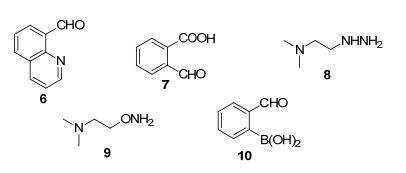
 图1
高活性成肟和成腙底物
Figure1.
Active substrates for oxime and hydrazone formation
图1
高活性成肟和成腙底物
Figure1.
Active substrates for oxime and hydrazone formation
引用本文:
蔡茂, 韩彦方, 章琪, 罗三中. 苯胺催化在生物大分子修饰和材料合成中的应用[J]. 有机化学,
2018, 38(1): 1-10.
doi:
10.6023/cjoc201708050

Citation: Cai Mao, Han Yanfang, Zhang Qi, Luo Sanzhong. Aniline Catalysis in Bioconjugations and Material Synthesis[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 1-10. doi: 10.6023/cjoc201708050
Citation: Cai Mao, Han Yanfang, Zhang Qi, Luo Sanzhong. Aniline Catalysis in Bioconjugations and Material Synthesis[J]. Chinese Journal of Organic Chemistry, 2018, 38(1): 1-10. doi: 10.6023/cjoc201708050
苯胺催化在生物大分子修饰和材料合成中的应用
摘要:
成肟或成腙的经典缩合反应具有较高的化学选择性,且底物易于合成,是一类简单但又重要的偶联反应,在生物分子修饰和材料合成中具有重要的应用价值.但该反应十分缓慢,而且往往需要在酸性溶液以及较高的底物浓度下进行,限制了其广泛应用.近年来的研究表明,苯胺作为亲核性催化剂可以显著加速成肟和成腙等缩合反应,初步解决了这类反应的速率问题,反应条件更为温和,生物兼容性好,已成为目前生物大分子修饰中最通用的偶联手段.本文详述了近年来芳胺催化剂的设计与发展、芳胺催化机制和构效关系以及高活性芳胺催化剂在生物大分子修饰和材料合成中的应用,并对未来的发展做了展望.
English
Aniline Catalysis in Bioconjugations and Material Synthesis
Abstract:
The oxime or hydrazone formation is a classic condensation reaction between aldehydes/ketones and hydroxyl amine or hydrazine. It is a simple, yet fundamental coupling reaction that has been widely applied in the ligations and conjugations of biomolecules and material synthesis. However, the reactions are usually sluggish and normally require acidic conditions with large excess of substrates to facilitate conversion, which limit their wide applications. Recent studies have shown that aniline as a nucleophilic catalyst can significantly accelerate the oxime/hydrazone formation reaction, preliminarily solving the reaction rate issue under mild and bio-compatible conditions. Therefore, it has been the most common coupling method in the modification of biological macromolecules. In this review, the design and development of aromatic amine catalyst in recent years as well as the catalytic mechanism and structure activity relationship are summarized. The application of highly active aniline catalysis in bioconjugations and material synthesis was also included together with a prospect for future development.
-
Key words:
- oxime
- / hydrazone
- / aniline catalysis
- / bioconjugation
- / material synthesis
-
高效可靠、专一性和兼容性强的偶联反应为生物大分子共价修饰和高分子材料的构建提供了强有力的工具, 一些代表性的例子包括Staudinger偶联反应、Cu催化的叠氮和炔烃的环加成反应(CuAAC)、环张力促进的叠氮和炔烃的环加成反应(SPAAC)以及逆电子Diels-Aleder反应(IEDDA)等[1].成腙成肟偶联反应是一类古老的反应, 早在19世纪末就分别由Meyer, Janny和Fischer[2]进行了广泛的研究.经典的醛/酮与胺类化合物缩合是一个可逆的过程, 所形成的亚胺(席夫碱)极不稳定; 而将亲核试剂改为烷基羟胺1或肼2, 所形成的肟4和腙5相对稳定(Eq. 1).该反应也表现出一定的特异性和生物兼容性; 反应的副产物只有水, 与其它偶联反应相比底物结构简单, 因此对生物体系干扰小; 而且反应条件温和, 能够在水相条件下进行.基于上述特性, 成肟成腙这一经典的偶联反应近年来在生物标记、分子探针、高分子材料和动态组合化学等方面受到了广泛的应用[3].但这一反应固有的缺陷也显而易见, 成肟成腙缩合反应的速率常数远远小于其它偶联反应, 例如Click反应、硫醇加成、Diels-Alder反应等[4].该反应通常需要至少一个反应底物过量或在酸性溶液中进行, 在中性条件下反应活性很低, 这也限制了其在生物体系中的应用.因此, 如何提高该偶联反应的反应速率并拓展其在生物和材料等领域的应用, 是近年来成肟成腙反应研究的重点与难点.通过设计特定的底物结构, 如Kool小组[5]在羰基或α-亲核试剂底物中引入酸性或碱性基团(6~9), Schmidt小组[6]通过在芳香醛酮的邻位引入硼酸(10), 可在中性条件下提高反应速率(图 1).
图1
高活性成肟和成腙底物
Figure1.
Active substrates for oxime and hydrazone formation
与设计特定底物结构相比, 芳胺催化的反应更具有普适性. 20世纪60年代, Jencks等[7]在合成缩氨脲时就使用苯胺作为亲核催化剂. 2006年, Dawson小组[8]首次将苯胺引入到生物体系当中催化成腙成肟的偶联反应, 在中性pH溶液以及低底物浓度条件下表现出明显的加速效应.芳胺作为温和的亲核试剂, 与生物体系中的许多官能团具有很好的兼容性, 这一催化体系因而受到广泛关注.本文详细综述了近年来芳胺催化剂的设计与发展、芳胺催化的机制和构效关系、以及高活性芳胺催化在生物大分子修饰和材料合成中的应用.
2 芳胺催化成肟和成腙反应
2.1 芳胺催化机理
Jencks[9]最早对成腙成肟反应机理进行了探讨并在随后的研究中得到证实, 该反应通常需要在酸性条件下进行, 反应机理如Scheme 1:首先α-亲核试剂进攻质子化羰基, 通过质子转移得到四面体中间体半缩醛胺12, 再经脱水得到目标产物.成肟成腙反应在不同pH下决速步不同[10], 在酸性条件下, 反应决速步为氨基的亲核进攻, 而在中性条件下为半缩醛胺的脱水反应.与羰基和伯胺形成的不稳定亚胺中间体相比, 肟和腙在水溶液中能够稳定存在.由于α-杂原子效应[11], 亚胺中氮原子邻位的氧原子或氮原子的电负性使得其碱性降低, 能够有效抑制质子化的过程, 从而减弱逆反应的发生, 使平衡有利于亚胺的生成.
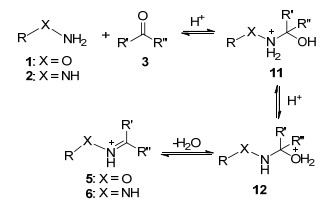 图式 1
成肟成腙反应机理
Scheme1.
Mechanism for the formation of oximes or hydrazones
图式 1
成肟成腙反应机理
Scheme1.
Mechanism for the formation of oximes or hydrazones
苯胺催化的机理(Scheme 2)主要分为两步[7], 亚胺中间体的产生(Ⅰ)和转氨化过程(Ⅱ).以对氯苯甲醛13为例, 第一步经苯胺亲核进攻得到四面体结构的中间体15, 后经脱水得到活性亚胺中间体17; 第二步为α-亲核试剂进攻亚胺中间体, 得到第二个四面体结构的中间体18或19, 最后苯胺消除得到目标产物(20或21).氮上孤对电子在芳环上的离域, 使芳胺成为一个好的离去基团.高活性的质子化芳胺-亚胺中间体17的形成是整个催化循环的关键, 该中间体能够迅速与α-亲核试剂反应得到肟或腙, 进而提高反应速率.
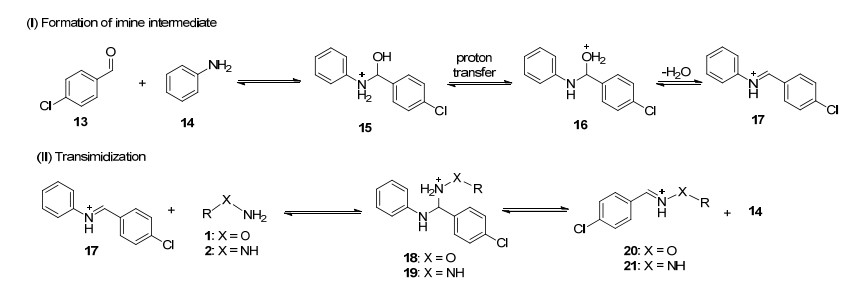 图式 2
苯胺催化的成肟成腙反应机理
Scheme2.
Mechanism for the aniline-catalyzed formation of oximes or hydrazones
图式 2
苯胺催化的成肟成腙反应机理
Scheme2.
Mechanism for the aniline-catalyzed formation of oximes or hydrazones
苯胺催化活性可以通过羰基和苯胺席夫碱共轭酸的pKa得到解释.质子化的羰基的pKa为-4~-10, 即使在pH=4.5的溶液中活性的亲电试剂浓度也非常低.继续降低溶液pH值虽能够提高亲电试剂浓度, 但质子化的亲核试剂与酸催化作用抵消, 因此只能在一定的pH范围催化缩合反应的进行.而在苯胺催化过程中, 苯胺共轭酸的pKa为4.6, 苯胺席夫碱共轭酸的pKa值为2.8, 相对于苯胺仅降低2个单位, 有利于高浓度的质子化亚胺的形成, 从而加速反应.除此之外, 形成苯胺席夫碱的平衡常数Keq较小(在酸性溶液中约为0.08 L• mol-1), 因此使用过量的催化剂不会影响目标产物的生成[9b].
最近, Yildiz小组[12]对苯胺催化的成肟反应进行了深入地探讨, 通过计算证实相对于苯胺催化, 酸催化的羰基活化能垒更高(分别为10.50和68.37 kJ/mol).该小组以乙醛22和甲基羟胺24为模板底物, 建立苯胺-亚胺的模型体系, 通过密度泛函理论计算, 对成肟反应过程中转氨化机理进行了研究(Scheme 3).作者认为在转氨化过程的三步中, 分子内质子转移得到中间体26为反应决速步.中间体26因质子化的氮原子与苯环相连, 能够稳定存在.通过计算也表明质子转移的活化能相较于非催化体系更低, 从理论上解释了苯胺在转氨化过程中的催化活性.
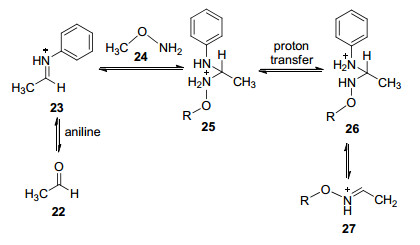 图式 3
苯胺-亚胺和烷氧羟胺成肟反应机理
Scheme3.
Mechanism for the formation of oximes from aniline-imine and alkoxyamines
图式 3
苯胺-亚胺和烷氧羟胺成肟反应机理
Scheme3.
Mechanism for the formation of oximes from aniline-imine and alkoxyamines
2.2 芳胺催化剂的设计与发展
Jencks等[7]在合成缩氨脲时发现, 相比于一般的酸催化剂, 苯胺及其取代衍生物的催化活性高达10~1000倍.作为亲核催化剂, 苯胺对成肟反应也具有相同的催化活性.对于中性条件下的成肟反应, 通常需要至少一个反应底物过量来提高反应速率.利用苯胺催化, Dawson小组[8a]实现了低浓度下未保护肽链的成肟偶联, 在pH=4.5时, 反应速率提高近400倍; 而肽链的成腙反应, 反应速率在pH=5.7的条件下提高70倍[8b].该小组首次在生物体系中通过芳胺催化剂实现了大分子的偶联, 并且大幅提高了反应速率.
然而苯胺的催化活性有限, 一般需要使用大过量的催化剂来提高反应速率.并且苯胺在水溶液中溶解度不高, 高浓度下表现出一定的生物毒性.为提高催化剂的活性和溶解度, 改善其在生物体系中的兼容性, 通过在苯环中引入不同取代基并改变其相对位置, 或者设计新的催化骨架, 可以得到一系列高催化活性的芳胺催化剂.
除苯胺外, Dawson等[13]也利用对甲氧基苯胺(28)作为亲核催化剂(图 2), 在pH=7的中性条件下, 使肽链的成肟偶联反应速率提高40倍. 2011年, Bane小组[14]以3-酰基酪氨酸37和香豆素肼38为模板反应, 利用4-氨基苯丙氨酸29(图 2)催化成腙反应的进行, 实现对温度与pH敏感α-微管蛋白的荧光标记(Eq. 2). 4-氨基苯丙氨酸29与苯胺具有相近的催化活性, 因其作为两性离子不易使蛋白质变性, 表现出更好的生物兼容性, 使得标记后的微管蛋白具有更高的聚合活性.与4-氨基苯丙氨酸(29)相比, 苯丙氨酸在反应中催化效果很差, 这也说明芳胺相对于脂肪族类氨基催化剂具有更高的活性.
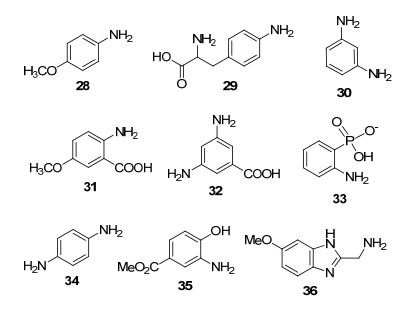 图2
高活性芳胺催化剂
Figure2.
Highly active aromatic amine catalysts
图2
高活性芳胺催化剂
Figure2.
Highly active aromatic amine catalysts
2013年, Distefano小组[15]利用间苯二胺30(图 2)催化的醛成肟反应, 实现了对细胞提取物中蛋白质的纯化和聚乙二醇化以及睫状神经营养因子的荧光标记(Scheme 4).相对于苯胺, 间苯二胺有更高的催化活性, 在水中的溶解度较大, 并且间苯二胺催化的反应速率与催化剂的浓度成线性关系, 不断提高催化剂浓度至900 mmol•L-1可使反应速率常数与苯胺催化相比提高近20倍.
 图式 4
蛋白质的纯化和选择性标记
Scheme4.
Purification and selective labeling of protein
图式 4
蛋白质的纯化和选择性标记
Scheme4.
Purification and selective labeling of protein
随后, Kool小组[16]筛选氨茴酸和氨基苯甲酸衍生物时发现, 5-甲氧基氨茴酸(5MA, 31)以及3, 5-二氨基苯甲酸(32, 图 2)具有较高的溶解性和催化活性, 其二级反应常数最高为苯胺催化的6倍.作者认为氨茴酸高的催化活性是由于在亚胺和腙的形成过程中, 邻位羧基会发生分子内质子转移.改变酸性基团得到的邻氨基苯膦酸(33, 图 2), 是目前活性最高的芳胺催化剂, 适用于各种醛和酮底物[17]. Baca小组[18]利用对苯二胺34(图 2), 实现了T3蛋白质的聚乙二醇化.在一定的酸性pH范围和低温、低浓度下, 对苯二胺的催化活性均高于苯胺.肽链中的醛基由高碘酸钠氧化得到, 芳胺催化下与羟胺发生成肟反应, 可以实现在特定位点的聚乙二醇修饰(Scheme 5).
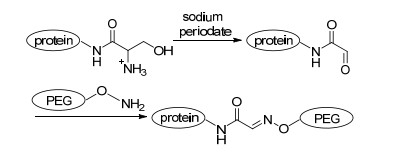 图式 5
醛基的引入和PEG偶联
Scheme5.
Introduction of aldehyde moiety and oxime ligation with PEG
图式 5
醛基的引入和PEG偶联
Scheme5.
Introduction of aldehyde moiety and oxime ligation with PEG
2015年, Kool小组[19]通过设计新的催化剂骨架, 得到了高催化活性的2-氨基-3-羟基苯甲酸甲酯(35, 图 2).作者还利用L-组氨酸结构设计了2-氨甲基苯并咪唑催化剂, 5-甲氧基取代的催化剂36具有高的催化活性(图 2).该类催化剂的底物适用范围广, 可以实现各类芳香醛与苯肼40的成腙偶联; 其二级反应速率常数可达42 L•mol-1•s-1, 对于大位阻的酮底物47~49也表现出一定的催化活性(Eq. 3).
2.3 芳胺催化构效关系
2.3.1 催化剂的邻位质子效应
伯胺与醛酮的缩合反应, 往往表现出明显的酸添加剂效应[20]. Hine等[21]研究发现N, N-二甲基乙二胺-质子酸二元体系与醛酮反应形成亚胺的速率比普通伯胺快得多, 其中质子化叔胺通过分子内酸催化, 促进半缩醛胺转化为亚胺.基于此, Kool等设计发展了双功能的芳香伯胺催化体系.以4-肼基-7-硝基-2, 1, 3-苯并氧杂二唑(50)与4-硝基苯甲醛(51) (Eq. 4)为模板底物, 改变氨茴酸中羧基与氨基的相对位置, 研究发现仅处于邻位的芳胺催化剂具有高的催化活性, 且含取代基的苯甲酸的催化活性远小于氨茴酸.因此, 作者认为氨茴酸中羧酸作为分子内酸催化剂, 与半缩醛胺形成八元环状过渡态, 催化决速步中亚胺的形成(Eq. 4).此外, 在羧基的邻位存在甲基时, 芳胺催化剂的活性大大降低, 也从侧面印证了羧酸参与了过渡态的形成.
Kool和Crisalli等[17]利用相同的模型反应, 系统研究了苯胺邻位活性质子酸基团pKa对催化活性的影响(Eq. 4).作者认为, 邻位基团在中性环境下的pKa越大, 则质子酸浓度越大, 催化活性越高.对于pKa较小的2-氨基苯磺酸(54)的催化活性较低, 而5-(2氨基苯基)四唑(55)与苯胺具有相近的pKa值和催化活性. 2-氨基苯膦酸(33)的pKa值为7.29, 与生理条件相近, 催化活性远大于氨茴酸, 而继续增大取代基的pKa值则催化活性大大降低, 如2-氨基苯羟肟酸(56)和2-氨基苯磺酰胺(57).由此可知, 邻位质子酸的pKa值越接近中性pH值, 催化活性越高.
Kool小组[19]通过设计新的催化剂骨架, 发现邻氨基苯酚表现出较好的催化活性(Eq. 3).当氨基与羟基不处于邻位时, 催化活性与苯胺相近, 而2-氨基-3-羟基苯甲酸甲酯(35)的二级反应速率常数为苯胺的7倍.作者认为在决速步形成的七元环过渡态, 与邻位质子酸基团参与的八元环过渡态相比(Scheme 5), 在构象上更为有利, 因而可加速反应进行.进一步研究表明脂肪型的伯胺催化剂36也表现出与氨基苯酚类似的活性(Eq. 5).
2.3.2 芳环的取代基效应
Kool小组[16]研究发现, 给电子基团(甲氧基、甲基)使氨茴酸的催化活性提高, 而吸电子基团(如卤素、硝基)使其催化活性降低.同样, 2-氨基苯磺酸虽然有邻位质子效应, 但由于磺酸基团强的吸电子能力, 使得氨基的亲核型减弱, 基本不具有催化活性[17]. Baca小组[18]发现对位取代的芳胺催化剂活性更高, 并且氨基、羟基和甲氧基取代的苯胺活性依次递减, 这可以由对应的苯胺中氨基亲核性降低得到解释.在提高氨茴酸催化活性的过程中, 氨基或羧基的邻位有甲基取代基时催化剂的活性大大降低, 并且对位取代的芳胺催化剂活性一般高于邻位取代.对于催化剂的稳定性, 由于给电子基团使得苯环的电子密度增大, 在空气中易被氧化.如4-甲氧基苯胺(28)催化反应时在空气中放置会得到棕色的氧化物沉淀, 5-甲氧基-2-氨基苯膦酸虽具有高的催化活性, 但在不脱氧的溶液中也会失活, 只能使用催化活性略低的甲基取代的芳胺.因此, 对于芳胺催化剂的优化与筛选还需考虑位阻效应, 催化剂稳定性和兼容性以及溶解性等因素.
表 1汇总了目前发展的芳胺催化的成肟和成腙反应体系[22], 不同芳胺的催化活性具有明显差异, 也取决于反应体系.如间二苯胺, 对于T3蛋白的聚乙二醇化几乎不具有催化活性, 一般会抑制成肟反应的进行, 但却适用于α-微管蛋白体系.间二苯胺和对苯二胺易被氧化不稳定, 限制了其广泛用.而氨基取代的苯膦酸和氨茴酸催化活性高, 适用于各种成腙成肟的偶联反应.
 表 1
用于成肟成腙反应的亲核催化剂概述a
Table 1.
Overview of nucleophilic catalysts used in oxime and hydrazoneformation reaction
表 1
用于成肟成腙反应的亲核催化剂概述a
Table 1.
Overview of nucleophilic catalysts used in oxime and hydrazoneformation reaction
Catalyst Fragment 1 Fragment 2 Ratea/(L•mol-1•s-1) Property 


1.4±0.4 新的催化剂骨架, 高反应活性 


1.2±0.09 适用于酮和芳香醛 


0.19b 两个氨基具有不同的pKa值, 在较大的pH范围都具有高的催化活性, 高水溶性 


0.142 目前最高活性的芳胺催化剂, 底物适用范围广 


0.11 高水溶性, 适用于芳香醛和脂肪族酮 

Citral 41.5±1.2b 高水溶性, 易氧化成醌 


— 高的生物兼容性, 催化活性稍低于苯胺 aReactions were performed in phosphate buffer, 1 mmol•L-1 catalyst, pH 7.0, at r.t.; bThe concentration of catalyst were respectively 2 and 50 mmol•L-1. 3 芳胺催化成肟和成腙反应的应用
3.1 生物大分子修饰
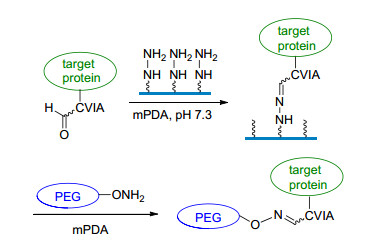
早在2008年, Dirksen等[13]就实现了未保护肽链的荧光标记. Dawson小组[23]利用苯胺催化biotin-羟胺的成肟反应, 通过带有荧光基团的streptavidin与biotin特异性结合, 实现了对细胞表面糖蛋白的特异性标记, 而含有唾液酸的细胞表面糖蛋白, 可以通过高碘酸氧化引入醛基(图 3).在低的底物浓度和温度下, 苯胺催化能够专一性地对细胞表面进行荧光标记.
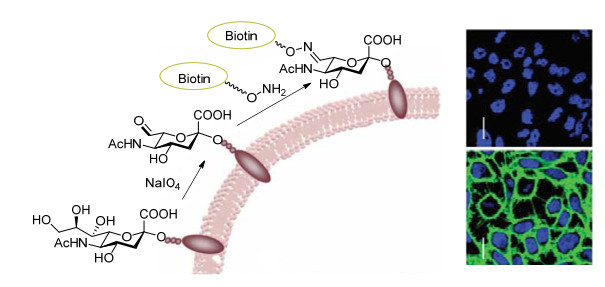 图3
通过苯胺催化的成肟反应标记细胞表面糖蛋白[23]
Figure3.
Labeling cell-surface glycoproteins by aniline-catalyzed oxime ligation
图3
通过苯胺催化的成肟反应标记细胞表面糖蛋白[23]
Figure3.
Labeling cell-surface glycoproteins by aniline-catalyzed oxime ligation
Meijler及其同事[24]首次实现了细胞内细菌受体的活细胞标记和成像, 作者利用两步的标记方法, 首先利用活性的群体感应信号分子类似物异硫氰酸盐ITC-12与转录激活子LasR结合, 再将带有荧光标记BODIPY的烷氧基氨基通过共价键连接(Scheme 6).而苯胺催化剂的使用, 大大缩短了标记时间, 第一次成功实现了绿脓杆菌群体感应受体在活细胞内的荧光标记与成像.同时作者发现LasR分布在细胞的两端而并非整个细胞质膜(Scheme 6).
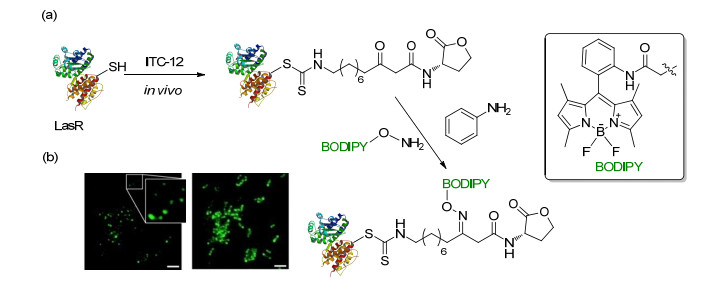 图式 6
利用苯胺催化的成肟反应实现群体感应受体标记[24]
Scheme6.
QS receptor labeling using aniline-catalyzed oxime chemistry
图式 6
利用苯胺催化的成肟反应实现群体感应受体标记[24]
Scheme6.
QS receptor labeling using aniline-catalyzed oxime chemistry
Cairo及其同事[25]也报道了高碘酸钠与苯胺催化结合的偶联反应, 实现对细胞中的糖复合物的检测.与Dawson小组不同, 作者使用4-肼基-7-硝基-2, 1, 3-苯并氧杂二唑(50) (Eq. 4)原位生成苯肼, 其本身即为荧光分子且具有较高的荧光量子产率, 不需要再加入连有荧光标记的streptavidin进行检测成像. Risse小组[26]在研究对温度和酸敏感的人体亚硫酸盐氧化酶时, 利用对甲氧基苯胺催化的成肟反应, 在温和条件下实现了对非天然氨基酸的定点自旋标记.
3.2 生物检测
正电子发射计算机断层显像(PET)是作为医药和生物的重要成像手段, 是利用放射性同位素18F来实现的, 而成像的先决条件就是在成像前引入PET示踪物.苯胺催化的成腙成肟反应能够在生理条件下进行, 具有高的化学选择性, 并且反应过程中不需要对生物分子进行保护, 因此是非常有效的PET示踪物合成手段.通过设计不同氟化的醛结构, 可以实现对于肽链和蛋白质的放射性标记(图 4).通过对生物体内天然氨基酸的标记, 有望实现对疾病的诊断以及生理变化的检测[27].
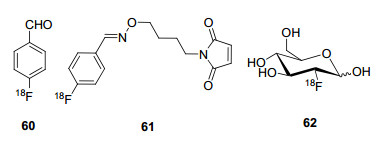 图4
用于肽链标记的含18F醛
Figure4.
18F-containing aldehydes for peptide labeling
图4
用于肽链标记的含18F醛
Figure4.
18F-containing aldehydes for peptide labeling
芳胺催化剂也作为分子探针实现了特定分子的检测. Kool小组[28]通过荧光腙染料, 利用该组设计的高活性芳胺催化剂5-甲氧基氨茴酸(Eq. 6), 实现了对细胞内绝大部分烷基醛的检测.其原理在于将荧光标记分子与带有荧光淬灭结构的醛结合来得到黑色的腙试剂, 在芳胺催化下与待检测的醛反应释放淬灭剂发出荧光, 首次实现了在活细胞内对醛总量的检测.
3.3 生物组织样本修复
在生物正交反应中, 具有良好兼容性的断键反应也得到了不断的发展和应用[29].利用芳胺催化亚胺形成的可逆性, Kool小组实现了苯胺催化的生物组织样本修复.对于一般的临床组织标本, 都是通过福尔马林固定石蜡包埋制备, 但甲醛会与生物分子反应, 不利于检测分析.通常的处理办法是升温, 但核酸分子会发生分解. 2015年, Kool小组[30]利用芳胺催化剂实现了甲醛与单个核苷酸分子加成的逆过程催化.对于RNA寡核苷酸链以及福尔马林固定石蜡包埋RNA的甲醛加成物的去除, 5MA具有高的催化活性, 核酸产率远远高于传统的核酸提取方法.在对催化机理的探讨中, 作者认为5MA在反应过程中同时作为酸催化剂和亲核催化剂(Scheme 7).在温和条件下实现生物分子与甲醛加成的逆反应催化, 可应用于数量庞大的生物组织样本的结构修复以及研究分析.
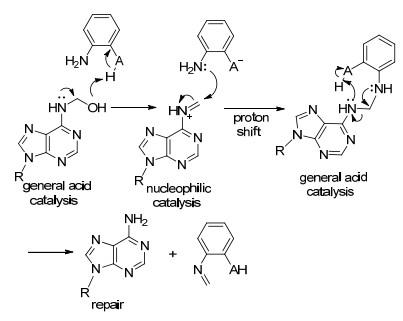 图式 7
苯胺催化半缩醛胺逆过程
Scheme7.
Aniline catalyzed retro reaction of hemiaminals
图式 7
苯胺催化半缩醛胺逆过程
Scheme7.
Aniline catalyzed retro reaction of hemiaminals
3.4 材料合成
超分子凝胶在石油化工、医疗保健和组织工程学领域均有广泛的应用前景, 而这些处于亚稳态的材料的特性能够通过催化剂进行调控. 2013年, Eelkema小组[31]报道了利用苯胺催化的成腙反应实现对超分子凝胶力学性质以及结构的调控.作者根据之前的环己烷-氨基酸链接的水凝胶结构, 设计了水溶性的三腙63和醛64在原位形成腙65 (Scheme 8).从图中可以看出得到的水凝胶分子再通过氢键和疏水相互作用自组装得到纤维结构.酸或苯胺催化剂可从动力学上实现反应的调控, 通过加快反应速率得到稠密的均相网状支纤维结构, 并增强机械强度.苯胺催化剂可以在中性条件下得到与酸催化相近的结果, 这也扩大了其在超分子材料合成中的应用.
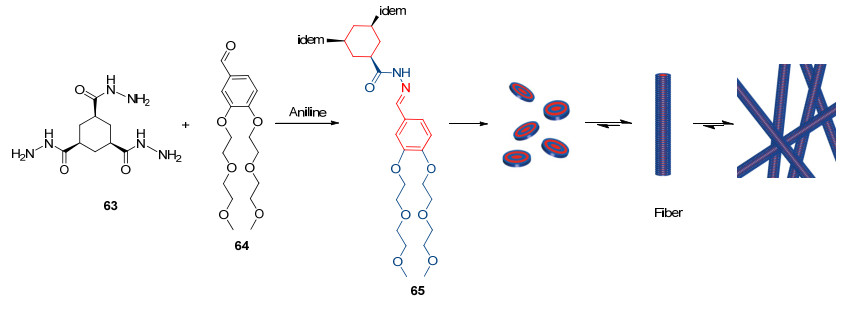 图式 8
催化剂控制的水凝胶合成
Scheme8.
Catalytic control over hydrogelator formation
图式 8
催化剂控制的水凝胶合成
Scheme8.
Catalytic control over hydrogelator formation
随后, Eelkema小组[32]受酶活化过程的启发, 通过化学信号活化催化剂, 实现了在时间上控制软材料的合成.作者设计了催化剂前体, 在与H2O2作用后释放出苯胺, 在特定时间成功实现聚合物凝胶以及上述超分子凝胶的合成(Scheme 9).该催化剂前体易于合成, 活化后的催化效果与苯胺相近.
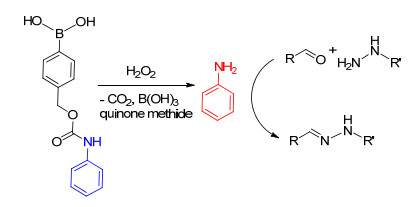 图式 9
被保护苯胺的活化以及催化成腙反应
Scheme9.
Activation of a protected aniline and subsequent catalysis of hydrozone formation
图式 9
被保护苯胺的活化以及催化成腙反应
Scheme9.
Activation of a protected aniline and subsequent catalysis of hydrozone formation
在超分子水凝胶性质的研究中, Eelkema等[33]发现通过改变醛或者酮的结构和亲水性可以调控弹性系数和网状结构等超分子性质.而在结构单元上可引入不同基团对胶体进行修饰, 如在醛上连接各种荧光探针可以实现超分子网状结构成像[33]. van Hest小组[34]利用苯胺催化的成肟反应(Scheme 10)可实现对高分子囊泡的表面修饰, 发展了一种新的动态高分子表面修饰方式.聚合物分子一端与肼相连, 另一端可修饰上不同的荧光剂基团66或67, 或者通过马来酰亚胺68与囊泡中的肽链连接.在苯胺催化下, 肼偶联的嵌段共聚物可与肼66和67以及其它共聚物如69进行表面官能团的交换实现荧光标记.
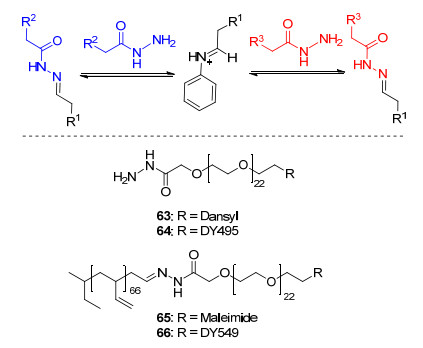 图式 10
苯胺催化聚合物分子修饰
Scheme10.
Aniline catalyzed derivatization of polymers
图式 10
苯胺催化聚合物分子修饰
Scheme10.
Aniline catalyzed derivatization of polymers
4 展望
综上所述, 芳胺催化的成腙成肟反应具有高的化学选择性与生物兼容性, 近10年来在生物标记与材料领域得到了广泛的关注和迅速的发展.但多数体系往往需要大过量和高浓度的苯胺催化剂, 无论是催化活性、反应的选择性还是反应的适用性都亟待进一步提高和改进, 设计发展新型的胺基识别与催化体系是值得开拓的研究方向.
-
-
[1]
(a) Li, Q. ; Dong, T. ; Liu, X. ; Lei, X. J. Am. Chem. Soc. 2013, 135, 4996.
(b) Oliveira, B. L. ; Guo, Z. ; Bernardes, G. J. L. Chem. Soc. Rev. 2017, 46, 4895.
(c) Saxon, E. ; Bertozzi, C. R. Science 2000, 287, 2007.
(d) Sletten, E. M. ; Bertozzi, C. R. Acc. Chem. Res. 2011, 44, 666.
(e) Rostovtsev, V. V. ; Green, L. G. ; Fokin, V. V. ; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596.
(f) Li, L. ; Zhang, Z. Molecules 2016, 21, 1393.
(g) Agard, N. J. ; Prescher, J. A. ; Bertozzi, C. R. J. Am. Chem. Soc. 2004, 126, 15046.
(h) Jewett, J. C. ; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272.
(i) Darko, A. ; Wallace, S. ; Dmitrenko, O. ; Machovina, M. M. ; Mehl, R. A. ; Chin, J. W. ; Fox, J. M. Chem. Sci. 2014, 5, 3770.
(j) Yang, M. ; Chen, P. Acta Chim. Sinica 2015, 73, 783(in Chinese).
(杨麦云, 陈鹏, 化学学报, 2015, 73, 783. )
(k) Xiong, D. C. ; Zhu, J. ; Han, M. J. ; Luo, H. X. ; Wang, C. ; Yu, Y. ; Ye, Y. ; Tai, G. ; Ye, X. S. Org. Biomol. Chem. 2015, 13, 3911. -
[2]
(a) Fisher, E. Ber. Dtsch. Chem. Ges. 1888, 21, 984.
(b) Janny, A. Ber. Dtsch. Chem. Ges. 1882, 15, 2778.
(c) Meyer, V.; Janny, A. Ber. Dtsch. Chem. Ges. 1882, 15. -
[3]
(a) Mahal, L. K.; Yarema, K. J.; Bertozzi, C. R. Science 1997, 276, 1125.
(b) Maxson, T.; Tietz, J. I.; Hudson, G. A.; Guo, X. R.; Tai, H. C.; Mitchell, D. A. J. Am. Chem. Soc. 2016, 138, 15157.
(c) Tang, L.; Yin, Q.; Xu, Y.; Zhou, Q.; Cai, K.; Yen, J.; Dobrucki, L. W.; Cheng, J. Chem. Sci. 2015, 6, 2182.
(d) Rosik, D.; Thibblin, A.; Antoni, G.; Honarvar, H.; Strand, J.; Selvaraju, R. K.; Altai, M.; Orlova, A.; Eriksson Karlstrom, A.; Tolmachev, V. Bioconjugate Chem. 2014, 25, 82.
(e) Collins, J.; Xiao, Z.; Müllner, M.; Connal, L. A. Polym. Chem. 2016, 7, 3812.
(f) Simpson, M. G.; Pittelkow, M.; Watson, S. P.; Sanders, J. K. Org. Biomol. Chem. 2010, 8, 1181. -
[4]
Saito, F.; Noda, H.; Bode, J. W. ACS Chem. Biol. 2015, 10, 1026. doi: 10.1021/cb5006728
-
[5]
Kool, E. T.; Park, D. H.; Crisalli, P. J. Am. Chem. Soc. 2013, 135, 17663. doi: 10.1021/ja407407h
-
[6]
Schmidt, P.; Stress, C.; Gillingham, D. Chem. Sci. 2015, 6, 3329. doi: 10.1039/C5SC00921A
-
[7]
Cordes, E. H.; Jencks, W. P. J. Am. Chem. Soc. 1962, 84, 826. doi: 10.1021/ja00864a030
-
[8]
(a) Dirksen, A.; Hackeng, T. M.; Dawson, P. E. Angew. Chem., Int. Ed. 2006, 45, 7581.
(b) Dirksen, A.; Dirksen, S.; Hackeng, T. M.; Dawson, P. E. J. Am. Chem. Soc. 2006, 128, 15602. -
[9]
(a) Jencks, W. P. J. Am. Chem. Soc. 1959, 81, 475.
(b) Cordes, E. H.; Jencks, W. P. J. Am. Chem. Soc. 1962, 84, 832.
(c) Sayer, J. M.; Pinsky, B.; Schonbrunn, A.; Washtien, W. J. Am. Chem. Soc. 1974, 96, 7998. -
[10]
(a) Malpica, A.; Calzadilla, M.; Cordova, T. J. Phys. Org. Chem. 2000, 13, 162.
(b) Malpica, A.; Calzadilla, M. J. Phys. Org. Chem. 2005, 18, 945. -
[11]
Sander, E. G.; Jencks, W. P. J. Am. Chem. Soc. 1968, 90, 6154. doi: 10.1021/ja01024a038
-
[12]
Kirmizialtin, S.; Yildiz, B. S.; Yildiz, I. J. Phys. Org. Chem. 2017, e3711. http://www.researchgate.net/publication/316745337_A_DFT-based_mechanistic_study_on_the_formation_of_oximes
-
[13]
Dirksen, A.; Dawson, P. E. Bioconjugate Chem. 2008, 19, 2543. doi: 10.1021/bc800310p
-
[14]
Blanden, A. R.; Mukherjee, K.; Dilek, O.; Loew, M.; Bane, S. L. Bioconjugate Chem. 2011, 22, 1954. doi: 10.1021/bc2001566
-
[15]
Rashidian, M.; Mahmoodi, M. M.; Shah, R.; Dozier, J. K.; Wagner, C. R.; Distefano, M. D. Bioconjugate Chem. 2013, 24, 333. doi: 10.1021/bc3004167
-
[16]
Crisalli, P.; Kool, E. T. J. Org. Chem. 2013, 78, 1184. doi: 10.1021/jo302746p
-
[17]
Crisalli, P.; Kool, E. T. Org. Lett. 2013, 15, 1646. doi: 10.1021/ol400427x
-
[18]
Wendeler, M.; Grinberg, L.; Wang, X.; Dawson, P. E.; Baca, M. Bioconjugate Chem. 2014, 25, 93. doi: 10.1021/bc400380f
-
[19]
Larsen, D.; Pittelkow, M.; Karmakar, S.; Kool, E. T. Org. Lett. 2015, 17, 274. doi: 10.1021/ol503372j
-
[20]
(a) Xu, C.; Zhang, L.; Luo, S. J. Org. Chem. 2014, 79, 11517.
(b) Xu, C.; Zhang, L.; Luo, S. Angew. Chem., Int. Ed. 2014, 53, 4149. -
[21]
Hine, J.; Li, W. S. J. Org. Chem. 1975, 40, 2622. doi: 10.1021/jo00906a010
-
[22]
Agten, S. M.; Dawson, P. E.; Hackeng, T. M. J. Pept. Sci. 2016, 22, 271. doi: 10.1002/psc.2874
-
[23]
Zeng, Y.; Ramya, T. N.; Dirksen, A.; Dawson, P. E.; Paulson, J. C. Nat. Methods 2009, 6, 207. http://www.ncbi.nlm.nih.gov/pubmed/19234450?dopt=AbstractPlus
-
[24]
Rayo, J.; Amara, N.; Krief, P.; Meijler, M. M. J. Am. Chem. Soc. 2011, 133, 7469. doi: 10.1021/ja200455d
-
[25]
Key, J. A.; Li, C.; Cairo, C. W. Bioconjugate Chem. 2012, 23, 363. doi: 10.1021/bc200276k
-
[26]
Hahn, A.; Reschke, S.; Leimkuhler, S.; Risse, T. J. Phys. Chem. B 2014, 118, 7077. doi: 10.1021/jp503471j
-
[27]
Li, X.-G.; Haaparanta, M.; Solin, O. J. Fluorine Chem. 2012, 143, 49. doi: 10.1016/j.jfluchem.2012.07.005
-
[28]
Yuen, L. H.; Saxena, N. S.; Park, H. S.; Weinberg, K.; Kool, E. T. ACS Chem. Biol. 2016, 11, 2312. doi: 10.1021/acschembio.6b00269
-
[29]
Li, J.; Chen, P. R. Nat. Chem. Biol. 2016, 12, 129. doi: 10.1038/nchembio.2024
-
[30]
Karmakar, S.; Harcourt, E. M.; Hewings, D. S.; Scherer, F.; Lovejoy, A. F.; Kurtz, D. M.; Ehrenschwender, T.; Barandun, L. J.; Roost, C.; Alizadeh, A. A.; Kool, E. T. Nat. Chem. 2015, 7, 752. doi: 10.1038/nchem.2307
-
[31]
Boekhoven, J.; Poolman, J. M.; Maity, C.; Li, F.; van der Mee, L.; Minkenberg, C. B.; Mendes, E.; van Esch, J. H.; Eelkema, R. Nat. Chem. 2013, 5, 433. doi: 10.1038/nchem.1617
-
[32]
Eelkema, R.; van Esch, J. H. Org. Biomol. Chem. 2014, 12, 6292. doi: 10.1039/C4OB01108B
-
[33]
(a) Trausel, F.; Versluis, F.; Maity, C.; Poolman, J. M.; Lovrak, M.; van Esch, J. H.; Eelkema, R. Acc. Chem. Res. 2016, 49, 1440.
(b) Poolman, J. M.; Maity, C.; Boekhoven, J.; van der Mee, L.; le Sage, V. A. A.; Groenewold, G. J. M.; van Kasteren, S. I.; Versluis, F.; van Esch, J. H.; Eelkema, R. J. Mater. Chem. B 2016, 4, 852. -
[34]
Brinkhuis, R. P.; de Graaf, F.; Hansen, M. B.; Visser, T. R.; Rutjes, F. P. J. T.; van Hest, J. C. M. Polym. Chem. 2013, 4, 1345. doi: 10.1039/C2PY20789C
-
[1]
-

图式 2 苯胺催化的成肟成腙反应机理
Scheme 2 Mechanism for the aniline-catalyzed formation of oximes or hydrazones

图式 3 苯胺-亚胺和烷氧羟胺成肟反应机理
Scheme 3 Mechanism for the formation of oximes from aniline-imine and alkoxyamines

图 3 通过苯胺催化的成肟反应标记细胞表面糖蛋白[23]
Figure 3 Labeling cell-surface glycoproteins by aniline-catalyzed oxime ligation

图式 6 利用苯胺催化的成肟反应实现群体感应受体标记[24]
Scheme 6 QS receptor labeling using aniline-catalyzed oxime chemistry

图式 9 被保护苯胺的活化以及催化成腙反应
Scheme 9 Activation of a protected aniline and subsequent catalysis of hydrozone formation
表 1 用于成肟成腙反应的亲核催化剂概述a
Table 1. Overview of nucleophilic catalysts used in oxime and hydrazoneformation reaction
Catalyst Fragment 1 Fragment 2 Ratea/(L•mol-1•s-1) Property 1.4±0.4 新的催化剂骨架, 高反应活性 1.2±0.09 适用于酮和芳香醛 0.19b 两个氨基具有不同的pKa值, 在较大的pH范围都具有高的催化活性, 高水溶性 0.142 目前最高活性的芳胺催化剂, 底物适用范围广 0.11 高水溶性, 适用于芳香醛和脂肪族酮 Citral 41.5±1.2b 高水溶性, 易氧化成醌 — 高的生物兼容性, 催化活性稍低于苯胺 aReactions were performed in phosphate buffer, 1 mmol•L-1 catalyst, pH 7.0, at r.t.; bThe concentration of catalyst were respectively 2 and 50 mmol•L-1.  下载: 导出CSV
下载: 导出CSV
-















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 35
- 文章访问数: 2831
- HTML全文浏览量: 648
 下载:
下载:
