 图式1
铜催化2-卤代芳基硫脲串联反应合成芳基氰胺
图式1.
Copper-catalyzed cascade reaction of 2-haloarylth-ioureas for the synthesis of arylcyanamides
图式1
铜催化2-卤代芳基硫脲串联反应合成芳基氰胺
图式1.
Copper-catalyzed cascade reaction of 2-haloarylth-ioureas for the synthesis of arylcyanamides
引用本文:
黎吉辉, 李正章, 张玉苍, 许文茸, 徐树英. 氰胺类化合物的合成及其应用研究进展[J]. 有机化学,
2017, 37(8): 1903-1915.
doi:
10.6023/cjoc201706003

Citation: Li Jihui, Li Zhengzhang, Zhang Yucang, Xu Wenrong, Xu Shuying. Progress on the Synthesis and Applications of Cyanamides[J]. Chinese Journal of Organic Chemistry, 2017, 37(8): 1903-1915. doi: 10.6023/cjoc201706003
Citation: Li Jihui, Li Zhengzhang, Zhang Yucang, Xu Wenrong, Xu Shuying. Progress on the Synthesis and Applications of Cyanamides[J]. Chinese Journal of Organic Chemistry, 2017, 37(8): 1903-1915. doi: 10.6023/cjoc201706003
氰胺类化合物的合成及其应用研究进展
English
Progress on the Synthesis and Applications of Cyanamides
Abstract:
Cyanamides are an important class of fine chemicals containing amino and cyano functionalities, which have been widely used for the synthesis of pharmaceuticals, agricultural chemicals, health products and materials, and attracted considerable attention from both organic synthetic chemists and medicinal chemists. Great advances in the synthesis and transformations of cyanamides were made, a diversity of synthetic methods and transformations of cyanamides were developed in the past two decades. In this paper, various synthetic methods and reactions of cyanamides are introduced comprehensively, their characteristics, rules, advantages and disadvantages are also summarized and discussed for the development of new synthetic methods and reactions of cyanamides.
-
Key words:
- cyanamides
- / synthetic methods
- / reactivities
- / nitrogen compounds
-
氰胺(R1R2NCN)为一类重要的二氮化合物, 具有多样性生物活性, 如杀虫活性、杀菌活性[1]、抗组织蛋白酶活性[2]和抗磷酸二酯酶4(PDE4) 活性[3]等, 广泛应用于农药和医药等领域.另外, 氰胺是一种重要的化工原料和有机合成药物中间体[4], 同时也是一种良好的金属配体[5].
由于氰胺在农药、医药、染料和有机合成等领域中都有广阔的应用前景, 引起了有机合成化学家和药物合成化学家的广泛关注.近20年来, 合成化学家们对氰胺的合成方法及其在有机合成中的应用展开全面研究, 取得了丰硕的研究成果.
最简单的氰胺为NH2CN, 是一种重要的有机化工中间体和医药原料, 可通过尿素在Ca(OH)2促进下高温脱水反应制备得到, 该方法已实现工业化[6].随着氰胺在不同领域应用的推广和深入, 对各种氰胺类化合物的需求也不断提高, 促进了氰胺类化合物合成方法的发展.近年来, 各种氰胺类化合物新的合成方法层出不穷, 特别是一些高效、低毒环保的合成方法.同时, 氰胺合成方法的发展有力地推动氰胺反应活性的研究, 一系列反应被发现并广泛应用于合成各种含氮类化合物, 拓展了氰胺在有机合成中的应用.本文主要综述了近20年来氰胺类化合物的合成方法及其在有机合成中的应用研究进展.
1 氰胺的合成
早期, 合成氰胺的方法主要基于NH-胺1和氰化溴(2)的亲核取代反应[7](Eq. 1) 及von Braun反应[8](Eq. 2).后来Parry等[9]又发展了固载化的胺和氰化溴的反应实现固相合成氰胺.这些方法原料简单易得、操作简便, 但存在比较突出的缺陷, 以剧毒的氰化溴为氰基化试剂, 且反应受电子和位阻效应影响严重, 位阻大或者贫电子的胺, 反应产率低.
由于氰化溴的剧毒性, 限制了上述合成方法的推广应用, 因此发展以低毒性化合物为原料的合成方法已成为氰胺合成亟待解决的问题.为此, 近十年来合成化学家们发展了各种以低毒性化合物为原料的合成方法.
2009年Satyanarayan等[10]以氨基二硫甲酸的铵盐6为起始原料, 通过“一锅煮”三步反应直接合成氰胺7 (Eq. 3).氨基二硫甲酸的铵盐首先在三乙胺和碘作用下发生还原脱硫反应生成异硫氰酸酯, 异硫氰酸酯接着和氨进行亲核加成和还原脱硫反应生成氰胺.该反应避免使用高毒性的氰基化合物为原料, 是较环保的合成方法, 但该反应只适用于合成一取代氰胺.
2010年, Punniyamurthy等[11]报道了CuSO4·5H2O催化2-碘芳基硫脲(8)和芳基碘(9)串联反应, 高效合成2-芳基硫代芳基氰胺(10) (Scheme 1, a).作者提出以下反应机理:首先, 2-卤代芳基硫脲在铜催化下进行分子内C—S偶联反应生成噻唑A.接着, 芳基碘和铜发生氧化加成反应, 并进一步和噻唑进行反应产生中间体B.最后B发生还原消除反应即可生成目标产物.几个月后, Patel课题组[12]又意外发现CuI催化2-卤代芳基硫脲(11)反应合成芳基氰胺类化合物(10) (Scheme 1, b).在配体协助下, CuI催化2-卤代芳基硫脲和碘代芳烃反应生成2-芳基硫代芳基氰胺.该催化体系对底物具有良好的兼容性, 2-碘代和2-溴代芳基硫脲都能适应反应.此外, 他们还发现, 在无配体条件下CuI催化2-卤代芳基硫脲的反应则直接发生脱硫反应生成2-卤代芳基氰胺12.
图式1
铜催化2-卤代芳基硫脲串联反应合成芳基氰胺
图式1.
Copper-catalyzed cascade reaction of 2-haloarylth-ioureas for the synthesis of arylcyanamides
2014年, Chen等[13]以毒性稍小的三甲基氰硅烷(TMSCN)为氰基化试剂, 发展了NaOCl/H2O促进二级胺1的亲电氰基化反应制备二取代氰胺3, 反应能够生成中等偏上的产率(Scheme 2). TMSCN先和NaOCl及H2O发生反应原位生成氰基氯(A), 接着和胺发生亲核取代反应产生目标产物.
 图式2
胺和TMSCN偶联反应合成氰胺
图式2.
Coupling reaction of amines and TMSCN for the synthesis of cyanamides
图式2
胺和TMSCN偶联反应合成氰胺
图式2.
Coupling reaction of amines and TMSCN for the synthesis of cyanamides
2014年Chien等[14]发展了氰14和羟胺15的两步一锅煮反应合制备一取代氰胺16, 反应生成51%~95%的产率(Eq. 4).羟胺和氰基化合物首先进行亲核加成反应生成脒肟, 接着在碱和磺酰氯的作用下发生Tiemann重排脱水生成氰胺.作者还发现咪肟的Tiemann重排和水解一锅煮反应, 实现对脲类化合物的合成.
2016年Morrill等[15]以胺1和三氯乙氰17为原料, 通过一锅煮两步反应实现对氰胺3的直接合成(Eq. 5).胺和三氯乙氰先进行亲核加成, 接着在在碱性条件下发生脱氯仿反应生成二取代氰胺.该反应可生成中等偏上的产率, 但底物适用范围窄, 只有二级胺适应于该反应.此外, 作者还将反应应用于合成具有抗PDE4活性的氰胺.
过渡金属催化多组分反应同样也已被应用于合成氰胺类化合物, 2001年Yamamoto课题组[16]发展了钯催化异氰18、三甲基硅叠氮19和烯丙醇酯20三组分反应高效合成氰胺21 (Scheme 3).反应可以实现一步构建多个化学键, 高产率生成芳基烯基氰胺.同时, 反应受底物的电子和位阻效应影响小, 且对氰基、酯基和氯等官能团都有良好的兼容性.基于实验结果, 作者提出了以下反应机理:烯丙醇酯、三甲基硅叠氮和钯催化剂先反应生成叠氮π-烯丙基钯A.叠氮π-烯丙基钯接着和异氰反应生成π-烯丙基钯络合物B, B发生类似Curtius重排的反应释放出氮气产生中间体C, C转化成互变异构体D(或E).最后, D(或者E)发生还原消除反应即可生成目标产物和钯催化剂.
 图式3
钯催化异氰、三甲基硅叠氮三组分反应合成氰胺
图式3.
Palladium-catalyzed three-component reaction of isocyanides, allyl carbonate and trimethylsilyl azide for the synthesis of cyanamides
图式3
钯催化异氰、三甲基硅叠氮三组分反应合成氰胺
图式3.
Palladium-catalyzed three-component reaction of isocyanides, allyl carbonate and trimethylsilyl azide for the synthesis of cyanamides
接下来, 他们以邻位炔基化的异氰苯22为原料, 在高温条件下, 成功将反应应用于合成N-氰基吲哚类化合物(Scheme 4)[17].该反应可以实现一步构建多个化学键直接合成吲哚类化合物.其反应过程和非邻位炔基的异氰反应相似, 经过多步反应产生中间体B, B接着发生连续的分子内碳碳三键氨基钯化和还原消除反应生成最终产物.
 图式4
钯催化邻炔基芳基异氰、烯丙醇酯和三甲基硅叠氮三组分反应合成N-氰基吲哚
图式4.
Palladium-catalyzed three-component reaction of 2-alkynylaryl isocyanides, allyl carbonate and trimethylsilyl azide for the synthesis of N-cyanoindoles
图式4
钯催化邻炔基芳基异氰、烯丙醇酯和三甲基硅叠氮三组分反应合成N-氰基吲哚
图式4.
Palladium-catalyzed three-component reaction of 2-alkynylaryl isocyanides, allyl carbonate and trimethylsilyl azide for the synthesis of N-cyanoindoles
后来Yamamoto课题组[18]又发现钯催化异氰24和三甲基硅叠氮(19)的反应也可以直接合成一取代氰胺25 (Scheme 5).该反应可应用于合成芳基氰胺和脂肪族氰胺, 生成中等偏高的产率, 对酯、氰基、炔基等官能团都具有良好的兼容性.反应可能经历以下反应历程:钯和TMSN3发生氧化加成反应产生钯络合物A, 再和异氰进行加成反应生成中间体B. B发生类似Curtius重排的反应释放出氮气产生C, C转化成互变异构体D(或E), D(或E)发生还原消除产生F, F最后在硅胶柱色谱纯化过程中脱去三甲基硅生成目标产物.
 图式5
钯催化异氰和三甲基硅叠氮反应合成氰胺
图式5.
Palladium-catalyzed reaction of isocyanides and trimethylsilyl azide for the synthesis of cyanamides
图式5
钯催化异氰和三甲基硅叠氮反应合成氰胺
图式5.
Palladium-catalyzed reaction of isocyanides and trimethylsilyl azide for the synthesis of cyanamides
除了上述构键氰胺官能团的合成方法, N, N-二取代氰胺还可通过NH-氰胺和碘化物的偶联反应快速合成.
2012年, Louie等[19]证实钯催化NH-氰胺26和卤代烃类27的C—N偶联反应可以简便合成二芳基氰胺28 (Eq. 6).芳基氰胺和脂肪族氰胺都适应反应, 生成中等偏上的产率.反应受卤代芳烃的位阻效应影响较大, 大位阻2-甲基苯基溴和2, 4, 6-三甲基苯基溴的反应没有任何的产物生成.
后来, Cui等[20]发现芳香氰胺(29)和六氟磷酸二芳基碘盐(30)可以在碱性无过渡金属条件下发生C—N偶联反应生成二取代氰胺31 (Scheme 6).反应在室温下进行, 条件温和, 但为了避免氰胺被氧化, 反应需要惰性气体的保护.另外, 作者还发现氰胺可以先和六氟磷酸二芳基碘盐进行反应生成芳基化氰胺, 芳基化氰胺接着在酸和铜共同作用下和三氟甲磺酸二芳基碘盐及H2O发生三组分反应实现一锅煮合成多取代脲类化合物32.
 图式6
氰胺和二芳基碘盐偶联反应合成氰胺和脲
图式6.
Cross coupling of NH-cyanamides and diaryliodoniums for the synthesis of cyanamides and ureas
图式6
氰胺和二芳基碘盐偶联反应合成氰胺和脲
图式6.
Cross coupling of NH-cyanamides and diaryliodoniums for the synthesis of cyanamides and ureas
最近, 我们课题组[21]证实NH-氰胺33和三氟甲磺酸二芳基碘盐34的C—N偶联反应可以在水溶液中实现, 高效合成二取代氰胺35 (Eq. 7).该反应操作间单, 不需要惰性气体的保护.此外, 底物适用范围广, 芳基氰胺和脂肪族氰胺都可以发生反应, 芳基氰胺的产率普遍比脂肪族氰胺的高.
2 氰胺的应用
氰胺含有氨基和氰基两种活性基团, 兼具亲核和亲电的反应性能, 易发生加成、取代、缩合等反应, 是一类多功能有机合成中间体, 可应用于合成多种含氮类化合物.下面我们将对氰胺在有机合成中的应用进行详细介绍.
2.1 胍及其衍生物的合成:
以氰胺为原料, 其和亲核试剂的加成反应已被应用于合成在医药、农药和材料等领域有广泛应用价值的胍及其衍生物.
早期, 氰胺和亲核试剂的加成反应主要应用于链状胍、异脲、硫代异脲的合成, 这类反应具有100%的原子经济性[22].后来, 合成化学家们将其推广到环胍及其衍生物的合成中, 其中Looper课题组[23]做了系统的研究工作. 2009年, 他们结合亲核加成和分子内的氢胺化反应, 发展了炔丙基氰胺36和胺37的串联反应直接合成2-氨基咪唑38 (Eq. 8).在La(OTf)3催化下, 胺和炔丙基氰胺首先发生亲核加成反应生成链状胍, 链状胍接着进行分子内炔氢氨化反应生成2-氨基咪唑. 2010年, 他们成功将反应拓展到2-硫基咪唑40和2-氧基咪唑42的合成[24].在碱促进作用下, 炔丙基氰胺即可以分别和硫醇39及醇41发生串联反应生成2-硫基咪唑(Eq. 8) 和2-氧基咪唑(Eq. 10).
2015年Fu等[25]同样基于氰胺的加成反应, 发展了铜催化氰胺43和邻卤代芳胺44的多米诺反应合成稠环胍45 (Eq. 11).氰胺首先和邻卤代芳烃发生C—N偶联反应, 接着再进行分子内连续的亲核加成和氢氨化反应即可实现对稠环胍的构建.
最近, Rassadin等[26]以氰胺46为原料, 发展了和吡啶N-氧47的无溶剂多米诺反应, 高效合成脲48 (Scheme 7).在甲磺酸的促进作用下, 氰胺和吡啶N-氧化物先发生亲核加成, 接着进行一系列重排反应生成N-(2-吡啶基)脲.该反应的原子利用率为100%, 且可在无溶剂条件下进行, 是一种绿色环保的脲类化合物的合成方法.
 图式7
氰胺和吡啶N-氧多米诺反应合成脲类化合物
图式7.
Domino reaction of cyanamides and pyridine N-oxides for the synthesis of ureas
图式7
氰胺和吡啶N-氧多米诺反应合成脲类化合物
图式7.
Domino reaction of cyanamides and pyridine N-oxides for the synthesis of ureas
除了氰胺的亲核加成反应, 近年来研究发现其自由基加成反应也可以实现, 并成功应用到多种胍类化合物的合成中.
2010年, Malacria和Lacôte等[27]证实N-酰基氰胺49在Bu3SnH/AIBN促进下引发自由基串联反应高效合成稠环胍50 (Scheme 8).在Bu3SnH/AIBN促进下, 叠氮基先生成氨基自由基, 接着发生串联的自由基加成和C—N偶联反应产生稠环胍.
 图式8
N-酰基氰胺自由基串联反应合成稠环胍
图式8.
Radical cascade reaction of N-acyl cyanamides for the synthesis of fused cyclic guanidines
图式8
N-酰基氰胺自由基串联反应合成稠环胍
图式8.
Radical cascade reaction of N-acyl cyanamides for the synthesis of fused cyclic guanidines
两年后, Malacria和Lacôte等[28]又发现N-酰基氰胺51和芳香二硫52在Hünig碱和光照条件作用下可发生自由基串联反应合成硫代异脲53 (Scheme 9).在光照条件下, 芳香二硫化合物和Hünig碱发生自由基反应生成硫醇自由基, 接着和氰基发生加成和后续的重排反应生成硫代异脲类化合物.此外, 基于该反应, 他们发展了N-酰基氰胺、二硫化合物和胺的一锅煮三组分反应, 实现对胍类化合物的直接合成.
 图式9
N-酰基氰胺和二硫化合物自由基串联反应合成异脲类化合物
图式9.
Radical cascade reaction of N-acyl cyanamides and diaryldisulfides for the synthesis of isothioureas.
图式9
N-酰基氰胺和二硫化合物自由基串联反应合成异脲类化合物
图式9.
Radical cascade reaction of N-acyl cyanamides and diaryldisulfides for the synthesis of isothioureas.
最近Neuville和Li等发展了铜催化氰胺54、脂肪胺55和硼酸56三组分反应高效合成N, N', N''-三取代胍57 (Scheme 10)[29].由于反应在氧气氛围下进行, 底物适用范围有一定的局限性且对官能团兼容性差.作者对反应机理进行了深入研究, 提出了以下反应机理:硼酸先和铜发生金属转移反应生成中间体A. A接着和氰胺进行配位, 并在碱作用下发生去质子化和重排生成B'. B'进行氧化、亲核加成和还原消除的串联反应生成目标产物.后来, 基于该三组分反应, 他们通过利用不同的氰胺为原料, 实现一锅煮反应直接合成2-氨基苯并咪唑和2-氨基喹啉[30].
 图式10
铜催化氰胺、胺和硼酸三组分反应合成胍
图式10.
Copper-catalyzed three-component reaction of cyanamides, amines and boronic acides for the synthesis of guanidines
图式10
铜催化氰胺、胺和硼酸三组分反应合成胍
图式10.
Copper-catalyzed three-component reaction of cyanamides, amines and boronic acides for the synthesis of guanidines
由于上述铜催化氰胺、胺和硼酸三组分反应的氧化反应条件对底物和官能团的兼容性较差, 且一些硼酸原料的热稳定差, 使得该反应的应用有一定的局限性.为了克服上述反应中存在缺点, 最近我们课题组以三氟甲磺酸二芳基碘盐58为芳基化试剂, 发展了铜催化其和氰胺54及胺55三组分反应高效构建胍59 (Eq. 12)[31].相交于之前发展的铜催化硼酸的三组分反应[30], 本反应具有更广泛的适用性.脂肪族和芳香族胺都能适应反应, 但芳香族胺的产率较低.杂环芳基碘盐同样也适用于本反应, 可用于合成芳杂环取代的胍类化合物.另外, 具有环张力的环丙基氰胺同样以较好的产率生成相应环丙基胍.然而, 该反应中二芳基碘盐仅有一个芳基参与反应, 反应原子经济性低.
2.2 2-氨基氮杂环的合成
过渡金属催化氰胺和炔的环加成反应是一类高效的有机反应, 具有很高的原子经济性, 倍受合成化学家的青睐, 广泛应用于各类环状含氮化合物的合成, 如广泛存在于药物分子中的2-氨基吡啶和2-氨基喹啉类化合物.
过渡金属催化氰胺和炔的环加成反应早在1978年就已经有报道[32].随着对其研究的深入, 近年来陆续有过渡金属催化氰胺和二炔的环加成反应的报道. 2004年Maryanoff等[33]报道了钴促进二取代氰胺61和二炔60的[2+2+2]环加成反应, 高效合成稠环氨基吡啶62 (Scheme 11).以CoCp(CO)2为促进剂, 反应在回流的1, 4-二氧六环溶剂中进行, 具有100%的原子经性.反应可能经历以下反应历程: CoCp(CO)2首先和二炔配位并释放出CO2生成络合物A, A接着发生炔氧化加成生成环钴二烯络合物B. B和氰胺发生类似Diels-Alder的环加成反应产生C, 最后C发生还原消除生成目标产物和钴催化剂.
 图式11
钴促进二炔和氰胺[2+2+2]环加成反应合成稠环2-氨基吡啶
图式11.
Cobalt-promoted [2+2+2] cycloaddition of bis-alkynes and cyanamides for the synthesis of fused 2-aminopyridines
图式11
钴促进二炔和氰胺[2+2+2]环加成反应合成稠环2-氨基吡啶
图式11.
Cobalt-promoted [2+2+2] cycloaddition of bis-alkynes and cyanamides for the synthesis of fused 2-aminopyridines
Louie等[34]证实以[Ni(cod)2]/IMes为催化剂, 可以实现二取代氰胺64和二炔63的[2+2+2]环加成催化反应得到65 (Eq. 13).反应可实现在低催化剂量和室温条件下进行, 条件比较温和, 对官能团具有良好的兼容性.反应具有良好的选择性, 不对称二炔反应生成的产物是氨基在较大基团邻位的稠环吡啶.此外, 作者还证实该催化体系还适用于氰胺和两分子单炔的反应[34a].
后来Wan课题组[35]和Louie课题组[36]又分别发现以廉价的铁为催化剂, 同样可以催化二取代氰胺67和二炔66的[2+2+2]环加成反应合成氨基稠环吡啶68 (Eq. 14). Louie等以FeCl2/L1为催化体系, 反应需要在加热条件下进行; 在该催化体系下, 不对称二炔的反应生成的产物主要是氨基在较小基团邻位的稠环吡啶. Wan和合作者则以FeI2/dppp为催化体系, 可实现反应在室温下进行; 该催化体系的区域选择性和FeCl2/L1的刚好互补, 不对称二炔反应的产物主要是氨基在较大基团邻位的稠环吡啶.
一年后, Louie课题组[37]还将铁催化体系应用到氰胺70和炔氰69的[2+2+2]环加成反应中, 实现对稠环2-氨基喹啉71的合成(Eq. 15).作者通过反应条件筛选优化, 找出最佳的催化体系为FeI2/i-PrPDAI, 反应可以在较低的温度下进行.
2015年, Takeuchi等[38]以铱为催化剂, 同样也可以高效催化二取代氰胺73和二炔72的[2+2+2]环加成反应, 反应适用范围广且对官能团的兼容性强, 可以很好地兼容酯基、羟基和羰基等官能团(Eq. 16).通过实验证实, 不对称二炔反应的区域选择性主要受电子效应控制, 二炔中富电子的一端容易和氰基中的碳相结合, 而缺电子的一端则和氰基中的氮相结合.此外, 作者还发现氰胺反应的产率普遍比氰反应[39]的高, 并通过理论计算对该现象进行解释.
最近, Ratovelomanana-Vidal和Michelet等[40]报道了Cp*Ru(CH3CN)3PF6催化α, ω-二炔75和二取代氰胺76的[2+2+2]环加成反应高效合成稠环2-氨基吡啶77 (Eq. 17).反应的条件温和, 对官能团具有良好的兼容性, 能兼容羟基、酯基、氰基和卤素等官能团.不对称二炔的反应生成很高的区域选择性, 主产物为位阻较小2-氨基吡啶类化合物.
除了和二炔的反应, 2015年Rassadin等[41]发现单炔78也可以和二取代氰胺79及2-甲基吡啶氧发生[2+2+1]环化反应生成2-氨基恶唑类化合物80 (Eq. 18).反应以Ph3PAuNTf2为催化剂和甲磺酸为促进剂, 在中等温度下进行, 生成中等偏上的产率.同时, 作者还发现了氰胺和2-甲基吡啶氧在甲基磺酸促进下反应生成脲类化合物.作者提出在金催化下炔先和2-甲基吡啶氧化物生成ɑ-氧金卡宾中间体IM-1, 接着和氰胺发生多步反应产生目标产物.
2.3 氰基化合物的合成
氰胺类化合物作为一类重要的亲电氰基化试剂, 已被广泛应用于合成具有高应用价值的氰基化合物.常见的亲电氰基化试剂有1-氰基苯并三唑[42]、1-氰基咪唑[43]、N-氰基丁二酰亚胺、1-氰基苯并咪唑[44]、N-氰基-N-苯基对甲苯磺酰胺(NCTS)[45]和二烷基氰胺[46]等, 其中N-氰基-N-苯基对甲苯磺酰胺(NCTS)具有毒性低、稳定和实用等优点, 近年来对其反应活性的研究异常活跃.
2011年, Beller和Anbarasan等[45a]以NCTS (81)为氰基化试剂, 发展了[{Rh(OH)(cod)2}]催化硼酸82的氰基化反应合成氰类化合物83 (Scheme 12).相较于金属氰化物的亲核氰基化反应, 该反应避免了使用剧毒试剂; 但反应中往往产生N-苯胺对苯磺酰胺副产物, 反应的原子经济性低下.结合已报道的铑催化芳基硼酸和不饱和键的加成反应机理, 作者推断以下反应过程:芳基硼酸和铑发生金属转移生成芳基铑A, A接着和NCTS配位产生络合物B.然后, B进行分子内的芳基转移产生芳基脒络合物C.最后, C发生还原消除生成芳基氰和氨基铑化物D, D在碱性条件和芳基硼酸反应再生成铑活性催化剂形成催化循环.同年, 他们又将NCTS应用至和格氏试剂的亲电氰基化反应中合成各种芳基氰, 反应中格氏试剂通过镁试剂和芳基溴化物的反应原位生成[45b]. 4年后, Gosmini等[45c]报道了CoBr2/Zn促进芳基溴和NCTS的氰基化反应.作者通过实验证实, CoBr2首先催化芳基溴先和Zn反应生成芳基锌试剂, 接着再催化芳基锌试剂和NCTS进行交叉偶联生成芳基氰.反应对酯基、烷硫基、氰基和酮羰基等活性官能团都有良好的兼容性.
 图式12
铑催化硼酸和NCTS氰基化反应合成氰
图式12.
Rhodium catalyzed cyanation of boronic acids with NCTS for the synthesis of nitriles
图式12
铑催化硼酸和NCTS氰基化反应合成氰
图式12.
Rhodium catalyzed cyanation of boronic acids with NCTS for the synthesis of nitriles
同样是在2011年, Wang等[45d]以NCTS为氰基化试剂, 在BF3·Et2催化下分别和吲哚及吡咯84发生C—H活化/氰基化反应直接合成氰基化合物85 (Eq. 19).吲哚化合物的反应选择性发生3-位上, 而吡咯的反应则主要发生在2-位上.相较于官能化芳烃的反应, 本方法直接使用简单易得的芳杂烃类化合物为原料, 减少对芳烃的官能化操作, 且具有较高的原子经济性.
2013年, Fu课题组[45e]将NCTS应用到芳烃(86)的导向氰基化反应中, 成功实现C—H活化直接合成芳基氰类化合物87 (Eq. 20).作者发现以肟醚、咪唑、吡啶和吡唑等作为导向基团, 在[RhCp*(CH3CN)3](SbF6)2的催化下都可以实现对导向基团邻位C(sp2)—H的活化反应合成直接邻位氰基化产物.该反应对官能团具有良好的兼容性, 羟基、碘、酯、烷硫基和环氧乙环等基团都能在反应中存活.几乎在同一时间, Anbarasan和合作者同样以NCTS为氰基化试剂, 发展了[Cp*RhCl2]2催化吡啶基导向的邻位C(sp2)—H活化氰基化反应合成2-(2-吡啶基)苯甲氰类化合物[45f].这两种C(sp2)—H活化氰基化反应的发现引起了合成化学家们对过渡金属催化芳烃和NCTS的直接氰基化反应研究的热潮.近四年来, 以NCTS为氰基化试剂, 铑催化不同基团导向的邻位C(sp2)—H活化反应陆续被发现, 如芳基磷酸酯邻位氰基化反应[45g]、N-酰基吲哚啉的7-氰基化反应、N-(2-嘧啶基)吲哚的2-氰基化反应[45h]、N-(2-吡啶基)吲哚的2-氰基化反应、N-(2-吡啶基)吡咯的2-氰基化反应[45i]和芳基咪唑并[1, 2-α]吡啶的双氰基化反应[45j]等.
除了作为氰基化试剂, 氰胺还被广泛用作氨基氰基化试剂, 和不饱键发生加成反应合成氰基取代的胺类化合物.
2016年Chien等[47]以铜为催化剂, 催化邻炔基芳香氰胺88的分子内氨基氰基化反应高效合成3-氰基吲哚89 (Scheme 13).作者提出了以下反应机理:铜先和末端炔反应生成炔基铜A, 接着和氰胺基发生CN基转移反应生成氰基亚烯基铜中间体B, 最后氨基和氰基亚烯基铜进行亲核加成和质子化反应生成目标产物.
 图式13
铜催化邻炔基芳香氰胺环化反应合成3-氰基吲哚
图式13.
Copper-catalyzed cyclization of o-alkynyl arylcyanamides for the synthesis of 3-cyanoindoles
图式13
铜催化邻炔基芳香氰胺环化反应合成3-氰基吲哚
图式13.
Copper-catalyzed cyclization of o-alkynyl arylcyanamides for the synthesis of 3-cyanoindoles
2014年Zeng课题组[48]报道了氰胺和苯炔前体的分子间氨基氰基化反应, 高效合成双官能分子2-氨基苯甲氰类化合物.在CsF的促进下, 2-(三甲基硅基)苯酚三氟甲磺酸酯(90)和芳基氰胺(91)反应生成2-氨基苯甲氰类化合物92 (Scheme 14).作者对反应过程进行探索, 并提出了以下反应机理: 2-(三甲基硅基)苯酚三氟甲磺酸酯在CsF的作用下脱掉三甲基硅基和三氟甲磺酸基生成苯炔, 苯炔接着和氰胺进行加成及后续的重排和质子化反应产生2-氨基苯甲氰.
 图式14
CsF促进2-(三甲基硅基)苯酚三氟甲磺酸酯和氰胺串联反应合成2-氨基苯甲氰类化合物
图式14.
CsF-promoted cascade reaction of cyanamides and (trimethylsilyl)aryl trifluoromethanesulfonate for the synthesis of 2-aminobenzonitriles
图式14
CsF促进2-(三甲基硅基)苯酚三氟甲磺酸酯和氰胺串联反应合成2-氨基苯甲氰类化合物
图式14.
CsF-promoted cascade reaction of cyanamides and (trimethylsilyl)aryl trifluoromethanesulfonate for the synthesis of 2-aminobenzonitriles
除了和炔的氨基氰基化反应, 氰胺还可以和烯烃发生氨基氰基化反应, 这类反应已被广泛应用于β-氰基胺类化合物的合成, 具有100%的原子经济性.
2014年Douglas等[49]报道了无金属Lewis酸促进的分子内双键氨基氰基化反应直接合成2-氰甲基吲哚啉(94) (Scheme 15).作者以[2-(N-对甲苯磺酰基)氰胺基苯基]烯烃(93)为原料, 在B(C6F5)3促进作用下, 实现对氰胺基C-CN的切断并和双键发生加成反应构建2-氰甲基吲哚啉.基于实验结果, 作者提出了以下反应机理:氰胺和B(C6F5)3配位生成中间体A, A发生烯烃对氰基碳的亲核进攻产生中间体C. C最后发生重排即可生成最终产物和促进剂B(C6F5)3.
 图式15
B(C6F5)3促进分子内烯烃氨基氰基化反应合成吲哚啉
图式15.
B(C6F5)3-promoted intramolecular aminocyanation of alkenes by cyanamides for the synthesis of indolines
图式15
B(C6F5)3促进分子内烯烃氨基氰基化反应合成吲哚啉
图式15.
B(C6F5)3-promoted intramolecular aminocyanation of alkenes by cyanamides for the synthesis of indolines
后来, Nakao和其合作者[50]又发展了钯催化分子内烯烃氨基氰基化反应.他们以N-酰基氰胺(95)为原料, 在CpPd(allyl)和BR43的共同作用下发生烯烃氨基氰基化反应合成吲哚啉96 (Scheme 16).基于实验结果, 作者对反应机理进行了推断: N-酰基氰胺和硼烷进行配位生成A, A的氰氨基接着和[Pd0]发生氧化加成生成C. C进行exo-trig顺式氨基钯化反应产生D. D最后发生还原消除反应和硼烷转移反应生成目标产物、[Pd0]催化剂和络合物A形成催化循环.
 图式16
钯催化分子内氨基氰基化反应合成吲哚啉
图式16.
Palladium-catalyzed intramolecular aminocyanation of alkenes for the synthesis of indolines
图式16
钯催化分子内氨基氰基化反应合成吲哚啉
图式16.
Palladium-catalyzed intramolecular aminocyanation of alkenes for the synthesis of indolines
2013年Wang等[51]发展了铑催化芳基乙烯97的分子内β-氰基化反应, 合成2-(2-氨基芳基)烯氰类化合物98 (Eq. 21).作者发现在[RhCl(COD)]2]/DPEphos催化下, 邻氰胺基芳基乙烯发生N—CN键断裂并和双键发生H和CN交换反应生成(2-氨基芳基)氰基乙烯类化合物.
2.4 脒及其衍生物的合成
脒为一类既是有多样性生物活性分子, 又是多功能的有机中间体.氰胺作为合成脒类化合物的一类重要前体, 已被广泛应用于合成各种脒类化合物.
2007年Malacria和合作者[52]报道了Bu3SnH/AIBN引发N-酰基氰胺99的分子内自由基串联反应高效构建四环喹唑啉100, 实现一步构建两个环骨架(Scheme 17).同时, 他们通过对反应条件的调整, 成功将该反应应用于合成天然生物碱luotonin A.作者基于底物考察的结果, 并通过密度泛函理论计算提出了以下反应机理: 2-碘代苄基氰胺和Bu3SnH/AIBN进行反应产生芳基自由基A, A发生碳氮叁键加成生成脒亚胺自由基B.活性中间体B进行6-endo-trig加成及芳化反应(烯基化合物发生的是还原反应), 或者[1, 5]原位取代和重排反应生成目标产物.
 图式17
N-酰基氰胺自由基串联反应合成四环稠合喹唑啉
图式17.
Radical cascade reaction of N-acyl cyanamides for the synthesis of tetracycle-fused quinazolines
图式17
N-酰基氰胺自由基串联反应合成四环稠合喹唑啉
图式17.
Radical cascade reaction of N-acyl cyanamides for the synthesis of tetracycle-fused quinazolines
几年后, 他们[53]又发展了N-酰基氰胺(101)的自由基串联反应合成三环喹唑啉102 (Scheme 18).作者对反应过程进行详细研究, 提出了以下反应机理: N-酰基氰胺和Bu3SnH/AIBN发生多步反应产生中间体A. A发生6-endo-trig加成生成B. B脱去自由基R3·生成中间体C. B和C之间进行自由基R3·迁移产生C和D. D最后发生自由基氢化生成目标产物.
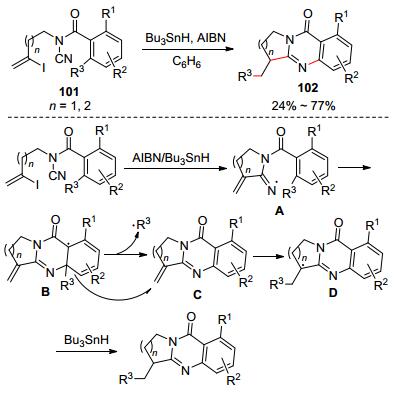 图式18
N-酰基氰胺自由基串联反应合成三环喹唑啉
图式18.
Radical cascade reaction of N-acyl cyanamides for the synthesis of tricycle-fused quinazolines
图式18
N-酰基氰胺自由基串联反应合成三环喹唑啉
图式18.
Radical cascade reaction of N-acyl cyanamides for the synthesis of tricycle-fused quinazolines
除了应用于环脒类化合物的合成, 通过氰胺反应合成链状脒类化合物也已有相关报道. 2012年, Larhed课题组[54]通过钯催化芳基三氟硼酸钾103和氰胺104的加成反应, 高效合成芳基脒类化合物105 (Scheme 19).他们以三氟醋酸钯为催化剂、6-甲基-2, 2'-二吡啶为配体和三氟乙酸为添加剂, 在微波促进作用下芳基三氟硼酸钾和氰胺发生亲核加成反应快速生成芳基脒.带供电子基团或者弱拉电子基团的芳基三氟硼酸钾都能参与反应生中等偏上的产率.基于实验结果, 作者对反应进行推断:二价钯络合物A和芳基三氟硼酸钾发生金属转移反应产生中间体B. B再和氰胺发生配体交换生成C, C进行碳氮三键的钯芳基插入反应产生D.最后, D进行质子化产生目标产物和钯催化剂.
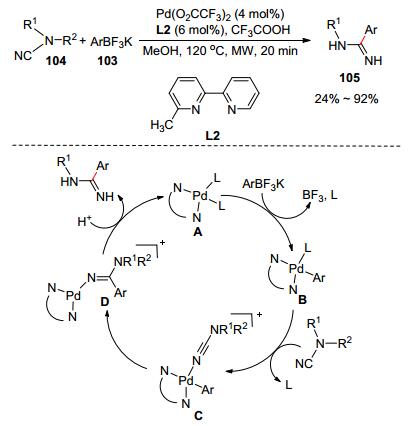 图式19
钯催化氰胺和芳基三氟硼酸钾加成反应合成芳基脒
图式19.
Palladium-catalyzed addition of cyanamides and potassium arlyltrifluoroborates for the synthesis of arylamidines
图式19
钯催化氰胺和芳基三氟硼酸钾加成反应合成芳基脒
图式19.
Palladium-catalyzed addition of cyanamides and potassium arlyltrifluoroborates for the synthesis of arylamidines
一年后, Larhed课题组[55]又证实了Pd(O2CCF3)2/6-甲基-2, 2'-二吡啶催化体系同样可以催化芳基甲酸(106)和氰胺107的脱羧加成反应, 生成芳香脒108 (Eq. 22).该反应对底物的适用范围有一定的局限性, 只有邻位带有供电子基团的芳香羧酸和脂肪氰胺适应于本反应.作者利用ESI-MS技术对反应过进行研究, 并通过密度泛函理论计算, 提出了类似芳基三氟硼酸钾和氰胺反应的机理(Scheme 19).芳基甲酸先和二价钯络合物进行配体交换生成羧基钯, 接着发生脱羧反应产生中间体B. B再进行配体交换、氰基的钯芳基插入反应和质子化串联反应即可生成目标产物.
类似地, 氰胺还可以应用于合成α-酮酰胺类化合物. Patel等[56]发展了钯催化氰胺109和α-酮酸110的串联反应高效合成α-酮酰胺类化合物111 (Scheme 20).在氧化条件下, Pd(O2CCF3)2催化α-酮酸和氰胺发生多步串联反应生成中等偏上产率的α-酮酰胺类化合物, 对卤素、氰基等官能团具有良好的兼容性.通过机理研究, 作者提出以下反应过程:氰胺和二价钯催化剂配位形成钯络合物A, 接着和α-酮酸氧化脱羧产生的酰基自由基在氧化条件下反应生成四价钯络合物B. B进行1, 2-位钯迁移生成C, 再进行还原消除反应生成中间体D和钯催化剂.最后, D在酸促进下发生水解反应生成目标产物.
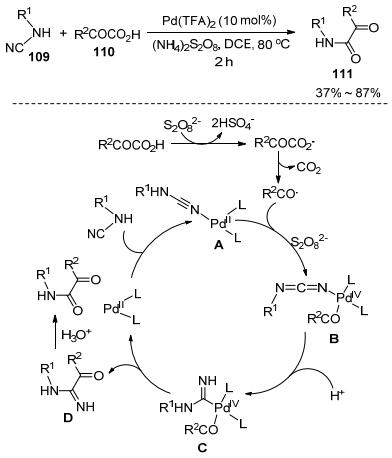 图式20
钯催化氰胺和α-羰基羧酸脱羧加成反应合成α-酮酰胺
图式20.
Palladium-catalyzed decarboxylative addition of cyanamides and α-oxocarboxylic acids for the synthesis of α-ketoamides
图式20
钯催化氰胺和α-羰基羧酸脱羧加成反应合成α-酮酰胺
图式20.
Palladium-catalyzed decarboxylative addition of cyanamides and α-oxocarboxylic acids for the synthesis of α-ketoamides
3 结论与展望
氰胺作为一种具有多样性生物活性的分子和多功能的精细化工中间体, 在医药、农药、材料和染料等领域都有广泛的应用, 激起了合成化学家对其合成方法和反应活性研究的热情.
氰胺类化合物的合成, 经历了由早期直接或者间接使用剧毒氰化溴原料到近些年来利用低毒性非氰化合物为原料的绿色发展.氰胺的合成方法大部分都是基于传统的两组分反应, 近年来通过过渡金属催化多组分反应合成多样性氰胺类化合物的方法也有报道.过渡金属催化多组分反应合成方法高效且具有良好的选择性, 能够实现对结构多样性氰胺类的合成, 但目前这类合成方法的研究还处于初级阶段.随着氰胺在药学和有机合成领域应用的推广, 对多样性氰胺类化合物有迫切的需求.因此, 发展低毒性原料的过渡金属催化多组分反应实现绿色合成结构多样性氰胺类化合物将是氰胺合成方法研究的一个重要方向.
随着氰胺合成方法研究的不断推进, 及大的促进了氰胺反应活性研究的发展.近20年来, 各种氰胺参与的反应已被发现, 高效合成各种含氮化合物, 如胍类化合物、2-氨基吡啶类化合物、脒类化合物和β-氰基胺类化合物等.这些反应中主要涉及氨基的亲电反应、氰基的亲核反应或者N-CN切断反应等, 氰胺仅作为单性反应试剂参与反应.氰胺为含有氨基和氰基双官能团试剂, 具有亲电性和亲核性, 可用作双性反应试剂, 但其作为双性反应试剂参与反应的研究工作鲜有报道, 所以以其作为双性合成子合成多样性含氮化合物的工作有待进一步挖掘, 也是氰胺反应活性亟待研究的重点问题.
-
-
[1]
Zhu, Y.; Loso, M. R.; Watson, G. B.; Sparks, T. C.; Rogers, R. B.; Huang, J. Z.; Gerwick, B. C.; Babcock, J. M.; Kelley, D.; Hegde, V. B.; Nugent, B. M.; Renga, J. M.; Denholm, I.; Gorman, K.; DeBoer, G. J.; Hasler, J.; Meade, T.; Thomas, J. D. J. Agric. Food Chem. 2011, 59, 2950. doi: 10.1021/jf102765x
-
[2]
(a) Lainé, D.; Palovich, M.; McCleland, B.; Petitjean, E.; Delhom, I.; Xie, H.; Deng, J.; Lin, G.; Davis, R.; Jolit, A.; Nevins, N.; Zhao, B.; Villa, J.; Schneck, J.; McDevitt, P.; Midgett, R.; Kmett, C.; Umbrecht, S.; Peck, B.; Davis, A. B.; Bettoun, D. ACS Med. Chem. Lett. 2011, 2, 142.
(b) Falgueyrat, J.-P.; Oballa, R. M.; Okamoto, O.; Wesolowski, G.; Aubin, Y.; Rydzewski, R. M.; Prasit, P.; Riendau, D.; Rodan, S. B.; Percival, M. D. J. Med. Chem. 2001, 44, 94. -
[3]
Feldman, P. L.; Brackeen, M. F.; Cowan, D. J.; Marron, B. E.; Schoenen, F. J.; Stafford, J. A.; Suh, E. M.; Domanico, P. L.; Rose, D.; Leesnitzer, M. A.; Brawley, E. S.; Strickland, A. B.; Vergese, M. W.; Connolly, K. M.; Bateman-Fite, R.; Noel, S. L.; Sekut, L.; Stimpson, S. A. J. Med. Chem. 1995, 38, 1505. doi: 10.1021/jm00009a011
-
[4]
(a) Larraufie, M. H.; Maestri, G.; Malacria, M.; Ollivier, C.; Fensterbank, L.; Lacote, E. Synthesis 2012, 44, 1279.
(b) Nekrasov, D. D. Russ. J. Org. Chem. 2004, 40, 1387. -
[5]
Crutchley, R. J. Coord. Chem. Rev. 2001, 219, 125.
-
[6]
Boatright, L. G.; Mackay, J. S. US 2721786, 1955[Chem. Abstr. 1956, 50, 21846].
-
[7]
(a) Nekrasov, D. D. Russ. J. Org. Chem. 2004, 40, 1387.
(b) Larraufie, M. H.; Maestri, G.; Malacria, M.; Ollivier, C.; Fensterbank, L.; Lacote, E. Synthesis 2012, 44, 1279. -
[8]
Braun, von J. Ber. Dtsch. Chem. Ges. 1907, 40, 3914. doi: 10.1002/(ISSN)1099-0682
-
[9]
Morgan, T.; Ray, N. C.; Parry, D. M. Org. Lett. 2002, 4, 597. doi: 10.1021/ol0172020
-
[10]
Nath, J.; Patel, B. K.; Jamir, L.; Sinha, U. B.; Satyanarayan, K. V. V. V. Green Chem. 2009, 11, 1503. doi: 10.1039/b914283p
-
[11]
Ramana, T.; Saha, P.; Das, M.; Punniyamurthy, T. Org. Lett. 2010, 12, 84. doi: 10.1021/ol9024088
-
[12]
Sahoo, S. K.; Jamir, L.; Guin, S.; Patel, B. K. Adv. Synth. Catal. 2010, 352, 2538. doi: 10.1002/adsc.v352:14/15
-
[13]
Zhu, C.; Xia, J.-B.; Chen, C. Org. Lett. 2014, 16, 247. doi: 10.1021/ol403245r
-
[14]
Lin, C.-C.; Hsieh, T.-H.; Liao, P.-Y.; Liao, Z.-Y.; Chang, C.-W.; Shih, Y.-C.; Yeh, W.-H.; Chien, T.-C. Org. Lett. 2014, 16, 892. doi: 10.1021/ol403645y
-
[15]
Ayres, J. N.; Ling, K. B.; Morrill, L. C. Org. Lett. 2016, 18, 5528. doi: 10.1021/acs.orglett.6b02775
-
[16]
Kamijo, S.; Jin, T.; Yamamoto, Y. J. Am. Chem. Soc. 2001, 123, 9453. doi: 10.1021/ja016355f
-
[17]
Kamijo, S.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 11940. doi: 10.1021/ja0272742
-
[18]
Kamijo, S.; Jin, T. Yamamoto, Y. Angew. Chem., Int. Ed. 2002, 41, 1780. doi: 10.1002/(ISSN)1521-3773
-
[19]
Stolley, R. M.; Guo, W.; Louie, J. Org. Lett. 2012, 14, 322. doi: 10.1021/ol203069p
-
[20]
Li, P.; Cheng, G.; Zhang, H.; Xu, X.; Gao, J.; Cui, X. J. Org. Chem. 2014, 79, 8156. doi: 10.1021/jo501334u
-
[21]
Li, J.; Zheng, X.; Li, W.; Zhou, W.; Zhu W.; Zhang, Y. New J. Chem. 2016, 40, 77. doi: 10.1039/C5NJ02153G
-
[22]
(a) Reddy, N. L.; Fan, W.; Magar, S. S.; Perlman, M. E.; Yost, E.; Zhang, L.; Berlove, D.; Fischer, J. B.; Burke-Howie, K.; Wolcott, T.; Durant, G. J. J. Med. Chem. 1998, 41, 3298.
(b) Snider, B. B.; O'Hare, S. M. Tetrahedron Lett. 2001, 42, 2455.
(c) Basterfield, S.; Rodman, F. B. S.; Tomecko J. W. Can. J. Res. 2011, 17, 390. -
[23]
Giles, R. L.; Sullivan, J. D.; Steiner, A. M.; Looper, R. E. Angew. Chem., Int. Ed. 2009, 48, 3116. doi: 10.1002/anie.v48:17
-
[24]
Giles, R. L.; Nkansah, R. A.; Looper, R. E. J. Org. Chem. 2010, 75, 261. doi: 10.1021/jo902326d
-
[25]
Lou, Z.; Wu, X.; Yang, H.; Zhu, C.; Fu, H. Adv. Synth. Catal. 2015, 357, 3961. doi: 10.1002/adsc.201500577
-
[26]
Rassadin, V. A.; Zimin, D. P.; Raskil'dina, G. Z.; Ivanov, A. Y.; Boyarskiy, V. P.; Zlotskiib S. S.; Kukushkin, V. Y. Green Chem. 2016, 18, 6630. doi: 10.1039/C6GC02556K
-
[27]
Larraufie, M.-H.; Ollivier, C.; Fensterbank, L.; Malacria, M.; Lacte, E. Angew. Chem., Int. Ed. 2010, 49, 2178. doi: 10.1002/anie.v49:12
-
[28]
Maestri, G.; Larraufie, M.-H.; Ollivier, C.; Malacria, M.; Fensterbank, L.; Lacôte, E. Org. Lett. 2012, 14, 5538. doi: 10.1021/ol3026439
-
[29]
Li, J.; Neuville, L. Org. Lett. 2013, 15, 6124. doi: 10.1021/ol4029622
-
[30]
Tran, L. Q.; Li, J.; Neuville, L. J. Org. Chem. 2015, 80, 6102. doi: 10.1021/acs.joc.5b00614
-
[31]
Li, J.; Wang, H.; Hou, Y.; Yu, W.; Xu, S.; Zhang, Y. Eur. J. Org. Chem. 2016, 2388.
-
[32]
Vollhardt, K. P. C.; Naiman, A. US 4328343, 1982[Chem. Abstr 1978, 90, 186806].
-
[33]
Boñaga, L. V. R.; Zhang H.-C.; Maryanoff B. E. Chem. Commun. 2004, 2394. doi: 10.1002/chin.200515140/full
-
[34]
(a) Stolley, R. M.; Maczka, M. T.; Louie J. Eur. J. Org. Chem. 2011, 3815.
(b) Kumar, P.; Prescher, S.; Louie, J. Angew. Chem., Int. Ed. 2011, 50, 10694. -
[35]
Wang, C.; Wang, D.; Xu, F.; Pan, B.; Wan, B. J. Org. Chem. 2013, 78, 3065. doi: 10.1021/jo400057t
-
[36]
Lane, T. K.; D'Souza, B. R.; Louie, J. J. Org. Chem. 2012, 77, 7555. doi: 10.1021/jo3012418
-
[37]
Lane, T. K.; Nguyen, M. H.; D'Souza, B. R.; Spahn, N. A.; Louie, J. Chem. Commun. 2013, 49, 7735. doi: 10.1039/c3cc44422h
-
[38]
Hashimoto, T.; Ishii, S.; Yano, R.; Miura, H.; Sakata, K.; Takeuchi, R. Adv. Synth. Catal. 2015, 357, 3901. doi: 10.1002/adsc.201500637
-
[39]
Onodera, G.; Shimizu, Y.; Kimura, J.; Kobayashi, J.; Ebihara, Y.; Kondo, K.; Sakata, K.; Takeuchi, R. J. Am. Chem. Soc. 2012, 134, 10515. doi: 10.1021/ja3028394
-
[40]
Ye, F.; Haddad, M.; Ratovelomanana-Vidal, V.; Michelet, V. Org. Lett. 2017, 19, 1104. doi: 10.1021/acs.orglett.7b00130
-
[41]
Rassadin, V. A.; Boyarskiy, V. P.; Kukushkin Y. V. Org. Lett. 2015, 17, 3502. doi: 10.1021/acs.orglett.5b01592
-
[42]
(a) Hughes, T. V.; Hammond, S. D.; Cava, M. P. J. Org. Chem. 1998, 63, 401.
(b) Hughes, T. V.; Cava, M. P. J. Org. Chem. 1999, 64, 313. -
[43]
Wu, Y.-Q.; Limburg, D. C.; Wilkinson, D. E.; Hamilton, G. S. Org. Lett. 2000, 2, 795. doi: 10.1021/ol0055263
-
[44]
Anbarasan, P.; Neumann, H.; Beller, M. Chem.-Eur. J. 2010, 16, 4725. doi: 10.1002/chem.201000086
-
[45]
(a) Anbarasan, P.; Neumann, H.; Beller, M. Angew. Chem. Int. Ed. 2011, 50, 519.
(b) Anbarasan, P.; Neumann, H.; Beller, M. Chem.-Eur. J. 2011, 17, 4217.
(c) Cai, Y.; Qian, X.; Rérat, A.; Auffrant, A.; Gosmini C. Adv. Synth. Catal. 2015, 357, 3419.
(d) Yang, Y.; Zhang, Y.; Wang, J. Org. Lett. 2011, 13, 5608.
(e) Gong, T.-J.; Xiao, B.; Cheng, W.-M.; Su, W.; Xu, J.; Liu, Z.-J.; Liu, L.; Fu, Y. J. Am. Chem. Soc. 2013, 135, 10630.
(f) Chaitanya, M.; Yadagiri, D.; Anbarasan, P. Org. Lett. 2013, 15, 4960.
(g) Gu, L.-J.; Jin, C.; Wang, R.; Ding, H.-Y. ChemCatChem 2014, 6, 1225.
(h) Mishra, N. K.; Jeong, T.; Sharma, S.; Shin, Y.; Han, S.; Park, J.; Oh, J. S.; Kwak, J. H.; Jung, Y. H.; Kima, I. S. Adv. Synth. Catal. 2015, 357, 1293.
(i) Chaitanya, M.; Anbarasan, P. J. Org. Chem. 2015, 80, 3695.
(j) Zhu, X.; Shen, X.J.; Tian, Z.-Y.; Lu, S.; Tian, L.-L.; Liu, W.-B.; Song, B.; Hao, X.-Q. J. Org. Chem. 2017, 82, 6022. -
[46]
Fukumoto, K.; Oya, T.; Itazaki, M.; Nakazawa, H. J. Am. Chem. Soc. 2009, 131, 38. doi: 10.1021/ja807896b
-
[47]
Liao, Z.-Y.; Liao, P.-Y.; Chien, T.-C. Chem. Commun. 2016, 52, 14404. doi: 10.1039/C6CC08601B
-
[48]
Rao, B.; Zeng, X. Org. Lett. 2014, 16, 314. doi: 10.1021/ol403346x
-
[49]
Pan, Z.; Pound, S. M.; Rondla, N. R.; Douglas, C. J. Angew. Chem., Int. Ed. 2014, 53, 5170.
-
[50]
Miyazaki, Y.; Ohta, N.; Semba, K.; Nakao, Y. J. Am. Chem. Soc. 2014, 136, 3732. doi: 10.1021/ja4122632
-
[51]
Wang, R.; Falck, J. R. Chem. Commun. 2013, 49, 6516. doi: 10.1039/c3cc43597k
-
[52]
(a) Servais, A.; Azzouz, M.; Lopes, D.; Courillon, C.; Malacria, M. Angew. Chem., Int. Ed. 2007, 46, 576.
(b) Beaume, A.; Christine Courillon, C.; Derat, E.; Malacria, M. Chem.-Eur. J. 2008, 14, 1238. -
[53]
Larrafie, M.-H.; Courillon, C.; Ollivier, C.; Lacôte, E.; Malacria, M.; Fensterbank, L. J. Am. Chem. Soc. 2010, 132, 4381. doi: 10.1021/ja910653k
-
[54]
Sävmarker, J.; Rydfjord, J.; Gising, J.; Odell, L. R.; Larhed, M. Org. Lett. 2012, 14, 2394. doi: 10.1021/ol300813c
-
[55]
Rydfjord, J.; Svensson, F.; Trejos, A.; Söjberg, P. J. R.; Sköld, C.; Sävmarker, J.; Odell, L. R.; Larhed, M. Chem.-Eur. J. 2013, 19, 13803. doi: 10.1002/chem.201301809
-
[56]
Guin, S.; Rout, S. K.; Gogoi, A.; Ali, W.; Patel, B. K. Adv. Synth. Catal. 2014, 46, 2559.
-
[1]
-

图式1 铜催化2-卤代芳基硫脲串联反应合成芳基氰胺
Scheme 1 Copper-catalyzed cascade reaction of 2-haloarylth-ioureas for the synthesis of arylcyanamides

图式2 胺和TMSCN偶联反应合成氰胺
Scheme 2 Coupling reaction of amines and TMSCN for the synthesis of cyanamides

图式3 钯催化异氰、三甲基硅叠氮三组分反应合成氰胺
Scheme 3 Palladium-catalyzed three-component reaction of isocyanides, allyl carbonate and trimethylsilyl azide for the synthesis of cyanamides

图式4 钯催化邻炔基芳基异氰、烯丙醇酯和三甲基硅叠氮三组分反应合成N-氰基吲哚
Scheme 4 Palladium-catalyzed three-component reaction of 2-alkynylaryl isocyanides, allyl carbonate and trimethylsilyl azide for the synthesis of N-cyanoindoles

图式5 钯催化异氰和三甲基硅叠氮反应合成氰胺
Scheme 5 Palladium-catalyzed reaction of isocyanides and trimethylsilyl azide for the synthesis of cyanamides

图式6 氰胺和二芳基碘盐偶联反应合成氰胺和脲
Scheme 6 Cross coupling of NH-cyanamides and diaryliodoniums for the synthesis of cyanamides and ureas

图式7 氰胺和吡啶N-氧多米诺反应合成脲类化合物
Scheme 7 Domino reaction of cyanamides and pyridine N-oxides for the synthesis of ureas

图式8 N-酰基氰胺自由基串联反应合成稠环胍
Scheme 8 Radical cascade reaction of N-acyl cyanamides for the synthesis of fused cyclic guanidines

图式9 N-酰基氰胺和二硫化合物自由基串联反应合成异脲类化合物
Scheme 9 Radical cascade reaction of N-acyl cyanamides and diaryldisulfides for the synthesis of isothioureas.

图式10 铜催化氰胺、胺和硼酸三组分反应合成胍
Scheme 10 Copper-catalyzed three-component reaction of cyanamides, amines and boronic acides for the synthesis of guanidines

图式11 钴促进二炔和氰胺[2+2+2]环加成反应合成稠环2-氨基吡啶
Scheme 11 Cobalt-promoted [2+2+2] cycloaddition of bis-alkynes and cyanamides for the synthesis of fused 2-aminopyridines

图式12 铑催化硼酸和NCTS氰基化反应合成氰
Scheme 12 Rhodium catalyzed cyanation of boronic acids with NCTS for the synthesis of nitriles

图式13 铜催化邻炔基芳香氰胺环化反应合成3-氰基吲哚
Scheme 13 Copper-catalyzed cyclization of o-alkynyl arylcyanamides for the synthesis of 3-cyanoindoles

图式14 CsF促进2-(三甲基硅基)苯酚三氟甲磺酸酯和氰胺串联反应合成2-氨基苯甲氰类化合物
Scheme 14 CsF-promoted cascade reaction of cyanamides and (trimethylsilyl)aryl trifluoromethanesulfonate for the synthesis of 2-aminobenzonitriles

图式15 B(C6F5)3促进分子内烯烃氨基氰基化反应合成吲哚啉
Scheme 15 B(C6F5)3-promoted intramolecular aminocyanation of alkenes by cyanamides for the synthesis of indolines

图式16 钯催化分子内氨基氰基化反应合成吲哚啉
Scheme 16 Palladium-catalyzed intramolecular aminocyanation of alkenes for the synthesis of indolines

图式17 N-酰基氰胺自由基串联反应合成四环稠合喹唑啉
Scheme 17 Radical cascade reaction of N-acyl cyanamides for the synthesis of tetracycle-fused quinazolines

图式18 N-酰基氰胺自由基串联反应合成三环喹唑啉
Scheme 18 Radical cascade reaction of N-acyl cyanamides for the synthesis of tricycle-fused quinazolines

图式19 钯催化氰胺和芳基三氟硼酸钾加成反应合成芳基脒
Scheme 19 Palladium-catalyzed addition of cyanamides and potassium arlyltrifluoroborates for the synthesis of arylamidines
-


 下载:
下载:



















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 82
- 文章访问数: 4747
- HTML全文浏览量: 1374
 下载:
下载:
