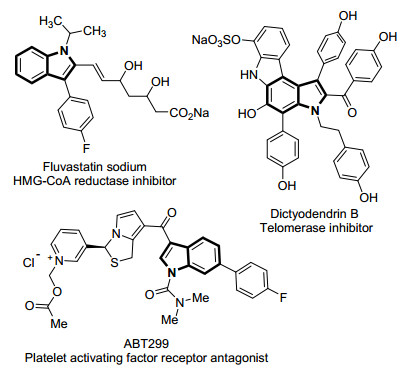
 图1
部分含有吲哚结构的药物或活性分子
Figure1.
Examples of drug and reactive molecules containing indole structure
图1
部分含有吲哚结构的药物或活性分子
Figure1.
Examples of drug and reactive molecules containing indole structure
引用本文:
骆钧飞, 徐星, 郑俊良. 吲哚C—H芳基化反应的研究进展[J]. 有机化学,
2018, 38(2): 363-377.
doi:
10.6023/cjoc201707031

Citation: Luo Junfei, Xu Xing, Zheng Junliang. Advance in C-H Arylation of Indoles[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 363-377. doi: 10.6023/cjoc201707031
Citation: Luo Junfei, Xu Xing, Zheng Junliang. Advance in C-H Arylation of Indoles[J]. Chinese Journal of Organic Chemistry, 2018, 38(2): 363-377. doi: 10.6023/cjoc201707031
吲哚C—H芳基化反应的研究进展
English
Advance in C-H Arylation of Indoles
Abstract:
The indoles motifs are widely found in the nature. One of the efficient strategy to access the indole derivatives is through the direct C-H functionalization of indole framework itself under transition-metal catalysis. Herein, the research advances on the transition-metal-catalyzed C-H arylation of indoles are reviewed.
-
Key words:
- transition-metal catalysis
- / C-H bond activation
- / indoles
- / arylation
-
吲哚类化合物是自然界中分布最广的杂环化合物之一, 其广泛的生物活性在抗癌药物、人类免疫缺陷病毒(HIV)以及糖尿病等领域备受关注[1].因此, 自从1866年Baeyer首次分离得到吲哚以来, 吲哚类化合物的合成方法研究就受到了有机合成化学家的广泛关注.已有大量的方法报道了通过各种各样的底物得到吲哚类化合物, 包括Fischer合成法[2]、Grandberg合成法[3]、Reissert合成法[4]、Leimgruber-Batcho合成法[5]、Madelung合成法[6]、Bischler合成法[7]、Bartoli合成法[8]、Nenitzescu合成法[9]、Gassman合成法[10]、Furstner合成法[11]、Fukuyama合成法[12]、Sugasawa合成法[13]等.近年来, 随着过渡金属催化C—H键活化的快速发展, 通过C—H键官能团化构建吲哚环的研究引起了化学家们的广泛兴趣, 许多新颖的方法学相继被报道[14].尽管上述传统的吲哚合成法以及近年来基于C—H活化构建吲哚环的方法可以得到不同的官能团取代的吲哚类化合物, 但是这些方法都需要预先引入官能团, 后进行环化反应得到吲哚环.因此对于底物的合成要求较高, 其他官能团的引入在很多情况下使得环化反应效率降低.
另一种吲哚类化合物合成的方法则是通过对吲哚骨架官能团化, 从而得到各种各样的吲哚衍生物.传统的方法是对吲哚进行C—H卤化, 然后通过经典的交叉偶联反应, 从而得到官能团取代的吲哚衍生物[15].近年来, 伴随着C—H键活化的蓬勃发展, 吲哚的直接C—H键官能团化的方法越来越受到有机化学家的青睐[16].吲哚直接C—H官能团化的策略可以快速有效地在吲哚骨架上引入特定的基团, 从而便捷地得到目标吲哚类化合物.该方法具有原子经济性以及反应步骤经济性的特点.因此, 发展新型的高选择性吲哚C—H官能团化的反应具有重要的意义.近年来, 大量新型有效的吲哚C—H键官能团化的方法学被报道, 然而目前关于这方面的综述仍然较少[17].联芳烃类化合物是许多天然产物、功能材料和化工产品的重要结构单元, 因此芳环C—C键的构建是目前过渡金属催化C—H键活化领域研究最多的反应类型之一(图 1).本文将根据吲哚不同位置的C—H键选择性的活化, 对近年来吲哚C—H键芳基化的方法进行了综述.
图1
部分含有吲哚结构的药物或活性分子
Figure1.
Examples of drug and reactive molecules containing indole structure
1 吲哚苯环骨架C—H芳基化
通常情况下, 吲哚的C3位置易于发生金属化作用, 从而使吲哚的C—H官能团化发生在C3位置.另一方面, 吲哚N原子很容易引入导向基, 进而导向吲哚C2位置的C—H活化.吲哚苯环骨架的电子云密度较之吡咯骨架小, 直接活化吲哚苯环骨架的C—H键较难进行, 方法较为欠缺.例如, 如果要在吲哚的C7位选择性的官能团化则需要先在C2位置处上引入一个取代基以阻止该位点的反应.此方法复杂, 且C2位上的取代基难以除去, 应用局限性大.直到2010年, Hartwig课题组[18]首次报道了用N-二乙基硅导向的Ir催化的C7位的吲哚C—H硼酸化, 硼酸化的产物进一步通过Suzuki-Miyaura偶联可得到C7-芳基吲哚(Scheme 2).
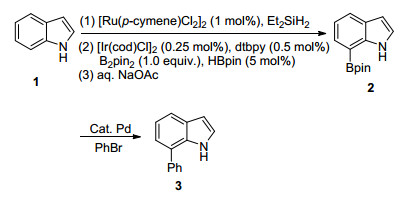 图式 1
一锅法制备C7-芳基取代的吲哚
Scheme1.
One-pot synthesis of C7-arylated indoles
图式 1
一锅法制备C7-芳基取代的吲哚
Scheme1.
One-pot synthesis of C7-arylated indoles
 图式 2
Cu催化的吲哚C6位芳基化
Scheme2.
Cu-catalyzed direct C6-arylation of indoles
图式 2
Cu催化的吲哚C6位芳基化
Scheme2.
Cu-catalyzed direct C6-arylation of indoles
2016年, 史壮志课题组[19]首次实现了Pd催化的吲哚C7位置的C—H芳基化(Eq. 1).该方法的关键在于合适的导向基N-P(O)tBu2, X衍射揭示底物中的导向基的O原子更靠近C7位置的C—H键, 从而确保了金属Pd与O原子配位后靠近C7位的C—H键, 从而高选择性地活化C7位C—H键.该反应具有很好的官能团兼容性, 吸电子基和给电子基取代的吲哚以及苯硼酸都能在该体系中反应得到相应的吲哚C7芳基化产物.
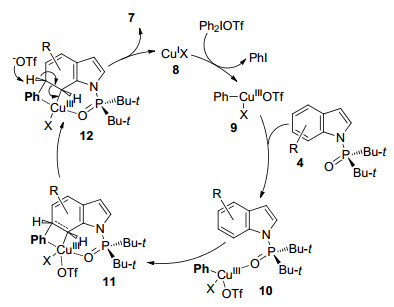
类似于吲哚C7位C—H键的官能团化, 吲哚C6位的C—H键活化的方法报道亦是寥寥无几.余金权和Baran课题组分别报道吲哚C6位的C—H烯化[20]和硼化[21]方法.然而, 在这两个方法中吲哚的吡咯核处都需要具有封闭基团以阻止吡咯骨架的反应, 因此局限性较大.史壮志等[22]在吲哚C7位C—H芳基化的研究基础上, 进一步在Cu催化下, 使用三氟甲磺酸二苯基碘鎓作为芳基化试剂, 实现了吲哚的C6位C—H键芳基化(Eq. 2).
基于Gaunt等[23]所报道的Cu催化的苯胺类衍生物间位的C—H键芳基化的机理, 作者提出了可能的反应机理:首先, CuO歧化作用生成的一价CuⅠ类物质8与三氟甲磺酸二苯基碘鎓反应形成三价CuⅢ中间体9. CuⅢ底物4中的N-P(O)tBu2导向基团配位后通过Heck型四元环过渡态11得到中间体12.最后, E2消除反应重新生成活性催化剂8和C6芳基化吲哚7 (Scheme 2).
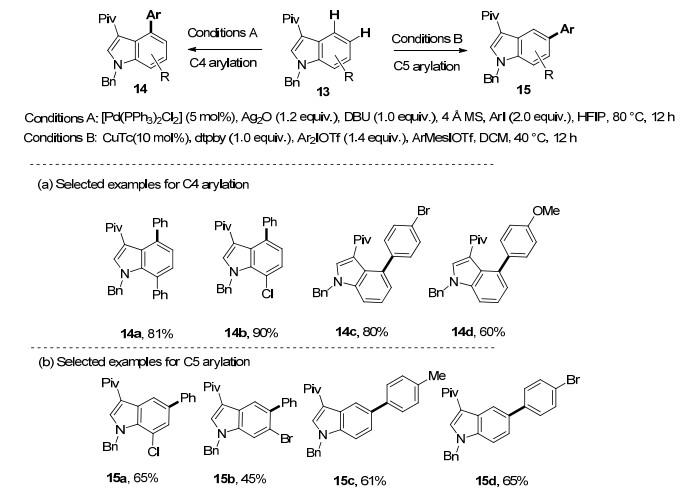
随后, 史壮志课题组通过进一步深入地研究, 实现了吲哚苯环骨架其余位置(C4和C5)的C—H键芳基化[24].基于特戊酰基可以导向多样的C—H键活化反应, 史壮志等认为, 如果在吲哚的C3位置引入特戊酰基, 该基团则可以潜在地选择性导向活化吲哚苯环骨架的C4和C5位置的C—H键.通过大量的筛选, 作者发现在Pd催化体系下, C3位的特戊酰基可以选择性地导向活化C4位置的C—H键, 从而使用碘苯衍生物作为芳基化试剂实现吲哚C4位C—H芳基化(Scheme 3).
 图式 3
吲哚C4和C5直接芳基化
Scheme3.
Direct arylation of indoles at C4 and C5 positions
图式 3
吲哚C4和C5直接芳基化
Scheme3.
Direct arylation of indoles at C4 and C5 positions
另一方面, 作者发现在Cu催化体系下, C3位的特戊酰基可以选择性地导向活化C5位置的C—H键, 从而使用三氟甲磺酸二苯基碘鎓作为芳基化试剂, 通过Heck型四元环过渡态实现吲哚C5位C—H芳基化(Scheme 3).特戊酰基可以通过Friedel-Crafts酰化方法选择性地安装在吲哚的C3位, 同时可以在对甲苯磺酸和二醇的存在下通过反Friedel-Crafts反应从产物中轻易地去除, 得到仅在C4或C5位芳基化的产物.此方法方便引入和去除导向基, 对吲哚芳基化的发展有重要意义.作者从吲哚出发, 氮原子用苄基保护, 吲哚C3位置引入特戊酰基.随后分别通过上述方法在吲哚C4和C5位置引入芳环得到化合物16和17.化合物16氮原子苄基脱保护再引入苯胺甲基基团得到SET7抑制剂SETin-1的前体化合物18.在化合物17的C3位置引入醛基甲酸基团则得到TPI-1抑制剂Tiplaxinin (19) (Scheme 4).
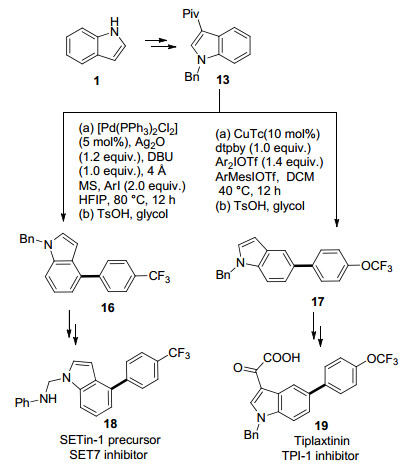 图式 4
吲哚C4和C5 C—H芳基化方法合成生物活性分子
Scheme4.
Bio-active molecules synthesis via C4 and C5 C—H arylation
图式 4
吲哚C4和C5 C—H芳基化方法合成生物活性分子
Scheme4.
Bio-active molecules synthesis via C4 and C5 C—H arylation
尽管吲哚苯环骨架C—H芳基化的方法报道较少, 但其他C—H官能团化的研究在最近几年取得了很大进展. 2016年, 马大为课题组[25]发现在Rh催化下, 在吲哚氮原子上引入特戊酰基作为导向基团可以促进吲哚C7位选择性C—H烯烃化(Eq. 3).在对底物研究过程中发现, 具有吸电子基团的底物反应缓慢, 产率较低.当C6位置有取代基时, 未观察到C7烯化产物, 而是得到C2烯化产物, 表明立体位阻效应极大地影响着该反应的区域选择性.同年, Chang课题组[26]报道了Ir催化的吲哚C7氨基化反应(Eq. 4).马大为[27]和Antonchick[28]等分别实现了Ir催化的吲哚C7位的磺酰胺化反应(Eqs. 5, 6).通过该方法马大为课题组成功制备微管蛋白解聚剂24和细胞增殖抑制剂25充血性心脏病药物, 产率分别达到92%和90%.此外, 王洪波和王亮等[29]通过一锅法反应在Rh催化下实现吲哚C2位和C7位同时烷基化(Eq. 7).该方法反应条件温和, 底物范围宽广和官能团耐受性良好.
类似于吲哚氮原子上的基团导向C7位置的C—H官能团化, 在吲哚的C3位引入导向基团可以选择性活化C4位置的C—H键. 2013年, 贾彦兴课题组[30]首次报道了Pd催化下的色氨酸C4位C—H烯基化(Eq. 8).该方法可应用于大量半萜类吲哚生物碱的合成.作者利用该方法实现了吲哚C4位的烯基化和胺环化的串联反应, 最终酯基水解得到Clavicipitic acid (31).史壮志课题组[31]通过在吲哚的C3位引入简单的乙酰基团作为导向基成功地实现了钯催化下的C4位氟代烷基化(Eq. 9).反应条件也同样适用于3-乙酰基苯并噻吩的C4位氟代烷基化. Prabhu课题组[32]通过在吲哚的C3位引入醛基, 在其导向下选择性地实现Ru催化下的吲哚C4位C—H烯基化(Eq. 10).最近, 贾彦兴课题组[33]同样使用了醛基导向基在铑催化下实现了吲哚衍生物C4位烯烃化(Eq. 11).基于上述方法该课题组成功完成了(-)-田麦角碱(37)和(-)-野麦角碱(38)的全合成.
一直以来对于吲哚C6位的C—H键官能化的报道非常少, 且只能观察到少量的反应性, 如Baran课题组[34]对于疣孢青霉原和烟曲霉毒素的全合成中提及配体控制的铱催化下可以实现N原子和C3位有取代基的吲哚衍生物的C6位活化, 但是选择性较差.近年来一些有机化学家们一直致力于该位点的C—H键活化, Frost教授课题组[35]使用C3位带有辅助配位导向的酯基并且N原子上有强配位导向的嘧啶基团的吲哚作为反应底物, 在Ru催化实现了吲哚C6选择性C—H键烷基化(Eq. 12), 最后, 作者从N-嘧啶基吲哚出发, 在该条件下通过一锅法反应, 成功地获得C3和C6位取代的产物, 但伴随C3位单取代产物生成, 产率一般.
2 吲哚C3位C—H芳基化
2.1 Pd催化吲哚C3位C—H芳基化
相比于吲哚苯环骨架的C—H键芳基化, 吲哚吡咯环上的C—H键芳基化反应的报道层出不穷.吲哚C3位是电子云密度最大的位点, 因此吲哚金属化作用容易发生在C3位置. 2007年, 何仁等[36]报道了三价磷酸作为配体的POPd催化剂, 以溴代苯作为芳基化试剂实现了吲哚C3位置的C—H芳基化(Eq. 13). Bellina和Rossi等[37]则使用了简单的Pd(OAc)2作为催化剂, 并与相转移催化剂BnBu3NCl共催化下高效地实现了吲哚C3位的C—H芳基化(Eq. 14).尽管作者尝试了不同配体, 但发现在没有配体的情况下得到最高产率.随后, Ackermann等[38]通过大量的配体筛选发现了一种对空气稳定的磷酸配体(HASPO), 可促进吲哚C3位的C—H芳基化反应(Eq. 15). Prim等[39]则使用了更为廉价的吡啶配体, 实现了吲哚C3位的C—H芳基化(Eq. 16).上述方法都是在均相催化体系下进行, 另一方面, Djalovitch等[40]使用了异相催化剂[Pd(NH3)4]2+/NaY实现了吲哚C3位的C—H芳基化(Eq. 17).尽管该方法的产率并不理想, 但是异相催化剂相比较与均相催化剂有着可回收或重复使用的优点.
该类反应普遍被接受的机理类似于Heck反应.首先Pd0与卤代烃氧化加成得到二价钯中间体48, 48与吲哚配位, 由于C3位置较大的电子云密度, 使得亲电取代发生在C3位得到50. 50在碱的作用下重新芳香化得到51, 最后还原消除重新得到Pd催化剂和C3芳基化吲哚产物52 (Scheme 5).
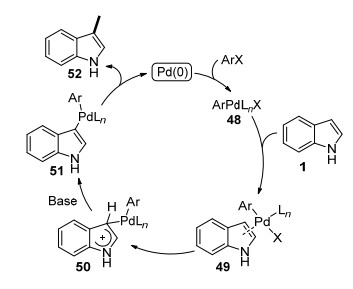 图式 5
Pd催化的吲哚C3位芳基化
Scheme5.
Pd-catalyzed C3-arylation of indoles
图式 5
Pd催化的吲哚C3位芳基化
Scheme5.
Pd-catalyzed C3-arylation of indoles
除了卤代芳烃作为芳基化试剂的方法外, 使用其他芳基化试剂实现吲哚C3位C—H芳基化的方法亦被较多的报道, 比如Larrosa课题组[41]使用了苯甲酸类衍生物, 通过脱羧偶联得到C3芳基吲哚(Eq. 18).陈保华等[42]则使用苯肼作为偶联试剂, 以空气为氧化剂实现吲哚C3位C—H芳基化(Eq. 19).邓国军等[43]使用了环己酮作为芳基化试剂, 通过脱氢作用-互变异构一锅反应得到C3芳基吲哚(Eq. 20).
C—H交叉脱氢偶联反应作为最有效、最经济的方法之一, 被众多有机化学家关注. 2007年, Fagnou课题组[44]首次通过活化双C—H键实现N-乙酰基保护的吲哚C3位的高选择性芳基化(Eq. 21).该方法使用廉价的苯作为芳基化试剂, 无需在任何偶联试剂预先官能团化, 极大地提高了反应经济性.由于苯环的C—H键较难活化, 过量的苯(30 equiv.)是该反应得以进行的保证, 作者认为苯环C—H键的活化可能的机理有亲电芳香金属化或者协同金属去质子化的过程.
相比较与苯环, 杂环芳香环的C—H键较易被活化, 因此, 吲哚与杂环芳香环的交叉偶联反应的方法报道较多.比如施章杰课题组[45]研究了吲哚C3位的自偶联方法(Eq. 22).李兴伟等[46]则使用了N-氧化吡啶作为偶联试剂实现了吲哚C3位于吡啶C2位的C—H/C—H偶联反应(Eq. 23). Itami等[47]同样使用了含氮杂环氧化物在Pd催化体系下完成与吲哚C3位的偶联反应(Eq. 24).游劲松[48]和Chupakhin课题组[49]分别使用了N-甲基咪唑衍生物和N-氧化咪唑衍生物作为芳基化试剂实现了吲哚C3位的芳基化(Eqs. 25, 26).
Schmalz和Fedorov等[50]将Pd催化的吲哚C3位C—H芳基化应用于allocolchicinoids化合物的合成(Scheme 6).该合成路线的关键步骤是吲哚衍生物72通过分子内的直接芳基偶联关环反应得到化合物73.最终通过脱保护, 脱水以及氨氢化反应得到allocolchici-noids化合物.
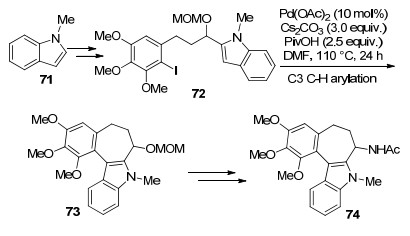 图式 6
Allocolchicinoids化合物合成
Scheme6.
Synthesis of allocolchicinoids via indole C3-arylation
图式 6
Allocolchicinoids化合物合成
Scheme6.
Synthesis of allocolchicinoids via indole C3-arylation
2.2 其他过渡金属催化吲哚C3位C—H芳基化
相比较于Pd金属催化的吲哚C—H键芳基化反应的研究, 其他过渡金属催化方法的研究较为欠缺. 2015年, Lee课题组[51]报道了在Rh催化体系下, 吲哚与1-重氮萘-2(1H)-酮反应得到3-芳基吲哚化合物(Eq. 27).该反应具有很好的选择性, 且产率高的特点.值得一提的是, 该反应在室温条件下就能进行, 而Pd催化体系下的反应往往需要在较高的反应温度下进行.反应可能经历的过程为, 重氮化合物75脱氮后, 与Rh催化剂形成Rh卡宾体77, 77与吲哚亲核加成得到两性离子78, 最后78去质子化以及Rh催化剂脱除得到产物79 (Scheme 7).
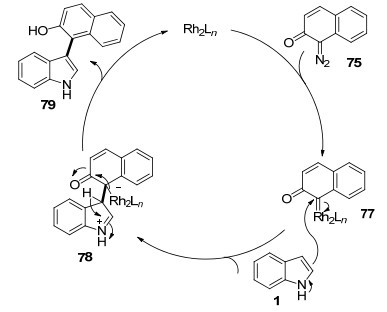 图式 7
Rh催化的吲哚C3位芳基化
Scheme7.
Rh-catalyzed C3-arylation of Indoles
图式 7
Rh催化的吲哚C3位芳基化
Scheme7.
Rh-catalyzed C3-arylation of Indoles
Lloyd-Jones课题组[52]研究了Au催化体系下吲哚与芳基硅烷的反应(Eq. 28).该反应具有反应条件温和、官能团兼容性好、实验操作简单的优点, 这些是大多数Pd催化体系下所不具有的.
相比较于Pd、Rh和Au, Cu的价格相对低廉催化, 因此广受青睐. 2008年, Gaunt课题组[53]报道了一种新型的Cu催化的吲哚C3位C—H键芳基化(Eq. 29).该方法可以在温和条件下进行, 其和Au催化的方法一样, 可以很好地与Pd催化的方法互补, 且相较于Au催化剂的昂贵, Cu催化剂更有经济优势.但是该方法使用二芳基碘鎓盐作为芳基化试剂, 其实际芳基化只需要一个芳香环, 而另一个芳香环则作为反应废料, 因此原子经济性不高.基于此, Greaney课题组[54]通过一锅法, 使用二芳基碘鎓盐作为芳基化试剂实现了吲哚C3位以及N位置的双芳基化反应(Eq. 30).
Gaunt课题组[55]通过多步的吲哚C—H官能团化的反应成功合成了复杂化合物Dictyodendrin B (Scheme 8).首先从4-溴吲哚88在铜催化下实现吲哚的C3位C—H芳基得到化合物89, 89进一步C2位C—H酰基化得到化合物90.随后利用之前Hartwig等[18]报道的一锅反应的方法实现了吲哚衍生物90的C7位的C—H芳基化得到多取代的吲哚衍生物91, 进一步的反应可得到端粒酶抑制剂dictyodendrin B.
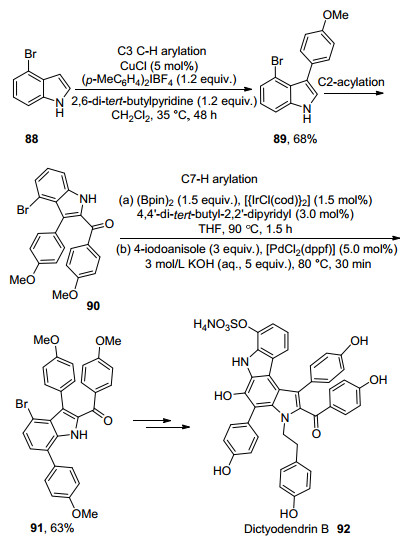 图式 8
Dictyodendrin B的合成
Scheme8.
Synthesis of dictyodendrin B
图式 8
Dictyodendrin B的合成
Scheme8.
Synthesis of dictyodendrin B
2.3 无过渡金属催化吲哚C3位C—H芳基化
无金属催化反应由于无重金属污染问题的存在一直备受有机化学家的关注. 2016年, 张应鹏和杨云裳等[56]使用了芳基重氮盐, 在若丹明催化下实现了吲哚C3位的C—H芳基化(Eq. 31).该方法只需在室温光照的条件下就能进行, 且官能团兼容性高.作者认为该反应可能经历的是自由基机理:芳基重氮盐与激发态若丹明作用生成苯自由基97和98, 苯自由基97与吲哚作用得到芳基化的吲哚自由基99, 99和98反应生成吲哚正离子100, 去质子化最终得到C3芳基化吲哚(Scheme 9). Wu等[57]则使用了更为安全的卤代烃作为芳基化试剂, 在叔丁基醇钾存在下实现了吲哚C3位的C—H芳基化(Eq. 32).该反应同时伴随着吲哚N-芳基化的副产物, 副产物的比例取决于底物的取代基类型.
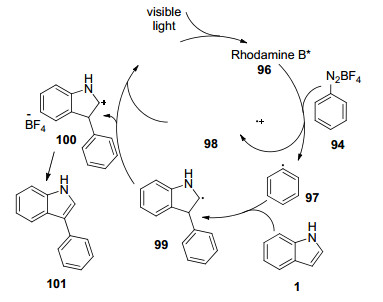 图式 9
无金属催化的吲哚C3位芳基化
Scheme9.
Metal-free C3-arylation of indoles
图式 9
无金属催化的吲哚C3位芳基化
Scheme9.
Metal-free C3-arylation of indoles
3 吲哚C2位C—H芳基化
3.1 Pd催化吲哚C2位C—H芳基化
在过去的几年中, 有机化学家对吲哚C2位的C—H键芳基化进行了大量地研究, 尤其是Pd催化的吲哚C2位C—H芳基化的方法相继被报道.较早的报道有Sames课题组[58]杂环卡宾作为配体, 使用溴苯或者碘苯实现了吲哚C2位的C—H芳基化(Eq. 33).该方法适用了各种唑类化合物的C—H芳基化. Bellina和Rossi等[59]则使用了简单易得的醋酸钯作为催化剂, 在碘化亚铜和碘苯存在下使吲哚C2位发生C—H芳基化(Eq. 34).但是该方法产率普遍较低. 2008年, Larrosa课题组[60]使用了银盐目的在于结合碘苯中的碘离子, 另外在邻硝基苯甲酸辅助下使用醋酸钯高效地实现了吲哚C2位C—H芳基化(Eq. 35).该方法在室温条件下就能进行, 吲哚N原子无保护或者甲基保护的底物均适用于该反应条件. Ren等[61]在此基础上, 使用了三氟甲酸银盐在Pd催化体系下实现了吲哚C3位C—H芳基化(Eq. 36).该方法的优点在于使用绿色的水作为反应溶剂, 而表面活性剂Tween 80在提高产率上起了很大的作用.最近, 薛丰田和姜超等[62]研究了降冰片烯作为瞬态的介体, Pd催化下的吲哚N原子无保护的C2芳基化反应(Eq. 37).该反应经历了Catellani型反应机理.
Albericio和Lavilla等[63]报道了Pd催化的色氨酸及其衍生物的C2位C—H芳基化反应(Eq. 38).色氨酸及其衍生物有着很好的生物活性, 比如化合物110b具有潜在的IL-8受体拮抗剂, 报道的方法需要较长的合成路线[64], 而通过C2位C—H芳基化的方法仅需一步即可.同时, C2位芳基化的色氨酸可用于标准固态肽链的合成.
Sanford课题组[65]以二芳基碘鎓盐作为芳基化试剂, 在Pd催化体系下, 室温条件即可实现吲哚C2位C—H芳基化(Eq. 39).而该反应的优点在于不需要当量的Ag作为添加剂. Olofsson和Backvall等[66]以氨基官能团化的介孔泡沫材料为载体的纳米钯(Pd0-AmP-MCF)作为催化剂, 二芳基碘鎓盐作为芳基化试剂以水作为溶剂高效地完成了吲哚C2芳基化.另一方面, 万颖和张炳森等[67]同样使用了二芳基碘鎓盐作为芳基化试剂, 在可重复使用的介孔材料固载Pd催化下实现吲哚C2芳基化.
2008年, 施章杰课题组[68]报道了室温下, 以苯硼酸作为芳基化试剂Pd催化吲哚C2位的直接C—H芳基化方法(Eq. 40).该方法只需要廉价的氧气作为氧化剂, 高效地将不同官能团取代的吲哚C2位C—H键转换位芳基-芳基C—C键.基于此研究, Cao课题组[69]将纳米钯包裹与MIL-101介孔材料中高效实现了苯硼酸与吲哚C2位的偶联反应.除此之外, 芳基三氟硼盐(ArBF3K)[70]、芳基硅氧烷(ArSi(OR)3)[71]、芳基磺酰氯(ArSO2Cl)[72]、芳基磺酰肼(ArSO2NHNH2)[73]均被用作芳基化试剂, 在Pd催化下与吲哚的C2位发生偶联反应.
2016年, Laha课题组[74]首次通过双C—H键活化在吲哚C2位实现芳基化(Eq. 41). N原子上不同官能团(Me、Bn、Ms、Ts)取代的吲哚均可在该反应条件下实现C2位C—H键向芳基-芳基C—C键的转换. N原子无保护基的吲哚不能发生在该条件下与苯发生偶联.
3.2 Rh催化吲哚C2位C—H芳基化
除了Pd金属, Rh催化吲哚C2位芳基化的研究也有不少报道.较早的有Sames课题组[75]报道的Rh催化下碘苯与吲哚C2的偶联(Eq. 42).作者认为该反应可能的机理如下:首先Rh催化剂114在反应条件下生成休眠态的催化剂115, 催化剂115与底物吲哚配位得到116, 该步骤是一个平衡态. 116通过C—H键金属化作用生成中间体117, 还原消除即得到最终C2芳基化吲哚和Rh化合物118, 118可与碘代苯、特戊酸铯反应重新得到催化剂115 (Scheme 10).
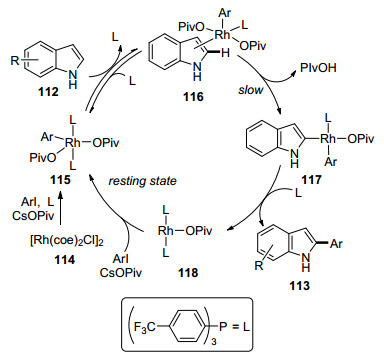 图式 10
RhⅢ催化的吲哚C2位芳基化
Scheme10.
RhⅢ-catalyzed C2-arylation of indoles
图式 10
RhⅢ催化的吲哚C2位芳基化
Scheme10.
RhⅢ-catalyzed C2-arylation of indoles
随后, 施章杰、李焕荣和徐立进等[76]在吲哚N原子上引入嘧啶导向基团, 该嘧啶基团可以引导Rh催化剂活化邻位的C—H键.从而使苯甲酸类衍生物通过脱羧偶联实现吲哚C2位的芳基化(Eq. 43).徐允河和Loh等[77a]、王亮和彭望明等[77]分别以芳基硼酸和芳基硅烷作为芳基化试剂, 以嘧啶作为导向基团, 在吲哚的C2位置引入了芳基(Eqs. 44, 45).另一方面, 崔孙良课题组[78]使用了N-甲氧基甲酰胺作为导向基团, 其N原子可配位Rh催化剂, 通过五元环中间体活化吲哚C2位置的C—H键, 最终实现吲哚C2位置的芳基化转换(Eq. 46).
3.3 Cu催化吲哚C2位C—H芳基化
2008年, Gaunt课题组[53]通过Cu催化剂, 以二苯基碘鎓盐作为芳基化试剂实现了高选择性的N-乙酰基吲哚C2位芳基化(Eq. 47).值得注意的是, 在类似条件下, 以N-甲基保护的吲哚以及N原子无保护基的吲哚为原料时, 该芳基化方法更倾向于C3位置.可能的原因是由于乙酰基的吸电子能力, 使得C3位置Cu金属化的吲哚发生迁移至C2位置得到中间体126, 最终中间体126经过还原消除得到产物123, 另外重新生成催化剂CuOTf (Scheme 11).
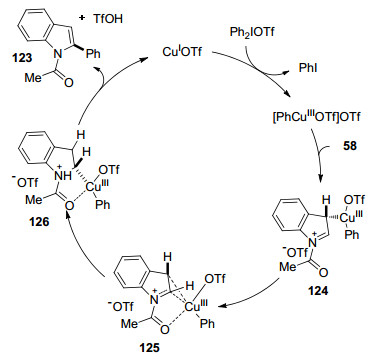 图式 11
C3至C2迁移的可能机理
Scheme11.
Proposed C3 to C2 migration of the C—Cu bond
图式 11
C3至C2迁移的可能机理
Scheme11.
Proposed C3 to C2 migration of the C—Cu bond
Hirano和Miura等[79]使用嘧啶作为导向基的策略, 在过量的Cu(OAc)2存在下, 通过双C—H键的活化成功构建了吲哚C2位的芳基-芳基C—C键(Eq. 48).该方法同时兼容给电子基以及吸电子基取代的吲哚衍生物, 同时苯噁唑、噁唑、苯并噻唑和苯并咪唑均可作为芳基化试剂.
3.4 其他过渡金属催化吲哚C2位C—H芳基化
Ru催化的吲哚芳基化方法鲜有报道, Pilarski课题组[80]率先报道了Ru催化的吲哚C2位芳基化的方法.该方法通过吲哚N原子上引入嘧啶导向基, 从而配位Ru催化剂实现吲哚C2位C—H键的活化, 最后在芳基硼酸试剂存在下完成C2位的芳基化反应(Eq. 49). Kapur等[81]则通过在N原子上引入吡啶官能团, 在类似条件下完成了吲哚C2位的芳基化过程(Eq. 50).与嘧啶导向基、吡啶导向基相比较更易引入以及芳基化后的除去.该类反应可能的机理是Ru(Ⅱ)催化剂配位导向基中的N原子, 进而活化吲哚C2位的C—H键形成Ru(Ⅱ)五元环中间体131.碱存在下金属转移作用使芳基硼酸中的芳基转移至Ru化合物生成132, 最终132还原消除得到吲哚C2位芳基化产物130以及零价Ru(0)催化剂. Ru(0)催化剂被Ag(Ⅰ)或者Cu(Ⅱ)氧化重新得到Ru(Ⅱ)催化剂(Scheme 12).
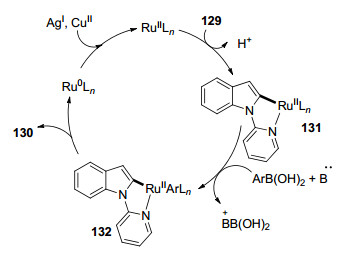 图式 12
Ru(Ⅱ)催化的吲哚C2位芳基化
Scheme12.
Ru(Ⅱ)-catalyzed C2-arylation of indoles
图式 12
Ru(Ⅱ)催化的吲哚C2位芳基化
Scheme12.
Ru(Ⅱ)-catalyzed C2-arylation of indoles
2017年, Punji课题组[82]首次实现了Ni催下吲哚C2位的芳基化反应(Eq. 51).该方法无需反应溶剂, 半小时即得到较高的产率.作者提出了两种可能的反应机理:首先催化剂133在LiN(SiMe3)2存在下转化为活性的Ni化合物134, 催化剂134在吡啶导向下C2位金属化得到Ni化合物135.在这之后有两种反应的可能性: (1) 135引发自由基反应, 与碘代苯反应生成三价Ni化合物136以及芳基自由基, 芳基自由基与136反应得到四价Ni化合物137. 137还原消除得到吲哚C2位芳基化产物130和催化剂133. (2) 135与碘代苯发生双分子氧化反应生成三价Ni化合物138和136.吲哚C2位芳基化产物130可通过还原消除化合物138得到, 并同时生成Ni催化剂139.最后Ni催化剂139与136反应可生成Ni化合物135以及Ni催化剂133 (Scheme 13).
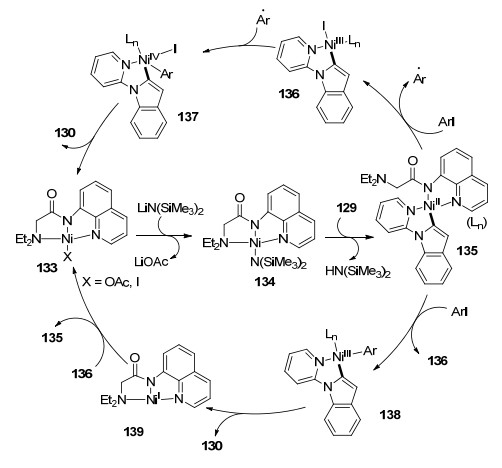 图式 13
Ni催化的吲哚C2位芳基化
Scheme13.
Ni-catalyzed C2-arylation of indoles
图式 13
Ni催化的吲哚C2位芳基化
Scheme13.
Ni-catalyzed C2-arylation of indoles
Co催化下的吲哚C2位芳基化亦有被报道, 宋毛平和牛俊龙等[83]通过在吲哚N原子上引入嘧啶或者吡啶基团, 在Co催化剂作用下, 以芳基硼酸作为芳基化试剂高选择性地构建了吲哚C2位芳基-芳基C—C键(Eq. 52).
作者提出该反应可能包含了自由基的过程.首先二价Mn离子在氧气存在下氧化成三价MnⅢ或者四价MnⅣ, 随后MnⅢ或者MnⅣ可氧化二价CoⅡ催化剂生成三价CoⅢ催化剂. CoⅢ催化剂与吡啶或者嘧啶导向基配位, 通过协同金属去质子化(CMD)过程得到五元Co环化合物142.另一方面, 芳基硼酸可在MnⅢ存在下生成芳基自由基, 该自由基可与三价Co环化合物142反应生成四价的Co环化合物143. 143通过还原消除反应即可得到吲哚C2位芳基化的产物141以及二价Co催化剂(Scheme 14).
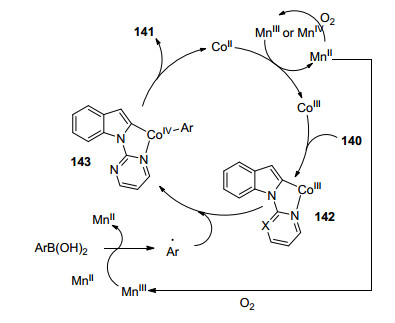 图式 14
Co催化的吲哚C2位芳基化
Scheme14.
Co-catalyzed C2-arylation of indoles
图式 14
Co催化的吲哚C2位芳基化
Scheme14.
Co-catalyzed C2-arylation of indoles
4 结论与展望
综上所述, 基于吲哚骨架C—H键官能团化的方法, 尤其是基于C—H键活化的吲哚芳基化反应的研究在近几年中取得了快速的发展, 为合成结构多样性的吲哚衍生物提供了一种快速有效的策略.迄今为止, 基于吲哚C2和C3位置的C—H键芳基化反应的研究较为成熟, 尤其是Pd金属催化的吲哚C2和C3位芳基化方法, 已有大量优秀的工作相继被报道.与此同时, 课题组首次报道了吲哚苯环骨架即C4—C7位置的C—H芳基化的方法.该方法的成功使得吲哚所有位置的C—H键均可实现直接芳基化的过程, 从而为快速有效地构建吲哚芳基化衍生物提供捷径.但是相对于吲哚C2和C3位置的C—H键芳基化的方法, 吲哚苯环骨架C—H键芳基化方法的研究很少, 因此深入研究发展吲哚苯环骨架的C—H键芳基化的反应具有很重要的意义.另一方面, 尽管吲哚C2和C3位的C—H键芳基化反应研究比较成功, 但是极大多数方法需要相对较为苛刻的反应条件, 其实用性仍有一定的局限, 并且所需催化剂一般都为昂贵的Pd和Rh催化剂.因此, 设计新型的廉价催化剂体系, 在相对温和条件下实现吲哚可控性的多位点C—H键芳基化, 使之能应用于天然产物、功能材料以及药物分子的合成中, 这仍然是该领域长期的目标.
-
-
[1]
(a) Curtin, M. L. ; Davidsen, S. K. ; Heyman, H. R. ; Garland, R. B. ; Sheppard, G. S. ; Florjancic, A. S. ; Xu, L. ; Carrera, G. M. ; Steinman, Jr., D. H. ; Trautmann, J. A. ; Albert, D. H. ; Magoc, T. J. ; Tapang, P. ; Rhein, D. A. ; Conway, R. G. ; Luo, G. ; Denissen, J. F. ; Marsh, K. C. ; Morgan, D. W. ; Summers, J. B. J. Med. Chem. 1998, 41, 74.
(b) Kochanowska-Karamyan, A. J. ; Hamann, M. T. Chem. Rev. 2010, 110, 4489.
(c) Alexandre, F. R. ; Amador, A. ; Bot, S. ; Caillet, C. ; Convard, T. ; Jakubik, J. ; Musiu, C. ; Poddesu, B. ; Vargiu, L. ; Liuzzi, M. ; Roland, A. ; Seifer, M. ; Standring, D. ; Storer, R. ; Dousson, C. B. J. Med. Chem. 2011, 54, 392.
(d) Chen, Y. -C. ; Xie, Z. -F. Chin. J. Org. Chem. 2012, 32, 462(in Chinese).
(陈永诚, 解正峰, 有机化学, 2012, 32, 462. )
(e) Dalpozzo, R. ; Bartoli, G. ; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247; -
[2]
Fischer, E.; Jourdan, F. Ber. Dtsch. Chem. Ges. 1883, 16, 2241. doi: 10.1002/(ISSN)1099-0682
-
[3]
Street, L. J.; Baker, R.; Castro, J. L.; Chambers, M. S.; Guiblin, A. R.; Hobbs, S. C.; Matassa, U. G.; Reeve, A. J.; Beer, M. S.; Middlemiss, D. N.; Noble, A. J.; Stanton, J. A.; Scholey, K.; Hargreaves, R. J. J. Med. Chem. 1993, 36, 1529. doi: 10.1021/jm00063a003
-
[4]
Reissert, A. Ber. Dtsch. Chem. Ges. 1897, 30, 1030. doi: 10.1002/(ISSN)1099-0682
-
[5]
Batcho, A. D.; Leimgruber, W. Org. Synth. 1985, 63, 214. doi: 10.15227/orgsyn.063.0214
-
[6]
Houlihan, W. J.; Parrino, V. A. Uike, Y. J. Org. Chem. 1981, 46, 4511. doi: 10.1021/jo00335a038
-
[7]
Nordlander, J. E.; Catalane, D. B.; Kotian, K. D.; Stevens, R. M.; Haky, J. E. J. Org. Chem. 1981, 46, 778. doi: 10.1021/jo00317a026
-
[8]
Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R. Tetrahedron Lett. 1989, 30, 2129. doi: 10.1016/S0040-4039(01)93730-X
-
[9]
Patrick, J. B.; Saunders, E. K. Tetrahedron Lett. 1979, 4009. http://www.sciencedirect.com/science/article/pii/S004040390186490X
-
[10]
Gassman, P. G.; Roos, J. J.; Lee, S. J. J. Org. Chem. 1984, 49, 717. doi: 10.1021/jo00178a033
-
[11]
Furstner, A.; Ernst, A. Tetrahedron 1995, 51, 773. doi: 10.1016/0040-4020(94)00987-6
-
[12]
Fukukyama, T.; Chen, X.; Peng, G. J. Am. Chem. Soc. 1994, 116, 3127. doi: 10.1021/ja00086a054
-
[13]
Sugasawa, T.; Adachi, M.; Sasakura, K.; Kitagawa, A. J. Org. Chem. 1979, 44, 578. doi: 10.1021/jo01318a021
-
[14]
(a) Stuart, D. R. ; Bertrand-Laperle, M. ; Burgess, K. M. N. Fagnou, K. J. Am. Chem. Soc. 2008, 130, 16474.
(b) Song, W. ; Ackermann, L. Chem. Commun. 2013, 49, 6638.
(c) Zhang, G. ; Yu, H. ; Qin, G. ; Huang, H. Chem. Commun. 2014, 50, 4331.
(d) Fan, Z. ; Song, S. ; Li, W. ; Geng, K. ; Xu, Y. ; Miao, Z. -H. ; Zhang, A. Org. Lett. 2015, 17, 310.
(e) Lu, B. -L; Li, X. -Y; Lin, Y. -M. Chin. J. Org. Chem. 2015, 35, 2275(in Chinese).
(卢贝丽, 李现艳, 林咏梅, 有机化学, 2015, 35, 2275. )
(f) Zhang, X. ; Guo, R. ; Zhao, X. Org. Chem. Front. 2015, 10, 1334.
(g) Fan, Z. ; Lu, H. ; Li, W. ; Geng, K. ; Zhang, A. Org. Biomol. Chem., 2017, 15, 5701. -
[15]
(a) Deng, H. ; Jung, J. -K. ; Liu, T. ; Kuntz, K. W. ; Snapper, M. L. ; Hoveyda, A. H. J. Am. Chem. Soc. 2003, 125, 9032.
(b) Nicolaou, K. C. ; Rao, P. B. ; Hao, J. ; Reddy, M. V. ; Rassias, G. ; Huang, X. ; Chen, D. Y. -K. ; Snyder, S. A. Angew. Chem., Int. Ed. 2003, 42, 1753.
(c) Nicolaou, K. C. ; Hao, J. ; Reddy, M. V. ; Rao, P. B. ; Rassias, G. ; Snyder, S. A. ; Huang, X. ; Chen, D. Y. -K. ; Brenzovich, W. E. ; Giuseppone, N. ; Giannakakou, P. ; O'Brate, A. J. Am. Chem. Soc. 2004, 126, 12897. -
[16]
See the most recent examples: (a) Petrini, M. Chem. Eur. J. 2017, 23, 16115.
(b) Tayu, M. ; Nomura, K. ; Kawachi, K. ; Higuchi, K. ; Saito, N. ; Kawasaki, T. Chem. -Eur. J. 2017, 23, 10925.
(c) Ni, J. -B. ; Zhao, H. -C. ; Zhang, A. Org. Lett. 2017, 19, 3159.
(d) Erdenebileg, U. ; Demissie, E. B. ; Hansen, J. H. Synlett 2017, 907.
(e) Borah, A. J. ; Shi, Z. -Z. Chem. Commun. 2017, 53, 3945.
(f) Leitch, J. A. ; McMullin, C. L. ; Mahon, M. F. ; Bhonoah, Y. ; Frost., C. G. ACS Catal. 2017, 7, 2616.
(g) Iagafarova, I. E. ; Vorobyeva, D. V. ; Loginov, D. A. ; Peregudov, A. S. ; Osipov, S. N. Eur. J. Org. Chem. 2017, 840.
(h) Zhao, Y. P. ; Sharma, U. K. ; Schroder, F. ; Sharma, N. ; Song, G. ; Van der Eycken, E. V. RSC Adv. 2017, 7, 32559. -
[17]
(a) Bandini, M. ; Melloni, A. ; Tommasi, S. ; Umani-Ronchi, A. Synlett 2005, 1199.
(b) Bandini, M. ; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608.
(c) Beck, E. M. ; Gaunt, M. J. Top. Curr. Chem. 2010, 292, 85.
(d) Cacchi, S. ; Fabrizi, G. Chem. Rev. 2011, 111, 215.
(e) Sandtorv, A. H. Adv. Synth. Catal. 2015, 357, 2403.
(f) Chen, J. -B. ; Jia, Y. -X. Org. Biomol. Chem. 2017. 15. 3550.
(g) Szatmari, I. ; Sas, J. ; Fulop, F. Curr. Org. Chem. 2017. 20. 2038. -
[18]
Robbins, D. W.; Boebel, T. A.; Hartwig, J. F. J. Am. Chem. Soc. 2010, 132, 4068; doi: 10.1021/ja1006405
-
[19]
Yang, Y. -Q.; Qiu, X. -D.; Zhao, Y.; Mu, Y. -C.; Shi, Z. -Z. J. Am. Chem. Soc. 2016, 138, 495. doi: 10.1021/jacs.5b11569
-
[20]
Yang, G.; Lindovska, P.; Zhu, D.; Kim, J.; Wang, P.; Tang, R. -Y.; Movassaghi, M.; Yu, J. -Q. J. Am. Chem. Soc. 2014, 136, 10807.
-
[21]
Feng, Y.; Holte, D.; Zoller, J.; Umemiya, S.; Simke, l. R.; Baran, P. S. J. Am. Chem. Soc. 2015, 137, 10160. doi: 10.1021/jacs.5b07154
-
[22]
Yang, Y. -Q.; Li, R. -R.; Zhao, Y.; Zhao, D. -B.; Shi, Z. -Z. J. Am. Chem. Soc. 2016, 138, 8734. doi: 10.1021/jacs.6b05777
-
[23]
(a) Phipps, R. J. ; Gaunt, M. J. Science 2009, 323, 1593.
(b) Chen, B. ; Hou, X. -L. ; Li, Y. -X. ; Wu, Y. -D. J. Am. Chem. Soc. 2011, 133, 7668. -
[24]
Yang, Y. -Q; Gao, P.; Zhao, Y.; Shi, Z. -Z. Angew. Chem., Int. Ed. 2017, 56, 3966. doi: 10.1002/anie.201612599
-
[25]
Xu, L. -T.; Zhang, C.; He, Y. -P.; Tan, L. -S.; Ma, D. -W. Angew. Chem., Int. Ed. 2016, 55, 321. doi: 10.1002/anie.201508117
-
[26]
Kim, Y.; Park, J.; Chang, S. Org. Lett. 2016, 18, 1892. doi: 10.1021/acs.orglett.6b00662
-
[27]
Xu, L. -T.; Tan, L. -S; Ma, D. -W. J. Org. Chem. 2016, 81, 10476. doi: 10.1021/acs.joc.6b01856
-
[28]
Song, Z.; Antonchick, A. P. Org. Biomol. Chem. 2016, 14, 4804. doi: 10.1039/C6OB00926C
-
[29]
Wang, L.; Li, Z.; Qu, X.; Peng, W. -M.; Hu, S. -Q, Wang, H. -B. Tetrahedron Lett. 2015, 56, 6214. doi: 10.1016/j.tetlet.2015.09.091
-
[30]
Liu, Q.; Li, Q.; Ma, Y.; Jia, Y. -X. Org. Lett. 2013, 15, 4528. doi: 10.1021/ol4020877
-
[31]
Borah, A. J.; Shi, Z. -Z. Chem. Commun. 2017, 53, 3945;
-
[32]
Lanke, V.; Prabhu, K. R. Org. Lett. 2013, 15, 6262. doi: 10.1021/ol4031149
-
[33]
Lv, J. -B; Wang, B; Yuan, K; Wang, Y; Jia, Y. -X. Org. Lett. 2017, 19, 3664. doi: 10.1021/acs.orglett.7b01681
-
[34]
Feng, Y.; Holte, D.; Zoller, J.; Umemiya, S.; Simke, L. R.; Baran, P. S. J. Am. Chem. Soc. 2015, 137, 10160. doi: 10.1021/jacs.5b07154
-
[35]
Leitch, J. A.; McMullin, C. L.; Mahon, M. F.; Bhonoah, Y.; Frost, C. G. ACS Catal. 2017, 7, 2616. doi: 10.1021/acscatal.7b00038
-
[36]
Zhang, Z. -Q.; Hu, Z. -Z.; Yu, Z. -X.; Lei, P.; Chi, H. -J; Wang, Y.; He, R. Tetrahedron Lett. 2007, 48, 2415. doi: 10.1016/j.tetlet.2007.01.173
-
[37]
Bellina, F.; Benelli, F.; Rossi, R. J. Org. Chem. 2008, 73, 5529. doi: 10.1021/jo8007572
-
[38]
Ackermann, L.; Barfeusser, S. Synlett 2009, 808.
-
[39]
Perato, S.; Large, B.; Lu, Q.; Gaucher, A.; Prim, D. ChemCatChem 2017, 9, 389. doi: 10.1002/cctc.v9.3
-
[40]
Cusati, G.; Djakovitch, L. Tetrahedron Lett. 2008, 49, 2499. doi: 10.1016/j.tetlet.2008.02.130
-
[41]
Cornella, J.; Lu, P.; Larrosa, I. Org. Lett. 2009, 11, 5506. doi: 10.1021/ol902304n
-
[42]
Chen, Y. -X.; Guo, S. -B.; Li, K. -L.; Qu, J. -P.; Yuan, H.; Hua, Q. -R.; Chen, B. -H. Adv. Synth. Catal. 2013, 355, 711. doi: 10.1002/adsc.201200997
-
[43]
Chen, S. -P.; Liao, Y. -F.; Zhao, F.; Qi, H. -R.; Liu, S. -W.; Deng, G. -J. Org. Lett. 2014, 16, 1618. doi: 10.1021/ol500231c
-
[44]
(a) Stuart, D. R. ; Villemure, E. ; Fagnou, K. J. Am. Chem. Soc. 2007, 129, 12072.
(b) Stuart, D. R. ; Fagnou, K. Science 2007, 316, 1172. -
[45]
Li, Y.; Wang, W. -H.; Yang, S. -D.; Li, B. -J.; Feng, C.; Shi, Z. -J. Chem. Commun. 2010, 46, 4553. doi: 10.1039/c0cc00486c
-
[46]
Gong, X.; Song, G. -Y.; Zhang, H.; Ling, X. -W. Org. Lett. 2011, 13, 1766. doi: 10.1021/ol200306y
-
[47]
Yamaguchi, A. D.; Mandal, D.; Yamaguchi, J.; Itami, K. Chem. Lett. 2011, 40, 555. doi: 10.1246/cl.2011.555
-
[48]
Wang, Z.; Li, K. -Z.; Zhao, D. -B.; Lan, J. -B.; You, J. -S.; Angew. Chem., Int. Ed. 2011, 50, 5365. doi: 10.1002/anie.201101416
-
[49]
Varaksin, M. V.; Utepova, I. A.; Chupakhin, O. N.; Charushin, V. N. J. Org. Chem. 2012, 77, 9087. doi: 10.1021/jo301618b
-
[50]
Sitnikov, N. S.; Kokisheva, A. S.; Fukin, G. K.; Neudcrfl, J. M.; Sutorius, H.; Prokop, A.; Fokin, V. V.; Schmalz, H. G.; Fedorov, A. Y. Eur. J. Org. Chem. 2014, 6481.
-
[51]
Baral, E. R.; Lee, Y. R.; Kim, S. H. Adv. Synth. Catal. 2015, 357, 2883. doi: 10.1002/adsc.v357.13
-
[52]
Cresswell, A. J.; Lloyd-Jones, G. C. Chem. -Eur. J. 2016, 22, 12641. doi: 10.1002/chem.v22.36
-
[53]
Phipps, R. J.; Grimster, N. P.; Gaunt, M. J. J. Am. Chem. Soc. 2008, 130, 8172. doi: 10.1021/ja801767s
-
[54]
Modha, S. G.; Greaney, M. F. J. Am. Chem. Soc. 2015, 137, 1416. doi: 10.1021/ja5124754
-
[55]
Pitts, A. K.; O'Hara, F.; Snell, R. H.; Gaunt, M. J. Angew. Chem., Int. Ed. 2015, 54, 5451. doi: 10.1002/anie.201500067
-
[56]
Zhang, Y. -P.; Feng, X. -L.; Yang, Y. -S.; Cao, B. -X. Tetrahedron Lett. 2016, 57, 2298. doi: 10.1016/j.tetlet.2016.04.051
-
[57]
Chen, J.; Wu, J. Angew. Chem., Int. Ed. 2017, 56, 3951. doi: 10.1002/anie.201612311
-
[58]
Toure, B. B.; Lane, B. S.; Sames, D. Org. Lett. 2006, 8, 1979. doi: 10.1021/ol053021c
-
[59]
Bellina, F.; Calandri, C.; Cauteruccio, S.; Rossi, R. Tetrahedron 2007, 63, 1970. doi: 10.1016/j.tet.2006.12.068
-
[60]
Lebrasseur, N.; Larrosa, I. J. Am. Chem. Soc. 2008, 130, 2926. doi: 10.1021/ja710731a
-
[61]
Xu, Z. -M.; Xu, Y. -L.; Lu, H. -F.; Yang, T.; Lin, X. -C.; Shao, L. -M.; Ren, F. Tetrahedron 2015, 71, 2616. doi: 10.1016/j.tet.2015.03.051
-
[62]
Gao, Y. -D.; Zhu, W. -Y.; Yin, L.; Dong, B.; Fu, J. -J.; Ye, Z. -W.; Xue, F. -T.; Jiang, C. Tetrahedron 2017, 58, 2213. doi: 10.1016/j.tetlet.2017.04.066
-
[63]
Preciado, S.; Mendive-Tapia, L.; Albericio, F.; Lavilla, R. J. Org. Chem. 2013, 78, 8129. doi: 10.1021/jo400961x
-
[64]
Paquet, J. -L.; Barth, M.; Pruneau, D.; Dodey, P. WO 2001038305, 2001.
-
[65]
Deprez, N. R.; Kalyani, D.; Krause, A.; Sanford, M. S. J. Am. Chem. Soc. 2006, 128, 4972. doi: 10.1021/ja060809x
-
[66]
Malmgren, J.; Nagendiran, A.; Tai, C. -W.; Backvall, J. E.; Olofsson, B. Chem. Eur. J. 2014, 20, 13531. doi: 10.1002/chem.v20.42
-
[67]
Duan, L. -L.; Fu, R.; Zhang, B. -S.; Shi, W.; Chen, S. -J.; Wan, Y. ACS Catal. 2016, 6, 1062. doi: 10.1021/acscatal.5b02147
-
[68]
Yang, S. -D.; Sun, C. -L.; Fang, Z.; Li, B. -J.; Li, Y. -Z.; Shi, Z. -J. Angew. Chem. 2008, 120, 1495. doi: 10.1002/(ISSN)1521-3757
-
[69]
Huang, Y. -B.; Ma, T.; Huang, P.; Wu, D. -S.; Lin, Z. -J.; Cao, R. ChemCatChem 2013, 5, 1877. doi: 10.1002/cctc.v5.7
-
[70]
Zhao, J. -L.; Zhang, Y. -H.; Cheng, K. J. Org. Chem. 2008, 73, 7428. doi: 10.1021/jo801371w
-
[71]
Liang, Z. -J.; Yao, B. -B.; Zhang, Y. -H. Org. Lett. 2010, 12, 3186.
-
[72]
Hfaiedh, A.; Yuan, K.; Ammar, H. B.; Hassine, B. B.; Soule, J. F.; Doucet, H. ChemSusChem 2015, 8, 1794. doi: 10.1002/cssc.201403429
-
[73]
Liu, C. -R.; Ding, L. -H.; Guo, G.; Liu, W. -W.; Yang, F. -L. Org. Biomol. Chem. 2016, 14, 2824. doi: 10.1039/C5OB02569A
-
[74]
Laha, J. K.; Bhimpuria, R. A.; Prajapati, D. V.; Dayal, N.; Sharma, S. Chem. Commun. 2016, 52, 4329. doi: 10.1039/C6CC00133E
-
[75]
Wang, X.; Lane, B. S.; Sames, D. J. Am. Chem. Soc. 2005, 127, 4996. doi: 10.1021/ja050279p
-
[76]
Zhang, L. -J.; Xue, X.; Xu, C. -H.; Pan, Y. -X.; Zhang, G.; Xu, L. -J.; Li, H. -R.; Shi, Z. -J. ChemCatChem 2014, 6, 3069. doi: 10.1002/cctc.201402534
-
[77]
(a) Lu, M. -Z. ; Lu, P. ; Xu, Y. -H. ; Loh, T. -P. Org. Lett. 2014, 16, 2614.
(b) Wang, L. ; Qu, X. ; Li, Z. ; Peng, W. -M. Tetrahedron Lett. 2015, 56, 3754. -
[78]
Zheng, J.; Zhang, Y.; Cui, S. -L. Org. Lett. 2014, 16, 3560. doi: 10.1021/ol5014312
-
[79]
Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2012, 51, 6993. doi: 10.1002/anie.201201491
-
[80]
Sollert, C.; Devaraj, K.; Orthaber, A.; Gates, P. J.; Pilarski, L. T. Chem. Eur. J. 2015, 21, 5258. doi: 10.1002/chem.201590053
-
[81]
Tiwari, V. K.; Kamal, N.; Kapur, M. Org. Lett. 2015, 17, 1766. doi: 10.1021/acs.orglett.5b00535
-
[82]
Jagtap, R. A.; Soni, V.; Punji, B. ChemSusChem 2017, 10, 2242. doi: 10.1002/cssc.201700321
-
[83]
Zhu, X.; Su, J. -H.; Du, C.; Wang, Z. -L.; Ren, C. -J.; Niu, J. -L.; Song, M. -P. Org. Lett. 2017, 19, 596. doi: 10.1021/acs.orglett.6b03746
-
[1]
-

图 1 部分含有吲哚结构的药物或活性分子
Figure 1 Examples of drug and reactive molecules containing indole structure

图式 4 吲哚C4和C5 C—H芳基化方法合成生物活性分子
Scheme 4 Bio-active molecules synthesis via C4 and C5 C—H arylation

图式 6 Allocolchicinoids化合物合成
Scheme 6 Synthesis of allocolchicinoids via indole C3-arylation
-


 下载:
下载:














 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 225
- 文章访问数: 8419
- HTML全文浏览量: 2717
 下载:
下载:
