 图式 1
Pd(OAc)2催化二芳基酮的邻位羟基化反应机理推测
Scheme1.
Proposed mechanism of Pd(OAc)2 catalyzed ortho-hydroxylation of diaryl ketones
图式 1
Pd(OAc)2催化二芳基酮的邻位羟基化反应机理推测
Scheme1.
Proposed mechanism of Pd(OAc)2 catalyzed ortho-hydroxylation of diaryl ketones
引用本文:
丁怀伟, 李娟, 郭庆辉, 肖琰. 过渡金属催化芳香烃C-H活化/C-O键偶联反应的研究进展[J]. 有机化学,
2017, 37(12): 3112-3129.
doi:
10.6023/cjoc201706006

Citation: Ding Huaiwei, Li Juan, Guo Qinghui, Xiao Yan. Research Progress on Transition-Metal-Catalyzed C-H Activation/C-O Coupling Reaction of Arenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3112-3129. doi: 10.6023/cjoc201706006
Citation: Ding Huaiwei, Li Juan, Guo Qinghui, Xiao Yan. Research Progress on Transition-Metal-Catalyzed C-H Activation/C-O Coupling Reaction of Arenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3112-3129. doi: 10.6023/cjoc201706006
过渡金属催化芳香烃C-H活化/C-O键偶联反应的研究进展
English
Research Progress on Transition-Metal-Catalyzed C-H Activation/C-O Coupling Reaction of Arenes
Abstract:
Transition metal catalyzed C-H activation/C-O coupling reaction of arenes serves as a powerful strategy for constructing phenols, aryl ethers and phenolic esters, and thus represents a hot topic in the field of organic chemistry. This strategy is effective and atom-economic as it allows people to construct C-O bond directly from arenes, avoiding pre-functionalization of arenes. Recent development on transition metal catalyzed C-H activation/C-O coupling reactions according to different species of transition metal catalysts is summarized. Possible mechanisms of typical protocols are explained and compared. Existing problems and limitations, and a brief outlook in this field is also presented.
-
Key words:
- C-H activation
- / C-O coupling
- / phenol
- / aryl ether
- / phenolic ester
-
C(sp2)—O键的构建在有机合成中有着不可或缺的作用, 是合成酚、醚和酯类化合物的重要手段.含C—O键的有机物不仅广泛地分布在天然活性产物中, 而且在药物设计中也占有重要地位[1]. Ullmann型偶联反应利用过渡金属为催化剂, 可以快速地构建C—C和C—X (X=O, N, S等)键[2]. Ullmann型C—O偶联反应采用预先卤代的芳香烃作为反应底物, 故这类反应会产生卤化氢副产物, 一般在碱性环境下进行, 对于复杂化合物的合成存在困难.随着Ullmann型C—O偶联反应催化体系发展的日趋成熟, 建立更加便捷和原子经济的金属催化体系来构建C—O键已逐渐成为有机化学领域的重点研究方向.近年来, C—H键活化反应为C—O偶联反应提供了新的思路, 并且已经有了很大的发展. C—H活化反应是指对C—H键直接官能团化的一类反应, 目前已成为构建C—C键以及C—X (X=Hal, O, N, S等)键的一种重要方法, 在有机合成和天然产物全合成中已有广泛的应用[3].除C—H活化反应外, 近年来发展的一些C—F键活化反应也能应用于构建C—O键[4].
和传统C—O偶联反应相比, C—H活化/C—O键偶联反应具有合成步骤更短、原子经济性更高等显著优点, 为酚类、芳基醚类和酚酯类化合物提供了丰富的合成方法. 2010年, Alonso和Pastor等[5]针对过渡金属催化的羟基化反应, 综述了以芳香烃和芳香卤代物为底物合成酚类化合物的方法.同年, Sanford等[6]综述了金属钯催化的多种C—H官能团化反应, 涉及到C(sp2)—H和C(sp3)—H的C—H活化/C—O偶联反应. 2011年, Ethaler和Company[7]综述了金属钯催化的芳香卤代物和芳香烃等底物的羟基化反应和烷氧基化反应. 2015年, Terent’ev等[8]综述了C(sp2)—H键和C(sp3)—H键的C—H活化/C—O偶联反应在合成醚类和酯类化合物中的应用.同年, Shi等[9]对过渡金属催化的C—H醚化反应进行了综述, 包括金属钯盐和金属铜盐催化的C(sp2)—H键和C(sp3)—H键的C—H醚化反应, 详细评述了构建C—O键合成醚类化合物的新方法.
C—H活化/C—O偶联反应发展迅速, 近年来出现了许多新方法, 取得了非常重要的进展, 例如新导向基团的出现实现间位和对位的C—H活化/C—O偶联反应等.但是, 目前还没有针对芳香烃C(sp2)—H活化反应/ C—O偶联反应合成酚类、醚类和酯类化合物的全面综述, 因此本文拟对这一领域的最新进展进行综述.根据文献调研, C—H活化/C—O偶联反应采用的过渡金属催化剂种类主要包括钯、铜、钌和钴等.其中钯和铜催化剂的应用较多, 而对于其它过渡金属的研究相对较少.不同催化体系具有不同的反应特点和催化机理, 因此本文将按照过渡金属催化剂的种类以及反应类型对过渡金属催化的芳香烃C—H活化/C—O键偶联反应分别进行综述.
1 钯催化的C—H活化/C—O偶联反应
1.1 C—H羟基化反应
C—H活化/C—O偶联反应最早可见于1990年, Fujiwara等 [10]报道了以Pd(OAc)2为催化剂, 在1.5 MPa氧气和一氧化碳的条件下, 以乙酸为溶剂进行苯的C—H键氧化反应(Eq. 1).该方法制备苯酚产率仅有2.3%, 同时反应条件比较苛刻, 需要在高压和高温(180 ℃)的条件下进行.近10年来, C—H活化/C—O偶联反应快速发展, 出现了很多反应高效、条件温和的合成方法.
2008年, Kim等 [11]报道了Pd(OAc)2催化2, 3-二苯基吡啶的邻位羟基化反应.该方法使用Oxone(过硫酸氢钾复合盐)作为氧化剂, 在PEG-3400/t-BuOH混合溶剂中进行吡啶环2-位芳基C—H羟基化反应(Eq. 2).该方法使用了多种双苯基吡啶类化合物作为反应底物进行酚类化合物的合成, 能够得到28%~76%收率的羟基化产物.
2009年, Yu等 [12]报道了以羧基为导向基团, Pd(OAc)2催化的苯甲酸类化合物的邻位C—H羟基化反应.该方法使用氧气作为氧化剂, 在二甲基乙酰胺(DMA)溶剂中合成邻羟基苯甲酸类化合物(Eq. 3).反应机理研究表明氧化剂氧气可能参与了羟基化产物的形成.向反应体系中加入苯醌能够明显地提升催化活性, 缩短反应时间.底物扩展实验表明, 芳烃环上无论连有供电子基团(如CH3, OCH3等)还是吸电子基团(如CF3, NO2, CN等), 都能顺利地转化为对应的酚类化合物.其中, 富电子芳香烃底物的收率更高, 分布在60%~80%之间.值得注意的是, 羟基化产物的收率随着氧气压力的增高可明显提高:当氧气提升至506 kPa时, 产率可以达到85%~95%.
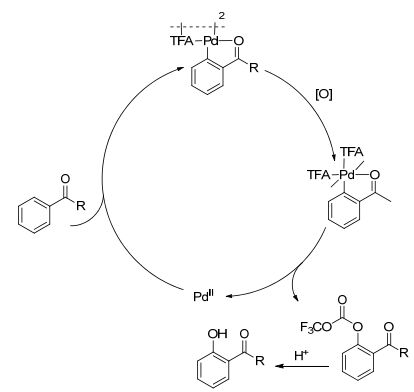
邻酰基苯酚类化合物是构成多种天然产物和药物分子的一种重要的结构单元[13]. 2012年, Rao课题组[14]报道了一种以羰基为导向基团的芳香烃类化合物的C—H羟基化反应.该课题组以二芳基酮类化合物为反应底物, Pd(OAc)2作为催化剂, K2S2O8为氧化剂, 在TFA/ TFAA(三氟乙酸/三氟乙酸酐)混合溶剂系统下制备邻羟基芳香酮类化合物(Eq. 4). TFA/TFAA混合溶剂系统在羟基化反应过程中扮演着双重的角色:一方面, 用于C—H键活化; 另一方面, 在反应过程中提供氧原子即作为羟基中氧原子的供源.同时, 该课题组发现TFA/ TFAA混合溶剂的比例能够影响反应速度:当比例接近9:1时最利于羟基化反应的进行.该课题组对反应机理进行了推测(Scheme 1): (1)钯催化剂与反应底物中羰基的氧原子进行螯合, 随后, 底物发生C—H键活化并形成PdⅡ二聚体复合物; (2) PdⅡ被氧化剂过硫酸钾氧化成PdⅣ; (3) PdⅣ复合物经还原消除后形成C—O键得到三氟乙酸酯, 同时PdⅣ被还原为PdⅡ; (4)三氟乙酸酯经水解得到羟基化产物.
图式 1
Pd(OAc)2催化二芳基酮的邻位羟基化反应机理推测
Scheme1.
Proposed mechanism of Pd(OAc)2 catalyzed ortho-hydroxylation of diaryl ketones
2012年, Dong课题组[15]以芳香酮或苯甲酸甲酯作为反应底物, 开发了2种反应体系用于C—H羟基化反应制备邻酰基苯酚化合物: (1)在DCE(二氯乙烷)溶剂中, Pd(TFA)2为催化剂, PhI(TFA)2为氧化剂(Eq. 5); (2)在三氟乙酸溶剂中, Pd(OAc)2作催化剂, K2S2O8作氧化剂(Eq. 6).在优化反应条件时发现, 当反应体系以PhI(OAc)2作氧化剂时, 在羰基的邻位有乙酰氧基取代物生成.该方法具有高位点选择性、较高的收率及催化剂的用量较少等优点.
同样以羰基作为导向基团, Kwong课题组[16]在2013年报道了一种在Pd(OAc)2催化下, 以PhI(OTFA)2为氧化剂的反应体系, 进行C—H羟基化反应制备各种取代的邻酰基苯酚的方法(Eqs. 7, 8).该方法中, 取代基不论是供电子基团还是吸电子基团的反应底物在反应体系中都能够得到很好的收率, 对应酚类化合物的产率分布在50%~88%.该反应体系具有反应条件温和、反应效率高等优点, 部分反应仅需2 h即可完成.
Yang课题组[17]于2013年报道了一种采用膦酰基R2(O)P作导向基团, 在Pd(TFA)2催化下, 以PhI(OTFA)2为氧化剂, CH3NO2为溶剂的反应体系中进行C—H羟基化进而制备酚类化合物的方法(Eq. 9).该反应体系条件温和, 在60 ℃发生反应. R2(O)P基团不仅可以作导向基团参与反应, 同时能够在羟基化产物中扮演着螯合基团的作用, 从而形成一类新型的P, O-双齿配体.
Jiao课题组[18]于2013年报道了以2-苯基吡啶类化合物为反应底物, 在氧气作为氧化剂条件下, PdCl2和N-羟基邻苯二甲酰亚胺(NHPI)共同催化2-苯基吡啶的C—H羟基化反应(Eq. 10).该方法利用自由基反应历程由钯盐和有机催化剂共同催化C(sp2)—H键官能团化.该反应在中性反应条件下进行, 同时氧气既作反应物又作为氧化剂.各种不同取代基的2-苯基吡啶类化合物都能够得到较高产率的对应的羟基化产物.该课题组对反应机理进行了推测(Scheme 2): (1) 2-苯基吡啶类化合物与催化剂PdCl2进行螯合后, 经C—H活化得到PdⅡ复合物; (2)邻苯二甲酰亚胺均裂形成PINO自由基(邻苯二甲酰亚胺-N-氧自由基)并触发苄基自由基的形成, 而苄基自由基在氧气的条件下形成过氧化自由基, 随后形成羟基自由基和苯甲醛(苯甲醛在氧气条件下被氧化成苯甲酸, 二者在GC/MS和TLC下都能被检测到); (3)羟基自由基与二价钯中间体结合形成PdⅢ复合物; (4) PdⅢ复合物发生还原消除反应形成C—O键, 得到羟基化产物.
 图式 2
PdCl2和NHPI共同催化2-苯基吡啶类化合物的羟基化反应机理推测
Scheme2.
Proposed mechanism of PdCl2 and NHPI co-catalyzed hydroxylation of 2-arylpyridines
图式 2
PdCl2和NHPI共同催化2-苯基吡啶类化合物的羟基化反应机理推测
Scheme2.
Proposed mechanism of PdCl2 and NHPI co-catalyzed hydroxylation of 2-arylpyridines
Patel课题组[19]于2013年报道了一种以2-苯基苯并噻唑类化合物作为底物, Pd(OAc)2作催化剂, 二羧酸碘苯(DIB)作氧化剂发生苯并噻唑类化合物的邻位羟基化的方法(Eq. 11).该反应以苯并噻唑为导向基团, 在醋酸中于110 ℃进行.该方法对底物的不同取代基都能够很好地兼容, 并得到较高收率的酚类产物.研究发现, 使用相同的底物和催化剂, 采用Oxone作为反应的氧化剂, 在TFA溶剂中也能够发生邻位羟基化.该课题组推测反应经历PdⅡ/PdⅣ的催化循环历程(Scheme 3): (1)二价钯催化剂与2-苯基苯并噻唑类底物结合得到钯复合物中间体; (2) PhI(OAc)2、AcOH与DIB进行氧化加成得到PdⅣ复合物中间体; (3)经历还原消除反应促使形成C—O键, 并生成邻位乙酰氧基化产物; PdⅡ催化剂释放从而进入下一个催化循环; (4)乙酰氧基化产物经历水解形成羟基, 得到邻位羟基化产物.
 图式 3
Pd(OAc)2催化2-苯基苯并噻唑类化合物的邻位羟基化反应机理推测
Scheme3.
Proposed mechanism of Pd(OAc)2-catalyzed ortho-hydroxylation of 2-arylbenzothiazole
图式 3
Pd(OAc)2催化2-苯基苯并噻唑类化合物的邻位羟基化反应机理推测
Scheme3.
Proposed mechanism of Pd(OAc)2-catalyzed ortho-hydroxylation of 2-arylbenzothiazole
苯并咪唑类化合物具有广泛的生物活性, 包括抗真菌、抗蠕虫、抗炎以及抗肿瘤等[20]. Nagesh课题组[21]于2014年通过对反应条件的一系列筛选, 发现以Pd(OAc)2作催化剂、Oxone为氧化剂, 在碳酸铯存在的条件下, 于N, N-二甲基甲酰胺(DMF)溶剂中, 2-苯基苯并咪唑类化合物能进行C—H活化发生邻位羟基化反应(Eq. 12).该反应体系中不仅能够得到较好收率的羟基化产物, 而且当以脂肪醇为溶剂时, 2-苯基苯并咪唑还能进行邻位烷氧基化反应生成芳基烷基醚类化合物(Eq. 13).
Dong课题组[22]于2015年提出了一种采用过氧叔丁醇(TBHP)作氧化剂, Pd(OAc)2催化的2-苯基吡啶类化合物邻位羟基化反应的方法(Eq. 14).该反应体系对吸电子基团和供电子基团都具有很好的耐受性, 能够获得较高产率的酚类化合物, 其产率分布在55%~88%之间.氮气氛围对目标化合物的合成并没有影响, 证明了过氧化叔丁醇氧化剂是羟基化产物中的氧原子唯一的来源.该课题组对此方法进行了反应机理的推测(Scheme 4): (1) 2-苯基吡啶与Pd(OAc)2在导向基团的邻位发生C—H键活化形成PdⅡ中间体; (2)过氧化叔丁醇均裂形成羟基自由基(HO•)和叔丁氧基自由基(t-BuO•); (3)从乙酸分子中离去一个氢自由基(H•)从而释放出乙酰氧基自由基(AcO•). PdⅡ中间体通过羟基自由基(HO•)和乙酰氧基自由基(AcO•)的氧化加成形成PdⅣ中间体; (4) PdⅣ中间体经过还原消除得到羟基化产物和再生的二价钯催化剂.
 图式 4
Pd(OAc)2催化2-苯基吡啶类化合物羟基化反应的机理推测
Scheme4.
Plausible mechanism of Pd(OAc)2-catalyzed hydroxylation of 2-arylpyridines
图式 4
Pd(OAc)2催化2-苯基吡啶类化合物羟基化反应的机理推测
Scheme4.
Plausible mechanism of Pd(OAc)2-catalyzed hydroxylation of 2-arylpyridines
2015年, Chakraborti课题组[23]报道了一种以Pd(OAc)2作催化剂、Na2S2O8为氧化剂、在1, 4-二氧六环溶剂中进行2-芳基苯并噁唑以及2-芳基苯并噻唑类化合物的C—H羟基化反应(Eq. 15).该反应利用了1, 4-二氧六环发生热分解能够形成羟基自由基的性质(Scheme 5) [24].对该方法涉及的反应机理推测如下(Scheme 6): (1) Pd(OAc)2与导向基团的不饱和氮原子结合形成络合物A; (2)络合物A发生C(sp2)—H键的活化形成络合物B, 接着络合物B发生去质子化脱去一分子HOAc形成环钯复合物C; (3) 1, 4-二氧六环与过硫酸根阴离子作用生成的羟基自由基和复合物C发生氧化加成得到PdⅣ复合物D或PdⅢ-PdⅢ复合物E; (4) D或E发生还原消除构建C—O键得到酚, 同时通过配体交换实现催化剂的循环利用.有意思的是, 利用该类导向基团能和金属催化剂形成金属配合物的特点, Wang和Ding课题组 [25]开发出一类新型的苯并噁唑铱(Ⅲ)配合物, 能够用于催化C—C和C—N键的形成, 实现苯胺和苯乙酮类化合物的烷基化反应.
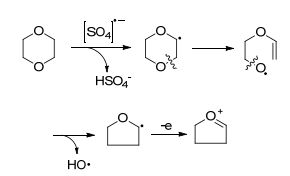 图式 5
二氧六环热分解为羟基自由基的过程
Scheme5.
Process of the generation of radical •OH from 1, 4-dioxane
图式 5
二氧六环热分解为羟基自由基的过程
Scheme5.
Process of the generation of radical •OH from 1, 4-dioxane
 图式 6
Pd(OAc)2催化2-芳基苯并噁(噻)唑羟基化反应的可能反应机理过程
Scheme6.
Plausible mechanism of Pd(OAc)2-catalyzed hydroxylation of benzothiazoles and benzoxazoles
图式 6
Pd(OAc)2催化2-芳基苯并噁(噻)唑羟基化反应的可能反应机理过程
Scheme6.
Plausible mechanism of Pd(OAc)2-catalyzed hydroxylation of benzothiazoles and benzoxazoles
2015年, Jiao课题组[26]报道了以肟甲醚类化合物作为底物, 钯催化的C—H羟基化反应(Eq. 16).反应以醋酸钯为催化剂, Oxone为氧化剂, 在DCE中进行.反应体系中PPh3或偶氮二羧酸二乙酯(DEAD)作为配体能够提高羟基化的效率.通常, 缺电子的芳香烃在C—H羟基化反应中活性较低, 而在该方法中, 肟甲醚类化合物进行C—H键活化/C—O偶联成酚反应时, 缺电子的底物收率却高达90%以上.另外, 该反应体系在中性环境中进行, 适用于酸敏感底物的羟基化反应.
2015年, Itoh课题组[27]报道以2-苯基吡啶类化合物为底物, PdCl2为催化剂, 35% H2O2为氧化剂, 在4-甲基-2-戊酮溶剂中于100 ℃进行羟基化反应(Eq. 17).底物扩展实验表明, 苯环上有对位取代基的底物更有利于羟基化反应的发生.
Guin小组[28]于2016年采用2-苯基吡啶类化合物为底物, 以Pd(CH3CN)2Cl2为催化剂, O2为氧化剂, 在正丁醛存在的条件下发生C—H羟基化反应(Eq. 18).该方法对底物取代基具有很好的耐受性:不论取代位置在苯环或吡啶环, 电子效应为吸电子基团或供电子基团都能取得较高的收率.在氩气氛围下, 反应没有得到目标羟基化产物, 因此氧气在反应中起着关键的作用.该课题组对反应机理进行了阐述(Scheme 7): (1) 2-苯基吡啶C—H键活化与钯催化剂通过邻位的螯合作用形成PdⅡ复合物; (2)正丁醛在氧气条件下被氧化成正丁酸并形成活化的过氧酰基自由基中间体; (3)自由基中间体与PdⅡ复合物发生氧化加成得到PdⅣ复合物; (4)经过还原消除得到羟基化产物以及再生的二价钯催化剂.
 图式 7
Pd(CH3CN)2Cl2催化的2-苯基吡啶类化合物的羟基化反应机理预测
Scheme7.
Proposed mechanism of Pd(CH3CN)2Cl2 catalyzed hydroxylation of 2-aryl pyridines
图式 7
Pd(CH3CN)2Cl2催化的2-苯基吡啶类化合物的羟基化反应机理预测
Scheme7.
Proposed mechanism of Pd(CH3CN)2Cl2 catalyzed hydroxylation of 2-aryl pyridines
Fan课题组[29]于2016年报道采用Pd(OAc)2作催化剂、TBHP作氧化剂以及碳酸铯存在的条件下, 由2-苯基苯酚类化合物制备二苯基双酚类化合物的方法(Eq. 19).芳香环上的取代基不论是吸电子基团(如F, Cl, Ph, CF3和COCH3等)还是供电子基团(如CH3, C2H5和OCH3等)均能发生该反应, 并能够得到较高产率的目标化合物, 其产率分布在58%~85%之间.
2015年, Yu课题组[30]首次报道了利用含氰基的导向基团进行间位的C—H活化/C—C偶联反应.这一方法很快被应用到C—H羟基化反应中. Sunoj课题组[31]于2016年首次报道了以氰基为导向基团, Pd(OAc)2催化芳烃类化合物的间位羟基化反应.在PhI(TFA)2为氧化剂, For-Gly-OH (N-甲酰基-甘氨酸)为配体的条件下, 在HFIP(六氟异丙醇)溶剂中, 取代芳烃在70 ℃进行芳烃间位羟基化反应生成对应的酚类化合物(Eq. 20).
同年, Bera和Matti课题组[32]报道了一种相似的催化体系实现导向基团间位的C—H羟基化反应.该反应体系利用磷酸酯键联接苯甲腈作为导向基团, 以Boc-Ala-OH (N-叔丁氧基羰基丙氨酸)为配体, 在80 ℃发生间位的C—H羟基化反应(Eq. 21).该课题组还发现, 利用Ac-Gly-OH (N-乙酰甘氨酸)作为配体时, 往反应体系中引入乙酸酐能发生了间位的C—H乙酰氧基化反应生成乙酸酯(Eq. 22).
1.2 C—H乙酰氧基化反应
2006年, Sanford课题组 [33]利用甲基肟醚作为导向基团, 以Pd(OAc)2作为催化剂进行芳烃C—H键的乙酰氧基化和甲氧基化反应, 分别合成芳基乙酸酯和芳基甲醚类化合物(Scheme 8).该课题组筛选了多种过氧化物氧化剂(oxone, K2S2O8, H2O2, m-CPBA, t-BuOOH), 最后选择oxone为最优的氧化剂.底物扩展实验表明, 卤素、氰基和三氟甲基等基团对反应体系均有较好的耐受性.
 图式 8
Pd(OAc)2催化的芳烃C—H乙酰氧基化和甲氧基化反应
Scheme8.
Pd(OAc)2 catalyzed C—H acetoxylation and methoxylation of arenes
图式 8
Pd(OAc)2催化的芳烃C—H乙酰氧基化和甲氧基化反应
Scheme8.
Pd(OAc)2 catalyzed C—H acetoxylation and methoxylation of arenes
2008年, Wang课题组[34]报道了一种以酰胺为导向基团, Pd(OAc)2为催化剂, K2S2O8为氧化剂, 于AcOH/ DCE (V:V=1:1)混合溶剂中在100 ℃实现芳胺类化合物的邻位C—H酰氧基化反应(Eq. 23).这种方法适用于卤素、甲氧基和乙酰基等多种取代基取代的底物, 收率最高可达93%.
2009年, Liang课题组 [35]利用双齿型的吡啶酰胺和喹啉-8-胺作为导向基团, 实现了芳香烃的选择性乙酰氧基化反应(Eqs. 24, 25).该反应使用Pd(OAc)2为催化剂, PhI(OAc)2为氧化剂, 在AcOH/Ac2O存在下, 以甲苯为溶剂, 在较高的温度下进行.结果表明, 喹啉-8-胺作为导向基团时收率更高.
2010年, Xu课题组 [36]报道了一种以嘧啶为导向基团的芳香烃选择性乙酰氧基化反应(Eq. 26).该反应以2-苯基嘧啶作为反应底物, Pd(OAc)2作催化剂, Cu(OTf)2为助催化剂, PhI(OAc)2作为氧化剂, 于AcOH/Ac2O混合溶剂中进行.该反应选择性较高, 主要生成单取代的芳基乙酸酯, 且反应条件对官能团的耐受性良好, 反应收率也较高.
2011年, Reddy课题组 [37]使用Pd(OAc)2为催化剂, PhI(OAc)2为氧化剂, 在AcOH/Ac2O混合物中, 实现了N-(2-苯甲酰基苯基)-苯甲酰胺的乙酰氧基化反应.使用甲醇作为溶剂时, 可以在相同条件下进行甲氧基化反应并且可以得到良好的收率(Scheme 9).
 图式 9
Pd(OAc)2催化的N-(2-苯甲酰基苯基)-苯甲酰胺的乙酰氧基化和甲氧基化反应
Scheme9.
Pd(OAc)2 catalyzed acetoxylation and methoxylation of N-(2-benzoylphenyl)benzamides
图式 9
Pd(OAc)2催化的N-(2-苯甲酰基苯基)-苯甲酰胺的乙酰氧基化和甲氧基化反应
Scheme9.
Pd(OAc)2 catalyzed acetoxylation and methoxylation of N-(2-benzoylphenyl)benzamides
2013年, Sanford课题组[38]报道了苯的乙酰氧基化反应, 该反应无需导向基团.反应以吡啶鎓为配体, Pd(OAc)2为催化剂, K2S2O8作为氧化剂, 在AcOH/Ac2O混合物中进行苯的乙酰化, 得到了71%的收率(Eq. 27).初步的研究证明, 吡啶鎓离子配体的关键作用是实现氧化剂K2S2O8的相转移.
Gevorgyan课题组[39]提出了利用(2-吡啶基)二异丙基甲硅烷基(PyDipSi)和(2-嘧啶基)二异丙基甲硅烷基(PyrDipSi)[40]作为可离去的导向基团进行乙酰氧基化反应. PyDipSi基团可用于芳烃的选择性乙酰氧基化或新戊酰氧基化反应.该导向基团的硼酸化可以制备取代的儿茶酚类化合物(Scheme 10).而PyrDipSi基团可实现双取代的乙酰氧基化反应, 继而可以用作间苯二酚的合成(Scheme 11).
 图式 10
(2-吡啶基)二异丙基甲硅烷基导向的乙酰氧基化反应和新戊酰氧基化反应
Scheme10.
(2-Pyridyl)diisopropylsilyl directed acetoxylation and pivaloxylation
图式 10
(2-吡啶基)二异丙基甲硅烷基导向的乙酰氧基化反应和新戊酰氧基化反应
Scheme10.
(2-Pyridyl)diisopropylsilyl directed acetoxylation and pivaloxylation
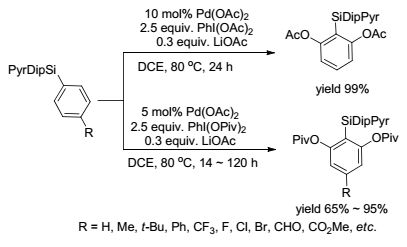 图式 11
(2-嘧啶基)二异丙基甲硅烷基导向的乙酰氧基化反应和新戊酰氧基化反应
Scheme11.
(2-Pyrimidyl)diisopropylsilyl directed acetoxylation and pivaloxylation
图式 11
(2-嘧啶基)二异丙基甲硅烷基导向的乙酰氧基化反应和新戊酰氧基化反应
Scheme11.
(2-Pyrimidyl)diisopropylsilyl directed acetoxylation and pivaloxylation
Yu课题组[41]在2014年采用酰胺键连接苯甲腈作为导向基团, 成功实现间位的C—H乙酰氧基化反应.反应体系采用Pd(OAc)2为催化剂, 利用Ac-Gly-OH为配体, PhI(OAc)2为氧化剂, 乙酸酐为酰基化试剂于HFIP溶剂中进行间位的C—H乙酰氧基化反应(Eq. 28).反应选择性高, 收率在32%~66%之间.
2016年, Li课题组[42]以N-对硝基苯磺酰基取代的酰胺键联接苯甲腈为导向基团, 采用和Yu课题组类似的反应条件, 实现了导向基团间位的C—H乙酰氧基化反应(Eq. 29).该导向基团可以在温和的条件下离去和再循环.某些底物能够生成双乙酰氧基化反应, 生成间苯二酚二乙酸酯类化合物.
Maiti课题组[43]于2015年报道了一种新颖的联苯甲硅烷基团, 首次实现芳环导向基团对位C—H键的有效官能团化.导向基团克服了电子效应和空间效应, 选择性地实现导向基团对位的C—H乙酰氧基化反应.该课题组利用联苯甲硅烷基为导向基团, Pd(OAc)2为催化剂, Piv-Ala-OH (N-特戊酰基-L-丙氨酸)为配体, PhI(OAc)2为氧化剂, 在HFIP中进行对位的C—H乙酰氧基化, 以较高的收率得到乙酸酯类化合物(Eq. 30).
1.3 C—H烷(芳)氧基化反应
2010年, Wang课题组[44]报道了Pd(OAc)2催化的苯甲酰胺类化合物的邻位烷氧基化反应.该反应体系利用N-甲氧基酰胺为导向基团, K2S2O8为氧化剂, 于脂肪醇和1, 4-环氧六环的混合溶剂中进行(Eq. 31).该反应在较低的温度下能够得到较好收率的芳基醚类产物.该反应条件对反应底物具有良好的耐受性, 具有给电子基团或吸电子基团的底物都可获得较高收率.另外, 除了伯醇可以得到相应的烷氧基化产物外, 仲醇(如异丙醇)也可以获得相应的产物, 但是具有较大位阻的叔醇(如叔丁醇)难以获得产物.
2014年, Fabis课题组[45]研究发现使用N-对甲苯磺酰酰胺甲酰基作为导向基团, Pd(OAc)2作为催化剂, 脂肪醇作为溶剂, 可以在温和的条件下进行芳香烃的烷氧基化反应, 反应收率分布在47%~95%.结构简单的脂肪醇, 如甲醇、乙醇和异丙醇等均能进行该类反应(Eq. 32).
2013年, Shi课题组[46]报道了一种以吡啶酰胺为导向基团, Pd(OAc)2为催化剂, PhI(OAc)2为氧化剂, 于脂肪醇中反应获得较高收率的邻位双烷氧基化产物的方法.该方法不仅适用于β-C—H键(n=0)烷氧基化, 也同样适用于γ-C—H (n=1)键烷氧基化(Eq. 33).底物拓展实验表明, 该方法不仅适用于给电子的取代基团(如Me, OMe), 也同样适用于吸电子的基团(如NO2, CF3, 卤素等), 但给电子基团取代的底物获得的产率明显高于吸电子基团取代的产物.另外, 该反应对各种类型的脂肪醇具有良好的耐受性.例如, 直链及支链的伯醇都能获得良好的收率; 以苄醇为底物也能够获得相应的苄基醚; 二级醇也能获得相应的产物, 但产率略低.
2013年, Sun课题组[47]报道了一种以偶氮基为导向基团, Pd(OAc)2催化的导向基团邻位的C—H烷氧基化, 可用于合成2-烷氧基二芳基偶氮类化合物.该反应利用PhI(OAc)2为氧化剂, 在80~100 ℃进行(Eq. 34).包括甲醇、乙醇、丙醇、异丁醇和异丙醇在内的多种脂肪醇均能发生该反应.该反应条件对底物官能团的耐受性良好, 一系列的具有给电子基和吸电子基的偶氮苯衍生物可直接被烷氧基化, 收率分布在35%~77%.该课题组推测反应经历PdⅡ/PdⅣ反应历程.
2014年, Sun课题组[48]报道了一种以Pd(OAc)2作为催化剂, PhI(OAc)2作氧化剂, 使用2-吡啶氧基作为导向基团的芳香烃C—H烷氧基化反应.该反应以脂肪醇为溶剂, 可得到较好的收率(Eq. 35).该反应在空气中进行, 条件温和, 对底物官能团的耐受性良好.甲醇、乙醇、丙醇和异丙醇均可以发生该类反应.
2014年, Sun课题组[49]报道了一种利用钯催化的N-亚硝基芳基胺的C—H烷氧基化反应.该反应使用Pd(CH3CN)2Cl2为催化剂, 以PhI(OAc)2作为氧化剂, 在脂肪醇中进行.反应条件温和, 在30 ℃即可反应, 反应收率最高可达89% (Eq. 36).导向基团N-亚硝基可容易地转化为其他官能团.
2014年, Kuang课题组[50]报道了一种2-苯基-1, 2, 3-三唑类化合物进行C—H烷氧基化反应的方法.该方法采用Pd(OAc)2作催化剂, 过硫酸钾作氧化剂, 在三氟乙酸存在的条件下, 于二氯乙烷溶剂中与脂肪醇发生C—H烷氧基化反应.不论供电子基团还是吸电子基团都能够表现出良好的兼容性, 而且具有较高的位点选择性(Eq. 37).机理研究表明反应经历PdⅡ/PdⅣ的循环过程.
2017年, Ji和Li课题组[51]以N-甲苯磺酰基苯甲酰胺和N-硝基苯磺酰基苯甲酰胺为底物, Pd(OAc)2为催化剂, PhI(OAc)2为氧化剂, 在TFA/TFE(三氟乙醇)混合溶剂中进行邻位的C—H三氟乙氧基化反应(Eq. 38).该反应条件温和, 于室温下就可得到相应的产物.对底物官能团的耐受性良好, 无论是吸电子基或给电子取代的底物都能获得中等至良好收率的产物.有意思的是, 反应产物能够在乙酰甘氨酸的作用下能够继续发生间位的C—H三氟乙氧基化反应生成二取代物.
2 铜催化的C—H活化/C—O偶联反应
2.1 C—H羟基化反应
2006年, Yu课题组[52]报道了2-苯基吡啶类化合物能发生C—H羟基化反应生成酚类化合物.该反应以Cu(OAc)2为催化剂, 氧气为氧化剂, 在乙腈溶剂中进行(Eq. 39).该反应底物苯环上的R基团可以为甲基、甲氧基、乙烯基和醛基, 其产率分布在43%~77%之间.反应机理研究表明该反应可能经历单电子转移反应历程.
 图式 12
2-苯基吡啶类化合物的羟基化反应
Scheme12.
Hydroxylation of 2-phenylpyridines
图式 12
2-苯基吡啶类化合物的羟基化反应
Scheme12.
Hydroxylation of 2-phenylpyridines
于2008年, Nagasawa课题组[53]报道了以苯甲酰苯胺类衍生物为反应底物, Cu(OTf)2作催化剂, 氧气为氧化剂, 以o-xylene为溶剂, 在140 ℃条件下合成2-苯基苯并噁唑类化合物的方法(Eq. 39).在该方法中R基不论是吸电子基团还是供电子基团都能够得到较高产率的目标化合物.
于2012年, Lei课题组[54]报道了一种氯化铜催化的以活性芳香烃为底物的C—H键羟基化反应(Eq. 40).该反应以氧气作为氧化剂, 在室温条件下进行.该课题组对反应机理进行了推测(Scheme 13): (1)催化剂CuCl2在叔丁醇钠的作用下转化为CuCl, 随后与叔丁醇钠反应形成活性的一价铜盐CuOBu-t; (2)底物ArH在叔丁醇钠的条件下发生了去质子化从而生成ArNa和叔丁醇; CuOBu-t与ArNa可能经历了金属转移反应生成ArCuⅠ复合物以及叔丁醇钠; (3) ArCuⅠ复合物被氧气氧化同时O—O键断裂形成含氧络合二铜复合物; (4)叔丁醇钠亲核进攻复合物, 这一过程中可能破坏了[Cu2(μ-O)2]2+络合中心, 因此生成CuⅢ中间体; (5)经历还原消除得到了ArONa, 经酸化得到羟基化产物.
 图式 13
氯化铜催化杂环芳香烃的羟基化反应机理推测
Scheme13.
Proposed reaction pathway for CuCl2 catalyzed hydroxylation of heterocycles
图式 13
氯化铜催化杂环芳香烃的羟基化反应机理推测
Scheme13.
Proposed reaction pathway for CuCl2 catalyzed hydroxylation of heterocycles
于2014年, Shi课题组[55]采用可移除的双配位基导向基团, 实现了苯甲酰胺类化合物的C—H羟基化反应.反应采用醋酸铜作催化剂, 在四丁基碘化铵(TBAI)存在下于DMF中进行(Eq. 41).该方法具有广泛的底物范围以及高收率等优点.其中R基团不论是供电子还是吸电子基团都能够得到不同收率的相应目标产物.同时该方法不仅可以应用于芳香烃底物的C—H羟基化反应, 也能将吡啶、噻吩和哒嗪等芳香杂环底物转化为对应的羟基化产物.该课题组对可能的反应机理进行了推测(Scheme 14): (1)在N, N-双配位基导向基团的作用下, 苯甲酰胺类化合物与二价铜盐催化剂络合形成CuⅡ-复合物; (2) C—H活化得到CuⅡ-芳基环状复合物; (3)通过歧化作用或者氧化作用得到N, N-螯合型CuⅢ-芳基中间体; (4)还原消除形成乙酰氧基化产物, 同时得到一价铜盐; (5)乙酰氧基化产物经由快速水解转化为羟基化产物; 一价铜盐被银盐氧化成二价铜盐进入下一个催化循环.
 图式 14
可移除双配位基促进铜催化芳香烃类化合物的羟基化反应机理推测
Scheme14.
Plausible reaction mechanism of copper-mediated hydroxylation of arenes directed by a removable bidentate group
图式 14
可移除双配位基促进铜催化芳香烃类化合物的羟基化反应机理推测
Scheme14.
Plausible reaction mechanism of copper-mediated hydroxylation of arenes directed by a removable bidentate group
2016年, Jana课题组[56]利用8-氨基喹啉类作为导向基团, 在水合醋酸铜的作用下, 以吡啶作配体, 在DMF与二甲基亚砜(DMSO)的混合溶剂中进行苯甲酰胺类化合物的邻位C—H羟基化反应(Eq. 42).有趣的是, 在同样的反应条件下, 如果缺少吡啶作配体, 有反应底物的二聚体生成.
2015年, Yu课题组[57]报道了以噁唑酰胺为导向基团, O2作为氧化剂, 在Cu(OAc)2和Na2CO3的作用下实现了邻位C—H羟基化反应(Eq. 43).研究表明往体系中加入一定量的水有助于反应的进行.该反应条件对底物的耐受性良好, 吸电子基和给电子基取代的底物都能获得较好的收率.邻位的卤素可直接被羟基化.对于间位取代的底物, 反应的区域选择性有利于生成空间位阻较低的产物.
2016年, Wang和Shi课题组 [58]报道了一种以二硫化物作为导向基团, CuI催化的C—H羟基化反应.该方法中, 采用硫酚类化合物和芳基硼酸类化合物作为反应的起始底物, 在碳酸铯与氧气的条件下, DMF溶剂中发生了C—H活化并迅速发生C—S偶联反应, 生成了2-苯硫基苯酚类化合物(Eq. 44).初步机理研究表明氧分子参与了羟基的形成.同时, 该方法可以用于喹啉类化合物的合成.
2.2 C—H烷氧基化反应
Cheng课题组[59]于2010年报道了以2-苯基吡啶类衍生物为反应底物, 在Cu(OAc)2作催化剂, O2作氧化剂的条件下, 与苯甲酸酐反应制备酚酯类化合物的方法(Eq. 45).该反应在甲苯中进行, 可以得到不同取代的苯甲酰化产物.通过分子动力学实验对反应机理进行了推测(Scheme 15): (1)苯甲酸酐与醋酸铜发生反应生成苯甲酸Cu(Ⅱ)盐; (2) Cu(Ⅱ)和2-苯基吡啶的苯环经C—H活化形成环状CuⅡ中间体A; (3)在Cu(Ⅱ)的存在下, A被氧化成CuⅢ中间体B; (4) B发生还原消除反应, 得到产物以及Cu(Ⅰ); (5) Cu(Ⅰ)盐被氧气氧化成Cu(Ⅱ), 开始下一催化循环.
 图式 15
铜催化的2-苯基吡啶类化合物酯化反应机理推测
Scheme15.
Plausible mechanism of copper-catalyzed ortho-acyloxylation of 2-aryl pyridines
图式 15
铜催化的2-苯基吡啶类化合物酯化反应机理推测
Scheme15.
Plausible mechanism of copper-catalyzed ortho-acyloxylation of 2-aryl pyridines
2013年, Gooen课题组[60]报道了2-苯基吡啶的C—H烷氧基化反应.以催化量的Cu(OAc)2作为催化剂, 银盐为氧化剂, 2-苯基吡啶类化合物与脂肪醇发生C—H活化/C—O偶联反应生成芳基烷基醚类化合物(Eq. 46). 2-苯基吡啶类底物在该催化体系下可以得到单或双取代的芳基烷基醚产物.芳基取代基不论是一系列的供电子基团还是吸电子基团, 反应都能够很好地进行.反应产率较高, 分布在41%~82%之间.同时, 吡唑、嘧啶、苯并喹啉等含氮杂环也能作为导向基团协助芳香烃的C—H活化反应.反应底物也可以是杂环类化合物如噻吩和苯并喹啉等.包括乙醇、正丁醇、叔丁醇和异丙醇等脂肪醇均能发生该反应.另外, 该反应体系不仅适用于C(sp2)—H键的烷氧基化反应, 同时2-苯基吡啶的类似物2-苄基吡啶作为反应底物在相同的反应条件下能发生苄位C(sp3)—H键的C—H烷氧化反应(Eq. 47).该课题组对反应机理进行了推测(Scheme 16): (1)在二价铜催化剂的存在下, 芳香烃底物经历C—H活化螯合形成CuⅡ中间体; (2)脂肪醇与三氟甲磺酸银反应转化成醇银盐, 将CuⅡ中间体氧化为CuⅢ中间体; (3)经还原消除得到烷氧化芳烃产物和一价铜盐; (4)一价铜盐在氧气的存在下被氧化为二价铜化合物.
 图式 16
芳香烃脱氢烷氧基化反应机理推测
Scheme16.
Proposed mechanism of dehydrogenative alkoxylation of arenes
图式 16
芳香烃脱氢烷氧基化反应机理推测
Scheme16.
Proposed mechanism of dehydrogenative alkoxylation of arenes
2013年, Stahl课题组[61]报道了N-(8-喹啉基)苯甲酰胺类化合物, 在二价铜盐Cu(OAc)2的作用下与甲醇发生C—H活化/C—O偶联反应(Eq. 48).在优化条件下, 能够得到中等产率的C—O偶联产物芳基甲醚类化合物, 其产率分布在54%~64%之间.
2013年, Daugulis课题组[62]报道了铜催化的苯甲酸衍生物的邻位烷氧基化和芳氧基化反应.该反应同样以喹啉-8-胺为导向基团, 采用经济的(CuOH)2CO3为催化剂, 空气为氧化剂, DMF为反应溶剂, K2CO3、四甲基胍或K3PO4为碱.多种酚类和脂肪醇均能进行该反应(Eq. 49).值得注意的是, 具有立体位阻的脂肪醇(如环丙基甲醇和三氟甲基甲醇等)亦能很好地进行反应.
2014年, Song课题组[63]报道了通过使用2-氨基-1-氧吡啶作为一种可离去的导向基团, 实现了苯甲酰胺的选择性芳氧基化反应.反应使用化学计量的Cu(OAc)2, 在Cs2CO3与4-二甲氨基吡啶(DMAP)的存在下进行.通过改变溶剂和试剂的投料量可以控制反应进行双取代反应或单取代反应.当邻二甲苯作为反应溶剂, Cs2CO3与DMAP的比例为2:1时, 可得到邻位单取代的产物(Scheme 17); 当反应以吡啶作为溶剂, Cs2CO3与DMAP的比例为1:1时, 可得到邻位双取代的产物(Eq. 50).
 图式 17
芳香烃的C—H芳氧基化反应
Scheme17.
C—H Aryloxylation of arenes
图式 17
芳香烃的C—H芳氧基化反应
Scheme17.
C—H Aryloxylation of arenes
同年, 该课题组[64]又报道了利用相同的取代基团, 以CuCl作为催化剂, 实现了取代芳烃的C—H烷氧基化反应(Eq. 42).该反应体系显示出对多种取代基, 如卤素、硝基、醚、烷氧基、酯和磺酰基等官能团的相容性.多种脂肪醇均可发生该类反应.
3 钌催化的C—H活化/C—O偶联反应
Ackermann课题组[65]于2012年报道了以苯甲酰胺类化合物为反应底物, 钌盐复合物([Ru(O2CMes)2-(p-cymene)])作为催化剂, PhI(OAc)2为氧化剂, 在TFA/TFAA混合溶剂系统中实现了N-取代苯甲酰胺类化合物的C—H羟基化反应(Eq. 51).该方法中催化剂的用量最低可至1.0 mol%, 仍能够高效地得到对应的羟基化产物.芳香环上的不同电子效应的取代基对反应条件都具有很好的耐受性.
同年, Ackermann课题组[66]报道了一种以羰基作导向基团, 钌盐复合物作催化剂, PhI(OAc)2为氧化剂, 在TFA/TFAA混合溶剂系统中制备羟基化芳香酮化合物的方法(Eq. 52).该方法具有底物范围广、化学位点选择性高的特点.该课题组以不同的官能团对反应条件进行了优化, 不论是吸电子基团还是供电子基团在反应中都具有很好的耐受性, 其产率分布在60%~85%之间.但富电子芳香烃底物较之缺电子底物更有利于C—H羟基化反应的发生.
Ackermann课题组[67]于2013年报道了利用另外一种钌盐复合物[RuCl2(p-cymene)]2作为催化剂, 以芳基氨基甲酸酯类化合物为底物, PhI(OTFA)2作氧化剂, 在二氯乙烷(DCE)溶剂中进行C—H羟基化反应(Eq. 53).同时, 该课题组发现同样的催化体系能够在苯甲醚类化合物的对位进行选择性羟基化反应(Eq. 54).
醛类化合物易被氧化为羧酸, C—H官能团化的强氧化条件可能会导致醛的过度氧化而降低目标产物的收率, 因此芳醛类化合物的C—H键活化反应是有机化学的一个难点.
2014年, Ackermann课题组[68]首次报道了一种醛基作导向基团的芳香烃的C—H羟基化反应.以钌盐复合物[RuCl2(p-cymene)]2作为催化剂, PhI(OTFA)2作氧化剂, 在DCE溶剂中可以将苯甲醛类化合物高效地转化为对应的羟基化产物(Eq. 55).该方法能够催化多种苯甲醛类化合物C—H键直接羟基化的反应, 同时获得的产物在不同条件下能够转化为多种杂环化合物.
Rao课题组[69]于2013年报道了利用[RuCl2(p-cymene)]2催化芳香胺衍生物的C—H羟基化反应.该方法采用取代的苯甲酰基作为可逆导向基团, K2S2O8为氧化剂, 在TFA/TFAA混合溶剂中进行单羟基化或者双羟基化反应(Eq. 56).在该反应体系中, 不同类型取代基的底物都具有很好的兼容性.
儿茶酚结构在天然产物和药物分子中是常见的结构骨架[70]. 2014年, Rao课题组[71]报道了钌盐复合物[RuCl2(p-cymene)]2催化氨基甲酸酯类化合物进行C—H羟基化反应的方法(Eq. 57).该方法具有选择性高, 官能团耐受性好, 操作简便, 产物单一等优点.当选择Pd(OAc)2代替[RuCl2(p-cymene)]2进行氨基甲酸酯类化合物C—H键羟基化反应时, 在室温条件下就能够进行, 但收率相对较低.利用该方法可以进行儿茶酚以及焦棓酸类化合物的克量级合成.
Hong课题组[72]于2015年报道了钌催化黄酮、色原酮和香豆素类衍生物的C—H键羟基化反应.黄酮类化合物5-位C—H键较更具亲核的3-位C—H键更容易选择性地进行羟基化.底物中如氟、氯、硝基和甲氧基等不同的取代基在该催化体系中都具有很好的耐受性(Eq. 58).萘醌和香豆素类化合物在该反应体系中也能顺利地进行羟基化反应(Eqs. 59, 60).该课题组并对该类反应的反应机理进行了推测(Scheme 18): (1)发生C—H活化, 底物导向基团和催化剂进行螯合形成二价钌五元环复合物; (2)在高价碘作用下钌被氧化成四价钌复合物; (3)经过还原消除得到5-三氟乙酰氧基-黄酮, 同时得到再生的二价钌催化剂; (4)三氟乙酸酯经水解得到羟基化产物.
 图式 18
钌催化黄酮类化合物羟基化反应的机理推测
Scheme18.
Proposed mechanistic pathway of Ru(Ⅱ)-catalyzed hydroxylation of flavones
图式 18
钌催化黄酮类化合物羟基化反应的机理推测
Scheme18.
Proposed mechanistic pathway of Ru(Ⅱ)-catalyzed hydroxylation of flavones
Rao课题组[73]于2016年报道了一种苯甲酰胺类化合物进行选择性羟基化反应的方法.通过不同的催化体系, 苯甲酰苯胺类化合物的两个芳香环能够分别进行选择性的邻位羟基化反应(Eqs. 61, 62).在[RuCl2(p-cymene)]2和K2S2O8的催化系统下, 在羰基的邻位发生了C—H羟基化反应.当选择Pd(OAc)2和K2S2O8的催化体系时, 则主要在苯胺基团的邻位发生C—H羟基化反应.反应机理研究表明, 在不同的催化系统下, 空间立体效应和电子效应控制着C—H羟基化反应的区域选择性.
2013年, Jeganmohan课题组[74]报道了以取代的乙酰苯胺为底物, 以钌盐复合物[RuCl2(p-cymene)]2作为催化剂, 以AgSbF6和(NH4)2S2O8为氧化剂, 在DCE溶剂中与羧酸实现了邻位的酰氧化反应(Eq. 63).反应在100 ℃进行, 收率最高可达81%.除乙酸外, 芳基甲酸也能发生该类反应.
4 钴催化的C—H活化/C—O偶联反应
相对上述所述过渡金属催化剂, 金属钴用于C—H活化/C—O偶联反应的报道较少. 2014年, Song课题组[75]报道了一种钴催化的取代芳烃和烯烃甲酰胺C(sp2)—H键的烷氧基化反应.该反应以2-氨基吡啶-1-氧化物为导向基团, 以Co(OAc)2•4H2O作为催化剂, 于空气中进行(Eqs. 64, 65).该合成方法简单, 适用于各种具有富电子和缺电子基团的苯甲酰胺衍生物作为底物.卤素、醚、甲氧基、三氟甲基和N, N-二甲基氨基等官能团在该反应体系中具有很好的耐受性.三氟甲基甲醇、甲氧基乙醇和苯甲醇等多种脂肪醇均能发生该类反应.
5 结论与展望
随着过渡金属催化的C—H活化反应的不断发展, C—H活化/C—O偶联反应已成为有机化学中构建C—O键合成芳基醚类和酚类化合物的一种有效途径, 并在有机合成中占有愈来愈重要的地位.种类繁多的催化体系为不同种类芳香烃的C—H活化/C—O偶联反应提供了条件.
过渡金属催化的C—H活化/C—O偶联反应虽然已经取得了重大的进展, 但仍然存在一些亟待改进和研究的科学问题: (1)反应均需要固定导向基团的协助, 单一催化体系的应用范围较为狭窄, 因此可逆导向基团和临时导向基团的持续开发仍然是未来的一个热点; (2)目前反应大多是在导向基团的邻位发生反应, 未来研究可能需要开发具有远程导向能力的导向基团, 如导向基团间位的C—H活化/C—O偶联反应; (3)部分反应体系还存在目标产物的收率偏低及C—H活化/C—O偶联反应机理的研究不够透彻等问题; (4)尽管过渡金属催化的碳氢键活化反应理论上具有更高的原子经济性, 但部分反应也有不可忽视的缺点, 如高的催化剂用量、较高的反应温度、对底物种类的限制性很强, 需要特定导向基团等, 不符合绿色化学的范畴, 并且目前没有将C—H活化反应进行工业化的例子.相信在不久的将来, 过渡金属催化的C—H活化/C—O偶联反应将得到更大的改善和突破, 成为有机化学中构建C—O键强有力的工具.
-
-
[1]
(a) Karioti, A.; Carta, F.; Supuran, C. T. Molecules 2016, 21, 1649;
(b) Yamaguchi, M. Synthetic Uses of Phenols in PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2009. -
[2]
(a) Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525.
(b) Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954.
(c) Chen, J.; Zhao, K.; Ge, B.; Xu, C.; Wang, D.; Ding, Y. Chem. Asian J. 2015, 10, 468.
(d) Lee, C.; Matunas, R. In Comprehensive Organometallic Chemistry III, Ed.: Crabtree, R. H., Elsvier, Oxford, 2007, p. 649. -
[3]
(a) Caro-Diaz, E. J. ; Urbano, M. ; Buzard, D. J. ; Jones, R. M. Bioorg. Med. Chem. Lett. 2016, 26, 5378.
(b) Gulías, M. ; Mascareñ as, J. L. Angew. Chem., Int. Ed., 2016, 55, 11000.
(c) Qiu, Y. ; Gao, S. Nat. Prod. Rep. 2016, 33, 562.
(d) Yamaguchi, J. ; Yamaguchi, A. D. ; Itami, K. Angew. Chem., Int. Ed. 2012, 51, 8960.
(e) McMurray, L. ; O'Hara, F. ; Gaunt, M. J. Chem. Soc. Rev. 2011, 40, 1885.
(f) Davies, H. M., Du, Bois J. ; Yu, J. Q. Chem. Soc. Rev. 2011, 40, 1855.
(g) Liu, Y. ; Liu, S. ; Xiao, Y. Beilstein J. Org. Chem. 2017, 13, 589.
(h) Liao, G. ; Shi, B. Acta Chim. Sinica 2015, 73, 1283 (in Chinese).
(廖港, 史炳锋, 化学学报, 2015, 73, 1283. )
(i) Zhu, Q. ; Wang, L. ; Xia, C. ; Liu, C. Chin. J. Org. Chem. 2016, 36, 2813 (in Chinese).
(朱庆, 王露, 夏春谷, 刘超, 有机化学, 2016, 36, 2813. )
(j) Chen, J. ; Zhao, K. ; Ge, B. ; Xu, C. ; Wang, D. ; Ding, Y. Chin. J. Chem. 2015, 33, 1015.
(k) Wang, D. ; Yu, X. ; Ge, B. ; Miao, H. ; Ding, Y. Chin. J. Org. Chem. 2015, 35, 676 (in Chinese).
(王大伟, 余晓丽, 葛冰洋, 苗红艳, 丁玉强, 有机化学, 2015, 35, 676. )
(l) Wang, D. ; Yu, X. ; Xu, X. ; Ge, B. ; Wang, X. ; Zhang, Y. Chem. Eur. J. 2016, 22, 8663.
(m) Wang, D. ; Ge, B. ; Li, L. ; Shan, J. ; Ding, Y. J. Org. Chem. 2014, 79, 8607.
(n) Ge, B. ; Wang, D. ; Dong, W. ; Ma, P. ; Li, Y. ; Ding, Y. Tetrahedron Lett. 2014, 55, 5443.
(o) Yu, X. ; Wang, D. S. ; Xu, Z. ; Yang, B. ; Wang, D. Org. Chem. Front. 2017, 4, 1011. -
[4]
Chen, J.; Zhao, K.; Ge, B.; Xu, C.; Wang, D.; Ding, Y. Chem. Asian J. 2015, 10, 468. doi: 10.1002/asia.v10.2
-
[5]
Alonso, D. A.; Nájera, C.; Pastor, I. M.; Yus, M. Chem.-Eur. J. 2010, 16, 5274. doi: 10.1002/chem.201000470
-
[6]
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147. doi: 10.1021/cr900184e
-
[7]
Enthaler, S.; Company, A. Chem. Soc. Rev. 2011, 40, 4912. doi: 10.1039/c1cs15085e
-
[8]
Krylov, I. B.; Vil', V. A.; Terent'ev, A. O. Beilstein. J. Org. Chem. 2015, 11, 92. doi: 10.3762/bjoc.11.13
-
[9]
Liu, B.; Shi, B.-F. Tetrahedron Lett. 2015, 56, 15. doi: 10.1016/j.tetlet.2014.11.039
-
[10]
Jintoku, T.; Nishimura, K.; Takaki, K.; Fujiwara, Y. Chem. Lett. 1990, 19, 1687. doi: 10.1246/cl.1990.1687
-
[11]
Kim, S. H.; Lee, H. S.; Kim, S. H.; Kim, J. N. Tetrahedron Lett. 2008, 49, 5863. doi: 10.1016/j.tetlet.2008.07.141
-
[12]
Zhang, Y. H.; Yu, J. Q. J. Am. Chem. Soc. 2009, 131, 14654. doi: 10.1021/ja907198n
-
[13]
Charest, M. G.; LernerC, D.; Brubaker, J. D.; Siegel, D. R.; Myers, A. G. Science 2005, 308, 395. doi: 10.1126/science.1109755
-
[14]
Shan, G.; Yang, X. L.; Ma, L.-L.; Rao, Y. Angew. Chem., Int. Ed. 2012, 51, 13070. doi: 10.1002/anie.201207458
-
[15]
Mo, F. Y.; Trzepkowski, L. J.; Dong, G. B. Angew. Chem., Int. Ed. 2012, 51, 13075. doi: 10.1002/anie.201207479
-
[16]
Choy, P. Y.; Kwong, F. Y. Org. Lett. 2013, 15, 270. doi: 10.1021/ol303088z
-
[17]
Zhang, H. Z.; Yi, H. M.; Wang, G. W.; Yang, B.; Yang, S. D. Org. Lett. 2013, 15, 6186. doi: 10.1021/ol403028a
-
[18]
Yan, Y. P.; Feng, P.; Zheng, Q. Z.; Liang, Y. F.; Lu, J. F.; Cui, Y. X.; Jiao. N. Angew. Chem., Int. Ed. 2013, 52, 5827. doi: 10.1002/anie.201300957
-
[19]
Banerjee, A.; Bera, A.; Guin, S.; Rout, S. K.; Patel, B. K. Tetrahedron 2013, 69, 2175. doi: 10.1016/j.tet.2012.12.067
-
[20]
Gurivendar, S.; Maninderjit, K.; Mohan, C. Int. Res. J. Pharm. 2013, 4, 1.
-
[21]
Kamal, A.; Srinivasulu, V.; Sathish, M.; Tangella, Y.; Nayak, V. L.; Rao, M. P. N.; Shankaraiah, N.; Nagesh, N. Asian J. Org. Chem. 2014, 3, 68. doi: 10.1002/ajoc.201300214
-
[22]
Dong, J. W.; Liu, P.; Sun, P.-P. J. Org. Chem. 2015, 80, 2925. doi: 10.1021/acs.joc.5b00167
-
[23]
Seth, K.; Nautiyal, M.; Purohit, P.; Parikh, N.; Chakraborti, A. K. Chem. Commun. 2015, 51, 191. doi: 10.1039/C4CC06864E
-
[24]
Yang, X.; Jasper, A. W.; Giri, B. R.; Kiefer, J. H.; Tranter, R. S. Phys. Chem. Chem. Phys. 2011, 13, 3686. doi: 10.1039/C0CP01541E
-
[25]
Wang, D.; Zhao, K.; Xu, C.; Miao, H.; Ding, Y. ACS Catal. 2014, 4, 3910. doi: 10.1021/cs5009909
-
[26]
Liang, Y. F.; Wang, X. Y.; Yuan, Y. Z.; Liang, Y. J.; Li, X. Y.; Jiao, N. ACS Catal. 2015, 5, 6148. doi: 10.1021/acscatal.5b01700
-
[27]
Yamaguchi, T.; Yamaguchi, E.; Tada, N.; Itoh, A. Adv. Synth. Catal. 2015, 357, 2017. doi: 10.1002/adsc.v357.9
-
[28]
Das, P.; Saha, D.; Saha, D.; Guin, J. ACS Catal. 2016, 6, 6050. doi: 10.1021/acscatal.6b01539
-
[29]
Duan, S. T.; Xu, Y. S.; Zhang, X. Y.; Fan, X. S. Chem. Commun. 2016, 52, 10529. doi: 10.1039/C6CC04756D
-
[30]
(a) Wang, X. C. ; Gong, W. ; Fang, L. Z. ; Zhu, R. Y. ; Li, S. ; Engle, K. M. ; Yu, J. Q. Nature 2015, 519, 334.
(b) Yuan, Y. ; Song, S. ; Jiao, N. Acta Chim. Sinica 2015, 73, 1231 (in Chinese).
(袁逸之, 宋颂, 焦宁, 化学学报, 2015, 73, 1231. ) -
[31]
Maji, A.; Bhaskararao, B.; Singha, S.; Sunoj, R. B.; Maiti, D. Chem. Sci. 2016, 7, 3147. doi: 10.1039/C5SC04060D
-
[32]
Bera, M.; Sahoo, S. K.; Maiti, D. ACS Catal. 2016, 6, 3575. doi: 10.1021/acscatal.6b00675
-
[33]
Desai, L. V.; Malik, H. A.; Sanford, M. S. Org. Lett. 2006, 8, 1141. doi: 10.1021/ol0530272
-
[34]
Wang, G. W.; Yuan, T. T.; Wu, X. L. J. Org. Chem. 2008, 73, 4717. doi: 10.1021/jo8003088
-
[35]
Gou, F.-R.; Wang, X. C.; Huo, P. F.; Bi, H. P.; Guan, Z. H.; Liang, Y. M. Org. Lett. 2009, 11, 5726. doi: 10.1021/ol902497k
-
[36]
Zheng, X.; Song, B.; Xu, B. Eur. J. Org. Chem. 2010, 4376.
-
[37]
Reddy, B. V. S.; Revathi, G.; Reddy, A. S.; Yadav, J. S. Tetrahedron Lett. 2011, 52, 5926. doi: 10.1016/j.tetlet.2011.08.098
-
[38]
Gary, J. B.; Cook, A. K.; Sanford, M. S. ACS Catal. 2013, 3, 700 doi: 10.1021/cs300786j
-
[39]
Chernyak, N.; Dudnik, A. S.; Huang, C.; Gevorgyan, V. J. Am. Chem. Soc. 2010, 132, 8270 doi: 10.1021/ja1033167
-
[40]
Gulevich, A. V.; Melkonyan, F. S.; Sarkar, D.; Gevorgyan, V. J. Am. Chem. Soc. 2012, 134, 5528. doi: 10.1021/ja3010545
-
[41]
Tang, R. Y.; Li, G.; Yu, J. Q. Nature 2014, 507, 215. doi: 10.1038/nature12963
-
[42]
Li, S.; Cai, L.; Ji, H.; Yang, L.; Li, G. Nat. Commun. 2016, 7, 10443. doi: 10.1038/ncomms10443
-
[43]
Bag, S.; Patra, T.; Modak, A.; Deb, A.; Maity, S.; Dutta, U.; Dey, A.; Kancherla, R.; Maji, A.; Hazra, A.; Bera, M.; Maiti, D. J. Am. Chem. Soc. 2015, 137, 11888. doi: 10.1021/jacs.5b06793
-
[44]
Wang, G. W.; Yuan, T. T. J. Org. Chem. 2010, 75, 476. doi: 10.1021/jo902139b
-
[45]
Péron, F.; Fossey, C.; Santos, J. S.; Cailly, T.; Fabis, F. Chem. Eur. J. 2014, 20, 7507. doi: 10.1002/chem.201303923
-
[46]
Chen, F.; Zhao, S.; Hu, F.; Chen, K.; Zhang, Q.; Zhang, S.; Shi, B. Chem. Sci. 2013, 4, 4187. doi: 10.1039/c3sc51993g
-
[47]
Yin, Z.; Jiang, X.; Sun, P. J. Org. Chem. 2013, 78, 10002. doi: 10.1021/jo401623j
-
[48]
Zhang, C.; Sun, P. J. Org. Chem. 2014, 79, 8457. doi: 10.1021/jo5014146
-
[49]
Gao, T.; Sun, P. J. Org. Chem. 2014, 79, 9888. doi: 10.1021/jo501902d
-
[50]
Shi, S.; Kuang, C. J. Org. Chem. 2014, 79, 6105. doi: 10.1021/jo5008306
-
[51]
Yang, L.; Li, S.; Cai, L.; Ding, Y.; Fu, L.; Cai, Z.; Ji, H.; Li, G. Org. Lett. 2017, 19, 2746. doi: 10.1021/acs.orglett.7b01103
-
[52]
Chen, X.; Hao, X. S.; Goodhue, C. E.; Yu, J. Q. J. Am. Chem. Soc. 2006, 128, 6790. doi: 10.1021/ja061715q
-
[53]
Ueda, S.; Nagasawa, H. Angew. Chem., Int. Ed. 2008, 47, 6411. doi: 10.1002/anie.v47:34
-
[54]
Liu, Q.; Wu, P.; Yang, Y. H.; Zeng, Z. Q.; Liu, J.; Yi, H.; Lei, A. W. Angew. Chem., Int. Ed. 2012, 51, 4666. doi: 10.1002/anie.201200750
-
[55]
Li, X.; Liu, Y. H.; Gu, W. J.; Li, B.; Chen, F. J.; Shi, B. F. Org. Lett. 2014, 16, 3904. doi: 10.1021/ol5016064
-
[56]
Singh, B. K.; Jana, R. J. Org. Chem. 2016, 81, 831. doi: 10.1021/acs.joc.5b02302
-
[57]
Sun, S. Z.; Shang, M.; Wang, H. L.; Lin, H. X.; Dai, H. X.; Yu, J. Q. J. Org. Chem. 2015, 80, 8843. doi: 10.1021/acs.joc.5b01351
-
[58]
Wang, D. W.; Yu, X.; Yao, W.; Hu, W. K.; Ge, C. Y.; Shi, X. D. Chem.-Eur. J. 2016, 22, 5543. doi: 10.1002/chem.201600597
-
[59]
Wang, W. H.; Luo, F.; Zhang, S. H.; Cheng, J. J. Org. Chem. 2010, 75, 2415. doi: 10.1021/jo1000719
-
[60]
Bhadra, S.; Matheis, C.; Katayev, D.; Goo en, L. J. Angew. Chem., Int. Ed. 2013, 52, 9279. doi: 10.1002/anie.201303702
-
[61]
Suess, A. M.; Ertem, M. Z.; Cramer, C. J.; Stahl, S.-S. J. Am. Chem. Soc. 2013, 135, 9797. doi: 10.1021/ja4026424
-
[62]
Roane, J.; Daugulis, O. Org. Lett. 2013, 15, 5842. doi: 10.1021/ol402904d
-
[63]
Hao, X.-Q.; Chen, L. J.; Ren, B.; Li, L. Y.; Yang, X. Y.; Gong, J. F.; Niu, J. L.; Song, M.-P. Org. Lett. 2014, 16, 1104. doi: 10.1021/ol500166d
-
[64]
Zhang, L. B.; Hao, X. Q.; Zhang, S.-K.; Liu, K.; Ren, B.; Gong, J. F.; Niu, J. L.; Song, M.-P. J. Org. Chem. 2014, 79, 10399. doi: 10.1021/jo502005j
-
[65]
Thirunavukkarasu, V. S.; Hubrich, J.; Ackermann, L. Org. Lett. 2012, 14, 4210. doi: 10.1021/ol3018819
-
[66]
Thirunavukkarasu, V. S.; Ackermann, L. Org. Lett. 2012, 14, 6206. doi: 10.1021/ol302956s
-
[67]
Liu, W. P.; Ackermann, L. Org. Lett. 2013, 15, 3484. doi: 10.1021/ol401535k
-
[68]
Yang, F. Z.; Rauch, K.; Kettelhoit, K.; Ackermann, L. Angew. Chem., Int. Ed. 2014, 53, 11285. doi: 10.1002/anie.201405647
-
[69]
Yang, X. L.; Shan, G.; Rao, Y. Org. Lett. 2013, 15, 2334. doi: 10.1021/ol400437a
-
[70]
Bedford, R. B.; Coles, S. J.; Hursthouse, M. B.; Limmert, M. E. Angew. Chem., Int. Ed. 2003, 42, 112. doi: 10.1002/(ISSN)1521-3773
-
[71]
Yang, X. L.; Sun, Y. H.; Chen, Z.; Rao, Y. Adv. Synth. Catal. 2014, 356, 1625. doi: 10.1002/adsc.v356.7
-
[72]
Kim, K.; Choe, H.; Jeong, Y.; Lee, J. H.; Hong, S. Org. Lett. 2015, 17, 2550. doi: 10.1021/acs.orglett.5b01138
-
[73]
Sun, Y. H.; Sun, T. Y.; Wu, Y. D.; Zhang, X. H.; Rao, Y. Chem. Sci. 2016, 7, 2229. doi: 10.1039/C5SC03905C
-
[74]
Padala, K.; Jeganmohan, M. Chem. Commun. 2013, 49, 9651. doi: 10.1039/c3cc45350b
-
[75]
Zhang, L. B.; Hao, X. Q.; Zhang, S.-K.; Liu, Z. J.; Zheng, X. X.; Gong, J. F.; Niu, J. L.; Song, M. P. Angew. Chem., Int. Ed. 2015, 54, 272. doi: 10.1002/anie.201409751
-
[1]
-

图式 1 Pd(OAc)2催化二芳基酮的邻位羟基化反应机理推测
Scheme 1 Proposed mechanism of Pd(OAc)2 catalyzed ortho-hydroxylation of diaryl ketones

图式 2 PdCl2和NHPI共同催化2-苯基吡啶类化合物的羟基化反应机理推测
Scheme 2 Proposed mechanism of PdCl2 and NHPI co-catalyzed hydroxylation of 2-arylpyridines

图式 3 Pd(OAc)2催化2-苯基苯并噻唑类化合物的邻位羟基化反应机理推测
Scheme 3 Proposed mechanism of Pd(OAc)2-catalyzed ortho-hydroxylation of 2-arylbenzothiazole

图式 4 Pd(OAc)2催化2-苯基吡啶类化合物羟基化反应的机理推测
Scheme 4 Plausible mechanism of Pd(OAc)2-catalyzed hydroxylation of 2-arylpyridines

图式 5 二氧六环热分解为羟基自由基的过程
Scheme 5 Process of the generation of radical •OH from 1, 4-dioxane

图式 6 Pd(OAc)2催化2-芳基苯并噁(噻)唑羟基化反应的可能反应机理过程
Scheme 6 Plausible mechanism of Pd(OAc)2-catalyzed hydroxylation of benzothiazoles and benzoxazoles

图式 7 Pd(CH3CN)2Cl2催化的2-苯基吡啶类化合物的羟基化反应机理预测
Scheme 7 Proposed mechanism of Pd(CH3CN)2Cl2 catalyzed hydroxylation of 2-aryl pyridines

图式 8 Pd(OAc)2催化的芳烃C—H乙酰氧基化和甲氧基化反应
Scheme 8 Pd(OAc)2 catalyzed C—H acetoxylation and methoxylation of arenes

图式 9 Pd(OAc)2催化的N-(2-苯甲酰基苯基)-苯甲酰胺的乙酰氧基化和甲氧基化反应
Scheme 9 Pd(OAc)2 catalyzed acetoxylation and methoxylation of N-(2-benzoylphenyl)benzamides

图式 10 (2-吡啶基)二异丙基甲硅烷基导向的乙酰氧基化反应和新戊酰氧基化反应
Scheme 10 (2-Pyridyl)diisopropylsilyl directed acetoxylation and pivaloxylation

图式 11 (2-嘧啶基)二异丙基甲硅烷基导向的乙酰氧基化反应和新戊酰氧基化反应
Scheme 11 (2-Pyrimidyl)diisopropylsilyl directed acetoxylation and pivaloxylation

图式 13 氯化铜催化杂环芳香烃的羟基化反应机理推测
Scheme 13 Proposed reaction pathway for CuCl2 catalyzed hydroxylation of heterocycles

图式 14 可移除双配位基促进铜催化芳香烃类化合物的羟基化反应机理推测
Scheme 14 Plausible reaction mechanism of copper-mediated hydroxylation of arenes directed by a removable bidentate group

图式 15 铜催化的2-苯基吡啶类化合物酯化反应机理推测
Scheme 15 Plausible mechanism of copper-catalyzed ortho-acyloxylation of 2-aryl pyridines

图式 16 芳香烃脱氢烷氧基化反应机理推测
Scheme 16 Proposed mechanism of dehydrogenative alkoxylation of arenes
-


 下载:
下载:

















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 54
- 文章访问数: 8653
- HTML全文浏览量: 1695
 下载:
下载:
