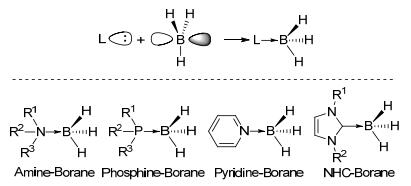
 图式1
稳定硼烷加合物的成键原理及主要类型
Scheme1.
Bond-forming principle and main classes of stable borane adducts
图式1
稳定硼烷加合物的成键原理及主要类型
Scheme1.
Bond-forming principle and main classes of stable borane adducts
引用本文:
杨吉民, 李子奇, 朱守非. 稳定硼烷加合物在有机硼化物合成中的应用研究进展[J]. 有机化学,
2017, 37(10): 2481-2497.
doi:
10.6023/cjoc201705034

Citation: Yang Jimin, Li Ziqi, Zhu Shoufei. Progresses on the Application of Stable Borane Adducts in the Synthesis of Organoborons[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2481-2497. doi: 10.6023/cjoc201705034
Citation: Yang Jimin, Li Ziqi, Zhu Shoufei. Progresses on the Application of Stable Borane Adducts in the Synthesis of Organoborons[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2481-2497. doi: 10.6023/cjoc201705034
稳定硼烷加合物在有机硼化物合成中的应用研究进展
摘要:
有机硼化合物在合成化学、材料科学、生命健康等领域都有广泛应用,因此有机硼化合物的合成一直是研究热点.目前,催化C-B键形成反应通常使用联硼酸频哪醇酯(B2Pin2)、频哪醇硼烷(HBPin)、儿茶酚硼烷(HBCat)等作为硼试剂.相比于传统的硼试剂,硼烷与胺、膦或N-杂环卡宾等强Lewis碱的加合物(统称为稳定硼烷加合物)具有易于合成、稳定性高、易操作等特点,其作为硼试剂参与的有机硼化合物的合成最近受到越来越多的关注,已被成功用于烯(炔)烃的硼氢化、C-H键硼化、卡宾对B-H键的插入、硼自由基串联环化、取代等反应中,为有机硼化合物的合成提供了新的思路和方法.以反应类型为线索,系统综述了稳定硼烷加合物在有机硼化物合成中的应用研究进展.
English
Progresses on the Application of Stable Borane Adducts in the Synthesis of Organoborons
Abstract:
Organoboron compounds are wildly used in organic synthesis, materials science, life and health science, etc. The development of synthetic methodologies of organoborons has therefore gained intense attention nowadays. Typically, Bis(pinacolato)diboron (B2Pin2), pinacolborane (HBpin) and catecholatoborane (HBCat) are predominantly used as boron reagents in catalytic C-B bond forming reactions. Different from the above traditional boron reagents, borane adducts with strong Lewis bases, such as amines, phosphines, and N-heterocyclic carbenes, are promising boron reagents because of their readily accessibility, relatively high stability, and easy operation. Moreover, the different chemical properties of these stable borane adducts towards the traditional boron reagents provide possibilities for development of new C-B bond formation reactions. The applications of the stable borane adducts as terminal boron reagents in hydroboration of alkenes or alkynes, C-H bond borylation, carbene insertion into B-H bonds, cascade cyclization initiated by boryl radicals and substitutions, which provide new methods for the preparation of organoborons are reviewed in this paper.
-
Key words:
- stable borane adduct
- / hydroboration
- / C—H bond borylation
- / B—H bond insertion
- / boryl radical
-
硼烷与胺、膦或氮杂环卡宾化合物等强Lewis碱形成的稳定配合物分别叫做胺硼烷加合物、膦硼烷加合物、吡啶硼烷加合物或氮杂环卡宾(NHC)硼烷加合物(Scheme 1).这些加合物在成键模式上与弱Lewis碱(如醚、硫醚等)与硼烷形成的配合物相似, 都是Lewis碱的孤对电子填充到硼烷的空p轨道上成键, 但是其稳定性更高, 可以统称为稳定硼烷加合物, 而且与后者在性质上也表现出很大的不同.由于硼烷的空p轨道被Lewis碱的孤对电子所填充, 所以相应的硼烷加合物具有较高的稳定性, 通常对水和空气都不敏感.另一方面, 由于配位键的存在, 使得硼烷加合物中的Lewis碱和硼烷之间形成了推拉电子的结构, 硼中心由sp2杂化变为类sp3杂化, 同时由平面结构变为类四面体结构, 硼中心的电子云密度明显提高.正是由于稳定硼烷加合物这种独特的电子结构, 使其具有不同于硼烷和硼酸酯的反应性, 同时还具有较高的稳定性, 在很多领域都有应用.胺硼烷加合物[1]是被研究得较多的一类硼试剂, 它在较低温度下可以用作温和的还原剂、储氢试剂、配体等.较高温度下胺硼烷可以释放出自由硼烷表现出硼烷的性质, 硼烷释放的温度可以被不同结构的胺控制, 所以胺硼烷也是一种可控释放硼烷的化合物.此外, 胺硼烷加合物作为氢给体被成功用于多种不饱和化合物的氢化反应中[2].吡啶硼烷加合物可以看成一类特殊的胺硼烷加合物, 两者性质比较类似.膦硼烷加合物[1]通常比胺硼烷更加稳定, 更难以分解, 它们易于制备, 被广泛用于有机磷化学, 如有机磷的分离、保护或活化.氮杂环卡宾硼烷加合物[3]相对更加稳定, 近些年对于它们的研究逐渐得到关注, 展现出了不同于胺和膦硼烷加合物的性质, 其作为还原剂、反应试剂、引发剂等在有机合成化学和高分子化学中展现出了巨大的潜力.
图式1
稳定硼烷加合物的成键原理及主要类型
Scheme1.
Bond-forming principle and main classes of stable borane adducts
有机硼化合物在合成化学、材料科学、医药等领域都有广泛应用, 因此有机硼化合物的合成一直是研究热点[4].目前, 催化碳-硼键形成反应通常使用联硼酸频哪醇酯(B2Pin2)、频哪醇硼烷(HBPin)、儿茶酚硼烷(HBCat)等作为硼试剂[5].一方面这些硼试剂通常比较容易水解, 稳定性欠佳; 另一方面, 这些硼试剂制备相对复杂[6], 如B2Pin2通常以强腐蚀性的三溴化硼作为起始原料, 通过多步转化制得, 产生较多污染性废物.近年来, 稳定硼烷加合物作为一种易得、稳定的硼试剂逐渐被用到了有机硼化合物的合成中, 并展现出一些独特优势, 受到了越来越多的关注.本文根据反应类型的不同, 对稳定硼烷加合物参与的有机硼化合物的合成进行了系统的梳理和总结, 阐述了反应的机理, 并对该领域未来发展进行了展望.
1 稳定硼烷加合物的合成
稳定硼烷加合物的合成非常方便(Scheme 2):实验室合成一般采用胺、膦或氮杂环卡宾与BH3•THF或BH3•SMe2反应[7], 取代弱Lewis碱生成稳定的硼烷加合物; 胺或膦的酸盐与NaBH4或LiBH4反应也可以生成相应的硼烷加合物[8], 工业上一般采用此方法大规模制备; 此外在适当反应条件下, 胺硼烷加合物或膦硼烷加合物可以与另一分子胺、膦或氮杂环卡宾发生交换反应生成相应的硼烷加合物[9].
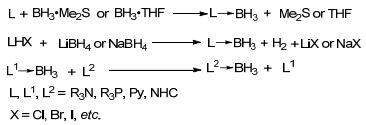 图式2
稳定硼烷加合物的合成方法
Scheme2.
Synthetic methods of stable borane adducts
图式2
稳定硼烷加合物的合成方法
Scheme2.
Synthetic methods of stable borane adducts
2 烯(炔)的硼氢化反应
烯(炔)的硼氢化反应是合成有机硼化合物的常用方法, 该反应通常使用BH3•THF、BH3•SMe2、9-borabi-cyclo[3.3.1]nonane (9-BBN)、HBpin和HBCat等高活性的硼试剂.稳定硼烷加合物在室温下通常不能与不饱和烃发生硼氢化反应.在早期的研究中, 通常在较高的温度下硼烷加合物发生解离, 解离后的自由硼烷再与不饱和烃发生硼氢化反应[10].近年来, 一些活化剂或催化剂促进的硼烷加合物与烯(炔)的硼氢化反应开始见诸报道.
2.1 碘或酸促进的硼氢化反应
2003年, Vedejs等[11]以I2为活化试剂(50 mol%)实现了首例高烯丙基胺1的硼烷加合物分子内硼氢化反应, 生成的环状有机硼2a和2b经氧化得到相应的醇3a和3b, 区域选择性主要受烯烃上的取代基R控制(Scheme 3).该反应用催化量的I2(10 mol%)同样可以顺利进行, 收率和选择性都较好.
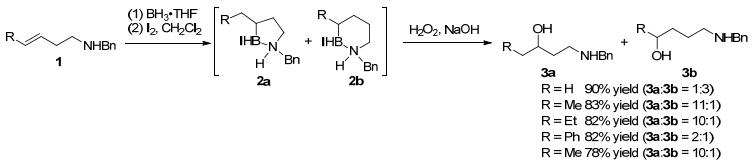 图式3
碘促进的高烯丙基胺-硼烷加合物的分子内硼氢化反应
Scheme3.
Iodine-promoted hydroboration of homoallylicamine-borane adducts
图式3
碘促进的高烯丙基胺-硼烷加合物的分子内硼氢化反应
Scheme3.
Iodine-promoted hydroboration of homoallylicamine-borane adducts
机理研究认为(Scheme 4):当体系中没有活化剂的时候, 如果要发生硼氢化反应, 需要双键进攻硼原子, 经由过渡态5断裂N—B配键生成6才能完成——然而这一过程在轨道匹配性上是不利的[12], 因而不能发生(Path a); 当体系加入活化试剂I2之后, 可以形成带有不稳定B—I键的中间体7, 随后双键对硼原子进攻, 经由共价型过渡态8或离子型过渡态9, 生成硼烷与烯烃结合的中间体10, 随后发生硼氢化得到目标产物(Path b和Path c).
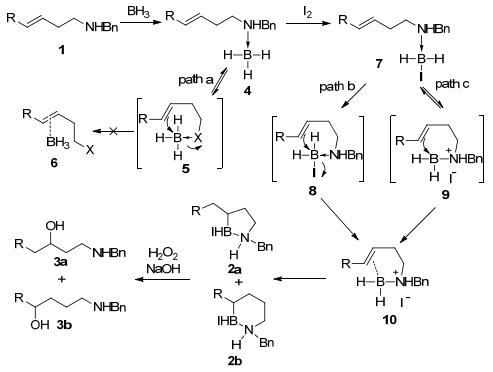 图式4
高烯丙基胺硼烷加合物的分子内硼氢化反应可能机理
Scheme4.
Proposed mechanisms for the intramolecular hydroboration of homoallylic amine-borane adducts
图式4
高烯丙基胺硼烷加合物的分子内硼氢化反应可能机理
Scheme4.
Proposed mechanisms for the intramolecular hydroboration of homoallylic amine-borane adducts
三氟甲磺酸能够有效地促进高烯丙基膦硼烷加合物11的硼氢化反应, 生成磷-硼杂环化合物12a和12b, 经氧化后生成相应的醇13a和13b, 反应的区域选择性比相应的高烯丙胺硼烷加合物高(Scheme 5).和I2的作用相似, 三氟甲磺酸通过和硼烷反应生成活性更高的离子型硼烷中间体, 促进了烯烃和硼烷的作用, 进而完成了硼氢化反应.
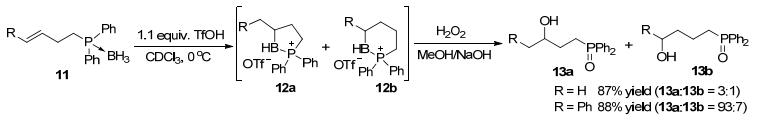 图式5
TfOH促进的高烯丙基膦硼烷加合物的分子内硼氢化反应
Scheme5.
TfOH-promotedintramolecular hydroboration of homoallylic phosphine-borane adducts
图式5
TfOH促进的高烯丙基膦硼烷加合物的分子内硼氢化反应
Scheme5.
TfOH-promotedintramolecular hydroboration of homoallylic phosphine-borane adducts
2005年, 以I2为活化剂, Vedejs等[13]实现了烯烃和吡啶硼烷加合物的分子间硼氢化反应(Scheme 6).一系列控制实验表明, 反应可能机理为:吡啶硼烷加合物与碘作用生成吡啶碘硼烷加合物14, 随后双键对其进行类SN2亲核进攻生成离子对中间体15, 最后经历四中心环状过渡态发生硼氢化反应生成16, 再经氧化得到相应的醇17.
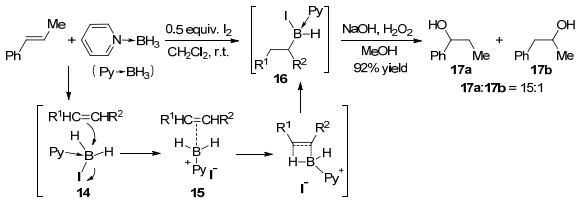 图式6
I2促进烯烃和吡啶硼烷加合物的分子间硼氢化反应
Scheme6.
I2-promoted intermolecular hydroboration of alkenes with pyridine borane adducts
图式6
I2促进烯烃和吡啶硼烷加合物的分子间硼氢化反应
Scheme6.
I2-promoted intermolecular hydroboration of alkenes with pyridine borane adducts
利用这个策略他们同样实现了硼烷加合物与炔烃的分子间硼氢化反应, 生成的烯基硼化合物18a和18b经氧化后生成相应的酮19a和19b.该反应的区域选择性受立体位阻影响, 当炔烃两端取代基大小相似时, 区域选择性很差(Scheme 7).
 图式7
I2促进炔烃和吡啶硼烷加合物的分子间硼氢化反应
Scheme7.
I2-promoted intermolecular hydroboration of alkynes with pyridine borane adduct
图式7
I2促进炔烃和吡啶硼烷加合物的分子间硼氢化反应
Scheme7.
I2-promoted intermolecular hydroboration of alkynes with pyridine borane adduct
I2活化吡啶硼烷加合物和烯烃的硼氢化反应在合成中有很好的潜在应用价值.硼氢化产物除了上文所展示的氧化为醇, 还可以进一步转化为在有机合成中常用的烷基频哪醇硼酸酯和三氟硼酸钾等有机硼试剂(Scheme 8)[14].
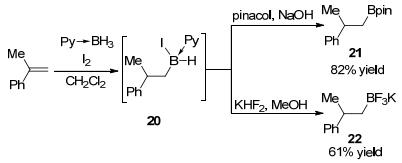 图式8
烷基频哪醇硼酸酯和三氟硼酸钾化合物的制备
Scheme8.
Preparation of alkylpinacolborate and potassium alkyltrifluoroborate
图式8
烷基频哪醇硼酸酯和三氟硼酸钾化合物的制备
Scheme8.
Preparation of alkylpinacolborate and potassium alkyltrifluoroborate
最近, Shenvi等以I2活化氨基硼烷加合物的分子内硼氢化反应为基础, 结合一个串联的烷基迁移反应, 方便地构筑了N-杂双环结构, 并成功合成了Indolizidine[15]和Lepadiformine[16]等天然产物(Scheme 9).
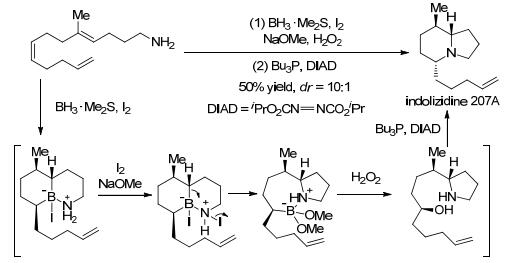 图式9
氨基硼烷加合物的硼氢化用于天然产物的合成
Scheme9.
Applications of hydroboration of amine-borane adducts in natural product synthesis
图式9
氨基硼烷加合物的硼氢化用于天然产物的合成
Scheme9.
Applications of hydroboration of amine-borane adducts in natural product synthesis
2012年, Vedejs等[17]报道了Tf2NH (Tf=CF3SO2)催化的氮杂环卡宾硼烷加合物(NHC-BH3)和烯烃的硼氢化反应(Scheme 10).没有催化剂参与时, 即使在剧烈的反应条件下, NHC-BH3和烯烃也不能够发生硼氢化反应, 但是当加入催化量的Tf2NH后, 可以在室温或低于室温的条件下生成二次硼氢化的产物.生成的二烷基的硼烷加合物稳定性较差, 不能柱层析分离, 但可被氧化生成相应的醇.他们提出了可能的反应机理: (1) NHC-BH3与强酸Tf2NH作用, 脱氢生成高活性硼鎓阳离子中间体23, 这才是真正的催化剂; (2)23和过量的烯烃连续发生两次硼氢化反应, 生成中间产物24; (3)24和NHC-BH3发生氢转移, 生成目标产物26a, 同时重新生成活性催化剂23.因为活性中间体24不含B—H键, 无法进行硼氢化反应, 所以卡宾硼烷加合物虽然含有三个B—H键, 但是只能得到二次硼氢化的产物.虽然该催化体系可以用于端烯和内烯的硼氢化, 但是由于硼鎓阳离子容易在烷基链上迁移(经由中间体25), 因此会生成迁移产物26b, 造成反应的区域选择性很难控制.
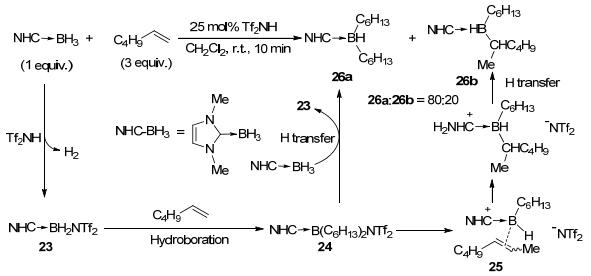 图式10
Tf2NH催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应
Scheme10.
Tf2NH-catalyzed hydroboration of alkenes with NHC-BH3
图式10
Tf2NH催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应
Scheme10.
Tf2NH-catalyzed hydroboration of alkenes with NHC-BH3
随后, Curran等[18]使用催化量的I2实现了NHC-BH3和烯烃的硼氢化反应, 给出中等或较高的收率(表 1).与Tf2NH催化剂相比, I2催化的硼氢化具有更广泛的底物适用范围, 可以生成稳定的可通过柱层析分离的单烷基硼烷加合物.此方法也可用于硼杂环化合物的合成, 比如与1, 5-环辛二烯反应合成硼杂双环化合物27, 而通过分子内的硼氢化反应可以合成硼杂并环化合物28(Scheme 11).
 表 1
催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应
Table 1.
I2-catalyzed hydroboration of alkenes with NHC-BH3
表 1
催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应
Table 1.
I2-catalyzed hydroboration of alkenes with NHC-BH3

Alkene Product Yield/% 

75 

68 

61 

50 

37 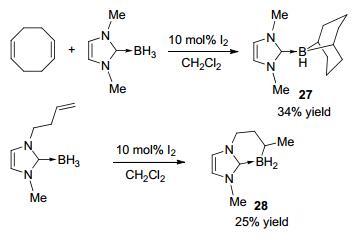 图式11
环状氮杂环卡宾硼烷的合成
Scheme11.
Synthesis of cyclic NHC-borane adducts
图式11
环状氮杂环卡宾硼烷的合成
Scheme11.
Synthesis of cyclic NHC-borane adducts
Curran等[19]提出了如Scheme 12所示反应机理, NHC-BH3与I2作用生成共价型的硼碘化合物29, 该化合物可以在溶液中长时间保存, 但对水、醇和烯烃等中性亲核试剂不稳定.硼碘化合物29在反应中起到了催化剂的作用, 它被烯烃进攻生成中间体或过渡态30, 随后发生快速硼氢化生成中间体31, 接下来与硼中心相连的碘再次被NHC-BH3上的氢取代, 经由桥连中间体32生成硼氢化产物, 同时重新生成活性催化剂29, 完成催化循环.
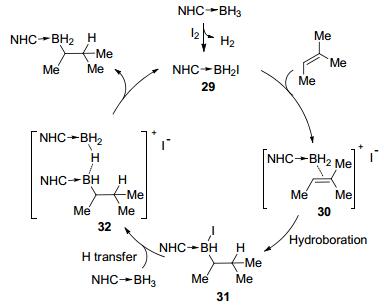 图式12
I2催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应可能机理
Scheme12.
Proposed mechanism of I2-catalyzed hydroboration of alkenes with NHC-BH3
图式12
I2催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应可能机理
Scheme12.
Proposed mechanism of I2-catalyzed hydroboration of alkenes with NHC-BH3
2013年, Curran等[20]报道了Tf2NH或I2促进的NHC-BH3和硅基取代炔烃的硼氢化反应(Scheme 13).比较有意思的是, 当底物为二硅基取代的炔烃时, 生成的是伴随着硅基迁移的1, 1-硼氢化产物33.该反应对于烷基硅基炔烃底物选择性较差, 生成的主要是多个二次硼氢化产物的异构体34a~34c, 对于苯基硅基炔烃底物主要得到单次硼氢化的产物35.
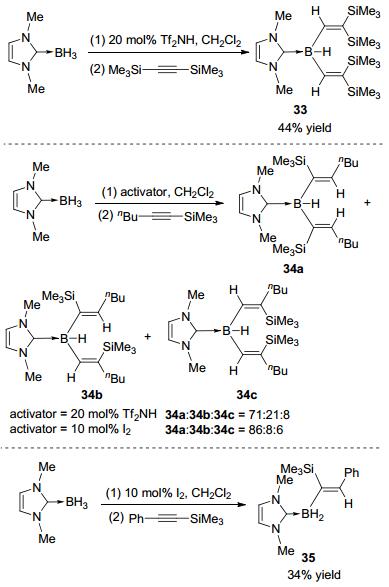 图式13
Tf2NH或I2促进的硅基取代的炔烃和氮杂环卡宾硼烷加合物的硼氢化反应
Scheme13.
Tf2NH/I2-promoted hydroborationof silylsub-stituted alkynes with NHC-BH3
图式13
Tf2NH或I2促进的硅基取代的炔烃和氮杂环卡宾硼烷加合物的硼氢化反应
Scheme13.
Tf2NH/I2-promoted hydroborationof silylsub-stituted alkynes with NHC-BH3
2.2 金属催化的硼氢化反应
相比于碘或酸促进的硼烷加合物的硼氢化反应, 金属催化的硼烷加合物硼氢化反应成功的例子较少. 2011年, Weller等[21]以Rh(PiBu2tBu)2BArF为催化剂实现了三甲胺硼烷加合物与叔丁基乙烯36的硼氢化反应(Scheme 14).他们认为反应机理为:催化剂与三甲胺硼烷生成中间体38, 随后铑对B—H键进行氧化加成生成中间体39, 再对烯烃进行迁移插入生成中间体40, 接下来进行还原消除生成中间体41, 最后与三甲胺硼烷发生交换生成反马氏硼氢化的产物37, 完成催化循环.中间体41得到了单晶衍射的确认.不过此催化体系只适用于叔丁基乙烯的底物.在随后的研究中, 他们发现Rh(xantphos)BArF[22]也可以催化此类反应, 并且进行了详细的机理实验, 认为还原消除的过程是反应的决速步.
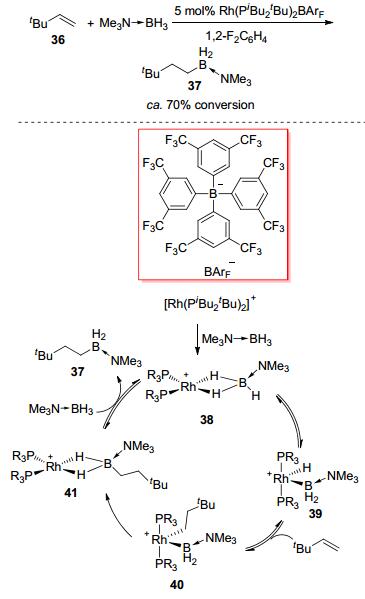 图式14
Rh催化的烯烃和三甲胺硼烷加合物的硼氢化反应
Scheme14.
Rh-catalyzed hydroboration of alkene with Me3N-borane adduct
图式14
Rh催化的烯烃和三甲胺硼烷加合物的硼氢化反应
Scheme14.
Rh-catalyzed hydroboration of alkene with Me3N-borane adduct
2012年, Parrain等[23]以离子型的Rh(Ⅰ)和手性双膦配体作催化剂实现了金属催化的氮杂环卡宾硼烷加合物的分子内硼氢化反应(Eq. 1).他们以烯丙基取代的氮杂环卡宾硼烷加合物42为底物, 能以最高94%的收率和98.4%ee的对映选择性得到五元环状含硼化合物43.
2014年, 史晓东等[24]报道了金催化的分子内的炔烃硼氢化反应(Scheme 15).他们采用氰基硼烷胺加合物44和三氮唑(TA)稳定的Au(Ⅰ)催化剂, 避免了催化剂的失活, 使得反应能顺利发生, 以中等或较高的收率合成了一系列含氮硼杂环化合物45.他们推测反应机理为金催化剂活化炔烃(经中间体46), 随后硼烷加合物的负氢对炔烃进攻生成烯基金中间体47进而生成目标产物.产物45中的氰基可以和LiAlH4或PhMgBr发生取代反应, 分别生成新的氮硼杂环化合物48或49.
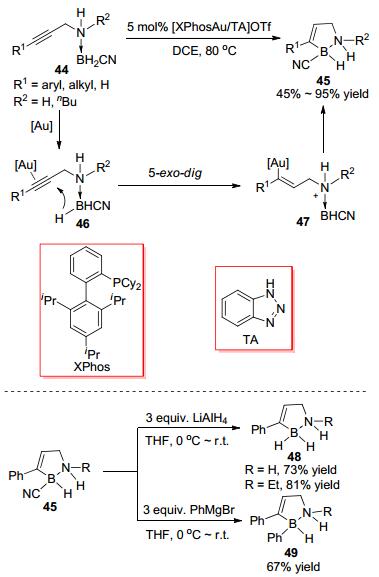 图式15
Au催化的炔烃和硼烷加合物的分子内硼氢化反应
Scheme15.
Au-catalyzed intramolecular hydroboration of alkynes withborane adducts
图式15
Au催化的炔烃和硼烷加合物的分子内硼氢化反应
Scheme15.
Au-catalyzed intramolecular hydroboration of alkynes withborane adducts
2016年, 史晓东等[25]使用金络合物和Cu(OTf)2作为催化剂实现了含炔基结构的氨基硼烷加合物50的分子内硼氢化反应, 合成了六元环状硼胺化合物51(Eq. 2).当不加入Cu(OTf)2时只能得到较差的转化率和选择性.他们认为Lewis酸催化剂Cu(OTf)2可以协助亚磷酸酯-三氮唑-Au络合物催化剂中三氮唑的离去, 从而生成活性的金催化剂, 促进了反应的进行.
2.3 硼烷加合物直接参与的硼氢化反应
在不添加活化试剂的条件下, 硼烷加合物还可以表现出亲核性, 与活泼炔烃发生硼氢化反应. 2014年, Curran等[26]报道了首例的苯炔的硼氢化反应(Scheme 16).由于苯炔需要现场制备, 生成苯炔所用的试剂与常用的硼试剂一般是不兼容的, 这就造成了苯炔硼氢化反应难以实现.而稳定的NHC-BH3可以在生成苯炔的环境下稳定存在, 因而可以完成硼氢化反应.例如, NHC-BH3可与和苯炔前体52在KF和冠醚存在下顺利反应生成NHC苯基硼烷加合物53(Scheme 16).产物53a可以与I2、Br2、NCS等反应生成相应的卤代硼烷加合物54, 进而水解为苯基硼酸; 也可以与Selectfluor作用生成稳定的NHC苯基二氟硼加合物55, 进而在钯催化下和溴苯发生交叉偶联反应, 以较高的收率生成相应的联苯化合物[27].
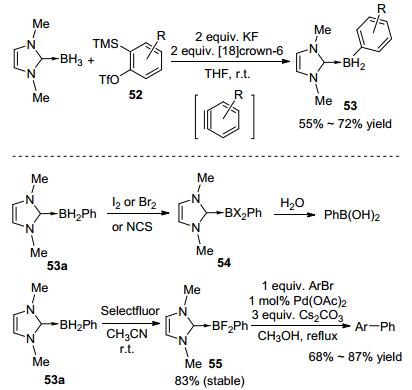 图式16
苯炔与氮杂环卡宾硼烷加合物的硼氢化反应
Scheme16.
Hydroboration of arynes with NHC-BH3
图式16
苯炔与氮杂环卡宾硼烷加合物的硼氢化反应
Scheme16.
Hydroboration of arynes with NHC-BH3
最近, Curran等[28]报道了NHC-BH3和乙炔二羧酸二甲酯的双硼氢化反应, 高效合成了NHC稳定的硼杂环丙烷化合物56(表 2).在这个反应中主要的副产物是1, 2-硼氢化反应生成的(E)-烯基硼烷加合物57.增大氮杂环卡宾氮上取代基的位阻可以提高硼杂环丙烷产物的选择性.化合物56和57不能互相转化.通过密度泛函理论(DFT)计算, 他们给出了可能的反应机理(Scheme 17): NHC-BH3首先经过渡态58将负氢反式选择性地转移至炔键生成两性离子中间体59, 随即溶剂THF与硼配位形成更稳定的四配位的硼鎓阳离子和乙烯基负离子的离子对中间体60, 随后THF配合的硼鎓阳离子将会从烯烃的阴离子π轨道方向加成, 其对应的过渡态为61.通过Oppenheimer分子动力学轨道模拟, 作者证明了由单一过渡态61可以同时得到硼杂环丙烷和烯烃硼烷两种产物:硼首先和烯烃的两端分别成键, 生成三元环中间体, 如果发生1, 2-氢迁移则得到硼杂环丙烷产物56, 如果发生C—B键的断裂, 则生成形式上单次硼氢化产物57.
表 2
由NHC-BH3和乙炔二羧酸二甲酯合成硼杂环丙烷化合物
Table 2.
Synthesis of NHC-boriranes by reactions of acetylenedicarboxylate esters with NHC-BH3

R 56:57 Yield/% 56 57 Me 33:67 19 35 iPr 52:48 14 29 2, 4, 6-Me3C6H2 81:19 31 10 2, 6-iPr2C6H3 86:14 80 5 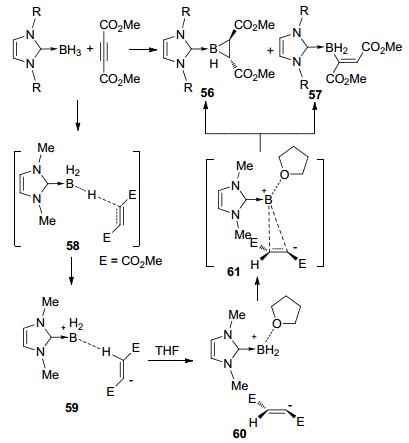 图式17
合成硼杂环丙烷反应机理
Scheme17.
Mechanism of synthesis of NHC-boriranes
图式17
合成硼杂环丙烷反应机理
Scheme17.
Mechanism of synthesis of NHC-boriranes
3 C—H键硼化
通过C—H键的活化/官能团化反应直接构筑C—B键是合成有机硼化物的有效途径, 相关研究引起了人们的关注, 成为了当前有机合成的研究热点之一[5d, 5e], 硼烷加合物参与的C—H键直接硼化反应目前也取得了一定的进展.
2009年, Vedejs等[29]报道了[Ph3C][B(C6H5)4]促进的苄胺硼烷62加合物分子内的C—H键硼化反应, 用nBu4NBH4还原淬灭反应后, 以中等的收率合成了含硼氮的五元杂环化合物63(Scheme 18).他们给出了可能的反应机理:强Lewis酸三苯基碳正离子作为氢受体与胺硼烷加合物作用, 得到具有强亲电性的硼鎓阳离子中间体64, 该中间体可与另一分子底物作用形成含有氢桥键的中间体65.随后64或65与芳环作用形成π中间体66, 进而生成Wheland中间体67, 经历过渡态68脱去H2得到稳定的化合物69, 最后被nBu4NBH4还原得到产物63.
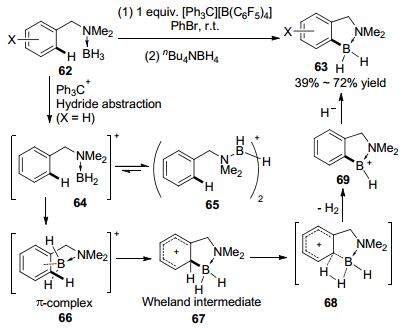 图式18
苄胺硼烷加合物的分子内芳基C—H键硼化反应
Scheme18.
Intramolecular aromatic C—H bond borylation of benzyl amine-boranes
图式18
苄胺硼烷加合物的分子内芳基C—H键硼化反应
Scheme18.
Intramolecular aromatic C—H bond borylation of benzyl amine-boranes
类似的, Stephan等[30]以[Ph3C][B(C6F5)4]为活化剂实现了氮杂环卡宾硼烷加合物的C—H键硼化反应(Scheme 19).在反应中加入1 equiv.的[Ph3C][B(C6F5)4], 氮杂环卡宾硼烷加合物70可以发生分子内的碳氢硼化得到产物71, 在130℃的反应温度下, 71可以继续发生分子内脱氢硼化生成具有平面结构的72.72可以与电子供体PPh3、Et3PO或DABCO相互作用形成相应的加合物73, 表现出经典的Lewis酸碱对的性质.当加入tBu3P时, 三分子的72可以发生脱氢偶联形成含有三个硼中心的正离子化合物74.这个方法也为合成平面型的硼鎓阳离子化合物提供了新的思路.
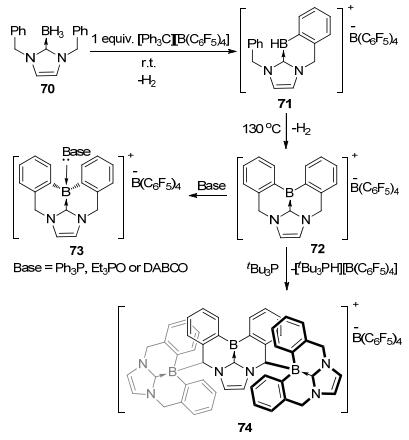 图式19
氮杂环卡宾硼烷加合物的C—H键硼化反应
Scheme19.
C—H bond borylation from NHC-borane
图式19
氮杂环卡宾硼烷加合物的C—H键硼化反应
Scheme19.
C—H bond borylation from NHC-borane
2011年, Vedejs等[31]以催化量的Tf2NH实现了脂肪胺硼烷加合物75的分子内C—H键硼化反应, 合成了B-N杂五元环状化合物76(Scheme 20).此反应需要相对较高的温度(>120℃).作者推测了反应机理:强酸Tf2NH作为氢受体活化硼烷加合物形成中间体77, 77也是催化循环中的真实催化剂, 其可与另一分子底物形成强亲电性硼鎓阳离子中间体78, 然后在较高的温度下离去一分子H2发生碳氢插入反应得到79或80, 随后与另一分子底物氢交换生成产物76, 并且重新生成活性催化剂77, 80也可被还原淬灭得到最终产物76.
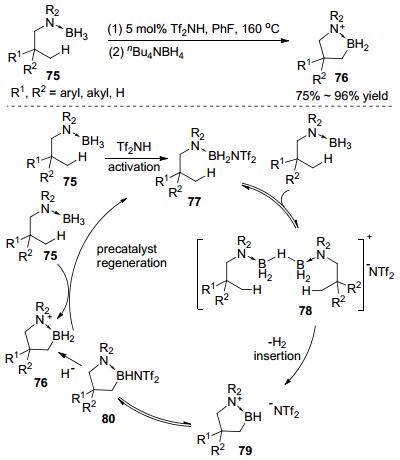 图式20
脂肪胺硼烷加合物的分子内C—H键硼化反应
Scheme20.
Intramolecular C—H bond borylation from benzyl alkyl amine-borane
图式20
脂肪胺硼烷加合物的分子内C—H键硼化反应
Scheme20.
Intramolecular C—H bond borylation from benzyl alkyl amine-borane
2013年, Vedejs等[32]以催化量的Tf2NH也实现了苄胺硼烷加合物的分子内芳烃的C—H键硼化反应(Eq. 3).与[Ph3C][B(C6H5)4]相比, 该反应需要较高的温度, 而反应的的选择性和收率有较大的提高.
2013年, Vedejs等[33]报道了Tf2NH促进的分子内膦硼烷加合物81的C—H键硼化反应(Scheme 21).此反应的中间体82可以被还原淬灭得到五元磷硼杂环化合物83, 也可以与KHF2作用得到邻苯酚三氟硼酸钾化合物84, 后者也被用到了Suzuki反应中.
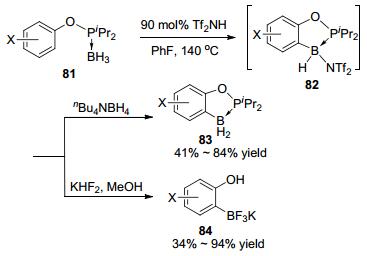 图式21
Tf2NH促进的膦硼烷加合物分子内的C—H键硼化反应
Scheme21.
Tf2NH-promoted intramolecular C—H borylation from phosphine-borane
图式21
Tf2NH促进的膦硼烷加合物分子内的C—H键硼化反应
Scheme21.
Tf2NH-promoted intramolecular C—H borylation from phosphine-borane
4 卡宾对B—H键的插入反应
金属催化的卡宾对碳-氢键及杂原子-氢键的插入反应具有反应活性高、选择性可控、条件温和、原子经济性好等特点, 是构筑碳-碳键及碳-杂原子键的一类简单高效的方法, 在有机合成中有着十分广泛的应用[34].硼烷一般表现出Lewis酸的性质, 是难以与卡宾进行插入反应的.而稳定的硼烷加合物由于硼的空轨道被一对电子填充, 显著提高了硼中心电子云密度, 使得B—H键插入反应得以实现.
1993年, Foucaud等[35]通过氯仿与氢氧化钠产生的二氯卡宾与硼烷保护的磷氮杂六元环状化合物作用时, 除了发生环丙烷化反应外, 意外地发现二氯卡宾还能对B—H键发生插入反应(Scheme 22).随后Carboni等[36]报道了二氯卡宾对不同的胺和膦硼烷加合物的插入反应, 除了单次B—H键的插入产物87, 还观察到了二次插入产物88和三次插入产物89.
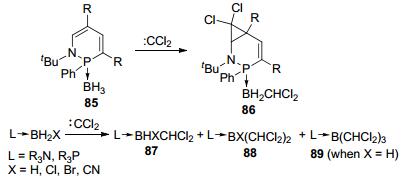 图式22
二氯卡宾对B—H键插入反应
Scheme22.
Insertion reaction ofdichlorocarbeneinto B—H bonds
图式22
二氯卡宾对B—H键插入反应
Scheme22.
Insertion reaction ofdichlorocarbeneinto B—H bonds
1996年, Imamoto等[37]报道了使用现场制备的高活性甲基或乙基钐卡宾对膦硼烷的B—H键插入反应.该反应具有很大的局限性, 除了要使用大过量的钐卡宾以外, 反应只能向硼原子上引入甲基或者乙基(Eq. 4).
催化卡宾对B—H键的插入反应直到近几年取得突破. 2013年, 朱守非、周其林等[38]报道了Cu催化的重氮酯或重氮酮90衍生的卡宾对胺硼烷加合物和膦硼烷加合物的B—H键插入反应, 在温和条件下以中等或很高的收率得到相应的α-酯基或α-羰基取代硼烷产物91(Eq. 5).他们还使用铜与手性螺环双噁唑啉配体94的络合物作为催化剂, 实现了α-重氮酯92对二甲基膦硼烷加合物93的不对称B—H键插入反应, 获得了很高的对映选择性(Eq. 6).重氮底物的酯基部分和硼烷加合物的Lewis碱结构对反应的对映选择性影响都很大. B—H键插入反应产物95a还能以高收率、高度构型保持地转化为手性硼酸频哪醇酯97这一重要的有机硼化合物(Scheme 23).当酯基存在时, 95a不能直接转化为频哪醇硼酸酯, 而是发生脱硼反应, 所以酯基需要被还原才可进行后续转化.随后工作中, 他们还实现了铜催化α-重氮酮98衍生的卡宾对B—H键的不对称插入反应[39], 取得了较高的收率和最高达83%ee的对映选择性(Eq. 7).
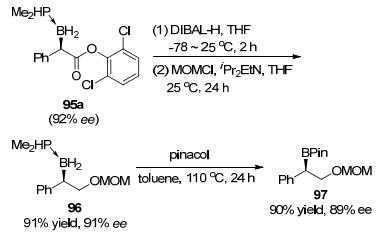 图式23
B—H键插入产物转化
Scheme23.
Transformation of B—H bond insertion product
图式23
B—H键插入产物转化
Scheme23.
Transformation of B—H bond insertion product
2013年, Curran等[40]报道了Rh(Ⅱ)催化的α-重氮羰基化合物100衍生的卡宾和氮杂环卡宾硼烷加合物101的插入反应, 以中等或较高的收率得到了一系列的α-硼烷羰基化合物102(Eq. 8).随后的工作中他们也实现了Rh(Ⅱ)催化的α-重氮酯103衍生的卡宾对膦和胺等硼烷加合物的插入反应, 同样获得了中等或较高的收率(Eq. 9)[41].
2015年, 徐明华等[42]报道了Rh(Ⅰ)催化的α-重氮酯和α-重氮酮卡宾对胺硼烷加合物的不对称B—H键插入反应.他们使用C1对称性的手性二环[2.2.2]辛二烯配体105与Rh(Ⅰ)的络合物作为催化剂, 顺利实现了上述转化, 以较高的收率和优异的对映选择性合成了手性α-硼烷羰基化合物106和107(Scheme 24).
图式24
Rh(Ⅰ)催化的卡宾对胺硼烷加合物的不对称B—H键插入反应
Scheme24.
Rhodium(Ⅰ)-catalyzed asymmetric B—H bond insertion of amine-borane adducts
最近, Gouverneur等[43]报道了Cu(Ⅰ)催化的α-三氟甲基重氮化合物108衍生的卡宾对B—H键的不对称插入反应, 合成了手性α-三氟甲基有机硼化合物110(Eq. 10).当以手性双二噁唑啉109为配体时, 可以得到最高81%ee的对映选择性.
金属催化的重氮卡宾对硼烷加合物的B—H键插入反应可以高效地构筑C—B键, 为有机硼化合物的合成提供了一个新的有效方法, 逐渐受到重视.但是, 使用重氮化合物作为卡宾前体进行B—H键插入反应也存在着明显的局限性.一方面, 重氮化合物通常具有较强的毒性和爆炸性, 限制了上述反应的实际应用.另一方面, 重氮化合物通常需要一个拉电子基团存在才能稳定, 这就限制了产物的类型.最近, 朱守非、周其林等[44]在催化B—H键插入反应研究上取得了重要进展, 部分解决了上述问题.他们以共轭的炔烃111为卡宾前体, 在铜或铑金属络合物催化下实现了B—H键插入反应, 为有机硼化合物的合成提供了新的方法(Eq. 11).该反应条件温和, 对于羰基烯炔和亚胺烯炔两类卡宾前体和胺基硼烷加合物的反应都能够给出近乎定量的收率.当使用手性双铑络合物催化剂时, 还能实现反应的不对称转化, 给出高达96%ee的对映选择性(Eq. 12).反应产物112a可以方便地转化为在有机合成中广泛应用的双芳基甲醇113、硼酸酯114和115等重要合成砌块(Scheme 25).动力学研究表明, 该反应对催化剂和炔烃底物是1级反应, 对硼烷加合物是0级反应, B—H键插入过程不是决速步. DFT计算表明金属卡宾的产生是决速步, B—H键插入经历了协同的机理, 通过三元环过渡态完成的(Scheme 26).
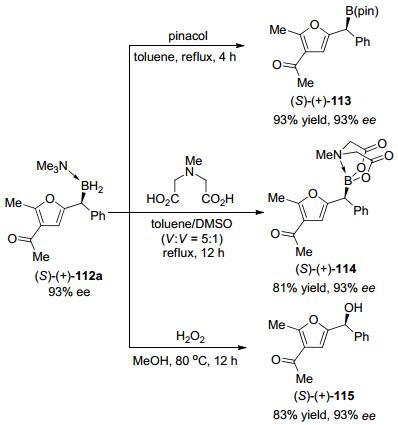 图式25
炔烃为卡宾前体的B—H键插入产物转化
Scheme25.
Transformations of B—H bond insertion product using alkynes as carbene precursors
图式25
炔烃为卡宾前体的B—H键插入产物转化
Scheme25.
Transformations of B—H bond insertion product using alkynes as carbene precursors
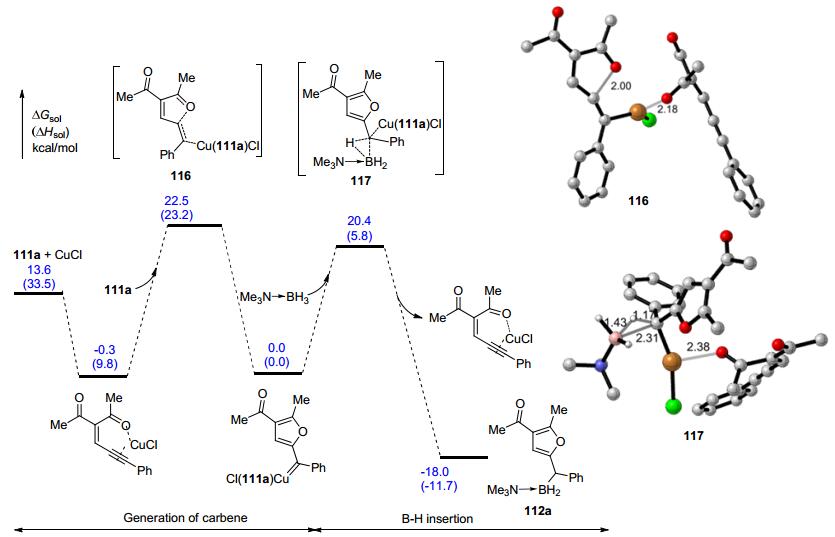 图式26
CuCl催化的炔烃为卡宾前体的B—H键插入反应吉布斯自由能曲线图
Scheme26.
Free energy profile for CuCl-catalyzed B—H bond insertion using alkynes as carbene precursors
图式26
CuCl催化的炔烃为卡宾前体的B—H键插入反应吉布斯自由能曲线图
Scheme26.
Free energy profile for CuCl-catalyzed B—H bond insertion using alkynes as carbene precursors
5 硼自由基参与的串联环化反应
硼的自由基化学一直备受关注[45].近些年来, 硼烷加合物的自由基中间体也逐渐得到认识[46], 并且用在了合成领域, 如:黄原酸酯化合物脱氧反应[47]、有机卤化物的脱卤反应[48]、缺电子烯烃的自由基聚合反应[49]、二硫化合物的均裂取代反应[50].最近, 稳定硼烷加合物的自由基中间体参与的有机硼化物的合成也取得了一些重要进展. 2017年, Curran和Taniguchi等[51]报道了氮杂环卡宾硼烷自由基参与的二炔化合物118的串联环化反应, 得到了多环结构的含硼化合物119(Scheme 27).他们认为反应机理为:氮杂环卡宾硼烷加合物在TBHN的引发下生成硼自由基中间体120, 随后对二炔底物自由基加成生成中间体121, 接着发生自由基环化反应生成自由基中间体122, 122可以与另一分子硼烷加合物发生攫氢反应生成产物119, 同时再生120.
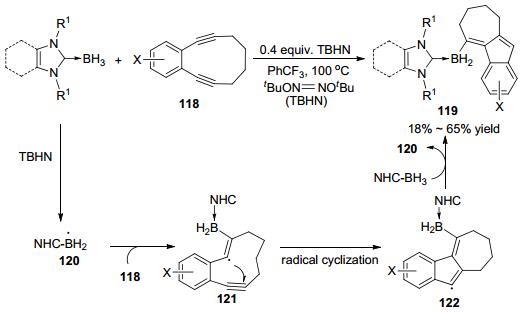 图式27
硼自由基参与的二炔环化反应
Scheme27.
Radical boryaltion/cyclization of diynes
图式27
硼自由基参与的二炔环化反应
Scheme27.
Radical boryaltion/cyclization of diynes
最近, 汪义丰等[52]报道了氮杂环卡宾硼烷自由基参与的1, 6-烯炔化合物123的环化反应, 得到了硼基团取代的六元环状化合物124和125(Scheme 28).两种产物的选择性主要受炔和烯烃上的取代基控制的.他们给出了可能的反应机理:氮杂环卡宾硼烷加合物和自由基引发剂反应生成硼自由基中间体120, 当底物是123a时, 120进攻苯基取代的双键生成中间体126, 随后发生对炔基的自由基进攻经6-exo-dig环化高对映选择性地得到127, 最后攫取硫醇上的氢原子生成目标产物124a, 生成的硫醇自由基可以继续攫取氮杂环卡宾硼烷加合物上的氢重新生成120.当底物为123b时, 120首先进攻苯基取代的炔键, 随后经关环、氢转移得到目标产物125b.
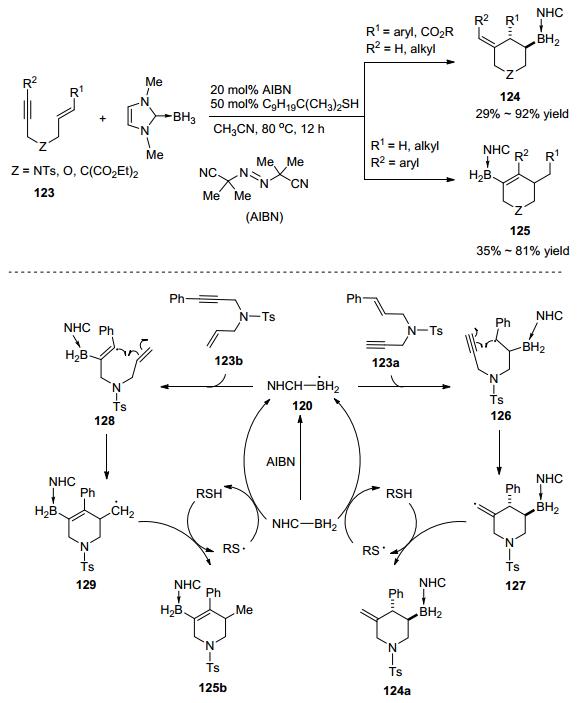 图式28
硼自由基参与的1, 6-烯炔环化反应
Scheme28.
Radical boryaltion/cyclization of 1, 6-enynes
图式28
硼自由基参与的1, 6-烯炔环化反应
Scheme28.
Radical boryaltion/cyclization of 1, 6-enynes
6 取代反应
NHC-硼烷加合物在碘或者HOTf的活化下生成活性较高的离子型硼烷加合物, 进而和亲核试剂发生取代反应.当使用氰基化合物作为亲核试剂时, 能够生成C—B键, 经水解生成硼取代酰胺(Scheme 29)[53]. NHC-碘硼烷加合物在还原剂联苯基锂存在下能够生成硼负离子, 进而和多种亲电试剂发生取代反应, 构筑C—B键(表 3)[19]. NHC-叠氮硼烷可以和炔烃发生[3+2]环加成生成硼取代的三氮唑[54], 由于没有涉及C—B键的形成, 在此不予展开, 感兴趣的读者可以参考相应文献.
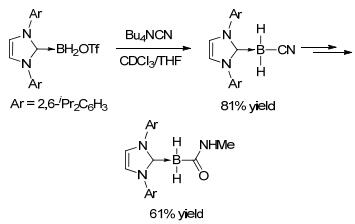 图式29
离子型NHC-硼烷加合物的亲核取代
Scheme29.
Nucleophilic substitution of ionic NHC-borane adducts
表 3
NHC-硼烷加合物与亲电试剂的取代反应
Table 3.
Reactions of electrophiles with NHC-borane adducts
图式29
离子型NHC-硼烷加合物的亲核取代
Scheme29.
Nucleophilic substitution of ionic NHC-borane adducts
表 3
NHC-硼烷加合物与亲电试剂的取代反应
Table 3.
Reactions of electrophiles with NHC-borane adducts

亲电试剂 产物 收率/% EtOAc 
39 nBuCHO 
45 

36 nBuCl 
46 

57 

51 7 结论与展望
有机硼化合物的合成一直是有机合成中的研究热点, 在近十年来稳定硼烷加合物作为一种硼试剂在有机硼化合物的合成中受到越来越多的关注, 并且表现出了独特的性质.它们大多易于制备、稳定性高、对水和空气都不敏感, 以至于常常表现出异于经典“硼烷化学”的性质.稳定硼烷加合物作为硼试剂参与的有机硼化合物的合成反应不断取得突破, 新的合成方法不断被开发出来.稳定硼烷加合物独特的化学性质也为C—B键形成反应提供了新的想象空间, 而且由其生成的有机硼化合物也逐渐得到应用.
但是还应看到, 稳定硼烷加合物为硼试剂参与的有机硼化物合成反应类型较少, 大多是在试剂量活化剂的条件下进行反应, 金属催化的反应类型较少, 反应选择性控制还是一个挑战性课题, 大多数稳定硼烷加合物参与的C—B成键反应的机理研究有待进一步深入.另一方面, 相关反应生成的有机硼烷加合物的转化方法也比较少, 在合成中的应用有待进一步发掘.上述问题随着研究的深入有希望得到解决, 因此, 我们有理由相信未来稳定硼烷加合物参与的有机硼化物合成反应及其产物的应用将会持续受到关注, 也必将为有机硼化学带来新的机遇.
-
-
[1]
(a) Carboni, B.; Monnier, L. Tetrahedron 1999, 55, 1197.
(b) Staubitz, A.; Robertson, A. P. M.; Sloan, M. E.; Manners, I. Chem. Rev. 2010, 110, 4023.
(c) Staubitz, A.; Robertson, A. P. M.; Manners, I. Chem. Rev. 2010, 110, 4079. -
[2]
For a recent review, see:(a) Yang, X.; Xie, Z.; He, J.; Yu, L. Chin. J. Org. Chem. 2015, 35, 603(in Chinese).
(阳香华, 谢珍茗, 何军, 余林, 有机化学, 2015, 35, 603.) For selected examples, see:
(b) Blaquiere, N.; Diallo-Garcia, S.; Gorelsky, S. I.; Black, D. A.; Fagnou, K. J. Am. Chem. Soc. 2008, 130, 14034.
(c) Shao, Z.; Fu, S.; Wei, M.; Zhou, S.; Liu, Q. Angew. Chem., Int. Ed. 2016, 55, 14653.
(d) Zhou, Q.; Zhang, L.; Meng, W.; Feng, X.; Yang, J.; Du, H. Org. Lett. 2016, 18, 5189.
(e) Li, S.; Meng, L.; Du, H. Org. Lett. 2017, 19, 2604. -
[3]
Curran, D. P.; Solovyev, A.; Brahmi, M. M.; Fensterbank, L.; Malacria, M.; Lacôte, E. Angew. Chem., Int. Ed. 2011, 50, 10294. doi: 10.1002/anie.201102717
-
[4]
For selected reviews, see:(a) Ramachandran, P. V., Brown, H. C. Organoboranes for Syntheses, ACS Symposium Series 783, American Chemical Society, Washington, DC, 2001.
(b) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
(c) Braunschweig, H.; Dewhurst, R. D.; Schneider, A. Chem. Rev. 2010, 110, 3924.
(d) Jäkle, F. Chem. Rev. 2010, 110, 3985.
(e) Dembitsky, V. M.; Quntar, A. A. A. A.; Srebnik, M. Chem. Rev. 2011, 111, 209.
(f) Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417.
(g) Leonori, D.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2015, 54, 1082. -
[5]
For reviews, see:(a) Burgess, K.; Ohlmeyer, M. J. Chem. Rev. 1991, 91, 1179.
(b) Beletskaya, I.; Moberg, C. Chem. Rev. 2006, 106, 2320.
(c) Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2010, 110, 890.
(d) Hartwig, J. F. Chem. Soc. Rev. 2011, 40, 1992.
(e) Ros, A.; Fernández, R.; Lassaletta, J. M. Chem. Soc. Rev. 2014, 43, 3229. -
[6]
(a) Welch, C. N.; Shore, G. S. Inorg. Chem. 1968, 7, 225.
(b) Brown, H. C.; Gupta, S. K. J. Am. Chem. Soc. 1975, 97, 5249. -
[7]
Zaidlewicz, M.; Brown, H. C.; Siebert, W. Advances in Boron Chemistry, The Royal Society of Chemistry, Cambridge, 1997, 171.
-
[8]
(a) Schaeffer, G. W.; Anderson, E. R. J. Am. Chem. Soc. 1949, 71, 2143.
(b) Nainan, K. C.; Ryschkewitsch, G. E. Inorg. Chem. 1969, 8, 2671. -
[9]
(a) Baldwin, A. R.; Washburn, R. M. J. Org. Chem. 1961, 26, 3549.
(b) Brahmi, M. M.; Monot, J.; Murr, M, D.; Curran, D. P.; Fensterbank, L.; Lacôte, E.; Malacria, M. J. Org. Chem. 2010, 75, 6983. -
[10]
(a) Brown, H. C.; Chandrasekharan, J. J. Am. Chem. Soc. 1984, 106, 1863.
(b) Kanth, J. V. B. Aldrichim. Acta 2002, 35, 57. -
[11]
Scheideman, M.; Shapland, P.; Vedejs, E. J. Am. Chem. Soc. 2003, 125, 10502. doi: 10.1021/ja034655m
-
[12]
Beak, P. Acc. Chem. Res. 1992, 25, 215. doi: 10.1021/ar00017a002
-
[13]
Clay, J. M.; Vedejs, E. J. Am. Chem. Soc. 2005, 127, 5766. doi: 10.1021/ja043743j
-
[14]
(a) Shapland, P.; Vedejs, E. J. Org. Chem. 2006, 71, 6666.
(b) Karatjas, A. G.; Vedejs, E. J. Org. Chem. 2008, 73, 9508.
(c) Scheideman, M.; Wang, G.; Vedejs, E. J. Am. Chem. Soc. 2008, 130, 8669. -
[15]
Pronin, S. V.; Tabor, M. G.; Jansen, D. J.; Shenvi, R. A. J. Am. Chem. Soc. 2012, 134, 2012. doi: 10.1021/ja211090n
-
[16]
Tabor, M. G.; Shenvi, R. A. Org. Lett. 2015, 17, 5776. doi: 10.1021/acs.orglett.5b02992
-
[17]
Prokofjevs, A.; Boussonnière, A.; Li, L.; Bonin, H.; Lacôte, E.; Curran, D. P.; Vedejs, E. J. Am. Chem. Soc. 2012, 134, 12281. doi: 10.1021/ja305061c
-
[18]
Pan, X.; Boussonnière, A.; Curran, D. P. J. Am. Chem. Soc. 2013, 135, 14433. doi: 10.1021/ja407678e
-
[19]
Monot, J.; Solovyev, A.; Bonin-Dubarle, H.; Derat, É.; Curran, D. P.; Robert, M.; Fensterbank, L.; Malacria, M.; Lacôte, E. Angew. Chem., Int. Ed. 2010, 49, 9166. doi: 10.1002/anie.201004215
-
[20]
Boussonnière, A.; Pan, X.; Geib, S. J.; Curran, D. P. Organometallics 2013, 32, 7445. doi: 10.1021/om400932g
-
[21]
Sewell, L. J.; Chaplin, A. B.; Weller, A. S. Dalton Trans. 2011, 40, 7499. doi: 10.1039/c1dt10819k
-
[22]
Johnson, H. C.; Torry-Harris, R.; Ortega, L.; Theron, R.; McIndoe, J. S.; Weller, A. S. Catal. Sci. Technol. 2014, 4, 3486. doi: 10.1039/C4CY00597J
-
[23]
Toure, M.; Chuzel, O.; Parrain, J. L. J. Am. Chem. Soc. 2012, 134, 17892. doi: 10.1021/ja309018f
-
[24]
Wang, Q.; Motika, S. E.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Angew. Chem., Int. Ed. 2014, 53, 5418. doi: 10.1002/anie.v53.21
-
[25]
Motika, S. E.; Wang, Q.; Akhmedov, N. G.; Wojtas, L.; Shi, X. Angew. Chem., Int. Ed. 2016, 55, 11582. doi: 10.1002/anie.201604986
-
[26]
Taniguchi, T.; Curran, D. P. Angew. Chem., Int. Ed. 2014, 53, 13150. doi: 10.1002/anie.201408345
-
[27]
Nerkar, S.; Curran, D. P. Org. Lett. 2015, 17, 3394. doi: 10.1021/acs.orglett.5b01101
-
[28]
McFadden, T. R.; Fang, C.; Geib, S. J.; Merling, E.; Liu, P.; Curran D. P. J. Am. Chem. Soc. 2017, 139, 1726. doi: 10.1021/jacs.6b09873
-
[29]
De Vries, T. S.; Prokofjevs, A.; Harvey, J. N.; Vedejs, E. J. Am. Chem. Soc. 2009, 131, 14679. doi: 10.1021/ja905369n
-
[30]
Farrell, J. M.; Stephan, D. W. Angew. Chem., Int. Ed. 2015, 54, 5214. doi: 10.1002/anie.v54.17
-
[31]
Prokofjevs, A.; Vedejs, E. J. Am. Chem. Soc. 2011, 133, 20056. doi: 10.1021/ja208093c
-
[32]
Prokofjevs, A.; Jermaks, J.; Borovika, A.; Kampf, J. W.; Vedejs, E. Organometallics 2013, 32, 6701. doi: 10.1021/om400651p
-
[33]
Cazorla, C.; De Vries, T. S.; Vedejs, E. Org. Lett. 2013, 15, 984. doi: 10.1021/ol303203m
-
[34]
(a) Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998.
(b) Dorwald, F. Z. Metal Carbenes in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 1999.
(c) Doyle, M. P. Chem. Rev. 1986, 86, 919.
(d) Doyle, M. P.; Forbes, D. C. Chem. Rev. 1998, 98, 911.
(e) Zhu, S.-F.; Zhou, Q.-L. Nat. Sci. Rev. 2014, 1, 580.
(f) Ford, A.; Miel, H.; Ring, A.; Slattery, C. N.; Maguire, A. R.; McKervey, M. A. Chem. Rev. 2015, 115, 9981. -
[35]
Bedel, C.; Foucaud, A. Tetrahedron Lett. 1993, 34, 311. doi: 10.1016/S0040-4039(00)60575-0
-
[36]
Monnier, L.; Delcros, J.-G.; Carboni, B. Tetrahedron 2000, 56, 6039. doi: 10.1016/S0040-4020(00)00565-2
-
[37]
Imamoto, T.; Yamanoi, Y. Chem. Lett. 1996, 25, 705. doi: 10.1246/cl.1996.705
-
[38]
Cheng, Q.-Q.; Zhu, S.-F.; Zhang, Y.-Z.; Xie, X.-L.; Zhou, Q.-L. J. Am. Chem. Soc. 2013, 135, 14094. doi: 10.1021/ja408306a
-
[39]
程清卿, 徐唤, 朱守非, 周其林, 化学学报, 2015, 73, 326. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmCheng, Q.-Q.; Xu, H.; Zhu, S.-F.; Zhou, Q.-L. Acta Chim. Sinica 2015, 73, 326(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[40]
Li, X.; Curran, D. P. J. Am. Chem. Soc. 2013, 135, 12076. doi: 10.1021/ja4056245
-
[41]
Allen, T. H.; Curran, D. P. J. Org. Chem. 2016, 81, 2094. doi: 10.1021/acs.joc.6b00091
-
[42]
Chen, D.; Zhang, X.; Qi, W.-Y.; Xu, B.; Xu, M.-H. J. Am. Chem. Soc. 2015, 137, 5268. doi: 10.1021/jacs.5b00892
-
[43]
Hyde, S.; Veliks, J.; Liégault, B.; Grassi, D.; Taillefer, M.; Gouverneur, V. Angew. Chem., Int. Ed. 2016, 55, 3785. doi: 10.1002/anie.201511954
-
[44]
Yang, J.-M.; Li, Z.-Q.; Li, M.-L.; He, Q.; Zhu, S.-F.; Zhou, Q.-L. J. Am. Chem. Soc. 2017, 139, 3784. doi: 10.1021/jacs.6b13168
-
[45]
For recent examples see:(a) Aramaki, Y.; Omiya, H.; Yamashita, M.; Nakabayashi, K.; Ohkoshi, S.; Nozaki, K. J. Am. Chem. Soc. 2012, 134, 19989. (b) Rosenthal, A. J.; Devillard, M.; Miqueu, K.; Bouhadir, G.; Bourissou, D. Angew. Chem., Int. Ed. 2015, 54, 9198. (c) Silva Valverde, M. F.; Schweyen, P.; Gisinger, D.; Bannenberg, T.; Freytag, M.; Kleeberg, C.; Tamm, M. Angew. Chem., Int. Ed. 2017, 56, 1135. doi: 10.1021/ja3094372
-
[46]
(a) Roberts, B. P. Chem. Soc. Rev. 1999, 28, 25.
(b) Rablen, P. R. J. Am. Chem. Soc. 1997, 119, 8350.
(c) Walton, J. C. Angew. Chem., Int. Ed. 2009, 48, 1726.
(d) Lalevée, J.; Blanchard, N.; Chany, A.-C.; Tehfe, M.-A.; Allonas, X.; Fouassier, J.-P. J. Phys. Org. Chem. 2009, 22, 986. -
[47]
(a) Barton, D. H. R.; Jacob, M. Tetrahedron Lett. 1998, 39, 1331.
(b) Ueng, S.-H.; Brahmi, M. M.; Derat, É.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Curran, D. P. J. Am. Chem. Soc. 2008, 130, 10082. -
[48]
(a) Pan, X.; Lalevée, J.; Lacôte, E.; Curran, D. P. Adv. Synth. Catal. 2013, 355, 3522.
(b) Ueng, S.-H.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Curran, D. P. Org. Biomol. Chem. 2011, 9, 3415.
(c) Pan, X.; Lacôte, E.; Lalevée, J.; Curran, D. P. J. Am. Chem. Soc. 2012, 134, 5669. -
[49]
(a) Telitel, S.; Schweizer, S.; Morlet-Savary, F.; Graff, B.; Tschamber, T.; Blanchard, N.; Fouassier, J. P.; Lelli, M.; Lacôte, E.; Lalevée, J. Macromolecules 2013, 46, 43.
(b) Lalevée, J.; Telitel, S.; Tehfe, M. A.; Fouassier, J. P.; Curran, D. P.; Lacôte, E. Angew. Chem., Int. Ed. 2012, 51, 5958. -
[50]
(a) Pan, X.; Vallet, A.-L.; Schweizer, S.; Dahbi, K.; Delpech, B.; Blanchard, N.; Graff, B.; Geib, S. J.; Curran, D. P.; Lalevée, J.; Lacôte, E. J. Am. Chem. Soc. 2013, 135, 10484.
(b) Telitel, S.; Vallet, A.-L.; Schweizer, S.; Delpech, B.; Blanchard, N.; Morlet-Savary, F.; Graff, B.; Curran, D. P.; Robert, M.; Lacôte, E.; Lalevée, J. J. Am. Chem. Soc. 2013, 135, 16938. -
[51]
Watanabe, T.; Hirose, D.; Curran, D. P.; Taniguchi, T. Chem.-Eur. J. 2017, 23, 5404. doi: 10.1002/chem.v23.23
-
[52]
Ren, S.-C.; Zhang, F.-L.; Qi, J.; Huang, Y.-S.; Xu, A.-Q.; Yan, H.-Y.; Wang, Y.-F. J. Am. Chem. Soc. 2017, 139, 6050. doi: 10.1021/jacs.7b01889
-
[53]
Solovyev, A.; Chu, Q.; Geib, S. J.; Fensterbank, L.; Malacria, M.; Lacôte, E.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 15072. doi: 10.1021/ja107025y
-
[54]
Merling, E.; Lamm, V.; Geib, S. J.; Lacôte, E.; Curran, D. P. Org. Lett. 2012, 14, 2690. doi: 10.1021/ol300851m
-
[1]
-

图式1 稳定硼烷加合物的成键原理及主要类型
Scheme 1 Bond-forming principle and main classes of stable borane adducts

图式3 碘促进的高烯丙基胺-硼烷加合物的分子内硼氢化反应
Scheme 3 Iodine-promoted hydroboration of homoallylicamine-borane adducts

图式4 高烯丙基胺硼烷加合物的分子内硼氢化反应可能机理
Scheme 4 Proposed mechanisms for the intramolecular hydroboration of homoallylic amine-borane adducts

图式5 TfOH促进的高烯丙基膦硼烷加合物的分子内硼氢化反应
Scheme 5 TfOH-promotedintramolecular hydroboration of homoallylic phosphine-borane adducts

图式6 I2促进烯烃和吡啶硼烷加合物的分子间硼氢化反应
Scheme 6 I2-promoted intermolecular hydroboration of alkenes with pyridine borane adducts

图式7 I2促进炔烃和吡啶硼烷加合物的分子间硼氢化反应
Scheme 7 I2-promoted intermolecular hydroboration of alkynes with pyridine borane adduct

图式8 烷基频哪醇硼酸酯和三氟硼酸钾化合物的制备
Scheme 8 Preparation of alkylpinacolborate and potassium alkyltrifluoroborate

图式9 氨基硼烷加合物的硼氢化用于天然产物的合成
Scheme 9 Applications of hydroboration of amine-borane adducts in natural product synthesis

图式10 Tf2NH催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应
Scheme 10 Tf2NH-catalyzed hydroboration of alkenes with NHC-BH3

图式12 I2催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应可能机理
Scheme 12 Proposed mechanism of I2-catalyzed hydroboration of alkenes with NHC-BH3

图式13 Tf2NH或I2促进的硅基取代的炔烃和氮杂环卡宾硼烷加合物的硼氢化反应
Scheme 13 Tf2NH/I2-promoted hydroborationof silylsub-stituted alkynes with NHC-BH3

图式14 Rh催化的烯烃和三甲胺硼烷加合物的硼氢化反应
Scheme 14 Rh-catalyzed hydroboration of alkene with Me3N-borane adduct

图式15 Au催化的炔烃和硼烷加合物的分子内硼氢化反应
Scheme 15 Au-catalyzed intramolecular hydroboration of alkynes withborane adducts

图式18 苄胺硼烷加合物的分子内芳基C—H键硼化反应
Scheme 18 Intramolecular aromatic C—H bond borylation of benzyl amine-boranes

图式20 脂肪胺硼烷加合物的分子内C—H键硼化反应
Scheme 20 Intramolecular C—H bond borylation from benzyl alkyl amine-borane

图式21 Tf2NH促进的膦硼烷加合物分子内的C—H键硼化反应
Scheme 21 Tf2NH-promoted intramolecular C—H borylation from phosphine-borane

图式24 Rh(Ⅰ)催化的卡宾对胺硼烷加合物的不对称B—H键插入反应
Scheme 24 Rhodium(Ⅰ)-catalyzed asymmetric B—H bond insertion of amine-borane adducts

图式25 炔烃为卡宾前体的B—H键插入产物转化
Scheme 25 Transformations of B—H bond insertion product using alkynes as carbene precursors

图式26 CuCl催化的炔烃为卡宾前体的B—H键插入反应吉布斯自由能曲线图
Scheme 26 Free energy profile for CuCl-catalyzed B—H bond insertion using alkynes as carbene precursors

图式29 离子型NHC-硼烷加合物的亲核取代
Scheme 29 Nucleophilic substitution of ionic NHC-borane adducts
表 1 催化的烯烃和氮杂环卡宾硼烷加合物的硼氢化反应
Table 1. I2-catalyzed hydroboration of alkenes with NHC-BH3
Alkene Product Yield/% 75 68 61 50 37  下载: 导出CSV
下载: 导出CSV
表 2 由NHC-BH3和乙炔二羧酸二甲酯合成硼杂环丙烷化合物
Table 2. Synthesis of NHC-boriranes by reactions of acetylenedicarboxylate esters with NHC-BH3
R 56:57 Yield/% 56 57 Me 33:67 19 35 iPr 52:48 14 29 2, 4, 6-Me3C6H2 81:19 31 10 2, 6-iPr2C6H3 86:14 80 5
下载: 导出CSV
表 3 NHC-硼烷加合物与亲电试剂的取代反应
Table 3. Reactions of electrophiles with NHC-borane adducts
亲电试剂 产物 收率/% EtOAc 39 nBuCHO 45 36 nBuCl 46 57 51
下载: 导出CSV
-





























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 119
- 文章访问数: 6258
- HTML全文浏览量: 2207
 下载:
下载:
