 图1
替诺福韦艾拉酚胺、替诺福韦酯、替诺福韦和索非布韦的结构
Figure1.
Structures of tenofovir alafenamide, tenofovir disoproxil, tenofovir and sofosbuvir
图1
替诺福韦艾拉酚胺、替诺福韦酯、替诺福韦和索非布韦的结构
Figure1.
Structures of tenofovir alafenamide, tenofovir disoproxil, tenofovir and sofosbuvir
引用本文:
聂飚, 金传飞, 钟文和, 任青云, 张英俊, 张霁. 磷酰胺酯前药策略及ProTide技术在药物研发中的应用与进展[J]. 有机化学,
2017, 37(11): 2818-2840.
doi:
10.6023/cjoc201705022

Citation: Nie Biao, Jin Chuanfei, Zhong Wenhe, Ren Qingyun, Zhang Yingjun, Zhang Ji. Application and Recent Progress of Phosphoramidate Prodrugs Strategies and ProTide Technology in Drug Discovery[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2818-2840. doi: 10.6023/cjoc201705022
Citation: Nie Biao, Jin Chuanfei, Zhong Wenhe, Ren Qingyun, Zhang Yingjun, Zhang Ji. Application and Recent Progress of Phosphoramidate Prodrugs Strategies and ProTide Technology in Drug Discovery[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2818-2840. doi: 10.6023/cjoc201705022
磷酰胺酯前药策略及ProTide技术在药物研发中的应用与进展
English
Application and Recent Progress of Phosphoramidate Prodrugs Strategies and ProTide Technology in Drug Discovery
Abstract:
This review provides the brief history for the development and advances of ProTide strategies in the field of antiviral and anticancer pharmaceutical development. A summary is provided for the synthesis of nucleoside phosphoramidates, recent breakthroughs in the stereoselective assembly of chiral prodrugs and its application. This technology is extremely useful and has simplified innovative drug design of phosphorous-containing prodrugs. Simplifying such structural modification may create a new avenue of competition in the invention of new medicines from the parent drug.
-
Key words:
- phosphoramidates
- / prodrugs
- / nucleoside and nucleotide
- / antiviral drug
- / phosphate and phosphonate
- / ProTide
-
从索非布韦(Sofosbuvir, 又译索磷布韦)到替诺福韦艾拉酚胺(Tenofovir Alafenamide), 引入磷酰胺酯前药策略实现治疗丙型肝炎病毒(HCV)和乙型肝炎病毒(HBV)的划时代突破
2016年11月11日, 美国食品药品监督管理局(FDA)批准了吉利德(Gilead Sciences)替诺福韦艾拉酚胺(Vemlidy®, Tenofovir Alafenamide, 1, 25 mg)用于治疗慢性乙型肝炎病毒(HBV)感染的上市申请. Vemlidy®是一种创新型、靶向性的替诺福韦(Tenofovir)磷酰胺酯(又译为氨基磷酸酯)前药(图 1), 与300 mg的替诺福韦酯Viread® (2)相比, 只需要少于十分之一的剂量就可达到类同的抗病毒功效.临床数据显示, Vemlidy®具有更大的血浆稳定性, 而且可更有效地将替诺福韦(3)递送到肝细胞, 所以可以使用更低的给药剂量, 导致血液中替诺福韦浓度更低[1].基于上述原因, 与Viread®相比, Vemlidy®有效改善了针对肾脏和骨骼的安全性参数. Vemlidy®也是在近十年内被批准用于治疗慢性乙型肝炎病毒的第一个药物[2].预测其将成为重磅炸弹药物(Blockbuster drug).
图1
替诺福韦艾拉酚胺、替诺福韦酯、替诺福韦和索非布韦的结构
Figure1.
Structures of tenofovir alafenamide, tenofovir disoproxil, tenofovir and sofosbuvir
在此之前, 2013年12月6日美国FDA批准吉利德公司抗丙型肝炎病毒(HCV)新药索非布韦(Sofosbuvir, 4)的上市申请, 它是含氟核苷酸类似物的磷酰胺酯前药, 也是一种有效的口服NS5B聚合酶抑制剂, 对丙肝病毒有很强抑制作用, 同时是治疗慢性丙型肝炎联合用药的重要组成药物(图 1)[2], 治愈率高达百分之90%, 上市第一年其全球销售额便超过100亿美元, 是名副其实的“重磅炸弹”药物[3].
磷酰胺酯和磷酸酯前药是密不可分的、相互关联的一类分子, 其水解或酶解即体内代谢的产物转化为有机磷酸(图 2)和单磷酸、二磷酸或三磷酸衍生物而发挥药效, 所以许多综述都将其合二为一进行讨论[4, 5].本文开始也就此做一些简单概述磷酸酯/磷酸前药的作用.核苷类药物在肿瘤、感染性疾病(病毒如HIV、HBV、HCV)等领域应用广泛.其作用靶点多为DNA聚合酶或RNA逆转录酶, 核苷类药物一般模拟天然核苷的结构, 竞争性地作用于酶活性中心, 嵌入正在合成的DNA或RNA链中, 干扰核酸代谢影响细胞增殖、分裂等.核苷类药物在细胞内经酶活化, 转化为磷酸, 其中单磷酸化是限速步骤, 因此通常在核苷类药物中引入磷酸或磷酸酯基团.早期设计的磷酸或磷酸酯衍生物因其磷-氧键稳定性较差, 极性大, 难以通过细胞膜等而限制了应用.为修饰改善此类药理化性质, 在药物设计中, 前药策略被广泛采用.通过磷酰胺酯前药技术将极性基团隐蔽来降低化合物极性, 增加亲脂性进而增加细胞透膜性.磷酰胺酯前药进入细胞后经特定酶代谢释放原型药物(图 2), 原型药物经磷酸化后发挥药效.毋容置疑, 长期以来, 在化合物中引入磷酸酯或磷酸及其钠盐作为前药在药物研究与开发中是一个极其重要的策略, 常见的基于磷酸前药策略而开发的药物[4](表 1).这样的前药策略主要的作用是: (1)通过改善药物的理化性质, 提高药物的传输速率和选择性; (2)提高药物的生物利用度; (3)对水溶性差的母体药物, 通过磷酸酯化提高药物的水溶性; (4)通过改变给药方式, 延长药物作用时间, 方便患者; (5)通过改善脂溶性, 提高细胞膜的穿透能力, 提高药效; (6)降低毒性, 提高治疗效果, 即提高药物的安全性[5, 6].
 图2
磷酰胺酯前药转化为磷酸前药
Figure2.
Phosphoramidate convert into phosphate prodrug
图2
磷酰胺酯前药转化为磷酸前药
Figure2.
Phosphoramidate convert into phosphate prodrug
 表 1
通过有机磷酸酯/有机磷酸及其钠盐前药改善母体药物的亲脂性和膜穿透能力[3b]
Table 1.
Phosphate and phosphonate prodrugs for improved lipophilicity or permeability
表 1
通过有机磷酸酯/有机磷酸及其钠盐前药改善母体药物的亲脂性和膜穿透能力[3b]
Table 1.
Phosphate and phosphonate prodrugs for improved lipophilicity or permeability
No. 药物名称 结构 前药策略 1 Adefovir dipivoxil
阿德福韦酯
(ⅰ)通过酯酶和磷酸二酯酶生物转化为阿德福韦
(ⅱ)使阿德福韦口服生物利用度从10%增加到阿德福韦酯的30%~45%2 Tenofovir disoproxil
替诺福韦酯
(ⅰ)通过酯酶和磷酸二酯酶生物转化为替诺福韦
(ⅱ)替诺福韦酯的口服生物利用度是替诺福韦的2.5倍3 Miproxifene phosphate
磷酸米泼昔芬
(ⅰ)通过碱性磷酸酯酶生物转化为米泼昔芬
(ⅱ) pH值为7.4时, 水溶性提高了近1000倍
(ⅲ)临床前大鼠和犬的生物利用度分别提高了28.8%和23.8%4 Fosamprenavir
福沙那韦钙
(ⅰ)通过碱性磷酸酯酶生物转化为福沙那韦
(ⅱ)水溶性提高了10倍以上
(ⅲ)更方便患者的服药剂量
(ⅳ)延长专利有效期5 Estramustine phosphate
雌二醇氮芥磷酯
(ⅰ)通过碱性磷酸酯酶生物转化为雌二醇氮芥
(ⅱ)注射剂和口服制剂均已上市6 Prednisolone phosphate
泼尼松龙磷酸钠
(ⅰ)通过碱性磷酸酯酶生物转化为泼尼松龙
(ⅱ)由于前药能开发出液体制剂, 改善儿童服用的顺应性7 Fludarabine phosphate
磷酸氟达拉滨
(ⅰ)通过碱性磷酸酯酶生物转化为氟达拉滨
(ⅱ)目前仅有注射剂上市8 Fosphenytoin
磷苯妥英
(ⅰ)通过碱性磷酸酯酶快速转化为苯妥英
(ⅱ)溶解度从苯妥英的25 μg/mL增加到磷苯妥英的140 mg/mL9 Fosfluconazole
福司氟康唑
(ⅰ)通过碱性磷酸酯酶快速转化为氟康唑
(ⅱ)不同规格的产品均能采用静脉给药
(ⅲ)显著提高水溶性(福司氟康唑溶解度为300 mg/mL)10 Phosphonooxymethyl
Propofol
(ⅰ)通过碱性磷酸酯酶快速转化为丙泊酚
(ⅱ)溶解度从丙泊酚的150 μg/mL显著提高到前药的500 mg/mL11 Propofol phosphate
丙泊酚磷酸酯
(ⅰ)通过碱性磷酸酯酶快速转化为丙泊酚
(ⅱ)显著增加了丙泊酚的水溶性例如, 雷公藤甲素又称雷公藤内酯或雷公藤内酯醇, 是从卫矛科植物雷公藤的根、叶、花及果实中提取的一种环氧二萜内酯化合物, 具有抗炎、抗癌及免疫抑制作用.雷公藤甲素与青蒿素、雷公藤红素、辣椒素和姜黄素, 是最有可能被开发成为现代药物的五种传统天然药用化合物之一[7], 不幸的是雷公藤甲素(5)的溶解度极差(17 μg/mL), 而通过三步化学合成转化成相应的磷酸二钠盐前药(6)后(图 3), 溶解度显著地提高到61 mg/mL, 更值得一提的是, 作为一个含有三个环氧丙烷单元的修饰天然产物, 其药物的稳定性也相当的不错[8].最近人体结肠癌动物模型实验结果表明, 通过腹腔注射, 剂量在每日给药低至0.3 mg/kg的条件下, 该磷酸二钠盐前药仍然能够有效地减少或消除肿瘤的生成; 另外, 通过口服方式给药, 剂量能控制在0.6和0.9 mg/kg之间, 在人体卵巢癌的动物摸型上也发现, 磷酸二钠盐前药有效, 具有良好的耐受性.
 图3
雷公藤内酯的结构和磷酸前药
Figure3.
Structures of triptolide and its phosphate prodrug
图3
雷公藤内酯的结构和磷酸前药
Figure3.
Structures of triptolide and its phosphate prodrug
苯并咪唑类驱虫药(Fenbendazole, 7)的磷酸盐前药8具有非常高的水溶性和稳定性(图 4), 应用时可直接加入到饮用水中[9].在猪体内的驱虫效果磷酸盐前药至少与原型药相当.进一步的药代实验显示, 前药给药时血浆中芬苯哒唑和其活性代谢产物浓度更高, 说明前药的使用剂量可以更低, 这样的话, 药物的毒性和副作用就会降低, 用药的安全窗就会更大.
 图4
水溶性和稳定性高的苯并咪唑类驱虫药的磷酸盐前药
Figure4.
Water-soluble and water-stable phosphate prodrugs of fenbendazole
图4
水溶性和稳定性高的苯并咪唑类驱虫药的磷酸盐前药
Figure4.
Water-soluble and water-stable phosphate prodrugs of fenbendazole
联芳基α-D-甘露糖苷9, 作为一类FimH拮抗剂, 可以开发用于治疗尿路感染.但由于其理化性质不理想, 主要是水溶性较差, 难以将其开发用于口服治疗. Ernst等[10]报道了将其修饰成磷酸盐前药10(图 5), 溶解度提高140倍; 在Caco-2细胞模型中, 前药不断被酶水解成活性成分, 形成亚饱和溶液, 在高浓度梯度下透过细胞膜.在体内模型中可以观察到Cmax更高并维持更长时间, 这样药物可以在尿液中保留更长时间发挥药效.
 图5
水溶性和稳定性高的磷酸盐前药
Figure5.
Water-soluble and highly stable phosphate prodrug
图5
水溶性和稳定性高的磷酸盐前药
Figure5.
Water-soluble and highly stable phosphate prodrug
克罗卡林(Cromakalim, 11)是一类钾通道开放剂, 通过降低眼内压用于治疗青光眼.由于克罗卡林水溶性有限, 给其临床应用带来了很大限制. Dosa等[11]报道了将其修饰成磷酸盐前药, 其中活性最好的化合物13 (Scheme 1), 增加水溶性的同时其化学稳定性会变得更好, 在正常血压的小鼠模型中一天一次给药降低眼内压的效果更明显.
 图式 1
制备克罗卡林水溶磷酸盐前药的方法
Scheme1.
Method to prepare water soluble prodrug of levcro-makalim
图式 1
制备克罗卡林水溶磷酸盐前药的方法
Scheme1.
Method to prepare water soluble prodrug of levcro-makalim
仔细地考查替诺福韦(TFV, 3)、替诺福韦酯(TDF, 2)和替诺福韦艾拉酚胺(TAF, 1)的结构差异, 不难发现替诺福韦酯(TDF)和替诺福韦艾拉酚胺是二次前药(前药的前药).许多前药并不是经过一步反应直接活化, 而是通过两步或多步反应来释放出母药的.显然替诺福韦含有磷酸基团, 极性太强, 转运能力弱, 不能进入血浆, 无法通过细胞膜进入有效作用位点.相反替诺福韦酯(TDF)和替诺福韦艾拉酚胺(TAF)的磷酸酯前药, 通过仔细的结构修饰进而显著地改善母体药物的物理化学性质, 如脂溶性的提高等, 不仅降低了药物的极性, 增加了药物的转运能力, 而且替诺福韦艾拉酚胺还能直接穿透细胞膜, 进入到淋巴细胞, 值得一提的是TAF在血液中更稳定, 但在T细胞中水解比TDF更快(图 6和表 2), 能有效到达作用位点, 对病毒的复制进行直接的攻击, 进而大大提高了药效, 极大地提高了生物利用度, 降低了毒副作用.显然替诺福韦艾拉酚胺中手性氨基酸(如L-丙氨酸)的引入、取代芳氧基的修饰是药效作用提高的关键.在过去短短的三年内, 从丙肝和乙肝两个“重磅炸弹”的横空出现, 并实现了在人类历史上征服和治愈丙肝病毒疾病划时代的、革命性的转变, 不难发现氧甲基磷酰胺酯和烷氧基磷酰胺酯前药策略在抗病毒药物研发中作用巨大, 功不可没.鉴于近年来已经有综述概述了磷酸酯前药的进展[12], 本文拟对这一类特殊的磷酸酯前药即近年来研究较多的磷酰胺酯前药的历史与发展状况做一评述, 以期起到“抛砖引玉”的效果.
 图6
三个药物在肠道、血浆和淋巴细胞之间的转运的过程图
Figure6.
Transporting of three drugs at gut, plasma and lymphoid cells
表 2
TFV, TDF和TAF的半衰期a
Table 2.
Half life of TFV, TDF and TAF
图6
三个药物在肠道、血浆和淋巴细胞之间的转运的过程图
Figure6.
Transporting of three drugs at gut, plasma and lymphoid cells
表 2
TFV, TDF和TAF的半衰期a
Table 2.
Half life of TFV, TDF and TAF
药物 T1/2/min 人血 T细胞摄取 TFV 稳定 稳定 TDF 0.4 71 TAF 90 28 a TDF:在血液中水解快, 而在T细胞中水解相对较慢; TAF:在血液中更稳定, 但在T细胞中水解比TDF更快. 1 ProTide技术的历史和发展简介
ProTide(源于PROdrug+nucleoTIDE)技术是20世纪90年代初由英国卡迪夫大学药学院的克里斯托弗·麦格根(Christopher McGuigan, 1958~2016)教授领导的团队首先发展起来的[13], 其技术核心就是通过前药的方式, 主要是由一个芳香取代基模体和一个氨基酸酯组成的结构以磷酰胺酯的形式将核苷类似物更加方便和有效地传输进入细胞內.研究的第一个突破是在1992年, McGuigan小组[13, 14]注意到芳氧基磷酸酯和烷氨基磷酸酯的功效. McGuigan等使用氯代磷酸酯由齐多夫定(AZT)制备磷酰胺酯. AZT的这些磷酸盐衍生物的抗HIV活性在一些情况下超过母体核苷的抗HIV活性.此外, 虽然齐多夫定在细胞系中几乎无活性(EC50 100 µmol•L-1), 但是取代的磷酰胺酯的活性高10倍(EC50 10 µmol•L-1).即磷酰胺酯更能够在细胞中保持活性.经过McGuigan和许多药物科学家的不断努力, 今天ProTide技术日趋完善, 据不完全统计, 目前至少有十个临床药物(包括上市的替诺福韦艾拉酚胺和索非布韦, 其余的见图 7)是采用该技术进行研发的, 其中的两个重磅炸弹药物已经革命化了抗丙肝病毒和乙肝病毒的治疗, 并深刻影响了未来小分子新药尤其是抗病毒和抗癌药物的研究进程[15].
 图7
进入临床试验的磷酰胺酯前药的化学结构
Figure7.
Chemical structures of nucleoside ProTide that under development in clinical trials
图7
进入临床试验的磷酰胺酯前药的化学结构
Figure7.
Chemical structures of nucleoside ProTide that under development in clinical trials
2 ProTides技术的基本修饰位点和透过细胞膜转运机制
ProTides技术的关键就是对核苷和核苷酸的母体结构在糖羟基的位置上用磷酸化的方式引入芳氧基和氨基酸(如L-丙氨酸)而形成磷酰胺酯, 即芳氧基磷酰胺三酯(Aryloxy phosphoramidate triesters), 广义而言, 在药物分子的游离羟基上引入这样的磷酰胺酯基团也称之为McGuigan的ProTide前药技术或策略[16].目前大部分处于临床阶段的核苷类前药都是应用该技术.其在体内经两步酶反应解离释放原型化合物, 首先酯基经酯酶水解, 在生理pH<7.4条件下带负电的羧基亲核进攻磷酸基团, 导致芳基离去形成不稳定的五元环中间态.水分子等亲核进攻该五元环使其开环, 生成磷酰胺代谢产物.接着第二种酶磷酰胺酶(组氨酸三联体核苷酸结合蛋白1, HINT-1)介导该代谢物的磷-氮(P-N)键的断裂, 释放单磷酸或磷酸酯原型化合物(图 8和Scheme 2).一般而言, 该类化合物一共有三个基本修饰点(图 9): (1)芳基取代基有卤素取代基或强吸电子基团, 离去能力好, 容易发生分子内亲核取代, 形成活泼的五元环内酯, 进而水解开环并在酶的催化下给出磷酸前药; (2)氨基酸侧链以直链为最佳选择, 其立体化学对生物活性至关重要(L构型比D构型的活性要好), α-氨基酸(如L-丙氨酸)效果最为明显; (3)酯基中以伯和仲烷基或苄基最为合适, 这种技术从这三方面入手, 由繁入简, 这样易于进行SAR构效关系研究, 发掘出药效最佳的临床药物, 因此越来越受到药物化学家的高度重视[17].
 图8
磷酰胺酯如何进入细胞传送磷酸前药的示意图
Figure8.
A general representation of how phosphoramidates deliver monophosphates into cells
图8
磷酰胺酯如何进入细胞传送磷酸前药的示意图
Figure8.
A general representation of how phosphoramidates deliver monophosphates into cells
 图式 2
芳氧基磷酰胺酯前药的作用方式
Scheme2.
Mode of action of aryloxyphosphoramidates or phosphonamidates
图式 2
芳氧基磷酰胺酯前药的作用方式
Scheme2.
Mode of action of aryloxyphosphoramidates or phosphonamidates
ProTide技术是一种最具有结构多样性的前药策略(图 9):氨基酸的类别(D/L)、手性(R/S)中心及羧酸酯取代基, 芳环上的修饰及其取代基, 磷原子上的手性(Sp/Rp)等结构变化既影响前药的脂水分配系数, 从而影响其吸收; 又影响水解酶的活性和特异性, 影响其释放速率.因此, 通过调整和优化这些非药效特异性的结构特征, 可以获得理想的磷酰胺酯前药, 由此奠定了ProTide技术的基石, 也正是有点石成金的ProTide技术, 才成就了索非布韦的传奇, 也焕发了替诺福韦艾拉酚胺的新生[17].
 图9
ProTide技术的基本修饰位点
Figure9.
Basic ProTide motif
图9
ProTide技术的基本修饰位点
Figure9.
Basic ProTide motif
例如氨基葡萄糖, 它是人体内合成的物质, 是形成软骨细胞的重要营养素, 也是健康关节软骨的天然组织成份.随着年龄的增长, 人体内的氨基葡萄糖的缺乏越来越严重, 关节软骨不断退化和磨损.氨基葡萄糖可以帮助修复和维护软骨, 并能刺激软骨细胞的生长. McGuigan等[18]发现一系列乙酰葡萄糖胺(Scheme 3, 23)氨基磷酸酯前药, 用于治疗骨关节炎(Osteoarthritis).但由于乙酰葡萄糖胺是一个大极性分子, 很难透过细胞膜, 通过在6-位引入氨基磷酸酯, 提高分子的脂溶性.在合成目标化合物过程中, 芳氧基团很容易被邻位羟基的参与作用下取代离去, 最终通过引入给电子的甲氧苯基才能得到目标化合物, 制备的一系列N-乙酰化氨基葡萄糖的磷酰胺24, 具有非常良好的抗骨质关节炎的保护作用.他们在测试中发现, 虽然氨基葡萄糖在10 mmol•L-1中显示活性, 但也观察到其毒性.值得强调的是, 在极低的浓度下(0.1 mmol•L-1), 一些氨基葡萄糖磷酰胺显示了很好的活性, 而没有观察到毒性, 这些化合物比氨基葡萄糖有>100倍的活性.值得强调的是, 在这个例子中构效关系研究(SAR)表明, 常被认为有利的带有L-丙氨酸磷酰胺酯活性不佳, 相反D-丙氨酸磷酰胺酯的活性更高.
 图式 3
N-乙酰化氨基葡萄糖和相应的磷酰胺酯
Scheme3.
Glucosamine and corresponding phosphoramidates of N-acetyl glucosamine
图式 3
N-乙酰化氨基葡萄糖和相应的磷酰胺酯
Scheme3.
Glucosamine and corresponding phosphoramidates of N-acetyl glucosamine
3 磷酰胺酯的合成及相关化学
核苷类磷酰胺酯前药的合成方法有以下三种方式(Scheme 4): (1)用磷酰氯29直接偶联核苷的游离羟基反应成酯[19]; (2)和二芳基亚磷酸酯发生单取代和氧化反应, 随后参与和氨基酸酯的氧化胺化[20]; (3)通过活化将氨基酸和核苷芳基酯进行偶联[21, 22].
 图式 4
制备核苷类磷酰胺酯前药的方法
Scheme4.
Methods to prepare phosphoramidates nucleoside prodrugs
图式 4
制备核苷类磷酰胺酯前药的方法
Scheme4.
Methods to prepare phosphoramidates nucleoside prodrugs
值得注意的是, 若考虑到磷(Ⅴ)中心的手性, 上过方法制备的氨基磷酸酯会产生大约1:1的非对映立体异构体, 这些异构体不可通过快速柱色谱分离.由于SP构型的和RP构型氨基磷酸酯体外活性的不同, 这也导致了发展光学纯的芳氧基氨基酸磷酰酯化试剂[23].关键的带有芳氧基和氨基酸酯的磷酰氯的制备如Scheme 5所示:从便宜易得的三氯氧磷出发, 在三乙胺的存在下, 首先和苯酚反应, 然后加入合适的氨基酸酯[24].
 图式 5
一般制备磷酰氯试剂的方法
Scheme5.
General method for preparation of phosphorochloridate reagent
图式 5
一般制备磷酰氯试剂的方法
Scheme5.
General method for preparation of phosphorochloridate reagent
带有芳氧基和氨基酸酯的磷酰氯37广泛应用于制备各种不同的烷氨基磷酸酯(Scheme 6).方法一是利用N-甲基咪唑(NMI)形成咪唑鎓盐来活化, 随后和核苷偶联[25], 相关的方法实例见图 10;方法二是利用叔丁基氯化镁除去核苷多糖5'位上的质子, 并随后与试剂37发生取代反应[26], 相关的方法实例见图 11.两种方法各有千秋, 但一般而言, 利用NMI方法收率较高, 使用也比较多.
 图式 6
制备核苷类烷氨基磷酸酯前药的两种途径
Scheme6.
Two pathways to generate phosphoramidate nucleosides prodrugs
图式 6
制备核苷类烷氨基磷酸酯前药的两种途径
Scheme6.
Two pathways to generate phosphoramidate nucleosides prodrugs
 图10
NMI活化方法制备的一些核苷类似物磷酰胺酯
Figure10.
Some phosphoramidates nucleoside prepared via NMI activation prodrugs
图10
NMI活化方法制备的一些核苷类似物磷酰胺酯
Figure10.
Some phosphoramidates nucleoside prepared via NMI activation prodrugs
 图11
t-BuMgCl方法制备的一些核苷类似物磷酰胺酯
Figure11.
Some phosphoramidate nucleosides prepared by t-BuMgCl method
图11
t-BuMgCl方法制备的一些核苷类似物磷酰胺酯
Figure11.
Some phosphoramidate nucleosides prepared by t-BuMgCl method
从上述两个方法中不难发现:磷酰胺酯化是发生在核苷中核糖的5位上的羟基, 值得一提的是如果糖上3位上有羟基, 则不可避免地有反应区域选择的问题, 以及双磷酰酯化和羟基保护与去保护等诸多问题, 造成了反应的收率偏低以及纯化的不易, 这些合成及工艺难点长久以来一直是困扰药物化学家的挑战之一(Scheme 7).
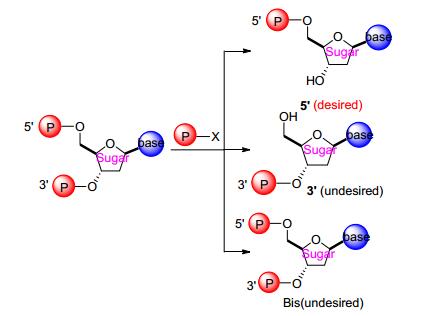 图式 7
核苷5位磷酸化和区域控制难题
Scheme7.
Nucleoside 5'-phosphorylation and regiocontrol challenge
图式 7
核苷5位磷酸化和区域控制难题
Scheme7.
Nucleoside 5'-phosphorylation and regiocontrol challenge
除上述两种方法以外, 为了解决原料中连接碱基所带来的反应物溶解度低下以及SAR构效关系研究的便利, Herdewijn等[27]报道了一种构建核苷类氨基磷酸酯前药的新方法(Eq. 1), 多糖氨基磷酸酯衍生物与核酸碱基通过Vorbrüggen反应以高收率得到保护的ProTide, 通过进一步选择性脱除乙酰基保护基得到目标分子.该方法的优势是可以快速引入不同的核酸碱基官能团, 构建ProTide化合物库.
磷酰胺酯核糖衍生物作为主要的基础原料与一些碱基通过Vorbrüggen偶联反应得到高产率的具有保护基的前药(Scheme 8).在糖单元上选择性水解乙酰基得到一系列目标前药[28].与传统方法相比较, 这种方法的优点是可以更灵活实现碱基部分的结构变化.
 图式 8
利用Vorbrüggen反应构建ProTide化合物
Scheme8.
Use of Vorbrüggen reaction for synthesis of ProTide
图式 8
利用Vorbrüggen反应构建ProTide化合物
Scheme8.
Use of Vorbrüggen reaction for synthesis of ProTide
2017年初, Merck制药的工艺化学家Simmons和Silverman领导的团队[29]从ProTide合成反应机理的分析和考虑出发, 终于找到了解决合成策略中带有普遍性、困扰合成化学界、并且近二十年来没有取得突破的两个主要挑战的答案: (1)缺乏高度有效专一、排它性地产生五价磷的非对映异构体(Sp vs Rp), 即不对称磷酸酯化的新反应; (2)对核苷上糖的多个具有类似化学活性的羟基进行单磷酸酯化的选择性识别.原来的传统烷氧基负离子方法中不可避免地产生高活性的氧负离子, 进而导致低下的区域选择性, 造成产物的多样性和分离纯化的困难.他们的新方法采用0.5 equiv.路易斯酸Me2AlCl活化手性磷酰胺酯(该试剂由Pharmasset制药首先研制开发), 与此同时采用pyridine碱摄取5位羟基上的质子.实验结果表明:这一方法具有高度的立体选择性和区域选择性, 核糖上3位上的羟基并不需要保护, 反应条件温和, 易于操作, 并且成功地应用于索非布韦(Sofosbuvir)、抗癌药物Acelarin和丙型肝炎病毒(HCV)临床药物INX-08189的合成上, 取得了一流的实验结果(Scheme 9). 不同于核苷类磷酰胺酯的制备, 烷基磷酰胺的制备一般都从相关的磷酸或磷酸酯前体通过下面三种方式来制备(Scheme 10): (1)从相关的磷酸出发, 经双氯化, 然后依次加入苯酚和氨基酸[31]; (2) DCC存在下, 从相关的磷酸出发, 和苯酚脱水偶联, 然后氯化, 氨基酸参与取代反应[31]; (3)从相关的磷酸二酯出发, 皂化为单磷酸, 然后和氨基酸发生偶联反应[32].
 图式 9
通过路易斯酸活化和碱助条件下实现磷酰胺酯化
Scheme9.
Lewis acid activation and base assistance for the ProTide synthesis
图式 9
通过路易斯酸活化和碱助条件下实现磷酰胺酯化
Scheme9.
Lewis acid activation and base assistance for the ProTide synthesis
 图式 10
从氨基酸制备核苷类磷酰胺酯前药的方法
Scheme10.
Methods to access phosphonamidates nucleoside prodrugs from amino acid
图式 10
从氨基酸制备核苷类磷酰胺酯前药的方法
Scheme10.
Methods to access phosphonamidates nucleoside prodrugs from amino acid
4 手性核苷磷酰胺酯前药合成方法的最新发展
如何合成手性磷化合物如磷酸酯或者磷酰胺酯, 并取得高立体选择性的结果, 是一个极具挑战性的难题.以往最有效的合成方法需要进行手性拆分或者使用化学计量的手性辅基.因此, 发展一种有效的方法立体选择性地高效合成手性磷酰胺酯就显得非常重要[33]. Ross等[34]在索非布韦磷酰胺酯前药的合成中, 引入了含有苯酚、五氟苯酚和光学活性氨基酸组成的手性磷(Sp)酰胺酯化新试剂68, 进而通过选择性的亲核取代, 有效和选择性地制备了单一的非对映异构体(99.7 de), 避免了一般在磷酰胺酯前药中出现的非对映异构体混合物的问题(Eq. 2), 在治疗丙肝病毒新药索非布韦工业化生产中做出了贡献, 该方法具有很好的实用性和可操作性.美中不足的是, 该反应采用了并不便宜易得的手性磷酰化试剂68, 并不是一个不对称催化的过程, 而且反应的产率也不是很理想(68%).
以不对称催化的方式合成磷-杂原子键的方法包括前手性底物的去对称化或者动态动力学不对称转化(DYKAT, Scheme 11), 这些方法的局限在于低催化效率和低选择性.除了磷原子中心的立体选择性以外, 另一个挑战在于核苷中5'-位和3'-位之间的化学选择性.自然界中, 酶可以通过一系列的催化模式进行选择性磷酰化. Ryan等[35]提出了五价的磷过渡态模型, 通过氢键的模式同时活化了亲核试剂和离去基团, 并对过渡态加以稳定.
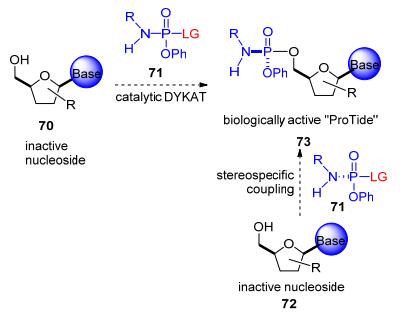 图式 11
具有生物活性磷酰胺酯的合成策略
Scheme11.
Synthetic strategies for biologically active ProTide
图式 11
具有生物活性磷酰胺酯的合成策略
Scheme11.
Synthetic strategies for biologically active ProTide
鉴于不对称磷酰胺酯化在新药研发中的重要作用以及现有方法的局限性, 施贵宝Tran和Eastgate领导的研发团队[36]决定尝试通过动态动力学拆分, 核苷酸磷酰胺酯前药76最初合成由磷酰氯74和核苷的烷氧基镁化物75反应生成.磷酰氯74存在1:1的非对映异构体, 而且不稳定, 在偶联反应条件下迅速降解, 这导致76的收率在柱层析后仅有27% (Eq. 3).另一种解决方案是采用对硝基苯酚PNP化合物74b, 它的稳定性好且能结晶.虽然这类反应中的转化率高, 然而由于种种原因这些措施都不理想, 不能选择性合成PNP的磷酸酯, 并且还需要额外的设备来拆分.另一种思路是将磷酰胺酸和核苷75在偶联试剂下成键, 如合成多肽时的偶联试剂.但制备出的磷酰胺酸结果大失所望, 它是一个粘稠的油状物, 实用价值不大.自动化筛选发现它的钙盐73可以结晶, 便于除去杂质和贮藏.在经过高通量筛选偶联试剂、活化试剂和辅助试剂, 发现加入手性助剂进行动态动力学拆分对于收率和选择性都有积极的影响.优化后, HATU作为偶联试剂, 奎宁作为手性助剂, 粗品收率提高到89%, 选择性为7:1 d.r., 获得了SP非对映体为优先的产物.该目标异构体在结晶时可高选择性地与苯甲醚形成共晶, 得到光学纯的活性药物组分, 这新条路线拥有稳定的中间体, 稳健的反应条件, 并不需柱层析, 相比之前的路线有质的提升(Scheme 12).施贵宝的工业化学家第一次完成了核苷类化合物的非对映选择性磷酸酯化反应, 在实用性、工业化方面又向前迈进了一大步.
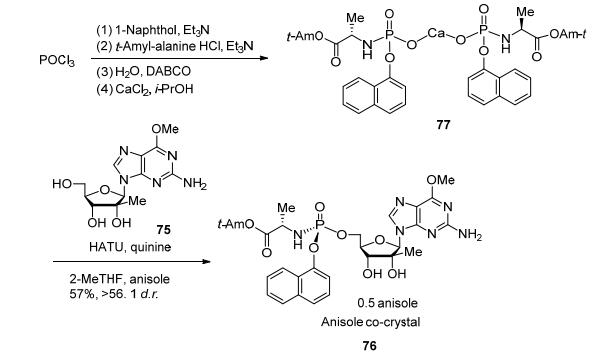 图式 12
通过动力学拆分实现非对映选择性核苷磷酰胺酯化的实用工艺
Scheme12.
Practical process for diastereoselective phosphorylation nucleoside via dynamic kinetic resolution
图式 12
通过动力学拆分实现非对映选择性核苷磷酰胺酯化的实用工艺
Scheme12.
Practical process for diastereoselective phosphorylation nucleoside via dynamic kinetic resolution
张万斌课题组[37]在2006年提出了利用手性叔胺催化不对称磷酰化反应的思路, 并进行手性磷酰胺类农药噻唑磷的高效合成方法开发.在大量实验和分析基础上, 他们最终设计合成了一类具有全新结构的手性双环咪唑骨架催化剂, 并将其成功地应用于不对称磷酰化反应, 实现了手性磷酰胺类化合物的首例不对称催化合成, 获得40%的对映选择性(Scheme 13).
 图式 13
通过动力学拆分首次催化对映选择性合成磷酰胺酯
Scheme13.
First catalytic enantioselective synthesis of phosphoramides via kinetic resolution
图式 13
通过动力学拆分首次催化对映选择性合成磷酰胺酯
Scheme13.
First catalytic enantioselective synthesis of phosphoramides via kinetic resolution
利用手性催化有效进行五价磷手性中心的建立一直以来都是悬而未决的难题.最好的方法都是利用拆解含有手性辅助基的原料来完成的. 2017年, Merck公司的DiRocco等[38]基于张万斌在手性双环咪唑骨架催化剂以及不对称磷酰化方面的工作, 并且在Jacobsen工作的启示下设计了具有C2对称性的手性双齿双环咪唑催化剂, 通过不同长度的碳脂肪链对催化剂进行调整, 以期通过减少高度有序过渡态的熵减效应来提高催化剂和底物的结合能力, 进而提高反应速率与选择性.设计并合成了一种在该类反应中效果更好的多功能催化剂85, 高立体选择性(d.r. 99:1)、高产率(92%)地实现了核苷类底物的不对称磷酰化, 并将其用于核苷类前药86 (MK-3682)的研发中(Eq. 4).在反应过程中, 磷酰氯底物先和催化剂作用, 再和核苷作用.由于氢键存在, 形成的手性环境对控制反应的立体选择性具有十分重要的影响.咪唑上的N可以作为碱协助攫取核苷中羟基位的H, 起到活化亲核试剂的作用, 而咪唑上的H以及苯环上的H会和P=O中的氧作用, 起到了稳定亲电试剂的作用.除此之外, 催化剂本身又是良好的离去基团.
该反应条件温和、操作简便、手性催化剂负载量较低, 具有极为优异的产率和选择性, 并且避免了传统上对其它羟基的保护和脱保护, 在磷手性化合物如磷酰胺类前药的合成中具有非常重要的意义.
5 核苷类磷酰胺酯药物的发展
核苷家族的氟化嘧啶类化合物是治疗固态肿瘤最为重要的化学药物, McGuigan等[39]采用ProTide策略合成了已有药物的磷酰胺酯前药(Eq. 5), 目的是避免核苷类化合物在转运中高度依赖转运体吸收以及高度依赖激酶介导活化的缺点.
McGuigan等[39]通过选用不同的芳香基、酯基和不同的氨基酸支链, 合成39个氟化嘧啶的磷酰胺酯前药, 并测试了其活性(Scheme 14), 发现所有磷酰胺前药对于腺嘧啶磷酸化酶具有耐受性从而避免失活, 这些磷酰胺酯前药在酸性和中性条件下以及血浆中稳定, 但是能在细胞内被羧肽酶活化, 实验发现部分化合物可以直接在细胞中检测到单磷酸化的核苷酸.部分化合物对于支原体感染的细胞也依然保持活性, 而氟脲嘧啶脱氧核苷啶(FUDR)则无此活性, 从这些前药对抗肿瘤细胞株抑制细胞增殖的活性数据来分析, 发现这类化合物能够成功地穿过细胞膜, 而且比母体核苷氟脲嘧啶脱氧核苷啶有更优越的性质.
 图式14
ProTide策略合成氟化嘧啶的磷酰胺酯前药
Scheme14.
Synthesis of fluorinated pyrimidine prodrug via phosphoramidate ProTide strategies
图式14
ProTide策略合成氟化嘧啶的磷酰胺酯前药
Scheme14.
Synthesis of fluorinated pyrimidine prodrug via phosphoramidate ProTide strategies
在合成和评价鸟嘌呤核苷及其L-甘氨酸酯的磷酰胺酯前药抗丙肝病毒的过程中[40], 发现鸟嘌呤核苷105的活性不如化合物104, 主要是由于其细胞透膜能力差, 并且在细胞内转化为活性核苷三磷酸酯的效率低, 而将化合物105采用Protide制成磷酰胺前药106后, 化合物的亲脂性增加, 而且可以避免第一步的磷酸化限速步骤, 能够有效地提高细胞内活性三磷酸酯的浓度, 该化合物106(图 12, INX-08189 or BMS-986094)曾处于临床阶段.
 图12
一些高效的抗丙肝病毒的鸟嘌呤核苷磷酰胺酯前药结构
Figure12.
Structures of potent anti-HCV guanosine nucleosides and ProTide
图12
一些高效的抗丙肝病毒的鸟嘌呤核苷磷酰胺酯前药结构
Figure12.
Structures of potent anti-HCV guanosine nucleosides and ProTide
需要注意的是, ProTide前药也并不总是获得正向效果, 例如, McGuigan采用ProTide策略, 希望得到化合物110的7-脱氮类似物磷酰胺前药112 (Scheme 15), 结果发现所得化合物抗丙肝病毒活性大为下降, 主要原因可能是化合物转化为主要的氨酰基中间体以及单磷酸酯的释放较慢, 研究发现, 在与Huh-7细胞一起孵育48 h后, 质谱发现化合物112仍然是最主要的峰, 也正因为转化速度较慢从而导致前药活性降低[41].
 图式 15
合成7-脱氮鸟嘌呤核苷类似物磷酰胺酯前药
Scheme15.
Synthesis of 7-deaza analogues of guanosine nucleoside phosphoramidates prodrug
图式 15
合成7-脱氮鸟嘌呤核苷类似物磷酰胺酯前药
Scheme15.
Synthesis of 7-deaza analogues of guanosine nucleoside phosphoramidates prodrug
2'-C-甲基胞苷113 (NM107)是HCV治疗领域第一个被报道的核苷酸NS5b聚合酶抑制剂, 而其缬氨酰酯前药114 (NM283)在HCV感染患者上则能有效降低病毒.研究人员采用ProTide技术合成了一系列2'-C-甲基胞啶的磷酰胺酯前药115[42](图 13), 并且测试了该类化合物在HuH7细胞系和几种肝细胞中的抗HCV复制子活性以及细胞毒性, 研究人员采用ProTide技术合成了一系列2'-C-甲基胞苷的磷酰胺前药(Scheme 16).同时也测试了该类前药化合物转化为核苷三磷酸的速度, 鉴于它的高活性, 系统的SAR研究最终给出了一些理想化合物, 它们在动物模型中给出高浓度的活性核苷三磷酸, 并且不具有由于酚类组成酶解所带来的毒性问题.
 图13
核苷酸NS5b聚合酶抑制剂和甲基胞啶磷酰胺酯前药
Figure13.
Nucleoside inhibitor of HCV NS5b polymerase and structures of phosphoramidate prodrugs of C-methylcytidine
图13
核苷酸NS5b聚合酶抑制剂和甲基胞啶磷酰胺酯前药
Figure13.
Nucleoside inhibitor of HCV NS5b polymerase and structures of phosphoramidate prodrugs of C-methylcytidine
 图式 16
合成甲基胞啶磷酰胺酯前药
Scheme16.
Synthesis of phosphoramidate prodrugs of C-methylcytidine
图式 16
合成甲基胞啶磷酰胺酯前药
Scheme16.
Synthesis of phosphoramidate prodrugs of C-methylcytidine
实验发现大部分化合物相对于母核化合物能大幅提高活性10倍到200倍左右, 这主要是由于在肝细胞中可以获得更高的三磷酸酯浓度.由于以往的磷酰胺前药可能释放一分子苯酚而可能带来各种毒性, 研究人员又进一步合成了亲脂性更强的磷酰胺单酯来避免引入苯酚基团.这类化合物中最好的是化合物122, 能够在各种属肝细胞中获得很高的三磷酸酯浓度, 从而提高其抗病毒活性, 同时又避免了毒性基团苯酚的引入(图 14).
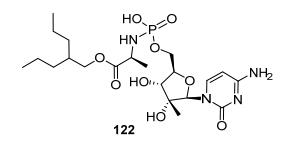 图14
高活性和亲脂性更强的磷酰胺酯前药
Figure14.
Highly potent and more lipophilic phosphoramidate prodrug
图14
高活性和亲脂性更强的磷酰胺酯前药
Figure14.
Highly potent and more lipophilic phosphoramidate prodrug
阿昔洛韦是一种广谱抗病毒药, 主要被报道用于治疗HSV-1和HSV-2, 但有报道磷酸化的阿昔洛韦可用于HIV-1逆转录酶的抑制剂. McGuigan等[43]采用ProTide技术合成了22个阿昔洛韦的磷酰胺酯前药(Scheme 17), 并且测试该类化合物的抗病毒活性.研究结果表明, 这些新合成的前药化合物相对母体阿昔洛韦来说, 对于HSV-1和HSV-2的活性没有很大提高, 但是这些化合物对于TK缺陷的HSV-1株仍有活性, 与之相反的是阿昔洛韦失活, 说明这类前药化合物避免了腺嘧啶激酶激活这一步骤.相对于HIV-1及HIV-2病毒株, 阿昔洛韦没有显示活性, 而这些前药化合物显示了微摩尔级别的抑制活性. McGuigan等还发现对于抗HSV而言, 氨基酸基团的改变是可以接受的, 而对于抗HIV而言, 这种变化则是不可以接受的, 仅仅只有L-丙氨酸以及苯丙氨酸有较好活性, 这些活性差异可能是由于底物的特异性或者分子内活化前药所需的酶含量不同所引起的.
 图式 17
采用ProTide技术合成阿昔洛韦的磷酰胺酯前药
Scheme17.
Application of phosphoramidate ProTide for acyclovir prodrug
图式 17
采用ProTide技术合成阿昔洛韦的磷酰胺酯前药
Scheme17.
Application of phosphoramidate ProTide for acyclovir prodrug
吉西他滨是癌症治疗中常用的核苷药物, 该药由于容易受癌症耐药株影响因此其疗效有限, 而加入磷酰胺酯基团则能够改善其对抗多种癌症耐药作用.研究人员合成了一系列吉西他滨磷酰胺酯前药128, 并且在一系列肿瘤细胞株中筛选其抗细胞活性.在所有化合物中发现化合物129 (图 15, NUC-1031)的体外活性最强[44].重要的是, 相对吉西他滨, 化合物129的活化明显不依赖于脱氧胞苷激酶以及核苷酸转运子, 并且能抵抗胞苷去氨酶介导的降解. 129在体内胰腺癌肿瘤模型中可以发现肿瘤体积的明显降低.新药Acelarin (NUC-1031)用于治疗胰腺癌和卵巢癌, 目前正开展临床三期试验, 有望成为抗癌新药.
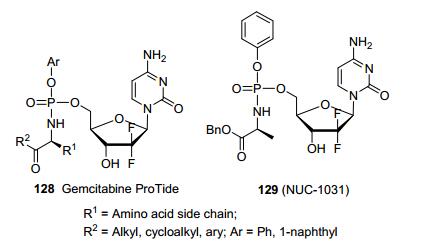 图15
利用ProTide技术合成吉西他滨的磷酰胺酯前药
Figure15.
Application of phosphoramidate ProTide for synthesis of gemcitabine prodrug
图15
利用ProTide技术合成吉西他滨的磷酰胺酯前药
Figure15.
Application of phosphoramidate ProTide for synthesis of gemcitabine prodrug
McGuigan等报道了一系列阿德福韦(Adefovir, 130a)和替诺福韦(Tenofovir, 130b)的磷酰胺酯前药化合物(Scheme 18), 该系列化合物的抗HIV活性显著优于两个原型药; 其中阿德福韦的磷酰胺酯前药活性是阿德福韦的600~1000倍, 而替诺福韦的磷酰胺酯前药是其活性的1000~2000倍.进一步的活性测试表明, 该系列化合物的抗HPV转化细胞增殖活性也优于原型药.在报道的化合物中, 含有5, 6, 7, 8-四氢萘基团的活性最好[45].
 图式 18
利用ProTide技术合成阿德福韦和替诺福韦的磷酰酯前药
Scheme18.
Application of phosphoramidate ProTide for Adefovir and Tenofovir prodrugs
图式 18
利用ProTide技术合成阿德福韦和替诺福韦的磷酰酯前药
Scheme18.
Application of phosphoramidate ProTide for Adefovir and Tenofovir prodrugs
Westwell等[46]设计合成了阿洛夫定(Alovudine 135)的磷酰胺酯前药136和137 (图 16), 在体外抗HIV活性和抗肿瘤活性均弱于原型药只在CEM/TK-肿瘤细胞系中抑制活性与原型药阿洛夫定135相当.
 图16
阿洛夫定的磷酰胺酯前药
Figure16.
Phosphoramidate ProTide for alovudine prodrug
图16
阿洛夫定的磷酰胺酯前药
Figure16.
Phosphoramidate ProTide for alovudine prodrug
Calenbergh等[47]合成了一系列3'-脱氧以及2', 3'-脱氧的胸腺嘧啶及腺嘌呤核苷, 活性测试结果显示这些化合物没有抗病毒活性; 而经过进一步修饰成磷酰胺酯前药143, 则显现出抗HIV活性(EC50=0.5~1.5 µmol•L-1), 说明原型药是由于较难通过细胞膜而没有表现出活性(Scheme 19).
 图式 19
具有抗HIV活性的apionucleoside前药
Scheme19.
Prodrug of apionucleoside family with anti-HIV activity
图式 19
具有抗HIV活性的apionucleoside前药
Scheme19.
Prodrug of apionucleoside family with anti-HIV activity
化合物144是结核分枝杆菌胸苷酸合成酶抑制活性最好的化合物(IC50=0.91 µmol•L-1)之一, 但该磷酸的极性使其很难穿透复杂的分枝杆菌的细胞壁.为此McGuigan等[48]通过ProTide技术将磷酸基团修饰成磷酰胺酯, 提高化合物的脂溶性, 使其更容易通过分枝杆菌的细胞壁.其磷酰胺酯前药150抗病毒活性和阿昔洛韦几乎一样, 是潜在的抗结核和抗病毒药物(Scheme 20).
 图式 20
合成抗结核和抗病毒药物的磷酰胺酯前药
Scheme20.
Synthesis of Phosphoramidate ProTide prodrug with anti-tubercular and anti-viral activity
图式 20
合成抗结核和抗病毒药物的磷酰胺酯前药
Scheme20.
Synthesis of Phosphoramidate ProTide prodrug with anti-tubercular and anti-viral activity
Jonckers等[49]发现化合物151是一类新颖的环状嘧啶核苷磷酰胺酯前药, 在Huh-7 HCV基因型1b细胞系中没有抗HCV活性, 是由于其产生非活性的代谢产物.相反, 通过在人源肝细胞孵化以及在其他药代实验中发现化合物151代谢生成对HCV NS5B具有强抑制活性的化合物.但相比于磷酰胺酯前药化合物152, 化合物151具有更优的性质(图 17).
 图17
抗丙型肝炎病毒的新颖环状嘧啶核苷磷酸酯前药
Figure17.
Anti-HCV phosphate ester prodrug from novel cyclic uridine nucleotide
图17
抗丙型肝炎病毒的新颖环状嘧啶核苷磷酸酯前药
Figure17.
Anti-HCV phosphate ester prodrug from novel cyclic uridine nucleotide
McGuigan等[50]设计合成并评价了大约50多个化合物4'-叠氮基胞嘧啶核苷的磷酰胺酯前药153的抗丙型肝炎病毒活性, 通过前药结构在酯、氨基酸和芳基部分的修饰变化, 这些丙型肝炎病毒抑制剂出现sub-μmol/L级别的高活性, 而且这类化合物在细胞复制过程中均无细胞毒(图 18).
 图18
吉利德抗埃博拉病毒磷酰胺酯(GS-5734)和一些磷酰胺酯前药
Figure18.
Gilead's ProTide GS-5734 against Ebola and some phosphoramidate prodrugs
图18
吉利德抗埃博拉病毒磷酰胺酯(GS-5734)和一些磷酰胺酯前药
Figure18.
Gilead's ProTide GS-5734 against Ebola and some phosphoramidate prodrugs
利巴韦林具有广泛的抗病毒活性, 利巴韦林磷酰胺酯前药的设计使具有生物活性的利巴韦林单磷酸盐传输进入细胞内, 一些化合物如154具有相似的抗病毒活性[51].
最近一系列有效的水痘一带状疱疹病毒(VZV)抑制剂2-氟衍生物155的合成和抗病毒活性已经被报道, 二环的核苷类似物就是其中之一[52].分子模型研究表明这些化合物与VZV编码的胸苷激酶的相互作用, McGuigan报道这些化合物的磷酰胺酯前药在组织细胞培养实验中氨基磷酸酯核苷前药抗VZV的有效性与母药相当, 但对于TK缺乏的VZV菌株则失去活性.
对抗肿瘤药Thymectacin进行结构修饰, McGuigan等[54]发现磷酰胺酯前药156对结肠癌和前列腺癌的体外药效有显著提高.这些化合物等比例存在两个非对映异构体, 在三种不同肿瘤细胞系的组织培养中进行评价, 发现Thymectacin胸腺嘧啶的母体中, 苯环上甲氧基亚氨基结构的改动能显著提高药效, 特别值得一提的是, 用苄基酯替代先导物中的甲基酯对结肠癌HT115细胞的药效提高了175倍.这个先导物可作为HT115细胞的微摩尔抑制剂, 可以进一步优化.
2013~2016年西非埃博拉病毒(EBOV)爆发是史上最大的全球公共卫生突发事件(超过28000例), 造成>11000人死亡, 其中有500名医护人员.在巨噬细胞作为临床诊断中, 吉利德重点筛查和优化确定了具有抗EBOV EC50=86 nmol•L-1的腺嘌呤C-核苷磷酰胺酯前药157 (GS-5734)[54]. SAR构效关系研究确定了1'-CN基团和C-连接的核碱基被暴露, 可以获得最佳抗EBOV药效和对宿主聚合酶的选择性. 2016年在Nature发表的研究[55]证实在非人灵长类EBOV挑战模型中, 在感染病毒后第3〜14 d每天给药(10 mg/kg)进行静脉注射治疗12 d后, 对病毒血症和死亡率有显着影响, 导致被感染治疗的动物有100%存活率.目前磷酰胺酯前药157正在临床Ⅱ研究中.
2'-氟-2'-脱氧鸟苷已经被报道在体内外抗流感病毒都有效, 修饰2'-氟-2'-脱氧鸟苷的类似物和相应的前药可作为潜在的抗流感病毒药物, 一种由鸟嘌呤和核糖组成的核糖苷前药158展示出好的抗病毒活性(EC99为12 µmol•L-1)[56].在两种不同的细胞测定中, 母体核苷没有抗病毒活性, 抗病毒结果得到了代谢实验的支持, 从而表明双前药概念的实用性.
最近制备的以溴夫定(Brivudine)为母体的磷酰胺酯前药160 (Eq. 6), McGuigan等[57]发现它对白血病细胞中的抑制效果比溴夫定提高了20倍.其中一些前药对抑制CEM和增殖表皮癌细胞HeLa细胞中的活性都比母体药物溴夫定要好, 而且对非致瘤性肺成纤维细胞的毒性很低, 几乎无法测量.
McGuigan等[58]合成一系列的以磷酰胺酯为形式的尿嘧啶苷(AraU)前药, 并且在病毒、白血病和固体肿瘤细胞系中测试其活性, 发现无论是母药还是前药都没有抗病毒活性, 也没有有效的抗肿瘤活性.研究代谢物释放的AraU单磷酸盐和分子模型研究表明, AraU L-丙氨酸磷酸盐衍生物可能不是氨基磷酸酯酶Hint-1的底物.尿嘧啶核苷酸类似物已经被证明抗病毒活性比较差, 评价抑制流行性感冒病毒RNA聚合酶的酶化实验显示, 有些尿嘧啶核苷酸三磷酸盐衍生物通过尿苷整合入病毒RNA从而有抑制病毒的作用.通过具有活性(单磷酸盐)尿嘧啶核苷酸类似物进入流行性感冒病毒感染的细胞实验, 发现有些化合物在MDCK细胞中(161a, EC99=49±38 µmol•L-1, 161b, EC99≥81 µmol•L-1)有活性, 然而有一些核苷酸类似物, 如2'-氟-2'-脱氧尿苷161b则没有活性[59](图 19).
 图19
磷酰胺酯为形式的尿嘧啶苷前药
Figure19.
Phosphoramidates prodrugs of AraU
图19
磷酰胺酯为形式的尿嘧啶苷前药
Figure19.
Phosphoramidates prodrugs of AraU
McGuigan等[60]合成了25个核苷类似物(Scheme 21)的磷酰二胺前药, 并测试了其抗癌或者抗病毒活性, 这类前药的优点主要是可以克服存在于其他磷酸酯前药的磷原子的手性问题, 因此能形成单一的立体异构体而不是非对应异构体混合物.活性测试发现ddA、ABC、和ACV的磷酰二胺前药对HIV-1和HIV-2的活性大幅提高, 而d4T前药则对不同的肿瘤株显示良好活性.与此相对的是, AICA前药衍生物则既无抗癌又无抗病毒活性.磷酰二胺转化为磷酸酯的机理如图 27所示, 使用羧肽酶Y的代谢研究表明, 如果化合物是缓慢地被代谢转化为活化形式, 则活性较低或者消失, 而如果化合物可以迅速被活化, 则活性提高.
 图式 21
磷酰二胺转化为磷酸酯前药的机理
Scheme21.
Putative bioactivation pathway of diamidate prodrugs
图式 21
磷酰二胺转化为磷酸酯前药的机理
Scheme21.
Putative bioactivation pathway of diamidate prodrugs
在HCV治疗领域, 已经有多个药物使用ProTide前药策略获得了活性好的化合物, 并且这些化合物目前都处于临床阶段或已上市.因为磷酰二胺可以设计成非手性形式, 从而避免磷酰胺前药中存在的合成难题.在合成得到的67个磷酰二胺前药化合物中[61](Scheme 22), 测试了其抗丙肝复制子活性, 显示所有磷酰二胺前药化合物在酸性、中性以及中等碱性和人血浆中都能稳定存在.羧肽酶Y能够活化这些化合物形成核苷酸氨基酸磷酸酯中间体, 而之前报道的芳氧基磷酰胺前药也可以形成类似中间体.在Huh7细胞系中发现大部分化合物的细胞毒性很低而其抗HCV活性可以达到nmol•L-1级别.其中8个化合物在大鼠肝脏中可以持续24 h检测到三磷酸酯浓度, 说明该类化合物具有很好的应用前景.
 图式 22
磷二酰胺酯作为有希望的磷酸前药
Scheme22.
Phosphorodiamidates as promising new phosphate prodrugs
图式 22
磷二酰胺酯作为有希望的磷酸前药
Scheme22.
Phosphorodiamidates as promising new phosphate prodrugs
最后, 值得强调的是2007年McGuigan等[62]首次通过ProTide技术还证明能够将无HCV抑制活性的核苷, 如4'-叠氮尿苷、4'-叠氮腺苷转化为高活性的磷酰胺酯前药(EC50: 0.22 µmol•L-1, Eq. 7).这使得药物研发工作者不得不重新审视一下过去被忽视、被认为是无生物活性的核苷母体化合物能否进行二次开发的可能性.
6 总结与展望
虽然磷酰胺酯前药的研究从20世纪90年代初就开始了, 但是直到近五年, 尤其是抗丙肝病毒的重磅炸弹药物索非布韦(2015年全球销售53亿美元, 复方全球销售139亿美元)和抗乙肝病毒的替诺福韦艾拉酚胺的获批上市后, 这一方法的独特优点以及临床研究出色的成果得到药物研发科学家的广泛重视和肯定.欣喜的是, 这方面的研究正处于一个繁荣的发展阶段[63]. 2016年在Nature发表的研究结果[55]证实在非人灵长类感染埃博拉病毒后, 静脉注射磷酰胺酯前药157 (GS-5734)治疗12天后被感染治疗的动物有100%存活率.值得强调指出, 近年来, 手性磷酰胺酯前药的不对称合成技术也取得了重大突破, 2017年Science上刊登了Merck制药采用多官能团催化剂进行立体选择性组建手性磷酰胺酯前药的新方法[38]; Nature Reviews上刊登了施贵宝制药的以机理为依据解决ProTide技术的挑战的新成果[36], 这些技术进展对新药设计与结构修饰改造, 包括挖掘老药新用等方面值得学习借鉴和推广, 相信核苷磷酰胺酯前药将对核苷药物的研发, 免除病人面对癌症和致命病毒如HIV、HBV、HCV、流感病毒, 登革热, 以及一些罕见的病毒如埃博拉病毒(EBOV)、诺如(Noro)、亨德拉(Hendra)、基孔肯雅(Chikungunya)病毒的困扰等方面有广泛的应用前景.这一领域合成新方法的涌现, 必将为新药研发和筛选提供一条崭新的途径和高效便利的手段, 推动药物研究的进一步发展.
后记 本文在写作过程中, 惊悉克里斯托弗·麦格根(Christopher McGuigan, 1958~2016)教授于去年过早地离开了这个他所热爱的世界.他开拓创新的ProTide技术造福了人类, 自己却因患癌症, 被病魔无情地夺走了生命.他研发的新药Acelarin (NUC-1031)用于治疗胰腺癌和卵巢癌, 目前正在临床三期研究试验中.谨以此文, 纪念这位英年早逝、在人类征服病毒道路上做出杰出贡献的药物化学家.
-
-
[1]
(a) Babusis, D.; Phan, T. K.; Lee, W. A.; Watkins, W. J.; Ray, A. S. Mol. Pharm. 2013, 10, 459.
(b) Lee, W. A.; He, G. X.; Eisenberg, E.; Cihlar, T.; Swaminathan, S.; Mulato, A.; Cundy, K. C. Antimicrob. Agents. Chemother. 2005, 49, 1898.
(c) Birkus, G.; Wang, R.; Liu, X. H.; Kutty, N.; MacArthur, H.; Cihlar, T.; Gibbs, C.; Swaminathan, S.; Lee, W.; McDermott, M. Antimicrob. Agents. Chemother. 2007, 51, 543. -
[2]
(a) Thornton, P. J.; Kadri, H.; Miccoli, A.; Mehellou, Y. J. Med. Chem. 2016, 59, 10400.
(b) Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447. -
[3]
(a) Schinazi, R. F. ; Shi, J. ; Whitaker, T. In Innovative Drug Synthesis, Eds. : Li, J. J. ; Johnson, D. S. ; Wiley, Hoboken, 2016, p. 61.
(b) Rautio, J. ; Kumpulainen, H. ; Heimbach, T. ; Oliyai, R. ; Oh, D. ; Jarvinen, T. ; Savolainen, J. Nat. Rev. Drug Discovery 2008, 7, 255.
(c) Zhang, J. ; Nie, B. ; Zhang, Y. J. Chin. J. Org. Chem. 2015, 35, 337 (in Chinese).
(张霁, 聂飚, 张英俊, 有机化学, 2015, 35, 337. ) -
[4]
(a) Hecker, S. J. ; Erion, M. D. J. Med. Chem. 2008, 51, 2328.
(b) Ji, X. ; Wang, J. ; Zhang, L. ; Zhao, L. X. ; Jiang, H. L. ; Liu, H. Acta Pharm. Sin. 2013, 48, 621 (in Chinese).
(姬勋, 王江, 张磊, 赵临襄, 蒋华良, 柳红, 药学学报, 2013, 48, 621. ) -
[5]
(a) Hostetler, K. Y. Antiviral Res. 2009, 82, A84.
(b) Parang, K. ; Wiebe, L. I. ; Knaus, E. E. Curr. Med. Chem. 2000, 7, 995.
(c) Li, W. B. ; Dong, F. H. ; Sun, C. J. Prog. Pharm. Sci. 2012, 36, 300 (in Chinese).
(李文保, 董芳华, 孙昌俊, 药学进展, 2012, 36, 300. ) -
[6]
Corson, T. W.; Crews, C. M. Cell 2007, 130, 769. doi: 10.1016/j.cell.2007.08.021
-
[7]
Patil, S.; Lis, L. G.; Schumacher, R. J.; Norris, B. J.; Morgan, M. L.; Cuellar, R. A. D.; Blazar, B. R.; Suryanarayanan, R.; Gurvich, V. J.; Georg, G. I. J. Med. Chem. 2015, 58, 9334. doi: 10.1021/acs.jmedchem.5b01329
-
[8]
Chassaing, C.; Berger, M.; Heckeroth, A.; Ilg, T.; Jaeger, M.; Kern, C.; Schmid, K.; Uphoff, M. J. Med. Chem. 2008, 51, 1111. doi: 10.1021/jm701456r
-
[9]
Kleeb, S.; Jiang, X. H.; Frei, P.; Sigl, A.; Bezencon, J.; Bamberger, K.; Schwardt, O.; Ernst, B. J. Med. Chem. 2016, 59, 3163. doi: 10.1021/acs.jmedchem.5b01923
-
[10]
Chowdhury, U. R.; Viker, K. B.; Stolz, K. L.; Holman, B. H.; Fautsch, M. P.; Dosa, P. I. J. Med. Chem. 2016, 59, 6221 doi: 10.1021/acs.jmedchem.6b00406
-
[11]
Mehellou, Y. C. Chem. Med. Chem. 2016, 11, 1114. doi: 10.1002/cmdc.v11.11
-
[12]
Pradere, U.; Garnier-Amblard, E. C.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2014, 114, 9154. doi: 10.1021/cr5002035
-
[13]
(a) McGuigan, C.; Pathirana, R. N.; Mahmood, N.; Hay, A. J. Bioorg. Med. Chem. Lett. 1992, 2, 701.
(b) Mehellou, Y.; Balzarini, J.; McGuigan, C. Chem. Med. Chem. 2009, 4, 1779. -
[14]
Cahard, D.; McGuigan, C.; Balzarini, J. Mini. Rev. Med. Chem. 2004, 4, 371. http://d.wanfangdata.com.cn/Periodical/jsjxb201411015
-
[15]
Maiti, M.; Persoons, L.; Andrei, G.; Snoeck, R.; Balzarini, J.; Herdewijn, P. Chem. Med. Chem. 2013, 8, 985 doi: 10.1002/cmdc.201300035
-
[16]
Piplani, M.; Rana, A. C.; Sharma, P. C. J. Pharm. Pharm. Sci. 2016, 19, 82. doi: 10.18433/J3X61S
-
[17]
McGuigan, C.; Serpi, M.; Bibbo, R.; Roberts, H.; Hughes, C.; Caterson, B.; Gibert, A. T.; Verson, C. R. A. J. Med. Chem. 2008, 51, 5807. doi: 10.1021/jm800594c
-
[18]
Zhou, L. H.; Zhang, H. W.; Tao, S. J.; Ehteshami, M.; Cho, J. H.; McBrayer, T. R.; Tharnish, P.; Whitaker, T.; Amblard, F.; Coats, S. J.; Schinazi, R. F. ACS Med. Chem. Lett. 2016, 7, 17. doi: 10.1021/acsmedchemlett.5b00402
-
[19]
Lioux, T.; Mauny, M. A.; Lamoureux, A.; Bascoul, N.; Hays, M.; Vernejoul, F.; Baudru, A. S.; Boularan, C.; Lopes-Vicente, J.; Qushair, G.; Tiraby, G. J. Med. Chem. 2016, 59, 10253. doi: 10.1021/acs.jmedchem.6b01300
-
[20]
Dabkowski, W.; Skrzypczynski, Z.; Michalski, J.; Piel, N.; McLaughlin, L. W.; Cramer, F. Nucleic Acids Res. 1984, 12, 9123. doi: 10.1093/nar/12.23.9123
-
[21]
(a) McGuigan, C.; Harris, S. A.; Daluge, S. M.; Gudmundsson, K. S.; McLean, E. W.; Burnette, T. C.; Marr, H.; Hazen, R.; Condreay, L. D.; Johnson, L.; De Clercq, E.; Balzarini, J. J. Med. Chem. 2005, 48, 3504.
(b) Sari, O.; Bassit, L.; Gavegnano, C.; McBrayer, T. R.; McCormick, L.; Cox, B.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Tetrahedron Lett. 2017, 58, 642.
(c) Zhou, L. H.; Zhang, H. W.; Tao, S. J.; Ehteshami, M.; Cho, J. H.; McBrayer, T. R.; Tharnish, P.; Whitaker, T.; Amblard, F.; Coats, S. J.; Schinazi, R. F. ACS Med. Chem. Lett. 2016, 7, 17. -
[22]
Cho, A.; Zhang, L. J.; Xu, J.; Lee, R.; Butler, T.; Metobo, S.; Aktoudianakis, V.; Lew, W.; Ye, H.; Clarke, M.; Doerffler, E.; Byun, D.; Wang, T.; Babusis, D.; Carey, A. C.; German, P.; Sauer, D.; Zhong, W. D.; Rossi, S.; Fenaux, M.; McHutchison, J. G.; Perry, J.; Feng, J.; Ray, A. S.; Kim, C. U. J. Med. Chem. 2014, 57, 1812. doi: 10.1021/jm400201a
-
[23]
(a) Kandil, S.; Balzarini, J.; Rat, S.; Brancale, A.; Westwell, A. D.; McGuigan, C. Bioorg. Med. Chem. Lett. 2016, 26, 5618.
(b) Mehellou, Y.; McGuigan, C.; Brancale, A.; Balzarini, J. Bioorg. Med. Chem. Lett. 2007, 17, 3666. -
[24]
Mahmoud, S.; Li, H.; McBrayer, T. R.; Bassit, L.; Hammad, S. F.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Nucleosides Nucleotides Nucleic Acids 2007, 36, 66. http://d.wanfangdata.com.cn/Periodical/jsjxb201411015
-
[25]
Gudmundsson, K. S.; Daluge, S. M.; Johnson, L. C.; Jansen, R.; Hazen, R.; Condreay, L. D.; McGuigan, C. Nucleosides Nucleotides Nucleic Acids 2003, 22, 1953. doi: 10.1081/NCN-120025242
-
[26]
Serpi, M.; Madela, K.; Pertusati, F.; Slusarczyk, M. Current Protocols in Nucleic Acid Chemistry, Wiley, New York, 2013, Chapter 15.
-
[27]
Vanheusden, V.; Herdewijn, P.; Van Calenbergh, S. J. Pharm. Belg. 2002, 41. http://d.wanfangdata.com.cn/Periodical/jsjxb201411015
-
[28]
Gao, L. J.; De Jonghe, S.; Herdewijn, P. Org. Lett. 2016, 18, 5816. doi: 10.1021/acs.orglett.6b02764
-
[29]
Simmons, B.; Liu, Z. Q.; Klapars, A.; Bellomo, A.; Silverman, S. M. Org. Lett. 2017, 19, 2218. doi: 10.1021/acs.orglett.7b00469
-
[30]
Liu, C.; Dumbre, S. G.; Pannecouque, C.; Huang, C. S.; Ptak, R. G.; Murray, M. G.; De Jonghe, S.; Herdewijn, P. J. Med. Chem. 2016, 59, 9513. doi: 10.1021/acs.jmedchem.6b01260
-
[31]
Pertusati, F.; Hinsinger, K.; Flynn, A. S.; Powell, N.; Tristram, A.; Balzarini, J.; McGuigan, C. Eur. J. Med. Chem. 2014, 78, 259. doi: 10.1016/j.ejmech.2014.03.051
-
[32]
(a) Celewicz, L.; Jozwiak, A.; Ruszkowski, P.; Laskowska, H.; Olejnik, A.; Czarnecka, A.; Hoffmann, M.; Hladon, B. Bioorg. Med. Chem. 2011, 19, 6375.
(b) Aliouane, L.; Rigaud, B.; Pfund, E.; Jean, L.; Renard, P. Y.; Lequeux, T. Tetrahedron Lett. 2011, 52, 259. -
[33]
Pertusati, F.; McGuigan, C. Chem. Commun. 2015, 51, 8070. doi: 10.1039/C5CC00448A
-
[34]
Ross, B. S.; Reddy, P. G.; Zhang, H. R.; Rachakonda, S.; Sofia, M. J. J. Org. Chem. 2011, 76, 8311. doi: 10.1021/jo201492m
-
[35]
Ryan, M.; Liu, T.; Dahlquist, F. W.; Griffith, O. H. Biochemistry 2001, 40, 9743. doi: 10.1021/bi010958m
-
[36]
(a) Eastgate, M. D.; Schmidt, M. A.; Fandrick, K. R. Nat. Rev. Chem. 2017, 1, 0016, DOI: 10.1038/s41570-017-0016.
(b) Tran, K.; Beutner, G. L.; Schmidt, M.; Janey, J.; Chen, K.; Rosso, V.; Eastgate, M. D. J. Org. Chem. 2015, 80, 4994. -
[37]
(a) Liu, S.; Zhang, Z. F.; Xie, F.; Butt, N. A.; Sun, L.; Zhang, W. B. Tetrahedron: Asymmetry 2012, 23, 329.
(b) Zhang, Z. F.; Xie, F.; Jia, J.; Zhang, W. B. J. Am. Chem. Soc. 2010, 132, 15939. -
[38]
DiRocco, D. A.; Ji, Y. N.; Sherer, E. C.; Klapars, A.; Reibarkh, M.; Dropinski, J.; Mathew, R.; Maligres, P.; Hyde, A. M.; Limanto, J.; Brunskill, A.; Ruck, R. T.; Campeau, L.-C.; Davies, I. W. Science 2017, 356, 426. doi: 10.1126/science.aam7936
-
[39]
McGuigan, C.; Murziani, P.; Slusarczyk, M.; Gonczy, B.; Vande Voorde, J.; Liekens, S.; Balzarini, J. J. Med. Chem. 2011, 54, 7247. doi: 10.1021/jm200815w
-
[40]
McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Brancale, A.; Zonta, N.; Chamberlain, S.; Vernachio, J.; Hutchins, J.; Hall, A.; Ames, B.; Gorovits, E.; Ganguly, B.; Kolykhalov, A.; Wang, J.; Muhammad, J.; Patti, J. M.; Henson, G. Bioorg. Med. Chem. Lett. 2010, 20, 4850. doi: 10.1016/j.bmcl.2010.06.094
-
[41]
Bourdin, C.; McGuigan, C.; Brancale, A.; Chamberlain, S.; Vernachio, J.; Hutchins, J.; Gorovits, E.; Kolykhalov, A.; Muhammad, J.; Patti, J.; Henson, G.; Bleiman, B.; Bryant, K. D.; Ganguly, B.; Hunley, D.; Obikhod, A.; Walters, C. R.; Wang, J.; Ramamurty, C. V. S.; Battina, S. K.; Rao, C. S. Bioorg. Med. Chem. Lett. 2013, 23, 2260. doi: 10.1016/j.bmcl.2012.12.004
-
[42]
Gardelli, C.; Attenni, B.; Donghi, M.; Meppen, M.; Pacini, B.; Harper, S.; Di Marco, A.; Fiore, F.; Giuliano, C.; Pucci, V.; Laufer, R.; Gennari, N.; Marcucci, I.; Leone, J. F.; Olsen, D. B.; MacCoss, M.; Rowley, M.; Narjes, F. J. Med. Chem. 2009, 52, 5394. doi: 10.1021/jm900447q
-
[43]
Derudas, M.; Carta, D.; Brancale, A.; Vanpouille, C.; Lisco, A.; Margolis, L.; Balzarini, J.; McGuigan, C. J. Med. Chem. 2009, 52, 5520. doi: 10.1021/jm9007856
-
[44]
Slusarczyk, M.; Lopez, M. H.; Balzarini, J.; Mason, M.; Jiang, W. G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. J. Med. Chem. 2014, 57, 1531. doi: 10.1021/jm401853a
-
[45]
Pertusati, F.; Hinsinger, K.; Flynn, A. S.; Powell, N.; Tristram, A.; Balzarini, J.; McGuigan, C. Eur. J. Med. Chem. 2014, 78, 259. doi: 10.1016/j.ejmech.2014.03.051
-
[46]
Velanguparackel, W.; Hamon, N.; Balzarini, J.; McGuigan, C.; Westwell, A. D. Bioorg. Med. Chem. Lett. 2014, 24, 2240. doi: 10.1016/j.bmcl.2014.03.092
-
[47]
Toti, K. S.; Derudas, M.; Pertusati, F.; Sinnaeve, D.; Van den Broeck, F.; Margamuljana, L.; Martins, J. C.; Herdewijn, P.; Balzarini, J.; McGuigan, C.; Van Calenbergh, S. J. Org. Chem. 2014, 79, 5097. doi: 10.1021/jo500659e
-
[48]
McGuigan, C.; Derudas, M.; Gonczy, B.; Hinsinger, K.; Kandil, S.; Pertusati, F.; Serpi, M.; Snoeck, R.; Andrei, G.; Balzarini, J.; McHugh, T. D.; Maitra, A.; Akorli, E.; Evangelopoulos, D.; Bhakta, S. Bioorg. Med. Chem. 2014, 22, 2816. doi: 10.1016/j.bmc.2014.02.056
-
[49]
Jonckers, T. H. M.; Tahri, A.; Vijgen, L.; Berke, J. M.; Lachau-Durand, S.; Stoops, B.; Snoeys, J.; Leclercq, L.; Tambuyzer, L.; Lin, T. I.; Simmen, K.; Raboisson, P. J. Med. Chem. 2016, 59, 5790. doi: 10.1021/acs.jmedchem.6b00382
-
[50]
McGuigan, C.; Kelleher, M. R.; Perrone, P.; Mulready, S.; Luoni, G.; Daverio, F.; Rajyaguru, S.; Le Pogam, S.; Najera, I.; Martin, J. A.; Klumpp, K.; Smith, D. B. Bioorg. Med. Chem. Lett. 2009, 19, 4250. doi: 10.1016/j.bmcl.2009.05.099
-
[51]
Derudas, M.; Brancale, A.; Naesens, L.; Neyts, J.; Balzarini, J.; McGuigan, C. Bioorg. Med. Chem. 2010, 18, 2748. doi: 10.1016/j.bmc.2010.02.015
-
[52]
Derudas, M.; Quintiliani, M.; Brancale, A.; Andrei, G.; Snoeck, R.; Balzarini, J.; McGuigan, C. Antivir. Chem. Chemother. 2010, 21, 15. doi: 10.3851/IMP1661
-
[53]
McGuigan, C.; Thiery, J. C.; Daverioa, F.; Jiang, W. G.; Davies, G.; Mason, M. Bioorg. Med. Chem. 2005, 13, 3219. doi: 10.1016/j.bmc.2005.02.041
-
[54]
Siegel, D.; Hui, H. C.; Doerffler, E.; Clarke, M. O.; Chun, K.; Zhang, L. J.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; Wang, Q.; Wolfe, L.; Jordan, R.; Soloveva, V.; Knox, J.; Perry, J.; Perron, M.; Stray, K. M.; Barauskas, O.; Feng, J. Y.; Xu, Y. L.; Lee, G.; Rheingold, A. L.; Ray, A. S.; Bannister, R.; Strickley, R.; Swa-minathan, S.; Lee, W. A.; Bavari, S.; Cihlar, T.; Lo, M. K.; Warren, T. K.; Mackman, R. L. J. Med. Chem. 2017, 60, 1648. doi: 10.1021/acs.jmedchem.6b01594
-
[55]
Warren, T. K.; Jordan, R.; Lo, M. K.; Ray, A. S.; Mackman, R. L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H. C.; Larson, N.; Strickley, R.; Wells, J.; Stuthman, K. S.; Van Tongeren, S. A.; Garza, N. L.; Donnelly, G.; Shurtleff, A. C.; Retterer, C. J.; Gharaibeh, D.; Zamani, R.; Kenny, T.; Eaton, B. P.; Grimes, E.; Welch, L. S.; Gomba, L.; Wilhelmsen, C. L.; Nichols, D. K.; Nuss, J. E.; Nagle, E. R.; Kugelman, J. R.; Palacios, G.; Doerffler, E.; Neville, S.; Carra, E.; Clarke, M. O.; Zhang, L. J.; Lew, W.; Ross, B.; Wang, Q.; Chun, K.; Wolfe, L.; Babusis, D.; Park, Y.; Stray, K. M.; Trancheva, I.; Feng, J. Y.; Barauskas, O.; Xu, Y. L; Wong, P.; Braun, M. R.; Flint, M.; McMullan, L. K.; Chen, S. S.; Fearns, R.; Swaminathan, S.; Mayers, D. L.; Spiropoulou, C. F.; Lee, W. A.; Nichol, S. T.; Cihlar, T.; Bavari, S. Nature 2016, 531, 381. doi: 10.1038/nature17180
-
[56]
Meneghesso, S.; Vanderlinden, E.; Brancale, A.; Balzarini, J.; Naesens, L.; McGuigan, C. Chem. Med. Chem. 2013, 8, 415. doi: 10.1002/cmdc.v8.3
-
[57]
Kandil, S.; Balzarini, J.; Rat, S.; Brancale, A.; Westwell, A. D.; McGuigan, C. Bioorg. Med. Chem. Lett. 2016, 26, 5618. doi: 10.1016/j.bmcl.2016.10.077
-
[58]
Mehellou, Y.; Valente, R.; Mottram, H.; Walsby, E.; Mills, K. I.; Balzarini, J.; McGuigan, C. Bioorg. Med. Chem. 2010, 18, 2439. doi: 10.1016/j.bmc.2010.02.059
-
[59]
Meneghesso, S.; Vanderlinden, E.; Stevaert, A.; McGuigan, C.; Balzarini, J.; Naesens, L. Antivir. Res. 2012, 94, 35. doi: 10.1016/j.antiviral.2012.01.007
-
[60]
McGuigan, C.; Bourdin, C.; Derudas, M.; Hamon, N.; Hinsinger, K.; Kandil, S.; Madela, K.; Meneghesso, S.; Pertusati, F.; Serpi, M.; Slusarczyk, M.; Chamberlain, S.; Kolykhalov, A.; Vernachio, J.; Vanpouille, C.; Introini, A.; Margolis, L.; Balzarini, J. Eur. J. Med. Chem. 2013, 70, 326. doi: 10.1016/j.ejmech.2013.09.047
-
[61]
McGuigan, C.; Madela, K.; Aljarah, M.; Bourdin, C.; Arrica, M.; Barrett, E.; Jones, S.; Kolykhalov, A.; Bleiman, B.; Bryant, K. D.; Ganguly, B.; Gorovits, E.; Henson, G.; Hunley, D.; Hutchins, J.; Muhammad, J.; Obikhod, A.; Patti, J.; Walters, C. R.; Wang, J.; Vernachio, J.; Ramamurty, C. V. S.; Battina, S. K.; Chamberlain, S. J. Med. Chem. 2011, 54, 8632. doi: 10.1021/jm2011673
-
[62]
Perrone, P.; Luoni, G. M.; Kelleher, M. R.; Daverio, F.; Angell, A.; Mulready, S.; Congiatu, C.; Rajyaguru, S.; Martin, J. A.; Leveque, V.; Pogam, S. L.; Najera, I.; Klumpp, K.; Smith, D. B.; McGuigan, C. J. Med. Chem. 2007, 50, 1840. doi: 10.1021/jm0613370
-
[63]
Osgerby, L.; Lai, Y. C.; Thornton, P. J.; Amalfitano, J.; Le Duff, C. S.; Jabeen, I.; Kadri, H.; Miccoli, A.; Tucker, J. H. R.; Muqit, M. M. K.; Mehellou, Y. J. Med. Chem. 2017, 60, 3518. doi: 10.1021/acs.jmedchem.6b01897
-
[1]
-

图 1 替诺福韦艾拉酚胺、替诺福韦酯、替诺福韦和索非布韦的结构
Figure 1 Structures of tenofovir alafenamide, tenofovir disoproxil, tenofovir and sofosbuvir

图 4 水溶性和稳定性高的苯并咪唑类驱虫药的磷酸盐前药
Figure 4 Water-soluble and water-stable phosphate prodrugs of fenbendazole

图式 1 制备克罗卡林水溶磷酸盐前药的方法
Scheme 1 Method to prepare water soluble prodrug of levcro-makalim
Reagents and conditions: (a) dibenzyl N, N-dimethylphosphorami-dite, tetrazole, CH2Cl2, CH3CN; (b) 30% H2O2, THF, 0 ℃; (c) TMSBr, DCM; (d) ion exchange using Na+-Dowex resin

图 6 三个药物在肠道、血浆和淋巴细胞之间的转运的过程图
Figure 6 Transporting of three drugs at gut, plasma and lymphoid cells
From International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy 2015, Washington, DC, Abst. # 6, J. M. Custodio et al., Gilead Science

图 7 进入临床试验的磷酰胺酯前药的化学结构
Figure 7 Chemical structures of nucleoside ProTide that under development in clinical trials

图 8 磷酰胺酯如何进入细胞传送磷酸前药的示意图
Figure 8 A general representation of how phosphoramidates deliver monophosphates into cells

图式 2 芳氧基磷酰胺酯前药的作用方式
Scheme 2 Mode of action of aryloxyphosphoramidates or phosphonamidates

图式 3 N-乙酰化氨基葡萄糖和相应的磷酰胺酯
Scheme 3 Glucosamine and corresponding phosphoramidates of N-acetyl glucosamine

图式 5 一般制备磷酰氯试剂的方法
Scheme 5 General method for preparation of phosphorochloridate reagent

图式 6 制备核苷类烷氨基磷酸酯前药的两种途径
Scheme 6 Two pathways to generate phosphoramidate nucleosides prodrugs

图 10 NMI活化方法制备的一些核苷类似物磷酰胺酯
Figure 10 Some phosphoramidates nucleoside prepared via NMI activation prodrugs

图 11 t-BuMgCl方法制备的一些核苷类似物磷酰胺酯
Figure 11 Some phosphoramidate nucleosides prepared by t-BuMgCl method

图式 8 利用Vorbrüggen反应构建ProTide化合物
Scheme 8 Use of Vorbrüggen reaction for synthesis of ProTide

图式 9 通过路易斯酸活化和碱助条件下实现磷酰胺酯化
Scheme 9 Lewis acid activation and base assistance for the ProTide synthesis

图式 10 从氨基酸制备核苷类磷酰胺酯前药的方法
Scheme 10 Methods to access phosphonamidates nucleoside prodrugs from amino acid

图式 12 通过动力学拆分实现非对映选择性核苷磷酰胺酯化的实用工艺
Scheme 12 Practical process for diastereoselective phosphorylation nucleoside via dynamic kinetic resolution

图式 13 通过动力学拆分首次催化对映选择性合成磷酰胺酯
Scheme 13 First catalytic enantioselective synthesis of phosphoramides via kinetic resolution

图式14 ProTide策略合成氟化嘧啶的磷酰胺酯前药
Scheme 14 Synthesis of fluorinated pyrimidine prodrug via phosphoramidate ProTide strategies
Reagents and conditions: (a) POCl3, Et3N, anhydrous Et2O, -78 ℃ for 1 h, then r.t. 1 h; (b) phenyl or 1-naphthyl phosphorodichloridate, Et3N, anhydrous DCM, -78 ℃, 1 ~ 3 h; (c) t-BuMgCl or NMI, anhydrous THF, r.t. 16~18 h.

图 12 一些高效的抗丙肝病毒的鸟嘌呤核苷磷酰胺酯前药结构
Figure 12 Structures of potent anti-HCV guanosine nucleosides and ProTide

图式 15 合成7-脱氮鸟嘌呤核苷类似物磷酰胺酯前药
Scheme 15 Synthesis of 7-deaza analogues of guanosine nucleoside phosphoramidates prodrug
Reagents and conditions: (a) anhyd. CH2Cl2, HBr (33% in acetic acid, 6.7 equiv.), 0 ℃ to r.t., 2 h; anhyd. CH3CN, KOH (3.0 equiv.), TDA-1 (0.2 equiv.), r.t., 1 h; (b) anhyd. CH2Cl2, BCl3 (10 equiv.), -78 ℃, 2 h then-20 ℃, 2 h, 30 min; (c) anhyd. MeOH, NaOMe (3.0 equiv.), reflux, overnight; (d) anhyd. THF, t-BuMgCl (1.2 equiv.), naphthyl L-alanine ester phospho-chloridate (1.2 equiv.), r.t., overnight.

图 13 核苷酸NS5b聚合酶抑制剂和甲基胞啶磷酰胺酯前药
Figure 13 Nucleoside inhibitor of HCV NS5b polymerase and structures of phosphoramidate prodrugs of C-methylcytidine

图式 16 合成甲基胞啶磷酰胺酯前药
Scheme 16 Synthesis of phosphoramidate prodrugs of C-methylcytidine
Reagents and conditions: (a) Et3N, DCM; (b) t-BuMgCl, dry THF; (c) acetone, pTsOH, 71%~87% yield; (d) t-BuMgCl, dry THF.

图 14 高活性和亲脂性更强的磷酰胺酯前药
Figure 14 Highly potent and more lipophilic phosphoramidate prodrug

图式 17 采用ProTide技术合成阿昔洛韦的磷酰胺酯前药
Scheme 17 Application of phosphoramidate ProTide for acyclovir prodrug
Reagent and conditions: (a) dimethylfornamide dimethyl acetal, anhydrous DMF, r.t., 1 d; (b) t-BuMgCl, THF, r.t., overnight or NMI, V(THF)/V(pyridine)=3/2, r.t., overnight; (c) 1-propanol, reflux, for 18 h or 2-propanol, reflux, 24~96 h.

图 15 利用ProTide技术合成吉西他滨的磷酰胺酯前药
Figure 15 Application of phosphoramidate ProTide for synthesis of gemcitabine prodrug

图式 18 利用ProTide技术合成阿德福韦和替诺福韦的磷酰酯前药
Scheme 18 Application of phosphoramidate ProTide for Adefovir and Tenofovir prodrugs

图式 19 具有抗HIV活性的apionucleoside前药
Scheme 19 Prodrug of apionucleoside family with anti-HIV activity

图式 20 合成抗结核和抗病毒药物的磷酰胺酯前药
Scheme 20 Synthesis of Phosphoramidate ProTide prodrug with anti-tubercular and anti-viral activity
Reagents and conditions: (a) octanoyl chloride, DIPEA, anhydrous CH2Cl2, 0 ℃ to 20℃, 2 h; (b) propargyloctanamide, Pd(PPh3)4, CuI, DIPEA, anhydrous DMF, 20 ℃; (c) 149, phosphorochlorides, NMI, anhydrous THF, 20 ℃, 12 h

图 17 抗丙型肝炎病毒的新颖环状嘧啶核苷磷酸酯前药
Figure 17 Anti-HCV phosphate ester prodrug from novel cyclic uridine nucleotide

图 18 吉利德抗埃博拉病毒磷酰胺酯(GS-5734)和一些磷酰胺酯前药
Figure 18 Gilead's ProTide GS-5734 against Ebola and some phosphoramidate prodrugs

图式 22 磷二酰胺酯作为有希望的磷酸前药
Scheme 22 Phosphorodiamidates as promising new phosphate prodrugs
Reagents and conditions: (a) Et3N (1 equiv.), POCl3 (1 equiv.), dry THF; (b) for symmetrical diamidates, amine (5 equiv.), Et3N (10 equiv.), dry DCM; for asymmetric diamidates, amine 1 (1 equiv.), Et3N (2 equiv.) then amine 2 (5 equiv.), Et3N (10 equiv.), dry DCM.
表 1 通过有机磷酸酯/有机磷酸及其钠盐前药改善母体药物的亲脂性和膜穿透能力[3b]
Table 1. Phosphate and phosphonate prodrugs for improved lipophilicity or permeability
No. 药物名称 结构 前药策略 1 Adefovir dipivoxil
阿德福韦酯(ⅰ)通过酯酶和磷酸二酯酶生物转化为阿德福韦
(ⅱ)使阿德福韦口服生物利用度从10%增加到阿德福韦酯的30%~45%2 Tenofovir disoproxil
替诺福韦酯(ⅰ)通过酯酶和磷酸二酯酶生物转化为替诺福韦
(ⅱ)替诺福韦酯的口服生物利用度是替诺福韦的2.5倍3 Miproxifene phosphate
磷酸米泼昔芬(ⅰ)通过碱性磷酸酯酶生物转化为米泼昔芬
(ⅱ) pH值为7.4时, 水溶性提高了近1000倍
(ⅲ)临床前大鼠和犬的生物利用度分别提高了28.8%和23.8%4 Fosamprenavir
福沙那韦钙(ⅰ)通过碱性磷酸酯酶生物转化为福沙那韦
(ⅱ)水溶性提高了10倍以上
(ⅲ)更方便患者的服药剂量
(ⅳ)延长专利有效期5 Estramustine phosphate
雌二醇氮芥磷酯(ⅰ)通过碱性磷酸酯酶生物转化为雌二醇氮芥
(ⅱ)注射剂和口服制剂均已上市6 Prednisolone phosphate
泼尼松龙磷酸钠(ⅰ)通过碱性磷酸酯酶生物转化为泼尼松龙
(ⅱ)由于前药能开发出液体制剂, 改善儿童服用的顺应性7 Fludarabine phosphate
磷酸氟达拉滨(ⅰ)通过碱性磷酸酯酶生物转化为氟达拉滨
(ⅱ)目前仅有注射剂上市8 Fosphenytoin
磷苯妥英(ⅰ)通过碱性磷酸酯酶快速转化为苯妥英
(ⅱ)溶解度从苯妥英的25 μg/mL增加到磷苯妥英的140 mg/mL9 Fosfluconazole
福司氟康唑(ⅰ)通过碱性磷酸酯酶快速转化为氟康唑
(ⅱ)不同规格的产品均能采用静脉给药
(ⅲ)显著提高水溶性(福司氟康唑溶解度为300 mg/mL)10 Phosphonooxymethyl
Propofol(ⅰ)通过碱性磷酸酯酶快速转化为丙泊酚
(ⅱ)溶解度从丙泊酚的150 μg/mL显著提高到前药的500 mg/mL11 Propofol phosphate
丙泊酚磷酸酯(ⅰ)通过碱性磷酸酯酶快速转化为丙泊酚
(ⅱ)显著增加了丙泊酚的水溶性 下载: 导出CSV
下载: 导出CSV
表 2 TFV, TDF和TAF的半衰期a
Table 2. Half life of TFV, TDF and TAF
药物 T1/2/min 人血 T细胞摄取 TFV 稳定 稳定 TDF 0.4 71 TAF 90 28 a TDF:在血液中水解快, 而在T细胞中水解相对较慢; TAF:在血液中更稳定, 但在T细胞中水解比TDF更快.
下载: 导出CSV
-










































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 333
- 文章访问数: 22298
- HTML全文浏览量: 4358
 下载:
下载:
