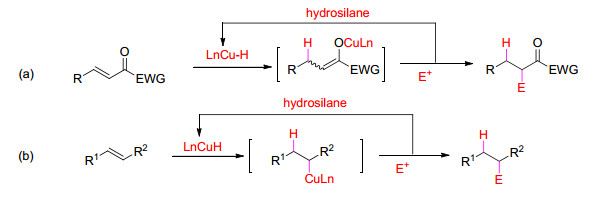
 图式 1
由Cu催化氢金属化反应引发的多米诺反应
Scheme1.
General domino reactions initialized by Cu-catalyzed hydrometallation
图式 1
由Cu催化氢金属化反应引发的多米诺反应
Scheme1.
General domino reactions initialized by Cu-catalyzed hydrometallation
引用本文:
梁婷婷, 姜岚, 干苗苗, 苏鑫, 李争宁. 铜催化对C-C不饱和键铜氢化引发的多米诺反应[J]. 有机化学,
2017, 37(12): 3096-3111.
doi:
10.6023/cjoc201706004

Citation: Liang Tingting, Jiang Lan, Gan Miaomiao, Su Xin, Li Zhengning. Domino Reactions Initialized by Copper-Catalyzed Hydrocupration of C-C Unsaturated Bonds[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3096-3111. doi: 10.6023/cjoc201706004
Citation: Liang Tingting, Jiang Lan, Gan Miaomiao, Su Xin, Li Zhengning. Domino Reactions Initialized by Copper-Catalyzed Hydrocupration of C-C Unsaturated Bonds[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3096-3111. doi: 10.6023/cjoc201706004
铜催化对C-C不饱和键铜氢化引发的多米诺反应
English
Domino Reactions Initialized by Copper-Catalyzed Hydrocupration of C-C Unsaturated Bonds
Abstract:
Compared with sequential reactions, domino reactions are more efficient and highly desirable in organic synthesis as fewer operational and purification procedures are involved, yielding the product with complexity in a more economic and environmently friendly manner. Domino reactions induced by copper-catalyzed hydrocupration of unsaturated C-C bonds in α, β-unsaturated ketones, α, β-unsaturated carboxylates, aryl alkenes, aliphatic alkenes and even alkynes are reviewed. A hydrosilane is used as a hydride source for CuH formation and the hydrocupration intermediates undergo subsequent addition to polar unsaturated bonds, e.g. carbonyls and imines, or proceed to substitution reactions, and finally, reaction involving two or more newly formed bonds proceed without purification of intermediate products or changing the operational conditions. Because of its simplicity and efficiency, this method is highly valuable in organic synthesis.
-
Key words:
- domino reaction
- / catalysis
- / copper
- / hydrocupration
- / reduction
- / alkene
-
按照Tietze的定义, 多米诺反应指在相同的反应条件下, 不需要重新加入其它试剂和催化剂就能连续进行的多步反应, 其中前一步反应的产物自发进行后续反应, 最终形成两个或多个化学键[1].广义的多米诺反应指加入催化剂或其它试剂引发的经过中间体连续形成多个化学键的反应.与分步反应相比, 多米诺反应不需要改变反应条件, 也不需要对中间产物进行分离纯化, 减少了操作过程和分离损失, 以高原子经济性和环境友好的方式生成最终产物, 在有机合成中更为高效, 受到高度关注[2~9].
尽管通过过渡金属特别是铜催化α, β-不饱和羰基化合物的共轭加成反应已有很久的历史[10~13], 但是用类似的反应还原α, β-不饱和羰基化合物中的C-C不饱和键则出现得比较晚[14~16].过渡金属催化共轭加成反应形成的金属烯醇中间体可与合适的亲电试剂继续反应, 从而进行多米诺反应.这已成为合成许多复杂结构化合物的有效方法.关于金属催化共轭还原α, β-不饱和羰基化合物[17~22]已有综述, 其中涉及到关键中间体金属氢化物对C-C不饱和键的氢金属化反应[11].铜化合物在这些反应中通常表现出比其它金属更优良的催化性能.除用于学术研究外, 铜催化剂因金属价格低, 在大规模应用中具有其它贵金属难有的吸引力. Chiu和Nishiyama等[23, 24]在2004年、2007年分别总结了由α, β-不饱和羰基化合物共轭还原引发的多米诺反应.考虑到这一领域的快速发展, 在此, 我们总结铜催化C-C不饱和键共轭还原引发的多米诺反应, 其中形成的烯醇中间体被亲电试剂捕获(Scheme 1, a); 以及对不带共轭吸电子基团的烯烃铜氢化引发的多米诺反应(Scheme 1, b).为便于更全面地理解这一领域的发展, 本文也包括2007年前发表的部分重要结果及少量其它过渡金属催化的结果.通常, 大多数铜催化剂包含配位的膦配体(包括手性配体), 以调整其催化活性和选择性.其中所用到的一些重要手性配体的结构如图 1所示.
图式 1
由Cu催化氢金属化反应引发的多米诺反应
Scheme1.
General domino reactions initialized by Cu-catalyzed hydrometallation
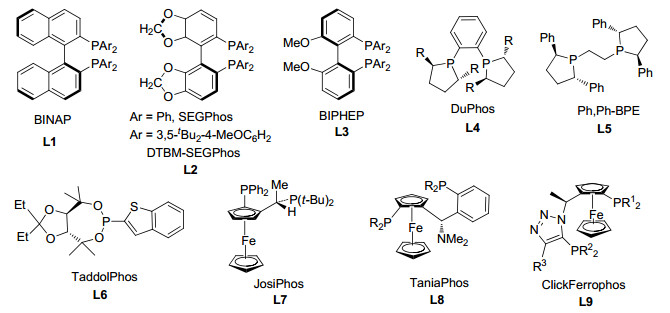 图1
Cu催化氢金属化引发多米诺反应中用到的手性配体
Figure1.
Some chiral ligands in Cu-catalyzed hydrometallation -induced domino reactions
图1
Cu催化氢金属化引发多米诺反应中用到的手性配体
Figure1.
Some chiral ligands in Cu-catalyzed hydrometallation -induced domino reactions
1 α, β-不饱和羰基化合物的反应
1.1 共轭还原/aldol反应
过渡金属催化α, β-不饱和羰基化合物的共轭还原反应代表着一类重要的反应, 由金属氢化物的产生、与极性C-C不饱和键加成以及金属氢化物的再生组成.铜配合物是还原被共轭羰基活化的C-C不饱和键的合适催化剂, 经共轭加成形成烯醇中间体, 后者水解后得到还原C-C不饱和键的产物.烯醇中间体也易于与极性不饱和键如羰基反应, 进行共轭还原/aldol反应.
首例过渡金属催化的还原/aldol加成反应可追踪至1987年, Revis和Hilty[25]采用氯化铑催化三甲基氢硅烷、甲基丙烯酸甲酯、丙酮间的反应, 以95%产率生成了β-硅氧基酯.氯化铑并不能催化硅烷基烯酮缩醛和丙酮间的aldol加成反应, 因而, 上述aldol加成反应经烯醇铑中间体进行.
2004年, Chiu等[26]报道了10 mol%的[CuHPPh3]6 (Stryker试剂)催化聚甲基氢硅氧烷(PMHS)、α, β-炔酮的还原/与分子内羰基的aldol加成多米诺反应, 形成五、六元环的环烯酮(Scheme 2), 同时伴随着一些过还原产物和脱水产物.对于某些底物的反应, 还伴随着还原/脱水产物.这些结果与使用化学计量[CuHPPh3]6还原剂的结果相似.然而, 由炔基不饱和羧酸酯仅生成简单还原的产物, 而不是期待的环化产物.
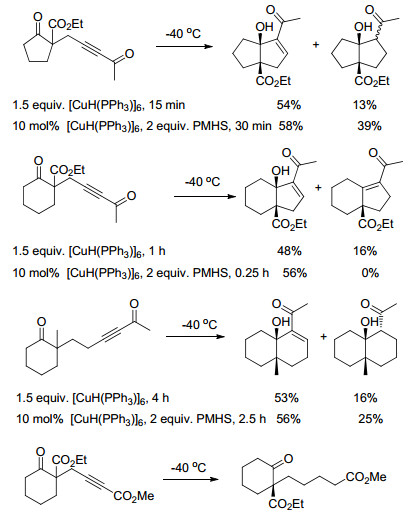 图式 2
Cu催化还原/分子内aldol加成反应
Scheme2.
Cu-catalyzed reductive intramolecular aldol reaction
图式 2
Cu催化还原/分子内aldol加成反应
Scheme2.
Cu-catalyzed reductive intramolecular aldol reaction
Lam课题组[27]在2005年报道了使用Cu(OAc)2•H2O-rac-L1或Cu-1, 1'-双(二苯基膦基)二茂铁(DPPF)作为催化剂和1, 1, 3, 3-四甲基二硅氧烷(TMDS)作为还原剂, 酮基-α, β-不饱和羧酸酯的共轭还原/分子内aldol加成反应(Eq.1, A=O).通过采用手性配体L1、L2和L3, 以49%~83% ee获得五元和六元环β-羟基内酯.然而, 需要高负载量的催化剂(5 mol%)和较长的反应时间(13~30 h, 室温), 才能得到较高的产率. Li等[28]发现使用Cu(Ⅰ)-2-(二苯基膦基)苯基醚(L10)作催化剂, 反应可在10 min内完成, 并得到更高的产率. Lam等[29]也报道了由α, β-不饱和酰胺生成β-羟基内酰胺的反应[Eq. 1, A=对甲氧苯基氨基(PMP-NH)], 产物dr值可达到19/1, 但反应产率中等(60%~70%), 这仅适于α, β-不饱和酰胺中仅含β-H或Me的底物.
2008年, Lipshutz等[30]报道了由Cu(OAc)2•H2O、手性配体L7和氢硅烷产生的催化剂促进酮基-α, β-不饱和酮与氢硅烷的多米诺反应, 以高产率得到三个连续的手性中心产物, 具有高的非对映和对映选择性. Z-和E-烯酮都能参加反应, 彼此产生的主要产物互为对映异构体(Eq. 2). β位带有空间位阻取代基的烯酮也能够进行反应.相比之下, 底物脂肪烯酮比芳基酮可得到更高ee值. 2009年, Riant等[31]实现了Cu-L8催化的对α, β-不饱和酯对称羰基的去对称化反应(Eq. 3).增加底物中酯基的位阻可将非对映体比值提高到100/0, ee高达95%.产生潜在的七和八元环的底物仅生成简单的共轭还原产物.该方法已经被应用于合成具有五个连续手性碳的三环化合物, 其中涉及到两次铜催化的多米诺反应, 目标产物难以通过其它方法得到(Scheme 3).
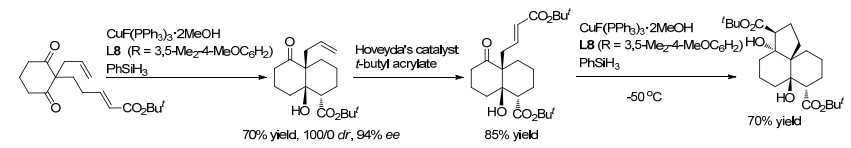 图式 3
经CuH催化还原/aldol加成环化多米诺反应构建多环化合物
Scheme3.
Construction of a polycyclic derivative via CuH-catalyzed domino reductive aldol cyclizations
图式 3
经CuH催化还原/aldol加成环化多米诺反应构建多环化合物
Scheme3.
Construction of a polycyclic derivative via CuH-catalyzed domino reductive aldol cyclizations
2012年, Chiu组[32]使用Cu(OAc)2•H2O-L8作催化剂和PhSiH3作还原剂, 实现了双羰基-α, β-不饱和硫酯类似的多米诺反应.由于硫原子和铜原子之间的强配位作用, α, β-不饱和硫酯的反应活性低于对应的O-羧酸酯, 需要更高的温度和更长的反应时间才能实现硫酯的高转化率.加入联吡啶可加速反应, 但并未改变非对映体和对映体选择性(Scheme 4), 这归因于其在环化后促进复分解的作用.六元环产物上的三个取代基为顺式位置关系.反应的立体选择性几乎不受不饱和硫酯中C-C双键构型影响.优化硫醇中烷基后发现对于酯中烷基大于乙基时, 还原环化产率降低10%~20%;一级烷基酯反应后产物的ee值比大位阻基团(t-Bu, Ph)的高, 其中使用苄基酯反应的ee值最高.五和六元环产物达到98/2 dr和大于90%的ee.产生潜在的七和八元环的底物只发生简单共轭加成反应.
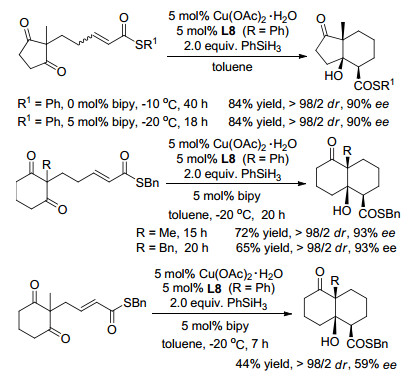 图式 4
Cu催化13-二羰基衍生物的还原/分子内aldol加成反应
Scheme4.
Cu-catalyzed reductive intramolecular aldol reaction of enethiolate derivatives of 1, 3-diones
图式 4
Cu催化13-二羰基衍生物的还原/分子内aldol加成反应
Scheme4.
Cu-catalyzed reductive intramolecular aldol reaction of enethiolate derivatives of 1, 3-diones
与分子内aldol反应相比, 在分子间aldol反应中烯醇铜表现出较低的活性和立体选择性. 2006年, Shibasaki等[33]报道了首例Cu催化的还原/分子间aldol反应.由CuOtBu-L1 (Ar=p-tolyl)催化苯乙酮、丙烯酸甲酯和过量氢硅烷反应(Eq. 4), 得到37%收率的aldol产物, 具有很低的dr值, 优势非对映体的ee值为30%, 同时得到副产物1-苯基乙醇.通过缓慢加入(EtO)3SiH可提高还原/aldol产物的收率, 但非对映选择性没有改善.由对称酮反应, 产物的ee值可达到80%. Li等[34]发现了Cu-L10催化取代苯乙酮与丙烯酸甲酯的共轭还原/aldol加成多米诺反应, 以80%~97%的分离产率得到产物, 具有anti-立体选择性.使用丙烯酸叔丁酯和体积大的配体4, 5-双(二苯基膦基)-9, 9-二甲基呫吨(Xantphos)可以将dr值提高到96/4. Shibasaki等[35]采用硼烷为还原剂, 苯乙酮与丙二烯基羧酸酯的反应可高产率进行, 得到α-加成产物或γ-加成产物.使用L2, 得到高γ选择性和99% ee; 采用L8则生成了α位加成产品(Scheme 5).
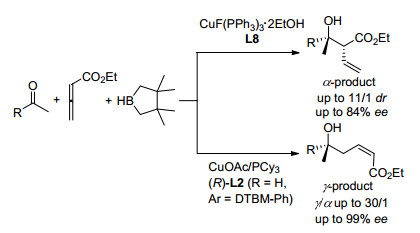 图式 5
Cu催化丙二烯酸酯的不对称还原/分子间aldol加成反应
Scheme5.
Cu-catalyzed asymmetric, reductive intermolecular aldol reaction of allenic ester
图式 5
Cu催化丙二烯酸酯的不对称还原/分子间aldol加成反应
Scheme5.
Cu-catalyzed asymmetric, reductive intermolecular aldol reaction of allenic ester
几乎与Shibasaki等的发现同时, Riant等[36]报道了CuF(PPh3)3•2MeOH-手性膦对Eq. 4中反应的催化性能.虽然Cu(Ⅰ)与单齿Feringa亚膦酸酯、三齿t-Bu-pybox(2, 6-双(4, 5-二氢-4-叔丁基噁唑-2-基)吡啶)和四齿Trost配体配位后仅表现出非常低的活性/或没有对映选择性, 但是Cu-双膦配合物显示出良好的催化化学选择性, 并具有一定的对映体选择性.就立体选择性而言, Taniaphos L8 (R=Ph)优于L1, L3 (R=Ph, 3, 5-(i-Pr)2C6H3)和L7 (R=3, 5-Me2C6H3).在Taniaphos L8存在下, 于-50 ℃反应, 产物的dr值为76/24、主要和次要异构体ee值分别为85% ee和94% ee.将L8中的R由Ph换为环己基, 反应的立体选择性进一步提高到92/8 dr和95% ee. Fukuzawa等[37]使用Cu(Ⅰ)-ClickFerrophos L9为催化剂, 也得到了类似的结果.然而, 对于醛的反应, 仅获得了中等的ee和低的dr.
Riant等[38]采用高活性CuF(PPh3)3•2MeOH-(S)-L1为催化剂, 实现了更有挑战的醛作为烯醇负离子捕获剂的多米诺反应(Eq. 5).通常在Cu催化下, 醛易于被还原.丙烯酸甲酯、苯甲醛和PhSiH3在室温下反应, 以89%化学选择性但低的立体选择性得到共轭还原/aldol加成产物, 反应的TON和TOF分别达到10000和40000 h-1.优化反应条件采用L8 (R=Ph)作配体后, 对环己基甲醛的反应, 可得到高化学选择性(醇<3%)和高对映体选择性(95% ee)的aldol加成产物, 但dr值仅为77/23.将L8中的R由苯基换为3, 5-二甲基-4-甲氧基苯基或环己基后, 苄醇成为主要产物.对于丙烯酸甲酯与芳基醛或环己烷甲醛的反应, 使用Cu-L8 (R=Ph)得到具有41/58~77/23 dr值和58%~96% ee的aldol加成产物.最近, Riant等[39]报道了Cu(IMes)(DBM) (IMes, 1, 3-二甲基-1, 3-二氢-2H-咪唑-2-亚基)对还原反应具有高反应性和化学选择性, 同样, 产物仅具有中等的dr值(对环己烷甲醛的反应为68/32 dr), 在-50 ℃下反应并没有改善dr值.低dr归因于在过渡状态中醛并不与烯醇铜配位.使用α, β-不饱和腈作为CuH受体进行还原/aldol加成反应, 得到92/8 dr, 但产率仅为54%.
对Eq. 1 (R1=Ph, R2=Me, A=PMP-NH, n=2)所示的低活性α, β-不饱和酰胺的反应, 采用Cu(OAc)2•H2O-BINAP[27]或DPPF[29]-TMDS催化体系, α, β-不饱和酰胺的转化率为77%~87%, 以低dr值(34/66~39/61)得到环状产物, 伴随着未环化的副产物.
1.2 共轭还原/aldol加成/内酯化多米诺反应
2014年, Li等[40]利用铜催化剂完成了马来酸酯或衣康酸酯、酮和氢硅烷之间的三步多米诺反应(Scheme 6).反应中, 马来酸酯或衣康酸酯作为不饱和羧酸酯以及烷氧基负离子受体双功能底物.该方法具有试剂适用性广泛[41]、反应物组合灵活、产率高的优点, 在构建含β-烷氧基酰基-γ-环内酯中具有一定的潜力.使用Et2Zn作为还原剂, 同一课题组也实现了马来酸酯的还原/aldol加成/内酯化反应.值得注意的是, 在Cu催化下, Et2Zn不是通常的烷基化试剂, 而是起还原剂的作用(Scheme 7)[42], 这样的例子并不常见[43].然而, 当使用富马酸酯时, 则发生烷基化/aldol加成/内酯化反应(Scheme 7).采用γ-和σ-酮羧酸酯作为双功能底物, Li等[44]通过对α, β-不饱和酯的铜氢化反应, 然后与γ-或β-酮羧酸反应完成三步多米诺反应(Eq. 6), 以高化学选择性、低非对映体选择性地得到γ-和δ-内酯.
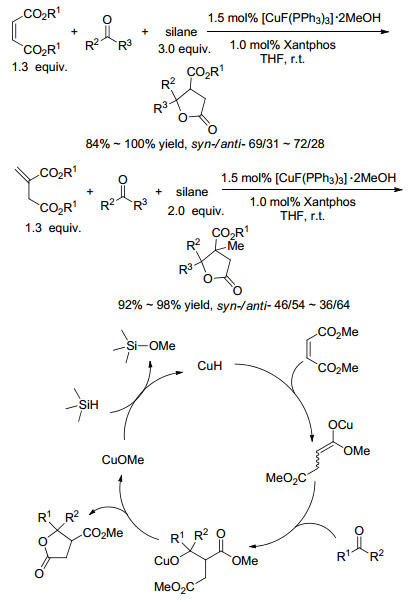 图式 6
Cu催化马来酸酯的还原/aldol加成/内酯化反应及机理
Scheme6.
Cu-catalyzed reductive aldol addition/lactonization reaction of maleate and the mechanism
图式 6
Cu催化马来酸酯的还原/aldol加成/内酯化反应及机理
Scheme6.
Cu-catalyzed reductive aldol addition/lactonization reaction of maleate and the mechanism
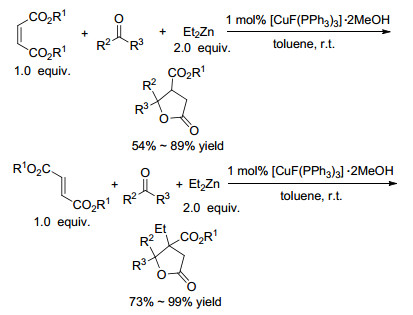 图式 7
Cu催化马来酸酯或富马酸酯与酮、Et2Zn的还原或烷基化/aldol加成/内酯化反应
Scheme7.
Cu-catalyzed reductive or alkylative aldol addition/lactonization reaction of maleate or fumarate with ketones and Et2Zn
图式 7
Cu催化马来酸酯或富马酸酯与酮、Et2Zn的还原或烷基化/aldol加成/内酯化反应
Scheme7.
Cu-catalyzed reductive or alkylative aldol addition/lactonization reaction of maleate or fumarate with ketones and Et2Zn
1.3 共轭还原/Mannich反应
作为亲电试剂, 亚胺对烯醇负离子的反应活性比羰基的低, 相应的反应较慢. 2008年, Shibasaki小组[45]报道了铜催化α, β-不饱和羧酸酯与亚胺的反应, 采用5 mol% CuOAc-PPh3为催化剂和(EtO)3SiH为还原剂, 对1-(4-氯苯基)乙酮亚胺与丙烯酸乙酯的反应得到60%产率和3/1 dr值的产物; 改用频哪醇硼烷(PinBH)作为还原剂, 反应的产率和dr值分别提高到92%和50/1 (Eq. 7).采用(R)-SEGPhos类配体(R)-L2 (R=Cl, Ar=Ph)和(EtO)3SiH还原剂, 可以实现更高的ee.在酸性条件下, 手性膦酰胺可以转化为β-氨基羧酸衍生物, 而不失去手性.该还原/Mannich反应克服了之前酮亚胺与乙烯酮缩醛反应生成β-氨基羧酸酯的不足, 后者仅适用于乙酸酯但不适用于丙酸酯的乙烯酮缩醛的反应[46]. Li等[47]报道了马来酸酯、未保护的亚胺和氢硅烷之间的多米诺反应, 以高产率和高dr值生成β-酯基-γ-内酰胺(Eq. 8).
1.4 共轭还原/共轭加成
由CuH对α, β-不饱和羰基加成产生的铜烯醇化物可与被吸电子基团活化的双键反应. 2009年, Lam等[48]报道了Cu(OAc)2•2H2O-SEGPhos催化双α, β-不饱和酯或甲基酮的反应, 产物中顺式异构体为优势产物; 而采用CuF(PPh3)3•2MeOH-(S)-L8-PMHS体系, 双α, β-不饱和芳基酮以高反式选择性形成环化产物, 收率中等(Scheme 8).
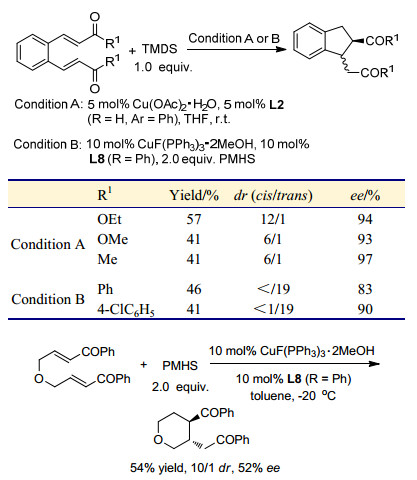 图式 8
Cu引发的还原/分子内Michael加成环化反应
Scheme8.
Intramolecular reductive Michael cyclization induced by Cu
图式 8
Cu引发的还原/分子内Michael加成环化反应
Scheme8.
Intramolecular reductive Michael cyclization induced by Cu
Li等[49]实现了衣康酸酯与Et2Zn的多米诺反应.在不存在羰基和亚胺等亲电子基团的情况下, Et2Zn与两分子的衣康酸酯进行两次连续共轭加成, 然后环化, 高产率地生成取代环戊酮(Scheme 9).使用氢硅烷替代Et2Zn, 可进行还原/共轭加成/环化反应.当其它亲电试剂, 例如醛、酮或亚胺存在时, 进行aldol加成或类似的反应.
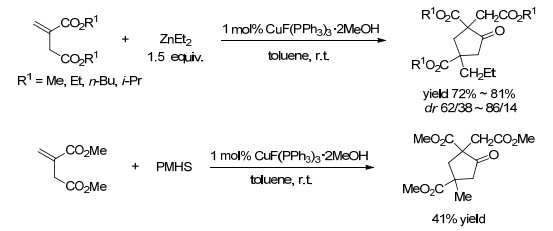 图式 9
衣康酸酯的二聚反应
Scheme9.
Dimerization of itaconate
图式 9
衣康酸酯的二聚反应
Scheme9.
Dimerization of itaconate
1.5 共轭还原/Ireland-Claisen重排等反应
在Morken和Miller[50]报道对Rh催化还原/Irreland-Claisen重排进行多米诺反应的基础上, 2016年, Chiu[51]实现了相应的Cu催化的高产率和非对映选择性还原/ Claisen重排反应(Scheme 10), 其中用到二乙氧基甲基氢硅烷(DEMS)还原剂和P(OEt)3配体.机理研究表明, 重排反应通过(E)-氢硅烷基乙烯酮缩醛中间体而不是烯醇Cu进行. (E)-丙烯酸-2-己烯酯与化学计量的[(Ph3P)CuH]6反应并不产生Claisen重排产物, 经后处理后得到简单的还原产物和其它副产物.由NMR观察到还原丙烯酸正己酯的主要产物为(E)-硅烷基乙烯酮缩醛, 该产物无法进行Claisen重排. Chiu的方法避免了采用贵金属, 也不需要Rh催化中所采用的对湿度高度敏感的Cl2MeSiH.
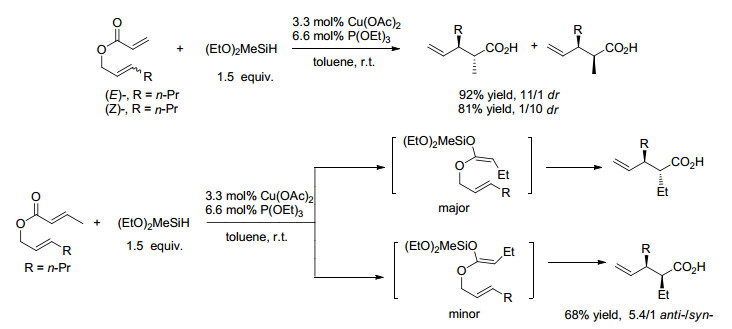 图式 10
Cu催化的还原Ireland-Claisen重排反应
Scheme10.
Cu-catalyzed reductive Ireland-Claisen rearrangement reactions
图式 10
Cu催化的还原Ireland-Claisen重排反应
Scheme10.
Cu-catalyzed reductive Ireland-Claisen rearrangement reactions
最近, Lipshutz等[52]采用CuH催化Morita-Baylis-Hillman (MBH)酯的SN2'反应.在L3 (Ar=苯基)存在下, 以高产率和立体选择性获得α, β-不饱和羧酸酯(Scheme 11, a); 使用MBH酮和手性L3 (Ar=3, 5-二甲基苯基)产生具有确定的烯烃几何形式的对映体富集的手性烯丙醇(Scheme 11, b).借助位阻作用和原位产生的三烷氧基硅离去基团, β位带有取代基的MBH酯可转化为特定立体构型的丙烯醇.
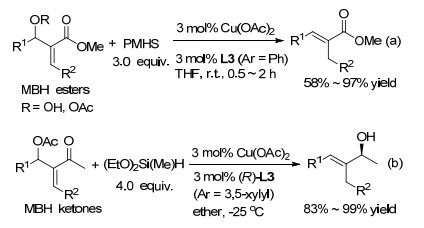 图式 11
Cu催化MBH酯、酮的还原反应及后继反应
Scheme11.
Cu-catalyzed reduction and followed reaction of MBH esters, ketones
图式 11
Cu催化MBH酯、酮的还原反应及后继反应
Scheme11.
Cu-catalyzed reduction and followed reaction of MBH esters, ketones
2 没有共轭吸电子基团的C―C键的反应
2.1 还原/aldol反应
尽管催化剂作用下, C=C键与N―H键[53, 54]和酰基―H键[55]的氢胺化和氢酰化已得到深入研究, 但在温和条件下对未活化的C=C键(即没有共轭吸电子基团)通过分子间氢金属化反应可追踪到2009年, Lam等[56]报道在醇的存在下, 配位的CuH可对映选择性地催化对芳基烯烃的还原反应, 这提供将未活化的C=C键作为潜在的烷基负离子前体的可能性.从此, 此类研究受到高度关注[57~59]. 2012年, Lam等[60]实现了手性膦-铜络合物催化喹啉-2-基乙烯与苯乙酮的还原/aldol反应, 以高马氏选择性得到非对映体的混合物及2-乙基喹啉(Scheme 12). Cu-L7和L8比Cu-L1、L3和L4表现出更高的对映体选择性.在产物的非对映选择性方面, Cu-L8表现更佳, 形成syn-优势异构体, 这归结于优先形成Z-构型N-烯丙基铜中间体.该催化剂适用于杂芳基乙烯、嘧啶基乙烯、噻唑基乙烯与链状、环状酮的反应.
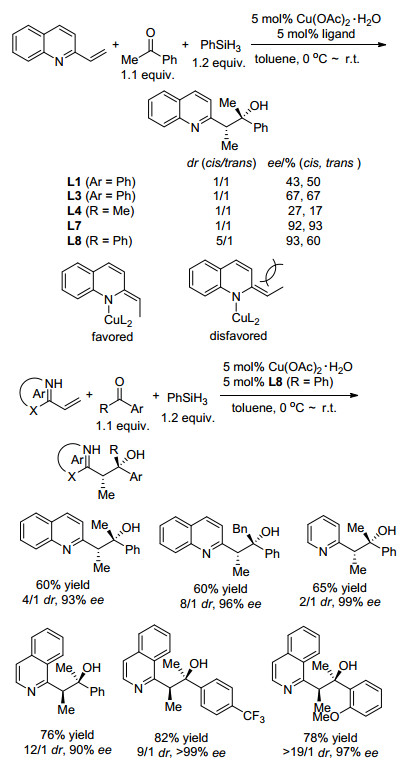 图图 12
Cu催化喹啉基乙烯与酮的对映体选择性还原/aldol加成反应
Scheme12.
Enantioselective Cu-catalyzed reductive aldolization of quinolin-2-yl ethene with a ketone
图图 12
Cu催化喹啉基乙烯与酮的对映体选择性还原/aldol加成反应
Scheme12.
Enantioselective Cu-catalyzed reductive aldolization of quinolin-2-yl ethene with a ketone
Buchwald等[61]在2016年报道了L5 (R=Ph)配位的Cu催化剂作用下的(多)不饱和烃的高对映选择性还原醛/ aldol反应(Eq. 9), CuH区域选择性地插入烯炔或双烯或苯乙烯的C=C键, 对映体选择性地形成炔丙基铜或类似物, 这一结构被相邻的C-C不饱和键所稳定.然而, 由苯乙烯参加的反应仅获得中等的dr值.催化剂可容忍包括酚羟基和羧基等官能团.反应在室温下进行, 具有很好的适用性, 可放大规模进行.
2.2 还原/Mannich反应
Lam等[62]发现以TMDS为还原剂, Cu-(S)-L2 DTBM-SEGPhos对N-杂芳基乙烯与N-Boc亚胺的反应具有良好的手性诱导能力(Eq. 10).在室温下, 以>3/1 dr和59%~94% ee值生成β-杂环胺. Boc保护基易于脱除得到手性胺.
2015年, Buchwald等[63]报道了Cu-(S, S)-L5 (R=Ph)催化亚胺官能化苯乙烯的还原/Mannich反应, 以优异的产率、高dr值和ee值得到了顺式苯并二氢吲哚(Eq. 11).醇添加剂对高产率至关重要, 这归因于L*Cu-N-二氢吲哚中间体的质子化和CuH的后续再生.另一方面, 烷基铜中间体在醇存在下也能被质子化.使用t-BuOD代替t-BuOH, 仅检测到痕量的芳基烷烃(<1%), 苯并二氢吲哚的收率也略有增加.这归结于同位素效应, t-BuOD对烷基铜的质子化比t-BuOH的慢.这种方法条件温和, 有良好的官能团耐受性, 非常有吸引力.
这一催化剂也适于温和条件下苯乙烯与亚胺的分子间反应(Eq. 12)[64].选用L5 (R=Ph)作为配体和N-膦酰基亚胺为底物对高转化率和反应选择性至关重要.在最佳化条件下, 使用N-磺酰基、N-芳基、或N-烷基亚胺为底物, L2或更小体积的L5 (R=Me, Et, i-Pr)为配体, 与4-苯基苯乙烯反应中仅得到较低收率的偶联产物.动力学实验表明L5 (R=Ph)的优势, Ph-BPE存在时, 亚胺还原速率比使用L2慢得多.同样的, 使用t-BuOH对于高产率显得很重要.计算表明, 使用L5 Ph-BPE时, 苯乙烯发生铜氢化的速率与使用DTBM-SEGPhos时的相当, 但比使用SEGPhos时的快180倍.因此, Ph-BPE配位的CuH催化剂可以抑制不期望的亚胺还原反应, 进行期望的铜氢化反应.由于广泛的官能化容忍性和杂环底物适用性, 这一方法可快速合成有两个连续立体中心的胺.就机理而言, Mannich反应中使用亚胺作为亲电试剂, 生成的Cu-N中间体与醇发生转金属化, 然后与硅烷反应生成CuH (Scheme 13).
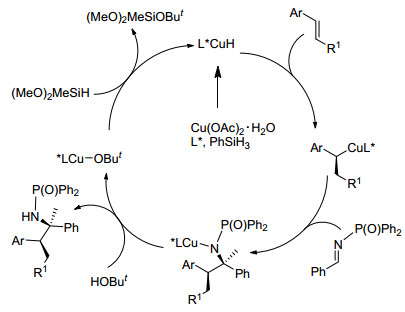 图式 13
Cu催化对亚胺还原/Mannich反应的机理
Scheme13.
Mechanism of Cu-catalyzed reductive Mannich reaction of imines
图式 13
Cu催化对亚胺还原/Mannich反应的机理
Scheme13.
Mechanism of Cu-catalyzed reductive Mannich reaction of imines
2016年, Buchwald等[65]报道了对端位联烯的化学选择性铜氢化和随后的Mannich反应.通过改变亚胺中的氮保护基, 从相同的联烯可获得支链或直链的高烯丙基胺加成物(Scheme 14).在Cu-1, 2-双(二环己基膦基)乙烷(DCyPE)作用下, 由N-苄基亚胺和N-2, 4-二甲氧基苄基亚胺得到支链加成物, 由N-二苯基膦酰基亚胺产生直链型产物.使用对映体纯(S, S)-L5配体, 直链型产物可获高ee值.密度泛函理论计算表明, 对联烯的铜氢化反应主要形成了热力学上有利的末端(E)-烯丙基铜中间体, 从而导致区域选择性反应.在烯丙基化过渡态中, 铜与N-膦酰基中的氧原子或N-苄基亚胺中的氮原子配位, 导致转移烯丙基片段中的末端碳和内部碳, 产生不同的异构体.
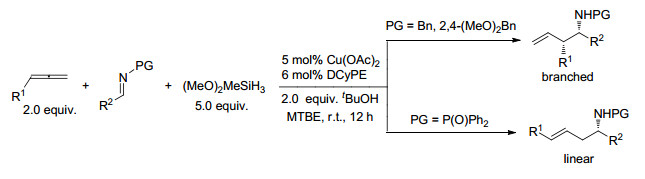 图式 14
丙二烯类化合物的还原/Mannich反应
Scheme14.
Reductive Mannich domino reaction of alllenes with imines
图式 14
丙二烯类化合物的还原/Mannich反应
Scheme14.
Reductive Mannich domino reaction of alllenes with imines
2.3 氢酰化
采用醛对C-C多重键进行氢酰化反应存在低选择性和低产率的问题, 而采用两种化合物分别为氢源和酰基源, 对C-C多重键进行氢酰化的方法则具有高选择性的优点.
1995年, Miura[66]报道以分子氢作为氢源, 苯甲酸酐作为酰化试剂, 叔胺作为碱进行苯乙烯的分子间氢苯甲酰化反应, 由[RhCl(cod)]2和膦配体产生的催化剂可使反应生成1, 2和1, 3-二苯基-1-丙酮的混合物.使用三苯基胂与阳离子型铑催化剂Rh(cod)2]BAr4F(ArF=3, 5-(CF3)2C6H3)配位的催化剂, 高区域选择性、高产率地得到支链氢酰化产物[67].钯催化下, 用酰氯和氢硅烷对联烯类化合物进行氢酰化反应也见报道, 高区域和立体选择性地形成α, β-不饱和酮[68].
在Cu(OAc)2-(S, S)-L4催化下, (取代)苯乙烯与氢硅烷和酸酐进行氢化反应, 在4 ℃下得到酮, 在40 ℃下可以进一步原位还原, 得到高对映选择性的顺式醇(Scheme 15)[69].使用酰氯作为烯醇化物捕获剂可产生β-羰基羧酸酯[70].
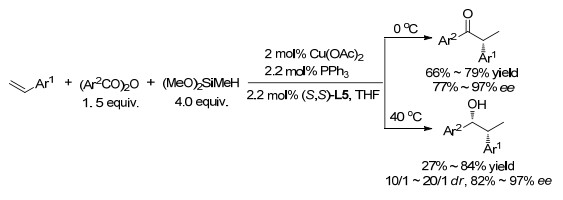 图式 15
芳基烯烃的氢酰化/还原反应
Scheme15.
Hydroacylation/reduction of aryl alkenes
图式 15
芳基烯烃的氢酰化/还原反应
Scheme15.
Hydroacylation/reduction of aryl alkenes
2.4 氢胺化
2013年, 两个课题组独立报道了铜催化苯乙烯与氢硅烷和亲电的O-苯甲酰羟胺的分子间区域选择性氢胺化反应. Hirano和Miura报道, 在1, 2-二氯乙烷中, 过量的LiOBut添加剂存在下, Cu(OAc)2-1, 2-双(二苯基膦基)苯(dppbz)催化苯乙烯与O-苯甲酰基-N, N-二乙基羟胺的反应[71].采用1.2 equiv.的胺化试剂、1.2 equiv.的PMHS和2.4 equiv.的LiOBut时, 产生具有马氏选择性的支链叔胺, 产率为41%.改用1, 2-双(3, 5-双(三氟甲基)苯基)膦基)苯(CF3-dppbz)配体, 产率提高到56%.作者指出配体的空间位阻和缺电子性质抑制了不期望的氢化铜中间体的聚集.优化氢硅烷、溶剂和LiOBut的量, 实现了99%的产率.该催化剂也适用于取代苯乙烯与N, N-二烷基羟胺衍生物的反应.使用10 mol% CuCl和10 mol%对映体纯的L4或L5组成的催化剂, 在四氢呋喃(THF)中反应, 产物的ee值达77%~94%.在后继的报道中, 底物可扩展至氧杂和氮杂双环烯烃, 高产率地形成氧杂和氮杂降冰片烯基和降冰片烷基胺, 具有中等到高的ee值[72].
2013年和2014年, Buchwald还分别报道了芳基烯烃[73]、脂族烯烃[73, 74]与N, N-二取代的O-苯甲酰羟胺(R2NOBz)和二乙氧基甲基氢硅烷(DEMS)的氢胺化反应.使用由Cu(OAc)2, L2和氢硅烷生成的催化剂, 芳基烯烃以马氏选择性生成β-手性胺(Scheme 16, a), 而1, 1-二取代的脂族烯烃以高反马氏区域选择性和高ee值生成胺(Scheme 16, b).该体系不需要Hirano和Miura的方法中所需的LiOBut添加剂.苯乙烯的β-碳上有一个或两个取代基可进行反应.然而, 当使用单烷基胺转移试剂[RN(H)O2CPh]时, 仅以低产率形成仲胺, 胺化试剂被其它反应消耗掉.在进一步工作中, 将胺转移试剂改为不太敏感的N-取代的O-4-二甲基氨基苯甲酰羟胺(RN(H)OBz-4-NMe2), 使用L2 (DBTM-SEGPhos)配体, 由萘乙烯可高区域、高对映选择性高产率地生成手性仲胺(Scheme 16, c)[75].这一体系适于包括碳水化合物、类固醇和氨基酸酯衍生物的多类胺转移试剂.
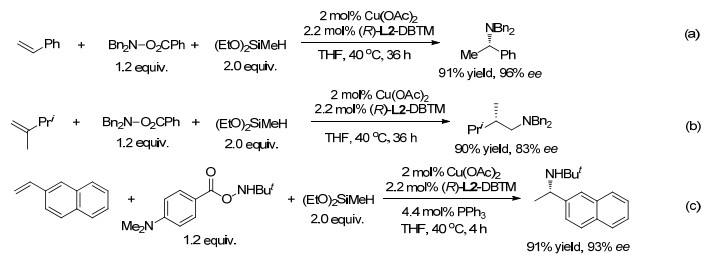 图式 16
Cu催化氢胺化反应形成手性胺
Scheme16.
Cu-catalyzed formation of chiral amines via reductive hydroamination
图式 16
Cu催化氢胺化反应形成手性胺
Scheme16.
Cu-catalyzed formation of chiral amines via reductive hydroamination
在另一篇论文中, Buchwald等[76]报道了乙烯基硅烷的氢胺化反应, 并获得了高区域和高对映选择性的氨基酸模拟物手性α-氨基硅烷(Scheme 17, a).马氏区域选择性可归结于乙烯基硅烷中的β-C的β-硅效应, 导致其带有部分正电荷, 通过超共轭稳定铜氢化产生的α-甲硅烷基烷基铜中间体. E-和Z-烯烃都产生相同的优势对映体, 但E-异构体比Z-异构体的反应活性更高, 并且产物的ee值更高.氘标记实验表明, E-和Z-异构体的铜氢化过程都具有非对映选择性, 这排除了反应过程中较不活泼的Z-异构体异构化为E-异构体的可能性. Buchwald等[77]还将CuH催化的氢胺化扩展至未被活化的内烯烃.由带有两个差别很小的脂族基的对称反式烯, 得到96%或更高ee的α-支链胺(Scheme 17, b).它甚至可以从非对称烯烃例如反-4-甲基-2-戊烯及反-4, 4-二甲基-2-戊烯以非常高的ee和>82/18 dr生成胺.在CuH催化中没有观察到由于β-H消除和迁移插入导致的内烯烃链“行走”现象, 这在Rh和Co催化中经常发生.然而, 需要使用过量的内烯烃以获得高产率. Hartwig等[78]使用化学计量的烯烃(Scheme 17, c), 对非对称的内烯烃进行了区域选择性氢胺化, 其中高烯丙基位置含有氧或氮取代基, 作者认为区域选择性受远程电负极性基团的电子诱导感应控制.采用N, N-二苄基-O-2, 4, 6-三氯苯甲酰基羟胺, 产物具有较高的区域选择性.同样, 使用由L2 DTBM-SEGPhos配位的铜催化剂, 可高对映体选择性地得到产物.
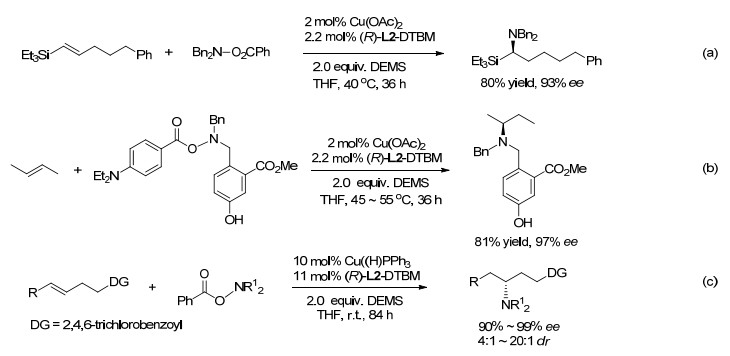 图式 17
Cu催化内烯烃的氢胺化反应
Scheme17.
Cu-catalyzed reductive hydroamination of internal alkenes
图式 17
Cu催化内烯烃的氢胺化反应
Scheme17.
Cu-catalyzed reductive hydroamination of internal alkenes
2016年, Buchwald等[79]报道了CuH催化烯丙基醇或酯或醚与氢硅烷和R2NOBz的高区域选择性氢胺化反应.反应涉及烷基铜的形成及其β-消除, 产生醇盐或羧酸盐以及富有对映体选择性的末端烯烃, 后者被铜氢化和胺化.醇盐或羧酸盐与氢硅烷再生CuH.值得注意的是, 带有β-烷氧基或类似物的三取代烯烃可以参与反应.其异常反应能力归因于加氢后再消除β-醇盐, 为产生稳定的Si―O键提供了条件, 因而, 整个反应在热力学上有利.也值得注意的是, 中间产物3, 3-烷基芳基取代的烯丙基酯以反马氏区域选择性产生接力产物.通过这一方法容易合成含有γ-手性中心的胺(Scheme 18, a).采用手性配体L1, L2或L3, 可以实现高对映体选择性反应.烯基环氧化物和烯基邻二醇缩酮进行插入/消除/氢胺化反应, 产生δ-手性胺(Scheme 18b和c).利用对不同类型双键优良的化学选择性及随后的化学选择性氢胺化, 可产生带两个不同N-取代基的手性二胺的任一立体异构体.
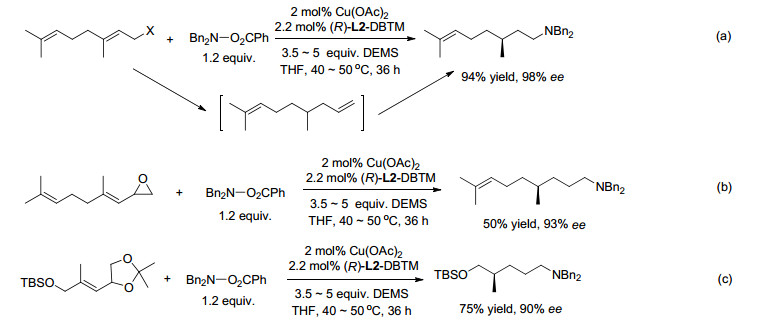 图式 18
通过Cu催化还原/氢酰化反应形成手性胺
Scheme18.
Cu-catalyzed formation of chiral amines via relay reductive hydroamination
图式 18
通过Cu催化还原/氢酰化反应形成手性胺
Scheme18.
Cu-catalyzed formation of chiral amines via relay reductive hydroamination
在铜催化剂存在下, 烯醛很容易还原成烯丙醇, 而烯酮易于以1, 4-加成的方式还原烯烃键.然而, 仔细选择配体可化学选择性和对映体选择性地对烯酮进行1, 2-还原, 产生烯丙基醇[80, 81]. Buchwald组[82]采用Cu-L2 DTBM-SEGPhos催化剂, 将烯醛/烯酮的1, 2-还原与氢胺化反应结合, 借助醛或酮羰基化合物的还原可以在比烯烃胺化反应更低的温度下进行, 快速地合成了氨基醇的立体异构体, 具有高的化学、区域、非对映体和对映体选择性, 产物的立体结构可预测.特别值得注意的, 利用该方法, Buchwald合成出了具有三个连续手性中心氨基醇的所有八种异构体.
在铜催化下对芳基炔烃铜氢化得到的中间体接着与Bn2NOBz反应产生烯胺(Scheme 19, a)[83].与L1 (Ar=Ph), Xantphos, L2 SEGPhos (R=H, Ar=Ph)或L2-DTBM-SEGPhos配位的Cu催化剂对于芳基内炔的反应有效, 按马氏区域选择性生成产物.在醇添加剂和对映体纯的L2-DTBM-SEGPhos存在下, 从芳基炔烃(甚至从末端炔烃)能高ee值地产生手性烷基胺(Scheme 19, b); 而在BINAP或Xantphos或L2存在下则形成烯胺. Buchwald将这一结果归因于Cu-L2-DTBM-SEGPhos独特的能力, 其CuH在炔烃的氢金属化反应中具有更高的反应性能, 醇对形成的烯基铜中间体进行质子化, 生成烯烃, 烯烃接着自发地进行了氢胺化反应.这一机理被下面的实验证实:无论加入或不加入醇类添加剂, 1, 2-二苯基乙炔的氢胺化产物(E)-N, N-二苄基-1, 2-二苯基乙烯胺在THF中与Cu(OAc)2-(R)-L2-DTBM-二乙氧基甲基氢硅烷在45 ℃共处18 h, 并不反应; 但是用(Z)-1, 2-二苯基乙烯代替(E)-N, N-二苄基-1, 2-二苯基乙烯胺, 则以97%产率生成N, N-二苄基-1, 2-二苯基乙胺.这一催化氢胺化反应适用于芳基末端炔烃和烷基末端炔烃, 分别形成马氏烷基胺和反马氏烷基胺.采用该方法, 可快速合成抗抑郁药duloxetine和fluoxetine、尿失禁药tolterodine和抗痴呆药物rivastigmine(图 2).
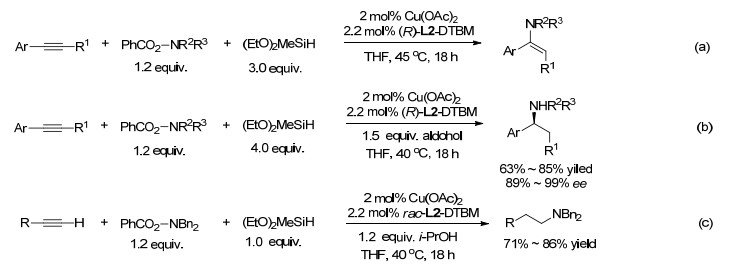 图式 19
Cu催化炔烃的氢胺化反应
Scheme19.
Cu-catalyzed reductive hydroamination of alkynes
图式 19
Cu催化炔烃的氢胺化反应
Scheme19.
Cu-catalyzed reductive hydroamination of alkynes
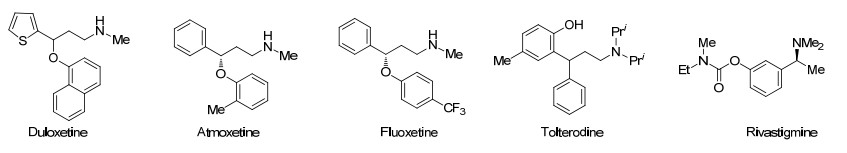 图2
通过Cu催化的氢胺化合成某些生物活性物质
Figure2.
Some bioactive compounds synthesized via Cu-catalyzed hydroamination
图2
通过Cu催化的氢胺化合成某些生物活性物质
Figure2.
Some bioactive compounds synthesized via Cu-catalyzed hydroamination
Hirano和Miura等[84]采用PMHS、O-苯甲酰羟胺与2-取代亚甲基环丙烷反应, 高选择性地生成环丙烷开环产物.由Cu(OAc)2、PMHS和CF3-dppbz配体产生的催化剂在室温下对反应有效, 以良好收率得到高烯丙胺, 开环仅发生在更拥挤的近端位置(Scheme 20).
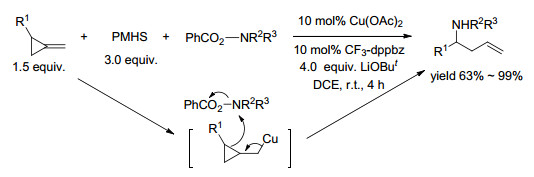 图式 20
Cu催化亚甲基环丙烷的氢胺化反应
Scheme20.
Cu-catalyzed reductive hydroamination of methylenecyclopropanes
图式 20
Cu催化亚甲基环丙烷的氢胺化反应
Scheme20.
Cu-catalyzed reductive hydroamination of methylenecyclopropanes
2.5 还原/C-C偶联反应
通过CuH催化, 对连有卤素的苯乙烯进行分子内加氢烷基化反应, 以47%~95%产率对映选择性地形成反式环丁烷、环戊烷、茚和六元N-和O-杂环(Eq. 13)[85].其关键在于通过使用甲醇锂产生CuH、溴离子作为离去基团及L2 (Ar=DTBM)作为配体.由烷氧基锂转金属化产生的烷氧基铜易于与氢硅烷反应, 再生CuH活性物质.该方法已被应用于5-羟色胺再摄取抑制剂paroxetine的合成(Eq. 14).
Semba、Sakaki和Nakao等[86]报道对芳基烯烃与芳基溴化物的催化铜氢反应和Pd催化反应, 生成了sp2-sp3偶联产物1, 1-二芳基烷烃. Buchwald等[87]使用L2 DTBM-SEGPhos配位的CuH催化反应, 对映选择性地得到1, 1-二芳基烷烃(Eq. 15).采用NaOTMS为碱对反应成功至关重要, 采用LiOTMS或KOTMS则没有产物生成. Buchwald发现加入BrettPhos作为第二种配体, 反应的收率有所提高; 采用CuOAc比用Cu(OAc)2可得到略高的收率和ee值.这一方法适于β-取代的芳基乙烯基和杂芳基溴化物的反应.就机理而言, 由烯烃和CuH产生烷基铜, 该中间体经转金属化, 形成同时联有烷基与芳基的Pd(Ⅱ)物种, 再经还原消除产生C-C偶联产物和联有配体的CuBr, 该铜盐与烷氧基负离子及氢硅烷反应后再生CuH催化物种.
基于醇锂对活化氢硅烷具有良好的效果, Buchwald等[88]报道了CuCl-(S, S)-L5催化芳基烯烃与烯丙基磷酸酯的高对映选择性SN2'反应(Eq. 16).其中用到LiOBut活化氢硅烷和消除形成磷酸盐. CuH催化的还原SN2'反应被扩展到乙烯基-B(pin) (pin=频哪醇)底物[89]. Cu-手性磺酸盐的N-杂环卡宾催化反应在dr值和ee值方面比使用相应的膦配合物表现出更优异的性能, 适于硼基烯与(E)-1, 2-二取代的烯丙基磷酸酯的反应, 经NaBrO3氧化产物中的C―B键, 以46%~91%的产率与98%的SN2'选择性、96/4的dr值和96% ee得到高烯丙醇(Eq. 17).
通过SIPr-CuOTf催化剂实现了末端芳基炔烃或烷基炔烃的氢烷基化, 在CsF存在下, 炔烃与(Me2HSi)2O和伯烷基/苄基三氟甲磺酸酯反应, 以反马氏区域选择性地生成(E)-烯烃(Scheme 21)[90]. SIPrCuF与氢硅烷的反应定量生成SIPrCuH, 该关键中间体在N2下在C6D6可稳定存在0.5 h. SIPrCuH与1.1 equiv.的炔烃在室温反应4 s、5 min, 生成烯基铜的收率分别为51%、95%; SIPrCuH与1.1 equiv.的R2OTf在4 s内完全反应, 但仅消耗0.5 equiv.的R2OTf, 生成还原产物R2H, 同时生成(IPrCu)2(μ-H)(OTf).这表明IPrCuH活性物种与R2OTf的反应速率远快于与炔烃的反应.双核化合物(NHC-Cu)2(μ-H)(OTf) (X=F, H, 烯基)与炔烃的高效氢烷基化有关(Scheme 21)[91].
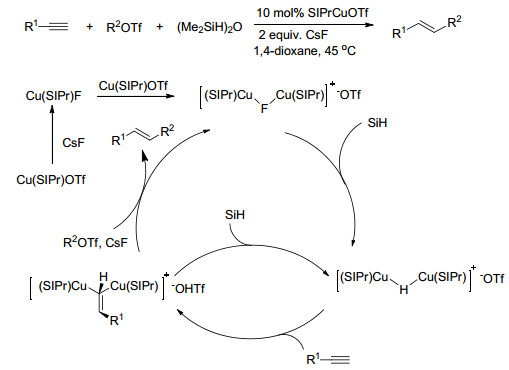 图式 21
Cu催化端基炔烃的氢烷基化反应及机理
Scheme21.
Cu-catalyzed hydroalkylation of terminal alkynes, and the mechanism
图式 21
Cu催化端基炔烃的氢烷基化反应及机理
Scheme21.
Cu-catalyzed hydroalkylation of terminal alkynes, and the mechanism
3 结论
烯烃铜氢化引发的多米诺反应在过去几十年得到快速发展.作为还原剂, 氢硅烷由于合适的反应性和低毒性, 已获得广泛应用.从铜氢化生成的烯醇化物或烷基金属化合物可以自发进行aldol缩合、Mannich反应或取代反应.通过仔细选择手性配体已经实现了许多高对映选择性反应.然而, 许多分子间反应仍然存在低非对映选择性的问题.实现多步多米诺反应和由Buchwald组开发的对不带吸电子基团的C-C不饱和键的反应代表这这一领域的最新发展方向.
-
-
[1]
Tietze, L. F. Chem. Rev. 1996, 96, 115. doi: 10.1021/cr950027e
-
[2]
Pellissier, H. Chem. Rev. 2013, 113, 442. doi: 10.1021/cr300271k
-
[3]
Saebang, Y.; Rukachaisirikul, V.; Kaeobamrung, J. Tetrahedron Lett. 2017, 58, 168. doi: 10.1016/j.tetlet.2016.12.006
-
[4]
Pellissier, H. Adv. Synth. Catal. 2015, 357, 2745. doi: 10.1002/adsc.v357.13
-
[5]
李雄武, 汪朝阳, 郑绿茵, 有机化学, 2006, 26, 1144. doi: 10.3321/j.issn:0253-2786.2006.08.024Li, X.-W.; Wang, C.-Y.; Zheng, L.-Y. Chin. J. Org. Chem. 2006, 26, 1144(in Chinese). doi: 10.3321/j.issn:0253-2786.2006.08.024
-
[6]
陈杰, 刘钦, 戴小鸯, 聂凛凛, 房辉辉, 吴小余, 有机化学, 2013, 33, 1. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201301002&dbname=CJFD&dbcode=CJFQChen, J.; Liu, Q.; Dai, X.-Y.; Nie, L.-L.; Fang, H.-H.; Wu, X.-Y. Chin. J. Org. Chem. 2013, 33, 1(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201301002&dbname=CJFD&dbcode=CJFQ
-
[7]
朱辉, 叶长青, 陈知远, 有机化学, 2015, 35, 2291. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201511004&dbname=CJFD&dbcode=CJFQZhu, H.; Ye, C.-Q.; Chen, Z.-Y. Chin. J. Org. Chem. 2015, 35, 2291(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201511004&dbname=CJFD&dbcode=CJFQ
-
[8]
冯亚栋, 张红, 程国林, 崔秀灵, 有机化学, 2014, 34, 1499. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201408005&dbname=CJFD&dbcode=CJFQFeng, Y.-D.; Zhang, H.; Cheng, G.-L.; Cui, X.-L. Chin. J. Org. Chem. 2014, 34, 1499(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201408005&dbname=CJFD&dbcode=CJFQ
-
[9]
Zhang, W.; Wang, Y.; Bai, C.; Wen, J.; Wang, N. Chin. J. Chem. 2015, 33, 401. doi: 10.1002/cjoc.v33.4
-
[10]
Schmid, T. E.; Drissi-Amraoui, S.; Crévisy, C.; Baslé, O.; Mauduit, M. Beilstein J. Org. Chem. 2015, 11, 2418. doi: 10.3762/bjoc.11.263
-
[11]
Phelan, J. P.; Ellman, J. A. Beilstein J. Org. Chem. 2016, 12, 1203. doi: 10.3762/bjoc.12.116
-
[12]
于艳飞, 单凤军, 李争宁, 姜岚, 有机化学, 2011, 31, 443. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201104004&dbname=CJFD&dbcode=CJFQYu, Y. F.; San, F.-J.; Li, Z.-N.; Jiang, L. Chin. J. Org. Chem. 2011, 31, 443(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201104004&dbname=CJFD&dbcode=CJFQ
-
[13]
唐凤翔, 叶久勇, 有机化学, 2015, 35, 1414. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201507004&dbname=CJFD&dbcode=CJFQTang, F.-X.; Ye, J.-Y. Chin. J. Org. Chem. 2015, 35, 1414(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201507004&dbname=CJFD&dbcode=CJFQ
-
[14]
Mori, A.; Fujita, A.; Nishihara, Y.; Hiyama, T. Chem. Commun. 1997, 2159. http://www.ncbi.nlm.nih.gov/pubmed/16703140
-
[15]
Jordan, A. J.; Lalic, G.; Sadighi, J. P. Chem. Rev. 2016, 116, 8318. doi: 10.1021/acs.chemrev.6b00366
-
[16]
方嫱, 隋耀宗, 虞景露, 谢琳洁, 杨黎耀, 吴俊文, 吴静, 杭州师范大学学报(自然科学版), 2015, 14, 1. doi: 10.3969/j.issn.1674-232X.2015.01.001Fang, Q.; Sui, Y.-Z.; Yu, J.-L.; Xie, L.-J.; Yaag, L.-Y.; Wu, J.-W.; Wu, J. J. Hangzhou Normal Univ. (Nat. Sci. Ed.) 2015, 14, 1(in Chinese). doi: 10.3969/j.issn.1674-232X.2015.01.001
-
[17]
Galestokova, Z.; Sebesta, R. Eur. J. Org. Chem. 2012, 6688.
-
[18]
Pellissier, H. Adv. Synth. Catal. 2016, 358, 2194. doi: 10.1002/adsc.v358.14
-
[19]
Li, Z.; Liu, G.; Pauline, C. Prog. Chem. 2008, 20, 1909. http://en.cnki.com.cn/article_en/cjfdtotal-hxjz200812007.htm
-
[20]
Lipshutz, B. H. Synlett 2009, 509.
-
[21]
Deutsch, C.; Krause, N.; Lipshutz, B. H. Chem. Rev. 2008, 108, 2916. doi: 10.1021/cr0684321
-
[22]
Rendler, S.; Oestreich, M. Angew. Chem., Int. Ed. 2007, 46, 498. doi: 10.1002/(ISSN)1521-3773
-
[23]
Chiu, P. Synthesis-Stuttgart 2004, 2210. http://hub.hku.hk/handle/10722/97502
-
[24]
Nishiyama, H.; Shiomi, T. In Metal Catalyzed Reductive C-C Bond Formation:A Departure from Preformed Organometallic Reagents, Ed.:Krische, M. J., Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, p. 105.
-
[25]
Revis, A.; Hilty, T. K. Tetrahedron Lett. 1987, 28, 4809. doi: 10.1016/S0040-4039(00)96631-0
-
[26]
Chiu, P.; Leung, S. K. Chem. Commun. 2004, 2308.
-
[27]
Lam, H. W.; Joensuu, P. M. Org. Lett. 2005, 7, 4225. doi: 10.1021/ol051649h
-
[28]
Zheng, A.; Jiang, L.; Li, Z. Chin. J. Chem. 2012, 30, 2587. doi: 10.1002/cjoc.201200523
-
[29]
Lam, H. W.; Murray, G. J.; Firth, J. D. Org. Lett. 2005, 7, 5743. doi: 10.1021/ol052599j
-
[30]
Lipshutz, B. H.; Amorelli, B.; Unger, J. B. J. Am. Chem. Soc. 2008, 130, 14378. doi: 10.1021/ja8045475
-
[31]
Deschamp, J.; Riant, O. Org. Lett. 2009, 11, 1217. doi: 10.1021/ol802879f
-
[32]
Ou, J.; Wong, W. T.; Chiu, P. Org. Biomol. Chem. 2012, 10, 5971. doi: 10.1039/c2ob25206f
-
[33]
Zhao, D.; Oisaki, K.; Kanai, M.; Shibasaki, M. Tetrahedron Lett. 2006, 47, 1403. doi: 10.1016/j.tetlet.2005.12.097
-
[34]
Li, Z.; Jiang, L.; Li, Z.; Chen, H. Chin. J. Chem. 2013, 31, 539. doi: 10.1002/cjoc.201300036
-
[35]
Zhao, D.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 14440. doi: 10.1021/ja0652565
-
[36]
Deschamp, J.; Chuzel, O.; Hannedouche, J.; Riant, O. Angew. Chem., Int. Ed. 2006, 45, 1292. doi: 10.1002/(ISSN)1521-3773
-
[37]
Kato, M.; Oki, H.; Ogata, K.; Fukuzawa, S.-I. Synlett 2009, 1299. doi: 10.1055/s-0029-1216724
-
[38]
Chuzel, O.; Deschamp, J.; Chausteur, C.; Riant, O. Org. Lett. 2006, 8, 5943. doi: 10.1021/ol062398v
-
[39]
Welle, A.; Diez-Gonzalez, S.; Tinant, B.; Nolan, S. P.; Riant, O. Org. Lett. 2006, 8, 6059. doi: 10.1021/ol062495o
-
[40]
Li, Z.; Zhang, Z.; Yuan, L.; Jiang, L.; Li, Z.; Li, Z. Synlett 2014, 25, 724. doi: 10.1055/s-00000083
-
[41]
Li, R.; Li, Z.; Jiang, L.; Li, Z. Chem. Pap. 2017, 71, 1825. doi: 10.1007/s11696-017-0175-y
-
[42]
Li, Z.; Li, R.; Gan, M.; Jiang, L.; Li, Z. Tetrahedron Lett. 2015, 56, 5541. doi: 10.1016/j.tetlet.2015.08.031
-
[43]
Yang, T.; Zhang, Y.; Cao, P.; Wang, M.; Li, L.; Li, D.; Liao, J. Tetrahedron 2016, 72, 2707. doi: 10.1016/j.tet.2015.12.062
-
[44]
Li, Z.; Wang, C.; Li, Z. Beilstein J. Org. Chem. 2015, 11, 213. doi: 10.3762/bjoc.11.23
-
[45]
Du, Y.; Xu, L.-W.; Shimizu, Y.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2008, 130, 16146. doi: 10.1021/ja8069727
-
[46]
Suto, Y.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 500. doi: 10.1021/ja068226a
-
[47]
Li, Z.; Feng, Y.; Li, Z.; Jiang, L. Synlett 2014, 25, 2899. doi: 10.1055/s-00000083
-
[48]
Oswald, C. L.; Peterson, J. A.; Lam, H. W. Org. Lett. 2009, 11, 4504. doi: 10.1021/ol901560r
-
[49]
Li, Z.; Li, R.; Jiang, L.; Li, Z. Molecules 2015, 20, 15023. doi: 10.3390/molecules200815023
-
[50]
Miller, S. P.; Morken, J. P. Org. Lett. 2002, 4, 2743. doi: 10.1021/ol026273b
-
[51]
Wong, K. C.; Ng, E.; Wong, W. T.; Chiu, P. Chem. Eur. J. 2016, 22, 3709. doi: 10.1002/chem.201504870
-
[52]
Linstadt, R. T. H.; Peterson, C. A.; Jette, C. I.; Boskovic, Z. V.; Lipshutz, B. H. Org. Lett. 2017, 19, 328. doi: 10.1021/acs.orglett.6b03464
-
[53]
Reznichenko, A. L.; Nawara-Hultzsch, A. J.; Hultzsch, K. C. In Stereoselective Formation of Amines, Eds.:Li, W.; Zhang, X., Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, p. 191.
-
[54]
Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L. J. Chem. Rev. 2015, 115, 2596. doi: 10.1021/cr300389u
-
[55]
Murphy, S. K.; Dong, V. M. Chem. Commun. 2014, 50, 13645. doi: 10.1039/C4CC02276A
-
[56]
Rupnicki, L.; Saxena, A.; Lam, H. W. J. Am. Chem. Soc. 2009, 131, 10386. doi: 10.1021/ja904365h
-
[57]
Soradova, Z.; Sebesta, R. ChemCatChem 2016, 8, 2581. doi: 10.1002/cctc.201600252
-
[58]
Pirnot, M. T.; Wang, Y. M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2016, 55, 48. doi: 10.1002/anie.201507594
-
[59]
Gribble, M. W.; Pirnot, M. T.; Bandar, J. S.; Liu, R. Y.; Buchwald, S. L. J. Am. Chem. Soc. 2017, 139, 2192. doi: 10.1021/jacs.6b13029
-
[60]
Saxena, A.; Choi, B.; Lam, H. W. J. Am. Chem. Soc. 2012, 134, 8428. doi: 10.1021/ja3036916
-
[61]
Yang, Y.; Perry, I. B.; Lu, G.; Liu, P.; Buchwald, S. L. Science 2016, 353, 144. doi: 10.1126/science.aaf7720
-
[62]
Choi, B.; Saxenaa, A.; Smith, J. J.; Churchill, G. H.; Lam, H. W. Synlett 2015, 26, 350. doi: 10.1055/s-0034-1379548?device=desktop
-
[63]
Ascic, E.; Buchwald, S. L. J. Am. Chem. Soc. 2015, 137, 4666. doi: 10.1021/jacs.5b02316
-
[64]
Yang, Y.; Perry, I. B.; Buchwald, S. L. J. Am. Chem. Soc. 2016, 138, 9787. doi: 10.1021/jacs.6b06299
-
[65]
Liu, R. Y.; Yang, Y.; Buchwald, S. L. Angew. Chem., Int. Ed. 2016, 55, 14077. doi: 10.1002/anie.v55.45
-
[66]
Kokubo, K.; Miura, M.; Nomura, M. Organometallics 1995, 14, 4521. doi: 10.1021/om00010a016
-
[67]
Hong, Y.-T.; Barchuk, A.; Krische, M. J. Angew. Chem., Int. Ed. 2006, 45, 6885. doi: 10.1002/(ISSN)1521-3773
-
[68]
Fujihara, T.; Tatsumi, K.; Terao, J.; Tsuji, Y. Org. Lett. 2013, 15, 2286. doi: 10.1021/ol400862k
-
[69]
Bandar, J. S.; Ascic, E.; Buchwald, S. L. J. Am. Chem. Soc. 2016, 138, 5821. doi: 10.1021/jacs.6b03086
-
[70]
Sato, K.; Isoda, M.; Ohata, S.; Morita, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Adv. Synth. Catal. 2012, 354, 510. doi: 10.1002/adsc.v354.2/3
-
[71]
Miki, Y.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2013, 52, 10830. doi: 10.1002/anie.201304365
-
[72]
Miki, Y.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2014, 16, 1498. doi: 10.1021/ol5003219
-
[73]
Zhu, S.; Niljianskul, N.; Buchwald, S. L. J. Am. Chem. Soc. 2013, 135, 15746. doi: 10.1021/ja4092819
-
[74]
Zhu, S.; Buchwald, S. L. J. Am. Chem. Soc. 2014, 136, 15913. doi: 10.1021/ja509786v
-
[75]
Niu, D.; Buchwald, S. L. J. Am. Chem. Soc. 2015, 137, 9716. doi: 10.1021/jacs.5b05446
-
[76]
Niljianskul, N.; Zhu, S.; Buchwald, S. L. Angew. Chem., Int. Ed. 2015, 54, 1638. doi: 10.1002/anie.201410326
-
[77]
Yang, Y.; Shi, S. L.; Niu, D. W.; Liu, P.; Buchwald, S. L. Science 2015, 349, 62. doi: 10.1126/science.aab3753
-
[78]
Xi, Y.; Butcher, T. W.; Zhang, J.; Hartwig, J. F. Angew. Chem., Int. Ed. 2016, 55, 776. doi: 10.1002/anie.201509235
-
[79]
Zhu, S. L.; Niljianskul, N.; Buchwald, S. L. Nat. Chem. 2016, 8, 144. doi: 10.1038/nchem.2418
-
[80]
Moser, R.; Boskovic, Z. V.; Crowe, C. S.; Lipshutz, B. H. J. Am. Chem. Soc. 2010, 132, 7852. doi: 10.1021/ja102689e
-
[81]
Mohr, J.; Oestreich, M. Angew. Chem., Int. Ed. 2016, 55, 12148. doi: 10.1002/anie.201606701
-
[82]
Shi, S.-L.; Wong, Z. L.; Buchwald, S. L. Nature 2016, 532, 353. doi: 10.1038/nature17191
-
[83]
Shi, S. L.; Buchwald, S. L. Nat. Chem. 2015, 7, 38. doi: 10.1038/nchem.2131
-
[84]
Nishikawa, D.; Sakae, R.; Miki, Y.; Hirano, K.; Miura, M. J. Org. Chem. 2016, 81, 12128. doi: 10.1021/acs.joc.6b02483
-
[85]
Wang, Y.-M.; Bruno, N. C.; Placeres, A. L.; Zhu, S.; Buchwald, S. L. J. Am. Chem. Soc. 2015, 137, 10524. doi: 10.1021/jacs.5b07061
-
[86]
Semba, K.; Ariyama, K.; Zheng, H.; Kameyama, R.; Sakaki, S.; Nakao, Y. Angew. Chem., Int. Ed. 2016, 55, 6275. doi: 10.1002/anie.201511975
-
[87]
Friis, S. D.; Pirnot, M. T.; Buchwald, S. L. J. Am. Chem. Soc. 2016, 138, 8372. doi: 10.1021/jacs.6b04566
-
[88]
Wang, Y.-M.; Buchwald, S. L. J. Am. Chem. Soc. 2016, 138, 5024. doi: 10.1021/jacs.6b02527
-
[89]
Lee, J.; Torker, S.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2017, 56, 821. doi: 10.1002/anie.201611444
-
[90]
Uehling, M. R.; Suess, A. M.; Lalic, G. J. Am. Chem. Soc. 2015, 137, 1424. doi: 10.1021/ja5124368
-
[91]
Suess, A. M.; Uehling, M. R.; Kaminsky, W.; Lalic, G. J. Am. Chem. Soc. 2015, 137, 7747. doi: 10.1021/jacs.5b03086
-
[1]
-

图式 1 由Cu催化氢金属化反应引发的多米诺反应
Scheme 1 General domino reactions initialized by Cu-catalyzed hydrometallation

图 1 Cu催化氢金属化引发多米诺反应中用到的手性配体
Figure 1 Some chiral ligands in Cu-catalyzed hydrometallation -induced domino reactions

图式 2 Cu催化还原/分子内aldol加成反应
Scheme 2 Cu-catalyzed reductive intramolecular aldol reaction

图式 3 经CuH催化还原/aldol加成环化多米诺反应构建多环化合物
Scheme 3 Construction of a polycyclic derivative via CuH-catalyzed domino reductive aldol cyclizations

图式 4 Cu催化13-二羰基衍生物的还原/分子内aldol加成反应
Scheme 4 Cu-catalyzed reductive intramolecular aldol reaction of enethiolate derivatives of 1, 3-diones

图式 5 Cu催化丙二烯酸酯的不对称还原/分子间aldol加成反应
Scheme 5 Cu-catalyzed asymmetric, reductive intermolecular aldol reaction of allenic ester

图式 6 Cu催化马来酸酯的还原/aldol加成/内酯化反应及机理
Scheme 6 Cu-catalyzed reductive aldol addition/lactonization reaction of maleate and the mechanism

图式 7 Cu催化马来酸酯或富马酸酯与酮、Et2Zn的还原或烷基化/aldol加成/内酯化反应
Scheme 7 Cu-catalyzed reductive or alkylative aldol addition/lactonization reaction of maleate or fumarate with ketones and Et2Zn

图式 8 Cu引发的还原/分子内Michael加成环化反应
Scheme 8 Intramolecular reductive Michael cyclization induced by Cu

图式 10 Cu催化的还原Ireland-Claisen重排反应
Scheme 10 Cu-catalyzed reductive Ireland-Claisen rearrangement reactions

图式 11 Cu催化MBH酯、酮的还原反应及后继反应
Scheme 11 Cu-catalyzed reduction and followed reaction of MBH esters, ketones

图 12 Cu催化喹啉基乙烯与酮的对映体选择性还原/aldol加成反应
Figure 12 Enantioselective Cu-catalyzed reductive aldolization of quinolin-2-yl ethene with a ketone

图式 13 Cu催化对亚胺还原/Mannich反应的机理
Scheme 13 Mechanism of Cu-catalyzed reductive Mannich reaction of imines

图式 14 丙二烯类化合物的还原/Mannich反应
Scheme 14 Reductive Mannich domino reaction of alllenes with imines

图式 16 Cu催化氢胺化反应形成手性胺
Scheme 16 Cu-catalyzed formation of chiral amines via reductive hydroamination

图式 17 Cu催化内烯烃的氢胺化反应
Scheme 17 Cu-catalyzed reductive hydroamination of internal alkenes

图式 18 通过Cu催化还原/氢酰化反应形成手性胺
Scheme 18 Cu-catalyzed formation of chiral amines via relay reductive hydroamination

图 2 通过Cu催化的氢胺化合成某些生物活性物质
Figure 2 Some bioactive compounds synthesized via Cu-catalyzed hydroamination

图式 20 Cu催化亚甲基环丙烷的氢胺化反应
Scheme 20 Cu-catalyzed reductive hydroamination of methylenecyclopropanes
-


 下载:
下载:






















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 40
- 文章访问数: 3448
- HTML全文浏览量: 773
 下载:
下载:
