; Shen, C.; Yao, J.; Yin, H.; Zhang, Y. Org. Lett. 2014, 16, 46. doi: 10.1021/ol402921w
-
[54]
Baba, K.; Tobisu, M.; Chatani, N. Angew. Chem., Int. Ed. 2013, 52, 11892. doi: 10.1002/anie.v52.45
-
[55]
Zhang, H.; Hu, R. -B.; Zhang, X. -Y.; Li, S. -X.; Yang, S. -D. Chem. Commun. 2014, 50, 4686. doi: 10.1039/C4CC01238K
-
[56]
Chary, B. C.; Kim, S.; Park, Y.; Kim, J.; Lee, P. H. Org. Lett. 2013, 15, 2692. doi: 10.1021/ol4009987
-
[57]
Chan, L. Y.; Cheong, L.; Kim, S. Org. Lett. 2013, 15, 2186. doi: 10.1021/ol400732q
-
[58]
杨军, 付婷, 龙洋, 周向葛, 有机化学, 2017, 37, 1111. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201705006&dbname=CJFD&dbcode=CJFQYang, J.; Fu, T.; Long, Y.; Zhou, X. Chin. J. Org. Chem. 2017, 37, 1111(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201705006&dbname=CJFD&dbcode=CJFQ
-
[1]
-

图式 4 Pd催化的醇羟基导向的邻位C—H活化反应
Scheme 4 Pd-catalyzed alcoholic hydroxyl-induced o-C—H activations

图式 7 Pd催化的CO作为碳源的羰基导向的邻位羧基化反应
Scheme 7 Pd-catalyzed carbonyl-induced o-carboxylation with CO as carbon source

图式 8 Cu催化的α-芳基羰基化合物的间位芳基化反应
Scheme 8 Cu-catalyzed m-arylation of α-aryl carbonyl compound

图式 11 Pd催化的1, 2, 3-三唑导向的邻位C—H键酰化反应
Scheme 11 Pd-catalyzed 1, 2, 3-azo-induced o-C—H acylation

图式 12 Cp*Rh催化的嘧啶基诱导的吲哚C-2位烯基化反应
Scheme 12 Cp*Rh-catalyzed pyrimidyl-induced C-2 alkenylation of indole

图式 13 Pipercyclobutanamide A全合成过程中的C—H活化步骤
Scheme 13 C—H activation steps in preparation of pipercyclobutanamide A

图式 17 Ag催化的三氮烯诱导的邻位三氟甲基反应
Scheme 17 Ag-catalyzed triazenyl-induced o-trifluoromethyla-tion

图式 18 Pd催化的N, N-二甲基氨基导向的邻位烯基化反应
Scheme 18 Pd-catalyzed N, N-dimethyl amino group-induced o-alkenylation

图式 19 Pd催化的胍基导向的邻位芳基化和烯基化反应
Scheme 19 Pd-catalyzed guanidyl-induced o-arylation and alkenylation

图式 21 硫醚诱导的非天然氨基酸的合成
Scheme 21 Thioether group-induced preparation of non-natural amino acid

图式 22 Pd催化的磺酰氨基诱导的C—H活化反应
Scheme 22 Pd-catalyzed sulfonamino group-induced C—H activations
-


 下载:
下载:


























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 429
- 文章访问数: 7814
- HTML全文浏览量: 2859
 下载:
下载:
