 图式 1
过渡金属催化烯丙基取代反应
Scheme1.
Transition metal-catalyzed allylic substitution reactions
图式 1
过渡金属催化烯丙基取代反应
Scheme1.
Transition metal-catalyzed allylic substitution reactions
引用本文:
邓颖颍, 杨文, 杨新, 杨定乔. 铱催化烯丙基酯的不对称烯丙基取代反应研究进展[J]. 有机化学,
2017, 37(12): 3039-3059.
doi:
10.6023/cjoc201704034

Citation: Deng Yingying, Yang Wen, Yang Xin, Yang Dingqiao. Progress in Iridium-Catalyzed Asymmetric Allylic Substitution Reactions with Allylic Esters[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3039-3059. doi: 10.6023/cjoc201704034
Citation: Deng Yingying, Yang Wen, Yang Xin, Yang Dingqiao. Progress in Iridium-Catalyzed Asymmetric Allylic Substitution Reactions with Allylic Esters[J]. Chinese Journal of Organic Chemistry, 2017, 37(12): 3039-3059. doi: 10.6023/cjoc201704034
铱催化烯丙基酯的不对称烯丙基取代反应研究进展
English
Progress in Iridium-Catalyzed Asymmetric Allylic Substitution Reactions with Allylic Esters
Abstract:
Iridium-catalyzed asymmetric allylic substitution reaction is one of the most important methods for the synthesis of chiral compounds. The recent research progress in iridium-catalyzed asymmetric allylic substitution reactions of allylic ester and its derivatives is reviewed with focus on the influences of the iridium catalysts, the substrate structures of allylic ester and its derivatives, the type of nucleophiles, the effects of solvents and additives on asymmetric substitution reaction. Moreover, the possible mechanisms are also discussed in this review.
-
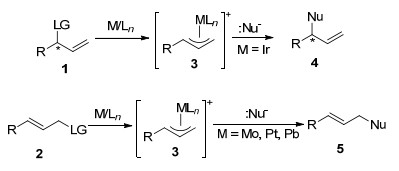
过渡金属催化不对称烯丙基取代反应是有机化学中重要的反应类型之一, 因为它们为构建C—C, C—N, C—O, C—S键和C—F键提供了很好的方法.虽然在过去的四十多年里, 人们已经对钯催化烯丙基取代反应进行了广泛的研究[1], 而其他过渡金属包括Mo[2], W[3], Fe[4], Ru[5], Ni[6], Rh[7], Pt[8], Cu[9]和Ir等也被发现能够很好地应用于催化此类反应.特别是铱催化烯丙基取代反应, 其反应条件温和, 立体化学选择性好, 因此被广泛地应用于合成光学活性的烯丙基胺、烯丙基醚(醇)、烯丙基硫化物和含氟烯丙基类型化合物.另外, 以钼、铂和钯等配合物为催化剂进行的取代反应, 亲核试剂(:Nu-)倾向于进攻π-烯丙基中间体3的烯丙基末端, 得到手性的直链产物5, 而以铱配合物为催化剂的烯丙基取代反应主要得到含支链的手性化合物4, 并且具有较高的立体化学选择性(Scheme 1)[10, 11].
图式 1
过渡金属催化烯丙基取代反应
Scheme1.
Transition metal-catalyzed allylic substitution reactions
由于烯丙基酯的反应活性相对烯丙基卤温和, 并且具有原料易得、制备简单等优点, 所以被广泛地应用于铱催化不对称烯丙基取代反应中.目前研究的比较多的烯丙基酯的代表性底物主要有烯丙基醋酸酯、烯丙基磷酸酯和烯丙基碳酸酯等[12].
1997年, Takeuchi等[13]首次报道了[Ir(COD)Cl]2 (COD=1, 5-环辛二烯)与配体P(OEt)3组成的配合物催化烯丙基醋酸酯与丙二酸酯钠盐的不对称烯丙基烷基化反应.随后, Janssen等[14]首次实现了[Ir(COD)Cl]2与双齿膦噁唑啉配体组成的配合物催化烯丙基烷基化反应.这些研究推动了铱催化烯丙基取代反应的发展, 激发了化学家们对于这类反应的研究兴趣.其中Hartwig, Helmchen, Alexakis, Carreira, Stoltz和游书力等[15~19]课题组都对铱催化烯丙基取代反应的研究做出了杰出的贡献.
本文综述了近几年来铱催化烯丙基酯衍生物与各种不同亲核试剂发生不对称取代反应的研究进展.同时还对烯丙基取代反应催化循环的部分可能反应机理进行了讨论.
1 碳亲核试剂
1.1 活泼亚甲基类碳亲核试剂
具有活泼亚甲基类型化合物是应用于铱催化烯丙基取代反应中最为常见的碳亲核试剂[20].
2008年, Eilbracht等[21]以[Ir(COD)Cl]2作为催化剂, 10a作为手性配体, 实现了γ-取代烯丙基碳酸甲酯6与二苯亚甲基甘氨酸乙酯7的不对称烯丙基取代反应(Eq. 1).研究表明:加入20 mol% 1, 4-二氮杂二环[2.2.2]辛烷(DABCO)对反应有利, 它不仅能活化催化剂, 而且能使底物7发生去质子化, 从而增强了底物7的亲核性.并获得较高的对映选择性.
同时他们还在同一种催化剂作用下, 分别研究了以氰基乙酸叔丁酯(11a)和氰基乙酸甲酯(11b)作为亲核试剂的烯丙基取代反应(Eq. 2).研究表明:加入不同的碱, 如六甲基二硅基胺基锂(LiHMDS), DABCO, KOH等, 均能获得中等的产率(60%~76%), dr值均接近1:1.
具有单氟化亚甲基单元的光学活性化合物, 例如氯法拉滨和双氟泼尼酯等药物.通过铱催化氟化亚甲基衍生物不对称烯丙基取代反应, 可得到含有单氟化亚甲基部分的支链烯丙基产物.这些单氟化烯丙基产物是用于合成具有生物活性化合物的非常重要的中间体.
2009年, 游书力和赵晓明课题组[22]以16a作为手性配体, 研究了铱催化γ-取代烯丙基碳酸甲酯6和氟代双(苯基磺酰基)甲烷(13)的烯丙基取代反应(Eq. 3).在最优反应条件下, γ-芳基烯丙基碳酸甲酯6的苯环上连有给电子基(如p-OCH3, m-OCH3, p-CH3和p-iBu)比连有吸电子基(如m-Cl, p-Br和p-CF3)的底物发生取代反应能够获得更高的产率.
2016年, 赵晓明课题组[23]继续研究了以氟化亚甲基衍生物作为亲核试剂的铱催化烯丙基取代反应.与之前研究不同的是, 他们以10b作为手性配体, 分别使用α-氟代丙二酸二甲酯(17a)和2-氟-3-氧代丁酸甲酯(17b)作为亲核试剂, 与γ-取代烯丙基碳酸甲酯6发生不对称烯丙基取代反应(Eq. 4).反应在常温下进行, 加入碱CsF比加入Cs2CO3能获得更高的产率.
2012年, 游书力课题组[24]研究了不同的N-芳基亚膦酰胺配体与铱形成配合物作为催化剂, 催化丙二酸二甲酯钠盐20不对称烯丙基烷基化反应(Eq. 5).研究表明:使用配体24a, 更有利于反应的进行.产物的产率高达99%, 主要的支链产物22的对映体过量值高达99% ee.同时支链产物22与直链产物23的产率比值>99:1.而使用配体25, 获得产物的产率和对映体过量值均较低(产率为30%, ee值为13%).
同年, 该课题组[25]还报道了[Ir(COD)Cl]2与配体10b组成的配合物为催化剂, 催化γ-取代烯丙基碳酸甲酯6与2-(4-硝基苯磺酰基)乙酸甲酯(26)发生烯丙基烷基化反应.得到的烷基化中间体27, 可继续发生去酰基化反应得到产物28 (Scheme 2), 或者发生Julia成烯反应得到产物29.
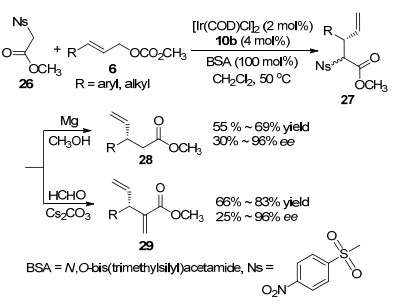 图式 2
铱催化26与6的烯丙基烷基化反应
Scheme2.
Ir-Catalyzed allylic alkylation reaction of 26 with 6
图式 2
铱催化26与6的烯丙基烷基化反应
Scheme2.
Ir-Catalyzed allylic alkylation reaction of 26 with 6
2016年, 钮大文课题组[26]报道了γ-取代烯丙基碳酸叔丁酯31和α-取代烯丙基碳酸叔丁酯(33a)分别与CH2(Bpin)2(双[(频哪醇)硼基]甲烷, 30)发生铱催化不对称烯丙基取代反应, 得到β-取代手性烯丙基硼酸酯产物32 (Eqs. 6, 7), 产率高达99%, ee值高达98%.研究表明:银盐作为添加剂对反应的产率和立体化学选择性有非常重要的影响.
1.2 次甲基类碳亲核试剂
如何构筑具有相邻的手性叔碳和季碳立体中心的化合物一直以来都是有机合成的难点和挑战[27].近年来人们利用前手性碳亲核试剂参与铱催化烯丙基取代反应, 成功地以高对映选择性和非对映选择性实现了具有相邻的手性叔碳和季碳立体中心化合物的构筑.
2013年, Hartwig等[28]报道了由[Ir(COD)Cl]2和亚磷酰胺配体10a组成的配合物与磷酸银盐组成的催化剂共同作用下, 以高的对映选择性和非对映选择性实现了吖内酯35的烯丙基烷基化反应(Eq. 8).研究表明:当没有加入磷酸银盐37时, 反应的非对映选择性较低(dr=7:1), 而加入磷酸银盐37后, 反应的非对映选择性显著提高(dr>20:1).
2014, 该课题组[29]继续使用10a作为手性配体, 研究了铱催化γ-取代烯丙基醋酸酯38与取代的5H-噁唑-4-酮39a~39d的不对称烯丙基取代反应(Eq. 9).反应需要加入碱Et2Zn, 使5H-噁唑-4-酮39的酸性α-H离去, 形成碳负离子作为亲核试剂, 发生取代反应获得高的非对映选择性.
同时该课题组还研究了γ-取代烯丙基碳酸叔丁酯31与取代的5H-噻唑-4-酮41a~41c的反应(Eq. 10).同样在该反应体系中, 加入碱Mg(NiPr2)2, 也能发生取代反应, 并获得高的非对映选择性.
2013年, Stoltz等分别报道了铱催化γ-取代烯丙基碳酸甲酯6与环状β-酮酸酯43[30]和开链的α-甲基-β-酮酸乙酯(46)[31]的不对称烯丙基取代反应.两种类型的反应都使用[Ir(COD)Cl]2与亚磷酰胺配体24a组成的配合物作为催化剂, 1, 5, 7-三氮杂二环[4.4.0]癸-5-烯(TBD)作为碱, 均以高区域选择性和对映选择性获得烯丙基烷基化取代产物.不同的是, 使用环状β-酮酸酯43作为底物的反应(Eq. 11), 需要加入LiBr作为添加剂, 而使用开链的α-甲基-β-酮酸乙酯46作为底物的反应(Eq. 12), 需要加入LiOBu-t作为额外的碱以促进反应的进行.
2016年, Stoltz等[32]报道了铱催化γ-芳基烯丙基碳酸甲酯6与环外α, β-不饱和丙二酸二甲酯(49)发生烯丙基烷基化反应.虽然使用配体24b和55均能催化反应的进行, 但是对于一些底物, 55比24b作为配体能获得更高的产率, 而且能更好地控制反应的区域选择性和对映选择性.烯丙基取代反应得到的烷基化中间体50不需要分离, 随后加入甲苯, 100 ℃条件下发生科普(Cope)重排得到产物51 (Scheme 3), 产率高达97%, ee值高达97%.同时该课题组还研究了以环内α, β-不饱和β-酮酸酯52作为亲核试剂的铱催化烯丙基烷基化反应(Scheme 3).
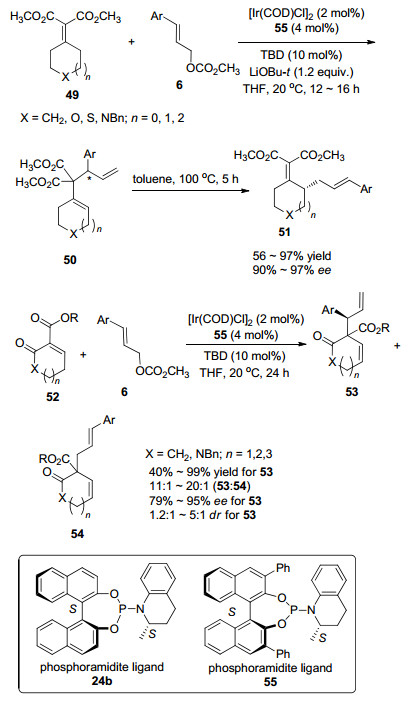 图式 3
铱催化α, β不饱和丙二酸酯49和酮酸酯52的不对称烯丙基烷基化反应
Scheme3.
Ir-catalyzed asymmetric allylic alkylation of α, β-unsaturated malonates 49 and ketoesters 52
图式 3
铱催化α, β不饱和丙二酸酯49和酮酸酯52的不对称烯丙基烷基化反应
Scheme3.
Ir-catalyzed asymmetric allylic alkylation of α, β-unsaturated malonates 49 and ketoesters 52
2016年, 钮大文课题组[33]以16b作为配体, 加入碱1, 8-二氮杂二环[5.4.0]十一碳-7-烯(DBU), 实现了铱催化γ-取代烯丙基碳酸叔丁酯31与亚胺衍生物56的烯丙基取代反应(Scheme 4).研究表明:反应首先通过亚胺56的α'-C选择性烯丙基化反应形成末端烯烃57, 随后末端烯烃57自发地进行2-aza-Cope重排, 最后得到产物58.
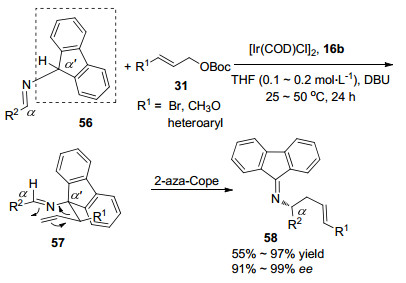 图式 4
铱催化亚胺衍生物56的烯丙基取代反应
Scheme4.
Iridium-catalyzed allylic substitution reaction of imine derivatives 56
图式 4
铱催化亚胺衍生物56的烯丙基取代反应
Scheme4.
Iridium-catalyzed allylic substitution reaction of imine derivatives 56
利用前手性碳亲核试剂参与烯丙基取代反应, 近年来取得了很大的进展, 除了上述提到Hartwig和Stoltz的工作之外, Carreira[34]和张万斌[35]等课题组都做出了出色的工作. 2013年, Carreira等[36]首次提出了在[Ir(COD)Cl]2与配体34a组成的催化剂和金鸡纳生物碱衍生的伯胺62的双重催化作用下, 支链烯丙基醇59与2-苯基丙醛60发生烯丙基取代反应(Eq. 13), 该方法成功构建了具有相邻叔碳和季碳立体中心的γ, δ-不饱和醛61. 2015年, 张万斌等[37]研究了吖内酯35作为前手性碳亲核试剂的钯催化烯丙基取代反应(Eq. 14), 与铱催化烯丙基取代反应不同的是, 该反应得到的产物为带有相邻季碳和叔碳立体中心的支链α-氨基酸产物64.
1.3 富电子芳烃类碳亲核试剂
2008年, 游书力课题组[38]以[Ir(COD)Cl]2与配体16a组成的配合物作为催化剂, 实现了吲哚衍生物65 C-3位上的烯丙基取代反应(Eq. 15).在最优的反应条件下, 对底物的普适性进行了考察, 发现带有给电子基的γ-芳基烯丙基碳酸甲酯发生取代反应, 能获得良好的产率(73%~85%)和对映体过量值(70%~92% ee).
2013年, 该课题组[39]继续对吲哚衍生物的铱催化烯丙基取代反应进行了研究.与之前不同的是, 他们使用[Ir(COD)Cl]2与自己设计合成的新型N-芳基亚磷酰胺配体70组成的配合物作为催化剂, 催化3-取代吲哚烯丙基碳酸甲酯68发生不对称烯丙基分子内取代反应(Eq. 16).
可能的反应机理见Scheme 5所示.首先, 铱(Ⅰ)催化剂与68发生氧化加成产生铱(Ⅲ)-π-烯丙基配合物71.然后, 在碱的作用下, 吲哚3位碳作为亲核试剂进攻铱(Ⅲ)-π-烯丙基部分, 形成螺环吲哚中间体72.中间体72接着转化成为中间体73, 最后五元杂环芳构化形成产物69.这种新型反应模式的发现, 不但更正了以前文献报道的吲哚分子内烷基化产物错误的结构, 而且提供了一条合成手性吲哚和吡咯稠环结构化合物的新思路.
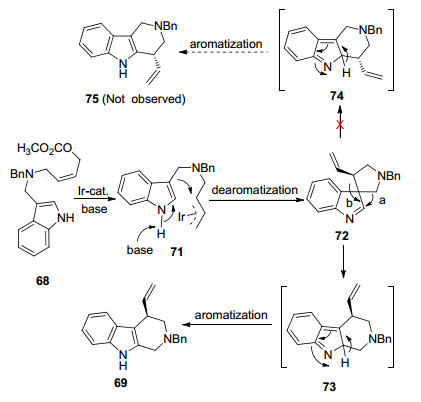 图式 5
3-吲哚烯丙基碳酸酯68分子内取代反应可能的反应机理
Scheme5.
Plausible mechanism of 3-indolyl allyl carbonates 68 intramolecular substitution reaction
图式 5
3-吲哚烯丙基碳酸酯68分子内取代反应可能的反应机理
Scheme5.
Plausible mechanism of 3-indolyl allyl carbonates 68 intramolecular substitution reaction
同时, 在相同催化体系下, 他们还研究了2-吡咯烯丙基碳酸甲酯76的不对称烯丙基分子内取代反应(Eq. 17).该反应生成双环产物77, 产率高达95%, ee值高达99%.
2015年, 游书力课题组[40]继续对2-吡咯烯丙基碳酸甲酯78的分子内烯丙基取代反应进行了研究.研究表明:使用与Eq. 17相同的催化剂, 当底物2-吡咯烯丙基碳酸甲酯78上的X为NTs, C(CO2CH3)2和C(CO2Et)2时, 便能以较高的产率和极佳的对映体过量值得到五元螺环2-氢吡咯产物79 (Eq. 18).
2011年, 该课题组[41]首次报道了铱催化苯酚烯丙基酯衍生物的分子内烯丙基取代反应.他们使用[Ir(COD)Cl]2与亚磷酰胺配体10b组成的配合物作为催化剂, Li2CO3作为碱, 对位取代的苯酚烯丙基甲酯衍生物80发生分子内烯丙基取代反应(Eq. 19), 生成螺环己二烯酮产物81, 均具有较高的产率和对映体过量值.
2012年, 该课题组[42]继续研究了间位取代的苯酚烯丙基酯衍生物的分子内烯丙基取代反应.与上述反应不同的是, 他们使用16a作为手性配体, 4-二甲氨基吡啶(DMAP)作为碱, 实现了间位取代的苯酚烯丙基酯衍生物82的铱催化分子内烯丙基取代反应, 生成四氢异喹啉产物83和84, 产率高达90%, ee值高达96% (Eq. 20).
2016年, 游书力课题组[43]报道了以N-P配体55作为手性配体, 铱催化2-取代萘酚烯丙基碳酸甲酯衍生物85a~85f的分子内烯丙基取代关环反应(Eq. 21).虽然使用[Ir(COD)Cl]2和[Ir(DBCOT)Cl]2 (DBCOT=二苯并环辛四烯)作为催化剂均能催化这一转化过程, 但是[Ir(DBCOT)Cl]2比[Ir(COD)Cl]2能更好地控制反应的对映选择性和非对映选择性.
2012年, Hartwig等[44]报道了环金属铱催化剂91a催化γ-取代烯丙基碳酸甲酯6与α-三甲基硅氧呋喃88发生不对称烯丙基取代反应(Eq. 22).在最优反应条件下, 带有吸电子基的γ-芳基烯丙基碳酸甲酯发生取代反应时, 获得的结果最佳, 生成的主要产物为3-取代丁烯酸内酯89, 产率达91%, ee值高达99%.
1.4 醛酮α-碳亲核试剂
醛酮类化合物α-位的烯丙基化反应一直是有机合成中的难点.通常采用的方法是直接将醛酮转化为烯醇醚或烯醇酯, 甚至烯胺来参与反应.
2014年, Hartwig等[45]报道了使用铱-肉桂基配合物95作为催化剂, 催化γ-取代烯丙基碳酸甲酯6与α-取代的环酮92发生不对称烯丙基取代反应(Eq. 23).研究表明:当分别加入Mg(OtBu)2, Ca(OiPr)2或Sr(OiPr)2作为添加剂时, 反应的产率和对映选择性均较低.但加入添加剂Ba(OtBu)2时, 反应的产率、非对映选择性和对映选择性均有显著的提高.
同年, 该课题组[46]还以[Ir(COD)Cl]2作为催化剂, 16b作为手性配体, 研究了由96作为亲核试剂的不对称烯丙基烷基化反应(Eq. 24).该反应的产物不仅在羰基的β位上有手性中心, 而且还能引入不同基团R, 从而使产物能够进一步地转化生成各种有价值的手性中间体.研究表明:在反应体系中加入KF和18-冠醚-6能促进反应顺利进行.
2015年, 该课题组[47]继续使用与Eq. 24相同的催化剂, 研究了γ-取代烯丙基碳酸甲酯6分别与亲核试剂98或100发生不对称烯丙基取代反应(Eqs. 25, 26).研究表明:这一反应需要加入KF或KOCH3, 但是他们对反应机理研究后, 发现这些添加剂不能活化烯醇三甲基硅烷(ROTMS), 而是作为一种碱来促进环金属Ir催化剂的形成.由烯丙基碳酸酯的氧化加成产生的碳酸根阴离子, 可能活化烯醇三甲基硅烷98, 以激发它们作为亲核试剂的反应活性, 与活化的烯丙基铱中间体发生亲核取代反应.
2014年, 该课题组[48]报道了[Ir(COD)Cl]2与配体16a组成的配合物作为催化剂, 催化γ-取代烯丙基碳酸三氯乙酯102 (OTroc=OCO2CH2CCl3)与亲核试剂103发生烯丙基取代反应(Eq. 27).亲核试剂103进攻102发生烯丙基取代反应, 生成产物104, 产物104还能够进一步的转化成为各种有价值的中间体.
2016年, Hartwig等[49]继续使用与Eq. 33相同的催化剂, 首次实现了三取代烯丙基磷酸二乙酯105与亲核试剂106发生烯丙基取代反应(Eq. 28).在最优的反应条件下, 底物105具有广泛的普适性, 能获得高产率和高对映体过量值的产物107.
同年, 该课题组[50]还报道了铱配合物111催化下, γ-取代烯丙基碳酸甲酯6与α-烷氧基酮108发生烯丙基取代反应(Scheme 6).研究表明:该反应的关键在于加入CuBr, α-烷氧基酮与Cu(Ⅰ)原位形成不稳定的环状螯合物Cu(Ⅰ)烯醇化物109, 随后再与γ-取代烯丙基碳酸甲酯6发生烯丙基取代反应.
 图式 6
铱催化α-烷氧基酮108的烯丙基取代反应
Scheme6.
Iridium-catalyzed allylic substituted reaction of α-alkoxy ketones 108
图式 6
铱催化α-烷氧基酮108的烯丙基取代反应
Scheme6.
Iridium-catalyzed allylic substituted reaction of α-alkoxy ketones 108
1.5 有机金属类碳亲核试剂
2007年, Alexakis等[51]报道了[Ir(COD)Cl]2与手性亚磷酰胺配体10b组成的配合物作为催化剂, 催化γ-取代烯丙基碳酸甲酯6与芳基锌试剂的不对称烯丙基取代反应(Eq. 29).在最优的反应条件下, 他们对底物γ-取代烯丙基碳酸甲酯6的结构进行了分析后发现, 当底物R=p-CH3OC6H4 (6)发生取代反应时, 产物112的区域选择性和对映选择性均较低, 而当底物R=p-ClC6H4 (6)发生取代反应时, 产物112的对映体过量值高达99.2% ee.
2009年, 该课题组[52]在上述反应研究的基础上, 还研究了铱催化醋酸-3-环己烯酯rac-114与芳基锌试剂的烯丙基取代反应(Eq. 30).研究表明:使用116作为手性N-P配体, 并加入TBD作为添加剂, 能够获得最佳的反应产率和对映选择性.
2015年Carreira等[53]报道了[Ir(COD)Cl]2与配体34b组成的配合物作为催化剂, 催化α-取代烯丙基碳酸叔丁酯33与一系列溴化烷基锌试剂117发生烯丙基烷基化反应(Eq. 31).研究表明:各种芳基、杂环芳基、烯基和炔基取代烯丙基碳酸叔丁酯33均能和溴化烷基锌试剂117反应, 获得的支链产物118, 产率达81%, 对映体过量值高达99% ee.与此同时, 在最优反应条件下, 一系列溴化烷基锌试剂117参与反应也能获得中等的收率(57%~78%)和较高的对映体过量值(80%~>99% ee).
1.6 α-甲基碳亲核试剂
2017年, 游书力课题组[54]首次报道了铱催化γ-取代烯丙基碳酸叔丁酯31与2-甲基吡啶119的烯丙基取代反应(Eq. 32).研究表明:反应需要加入BF3•OEt2, 它能与吡啶N原子结合从而增加α-甲基上氢的酸性, 并生成稳定的碳负离子作为亲核试剂与31发生取代反应.
1.7 其他的碳亲核试剂
2015年, Carreira等[55]报道了首例以121作为亲核试剂, 铱催化烯丙基取代反应.这一反应能够应用在两种不同的形式:当使用[Ir(COD)Cl]2与配体34a组成的配合物作为催化剂, 催化α-取代烯丙基碳酸叔丁酯33与121反应时(Eq. 33), 进行外消旋起始原料33的对映选择性动力学拆分, 所获得的产物(R)-122和(S)-33的对映体过量值均很高, 高达99% ee.当使用[Ir(COD)Cl]2与配体rac-34组成的配合物催化(S)-33与121反应时(Eq. 34), 进行具有光学活性的α-取代烯丙基碳酸叔丁酯(S)-33的立体烯丙基取代反应, 获得产物(S)-122.
2 含氮亲核试剂
2.1 氨作为氮亲核试剂
如果直接用氨作亲核试剂, 发生烯丙基氨基化反应比较困难, 因为氨可能会抑制催化剂的形成或代替手性配体与铱配合物形成非手性催化剂. 2007年, Hartwig等[56]首次报道了[Ir(COD)Cl]2与手性亚磷酰胺配体16b组成的配合物作为催化剂, 以氨作为亲核试剂, 成功地与γ-苯基烯丙基碳酸甲酯21发生不对称烯丙基氨基化反应(Eq. 35).反应过程中烯丙基碳酸酯完全转化, 生成双取代烯丙基胺124, 产率为93%, ee值高达99%.
2009年, 该课题组继续对γ-苯基烯丙基碳酸乙酯125与氨的反应进行了研究[57].与之前研究不同的是, 他们使用配合物127a作为手性催化剂, 催化γ-苯基烯丙基碳酸乙酯125与过量的氨(100 equiv.)发生不对称烯丙基氨基化反应, 然后用HCl酸化, 生成单取代烯丙基铵盐化合物126 (Eq. 36).研究表明:单取代烯丙基胺化合物的产率会随着氨的过量而增加.
同时该课题组还研究了“一锅法”合成手性N-烯丙基酰胺衍生物129的反应(Scheme 7).过量的氨123与γ-苯基烯丙基碳酸乙酯125发生不对称取代反应, 生成伯胺取代产物128, 加入酰基化试剂、三乙胺或K3PO4, 生成N-烯丙基酰胺129a~129c均具有中等的产率(63%~75%)和高对映体过量值(98% ee).
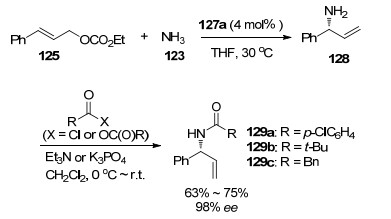 图式 7
“一锅法”合成手性N-烯丙基酰胺衍生物129
Scheme7.
One-pot synthesis of chiral N-allylamide derivatives 129
图式 7
“一锅法”合成手性N-烯丙基酰胺衍生物129
Scheme7.
One-pot synthesis of chiral N-allylamide derivatives 129
2.2 酰胺作为氮亲核试剂
由于直接使用氨作为亲核试剂的烯丙基氨基化反应较为困难, 酰胺作为一种氨等价物被广泛应用于烯丙基氨基化反应.
2007年, Hartwig等[56]还研究了[Ir(COD)Cl]2与亚磷酰胺配体134组成的配合物作为催化剂, 催化γ-取代烯丙基碳酸甲酯6与三氟乙酰胺钾盐130的烯丙基氨基化反应(Eq. 37), 所得产物131的产率和ee值均较高.
他们又研究了以LiNBoc2 (132)为亲核试剂, 铱催化不对称烯丙基取代反应(Eq. 38).在2006年Helmchen等[58]研究的基础上, 使用N-P配体134与[Ir(COD)Cl]2形成的络合物作为催化剂, 来催化这一反应, 实现了底物γ-取代烯丙基碳酸酯6的拓展.
2007年, Helmchen等[59]也报道了以酰胺类化合物作为亲核试剂的铱催化烯丙基氨基化反应.他们以[Ir(COD)Cl]2作为催化剂, 10b或16a作为手性配体, 催化γ-取代烯丙基碳酸甲酯6与酰胺类化合物135发生烯丙基氨基化反应(Eq. 39).需要加入8 mol% TBD来活化催化剂.
氨基甲酸酯作为一种酰胺类氮亲核试剂也能应用于铱催化的烯丙基氨基化反应中. 2009年, Hartwig等[60]使用环金属铱催化剂142, 研究了氨基甲酸叔丁酯138的铱催化烯丙基取代反应(Eq. 40).研究表明:反应不需要加入额外的碱.在对底物的不同结构进行分析时发现, 当烯丙基碳酸叔丁酯31上R1基团为环己基时, 反应没有区域选择性(55:45).对于其他芳基取代的烯丙基碳酸叔丁酯31, 可得到高区域选择性和对映选择性的产物139.
2.3 苯胺类氮亲核试剂
以苯胺作为氮亲核试剂的铱催化烯丙基取代反应也常见报道. 2007年, Hartwig等[61]研究了以苯胺143作为亲核试剂, 铱催化烯丙基氨基化反应(Eq. 42), 并首次提出了铱催化烯丙基氨基化反应可能的反应机理(Scheme 8). 3-苯基烯丙基碳酸甲酯21与配合物146可逆吸热加成, -OCO2CH3作为离去基团离去, 生成中间体148.苯胺143作为亲核试剂进攻中间体148, 生成烯丙基胺配合物147.再释放出取代产物144, 反应再进行下一次催化循环.
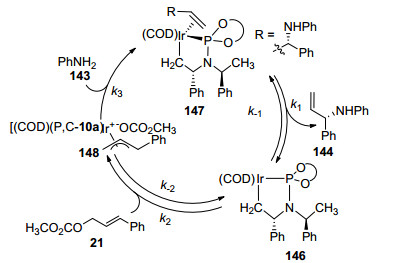 图式 8
铱催化21与苯胺143的烯丙基氨基化反应可能的反应机理
Scheme8.
Plausible mechanism of the iridium-catalyzed allylic amination of 21 with aniline 143
图式 8
铱催化21与苯胺143的烯丙基氨基化反应可能的反应机理
Scheme8.
Plausible mechanism of the iridium-catalyzed allylic amination of 21 with aniline 143
2012年, 游书力课题组[62]报道了以2-烯丙基苯胺149作为亲核试剂的铱催化烯丙基氨基化反应(Eq. 42).当使用[Ir(COD)Cl]2作为催化剂, 10b作为手性配体时, 反应无法获得目标产物.而使用相同的配体, 仅将催化剂换成[Ir(DBCOT)Cl]2时, 反应顺利进行, 并以高的产率、区域选择性和对映选择性获得主要的支链产物150.
2014年, 该课题组[63]继续使用与Eq. 42相同的催化剂和手性配体, 研究了2-炔基苯胺化合物152作为亲核试剂的不对称烯丙基氨基化反应, 氨基化中间体不需要分离, 便可继续在NaAuCl4•2H2O催化下主要生成2-取代N-烯丙基吲哚153 (Eq. 43).通过这种方法能够高效地合成吲哚2位上带有取代基(R2)的N-烯丙基吲哚化合物153和154.
但是, 当上述烯丙基氨基化反应结束后, 用硅胶柱色谱分离得到氨基化中间体, 随后向氨基化中间体中加入PdCl2和KI, 在80 ℃条件下继续发生关环反应, 形成吲哚2位和3位上带有取代基的N-烯丙基吲哚化合物155和156 (Eq. 44).
2.4 含氮杂环类氮亲核试剂
除了氨、酰胺和苯胺等常见的氮亲核试剂外, 以含氮杂环化合物作为氮亲核试剂也被广泛应用于铱催化烯丙基氨基化反应中.
2009年, Hartwig等[64]报道了分别以苯丙咪唑衍生物157a~157c、咪唑衍生物160a~160c和嘌呤衍生物163a~163b作为氮亲核试剂, 与γ-取代烯丙基碳酸甲酯6发生铱催化烯丙基氨基化反应(Eqs. 45~47).虽然使用配体16b或10a与[Ir(COD)Cl]2组成的配合物127a或91a均能催化这些反应的进行, 但是相比之下91a能够更好地控制反应的区域选择性和对映选择性, 以更高的产率和对映体过量值获得烯丙基氨基化产物.
2012年, 游书力课题组[65]报道了“一锅法”合成N-烯丙基吲哚的反应.他们继续以[Ir(DBCOT)Cl]2与配体10b组成的配合物作为催化剂, 催化γ-取代烯丙基碳酸甲酯6与二氢吲哚166发生烯丙基氨基化反应(Eq. 48).氨基化反应完成后, 继续向反应液中加入2, 3-二氯-5, 6-二氰对苯醌(DDQ)氧化, 反应30 min后, 便能以较高的产率(76%~92%)和高对映体过量值(82%~99% ee)获得主要的N-烯丙基吲哚产物167.
2014年, 该课题组[66]报道了首例铱催化2-取代吡啶烯丙基碳酸甲酯169和2-取代吡嗪烯丙基碳酸甲酯171分别发生分子内烯丙基取代反应.研究表明:当底物为2-取代吡啶烯丙基碳酸甲酯169时, 使用[Ir(COD)Cl]2与配体10b组成的配合物作为催化剂, 催化效果最佳, 得到2, 3-二氢吲嗪衍生物170.并且产率和对映体过量值均较高(Eq. 49).
当底物为2-取代吡嗪烯丙基碳酸甲酯171时, 使用[Ir(COD)Cl]2与配体24a组成的配合物作为催化剂, 结果最佳, 得到6, 7-二氢吡咯并[1, 2-a]吡嗪衍生物172产率高达95%, ee值高达97% (Eq. 50).
2015年, 游书力课题组[67]以10b作为配体, 研究了2-羟基吡啶及其衍生物173作为亲核试剂, 铱催化不对称烯丙基氨基化反应(Eq. 51).在最优反应条件下, 底物烯丙基碳酸甲酯上R1基团为烷基时, 产物的产率和区域选择性均较低, 而R1基团为芳基时, 产物的产率和对映选择性均较高.
2015年, 该课题组[68]首次报道了铱催化2-取代吡啶、吡嗪、喹啉和异喹啉烯丙基碳酸甲酯176的分子内不对称烯丙基取代反应(Eq. 52).在最优反应条件下, 通过分子内取代并关环, 能获得目标产物177, 产率高达99%, ee值高达99%.值得注意的是, 当进行2-取代吡嗪和2-取代喹啉烯丙基碳酸甲酯的分子内取代反应时, 使用N-P配体Me-THQphos 24a相比其它配体能够获得更好的结果.他们将这一事实归因于在活性铱催化剂形成过程中配体24a微妙的空间排列和特征C(sp2)—H键的活化.
他们还对分子内取代反应可能的反应机理进行了研究(Scheme 9).首先, 176与过渡金属Ir发生氧化加成产生π-烯丙基中间体178.随后通过喹啉上的N原子亲核取代形成喹啉鎓离子179, 随后发生去质子化, 最终得到脱芳构化产物177.他们认为喹啉鎓离子179是关键的中间体.
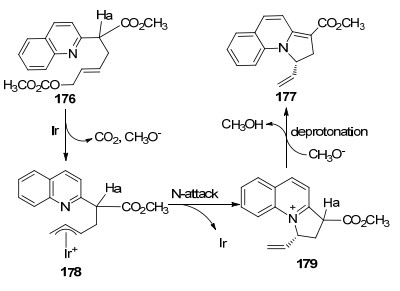 图式 9
分子内取代反应可能的反应机理
Scheme9.
Plausible mechanism of intramolecular substitution reaction
图式 9
分子内取代反应可能的反应机理
Scheme9.
Plausible mechanism of intramolecular substitution reaction
2016年, 该课题组[69]继续以24a作为配体, 实现了铱催化二溴取代吡咯衍生物180a, 180b的分子内烯丙基氨基化反应.其中Y=NPMB (PMB=对甲氧基苄基)的底物180b发生分子内取代关环反应获得的结果最佳, 得到的产物181b, 产率高达96%, 对映体过量值为96% ee (Eq. 53).
同年, 游书力课题组[70]还首次报道了使用氮杂环卡宾186作为手性配体的铱催化吲哚衍生的烯丙基碳酸甲酯182或吡咯衍生的烯丙基碳酸甲酯184的分子内烯丙基氨基化反应(Scheme 10).在最优的反应条件下, 底物R1为吸电子或给电子基团的吲哚衍生的烯丙基碳酸甲酯182发生分子内发生取代并关环反应均能获得较高的产率和对映体过量值.
 图式 10
铱催化分子内烯丙基氨基化反应
Scheme10.
Iridium-catalyzed intramolecular allylic amination reaction
图式 10
铱催化分子内烯丙基氨基化反应
Scheme10.
Iridium-catalyzed intramolecular allylic amination reaction
2017年, 该课题组[71]研究了含氮杂环化合物的分子内烯丙基氨基化反应, 报道了铱催化2-取代苯并噁唑、苯并噻唑和苯并咪唑烯丙基碳酸甲酯衍生物的分子内烯丙基取代反应.首先对2-取代苯并噁唑、苯并噻唑烯丙基碳酸甲酯衍生物187, 188分子内烯丙基取代反应进行了研究(Eq. 54), 发现只有使用N-P配体24a, 能够使底物完全转化.且当加入1 equiv. DBU作为碱时, 反应的对映选择性显著提高, 对映体过量值高达97% ee.
同样, 在铱的催化下, 使用底物2-取代苯并咪唑烯丙基碳酸甲酯衍生物191也能发生分子内烯丙基取代反应(Eq. 55).与Eq. 54反应不同的是此反应不需要加碱, 室温下便能获得较好的结果, 成环产物192的产率达81%, ee值高达96%.
2.5 其他的氮亲核试剂
胍类衍生物作为氮亲核试剂应用于铱催化烯丙基取代反应也有少量报道.胍基存在于许多天然产物和药物结构单元分子中, 并且具有一定的生物活性和药理活性.因此, 人们对该物质的研究比较感兴趣. 2009年, Miyabe等[72]报道了以带有两个Boc基团的胍类衍生物193作为亲核试剂, 在手性配体双噁唑啉基吡啶198 [PYBOX=双(1, 3-噁唑烷基-2-)吡啶]与[Ir(COD)Cl]2形成的手性催化剂作用下, 分别与γ-芳基烯丙基磷酸二乙酯194a和194b发生烯丙基氨基化反应, 生成单烯丙基取代产物195 (Scheme 11).然而在相同的催化体系下, 以带有三个Boc基团的胍类衍生物196作为亲核试剂, 与1-萘基取代烯丙基磷酸二乙酯194b发生反应, 则会生成双烯丙基取代产物197 (Scheme 11).
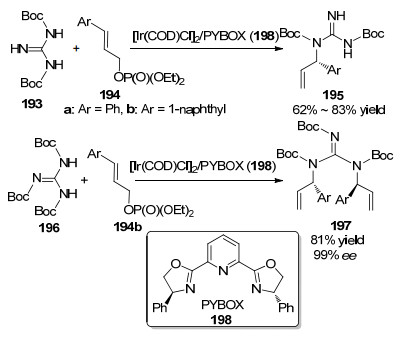 图式 11
铱催化胍类衍生物的烯丙基取代反应
Scheme11.
Iridium-catalyzed allylic substitution reactions of guanidine derivatives
图式 11
铱催化胍类衍生物的烯丙基取代反应
Scheme11.
Iridium-catalyzed allylic substitution reactions of guanidine derivatives
2011年, Helmchen等[73]尝试以[Ir(COD)Cl]2与亚磷酰胺配体10b或202a组成的配合物作为催化剂, 探讨了γ-取代烯丙基碳酸甲酯6与异羟肟酸衍生物199的烯丙基氨基化反应(Eq. 56).研究表明:反应需要加入碱TBD或者DBU.当使用10b作为配体时, 反应所需的时间短, 主要产物200的产率高达92%, ee值高达99%.同时获得产物200与产物201的产率比值高达99:1.
2013年, Lee等[74]报道了铱催化合成α, β-不饱和γ-氨基酯的反应.研究表明:胺类亲核试剂的选择对反应的影响很大.在[Ir(COD)Cl]2和外消旋Feringa’s亚磷酰胺配体rac-16组成的催化体系下, 以仲胺204作为亲核试剂, 发生取代反应生成取代产物205, 产率上升到99% (Eq. 57).而在相同的反应条件下, 将胺类亲核试剂换成烯丙基胺206, 则反应生成3, 4-二取代噁唑烷-2-酮衍生物207 (Eq. 58), 产率只上升到83%.值得注意的是, 催化剂需要在丙胺中活化20 min.
2014年, Helmchen等[75]报道了铱催化下, 以邻溴苯甲胺208作为亲核试剂与γ-取代烯丙基碳酸甲酯6发生烯丙基氨基化反应(Eq. 59).虽然使用10a和202b作为手性配体均可催化这一转化过程, 但是对于一些底物, 使用配体202b所得到的结果更佳, 主要的产物209的产率达86%, ee值高达98%.
2015年, Evans等[76]报道了[Ir(COD)Cl]2与亚磷酰胺配体94b组成的配合物作为催化剂, 催化氮叶立德S, S-二苯基硫亚胺212与γ-取代烯丙基碳酸酯213的反应(Eq. 60).他们发现:在该反应体系中加入Cs2CO3能够大大提高N-烯丙基硫亚胺214的产率和ee值.
3 含氧亲核试剂
2007年, Uozumi等[77]报道了以苯酚216作为亲核试剂的铱催化不对称烯丙基醚化反应.他们研究了不同的磷酸二亚磷酰胺配体对反应的影响, 发现使用配体219, 能够得到S构型的产物(S)-217, 具有很好的区域选择性, 但是产率仅为30%, ee值仅为16% (Scheme 12).使用配体220, 能够以中等的对映选择性和非对映选择性获得R构型的产物(R)-217 (Scheme 12).对映体过量值有显著提高, 上升到74% ee.
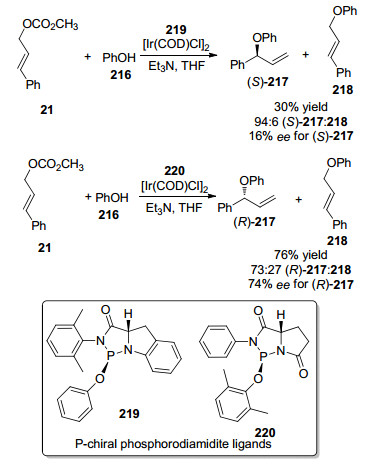 图式 12
铱催化烯丙基醚化反应
Scheme12.
Iridium-catalyzed allylic etherification
图式 12
铱催化烯丙基醚化反应
Scheme12.
Iridium-catalyzed allylic etherification
2012年, 游书力课题组[78]也研究了苯酚衍生物作为亲核试剂的铱催化醚化反应, 他们以2-乙烯基苯酚221作为亲核试剂与γ-取代烯丙基碳酸甲酯6发生铱催化烯丙基醚化反应(Eq. 61).不同的碱对反应的产率和区域选择性影响很大, 但对反应的对映选择性影响不大.当加入有机碱DBU时, 产率仅为10%, 反应没有区域选择性(53:47).加入碱Et3N时, 反应无法获得目标产物.而只有加入无机碱Cs2CO3或K3PO4时, 反应能高产率和高区域选择性地获得目标产物222.对映体过量值上升到95% ee.
2014年, 该课题组[79]报道了[Ir(DBCOT)Cl]2分别与亚磷酰胺配体16b和10a组成的环金属铱催化剂228a和229a, 催化碱金属羧酸盐225与γ-取代烯丙基磷酸酯224发生烯丙基酯化反应(Eq. 62).研究表明:催化剂228a催化效果最佳, 反应在室温下进行, 得到主要的烯丙基酯支链产物226, 产率达90%, ee值高达99%.羧酸盐中的阳离子对反应的选择性和速率有一定的影响, 其中Cs+和K+比Na+和Li+反应的速率更快.
2014年, Feringa等[80]首次实现了铱催化230酰胺羰基上的氧原子作为亲核试剂发生分子内烯丙基取代反应(Eq. 63).研究表明:加入Cs2CO3, K2CO3等无机碱会降低反应的速率, 而加入有机碱DABCO, 不仅能够加快反应的速率, 而且能够得到苯并噁嗪衍生物231, 产率高达93%, 对映体过量值高达97% ee.
2014年, 赵晓明课题组[81]报道了铱催化γ-取代烯丙基碳酸甲酯6与脂肪胺233发生烯丙基取代反应(Eq. 64), 却得到了烯丙基氨基甲酸酯产物234.研究表明:只有使用K3PO4作为碱, 二甲亚砜(DMSO)作为溶剂时, 反应才能够以高产率和对映体过量值获得主要产物234.
他们还提出了可能的反应机理(Scheme 13), 首先铱配合物与取代烯丙基碳酸甲酯6反应, 形成π-烯丙基铱中间体237, 并释放出甲氧基负离子和CO2, 生成的CO2迅速与脂肪胺233发生氨基化反应, 形成N-烷基氨基甲酸盐负离子238, 随后亲核进攻中间体237生成产物234.中间体237也可能与脂肪胺233结合生成副产物235, 并释放出铱配合物, 继续进行下一个催化循环.
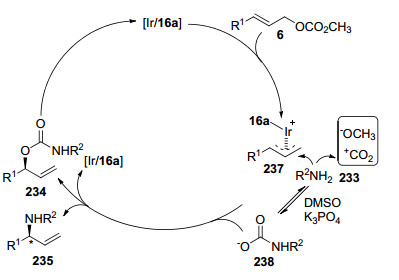 图式 13
铱催化6与脂肪胺233的烯丙基取代反应可能的反应机理
Scheme13.
Plausible mechanism of the iridium-catalyzed allylic substitution of 6 with aliphatic amine 233
图式 13
铱催化6与脂肪胺233的烯丙基取代反应可能的反应机理
Scheme13.
Plausible mechanism of the iridium-catalyzed allylic substitution of 6 with aliphatic amine 233
同年, 该课题组[82]使用与Eq. 78相同的催化剂, 实现了直接利用二氧化碳参与反应, 并形成氧亲核试剂, 进行铱催化的烯丙基取代反应, 最终也能获得烯丙基氨基甲酸酯产物234 (Eq. 65).该方法解决了邻位取代的苯基烯丙基底物通常导致ee值和收率降低的难题, 即所谓的邻位负效应.
除了使用苯酚、羧酸盐等有机含氧化合物作为氧亲核试剂, 无机含氧酸盐也能作为氧亲核试剂应用于铱催化烯丙基取代反应. 2011年, Helmchen等[83]以KHCO3作为亲核试剂, 实现了铱催化烯丙基羟基化反应(Eq. 66).研究表明:使用二甲基甲酰胺DMF/H2O (V:V=10:1)作溶剂时, 不需要加入18-冠醚-6作为添加剂, 并且能够提高反应的速率.由于以往所使用的[Ir(COD)Cl]2与亚磷酰胺配体10b所组成的催化剂244对水敏感, 于是他们使用[Ir(DBCOT)Cl]2与配体16a、10b制备新的铱催化剂228b和229b.结果表明:这种催化剂在水和空气中比较稳定, 并且能够催化反应获得主要的烯丙基醇产物242, 产率达95%, ee值达95%.
4 含硫亲核试剂
由于Na2SO3、Na2S、磺酸等含硫化合物廉价易得, 故将这类化合物应用于铱催化烯丙基取代反应引起了化学工作者们的兴趣.
2010年, 游书力课题组[84]报道了[Ir(COD)Cl]2与配体16a组成的配合物作为催化剂, 催化γ-取代烯丙基亚磺酸酯245发生分子内烯丙基取代和异构化串联反应(Scheme 14).研究表明, 生成的产物三取代乙烯基砜247是通过烯丙基砜246异构化得到的.反应必须加入1.0 equiv DBU, 随着DBU加入量减少反应的转化率显著降低, 当不加入DBU时无法得到产物247.他们对反应机理研究后发现, DBU催化异构化反应的发生.
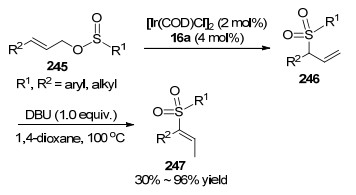 图式 14
铱催化烯丙基亚磺酸酯245的分子内烯丙基取代和异构化串联反应
Scheme14.
Tandem Ir-catalyzed intramolecular allylic substitution reaction of allyl sulfinate 245 and Isomerization
图式 14
铱催化烯丙基亚磺酸酯245的分子内烯丙基取代和异构化串联反应
Scheme14.
Tandem Ir-catalyzed intramolecular allylic substitution reaction of allyl sulfinate 245 and Isomerization
具有手性碳-硫键的有机含硫化合物在有机催化和药物化学中具有重要的作用.在过渡金属催化下, 以含硫化合物作为亲核试剂发生不对称烯丙基取代反应是合成手性含硫化合物的直接方法.
2010年, Hartwig等[85]报道了以烷基或芳基亚磺酸钠248作为亲核试剂的铱催化不对称烯丙基取代反应(Eq. 67).研究表明:他们使用环金属配合物127b作为催化剂, 催化γ-取代烯丙基碳酸甲酯6与烷基、芳基亚磺酸钠248发生反应, 均能获得令人满意的结果.得到主要的烯丙基砜产物249的产率高达99%, ee值高达98%.
类似地, 2014年, 赵晓明课题组[86]继续使用与Eq. 67相同的催化剂, 实现了γ-取代烯丙基碳酸甲酯6与Na2SO3 (251)的烯丙基取代反应(Eq. 68).研究表明:在25 ℃下, 用THF/H2O (V:V=4:1)作混合溶剂时, 取代反应结果最佳.他们还发现减少混合溶剂(THF/H2O)中水的含量会使产物252和253的总产率明显下降, 水的减少, 导致Na2SO3的溶解量也减少.值得注意的是:由于有机氟化合物在药物分子结构中的重要性, 在苯环上带有氟原子的252a和252b都有望用于合成单氟化头孢磺啶.
以硫酚作为含硫亲核试剂的铱催化烯丙基取代反应也有报道. 2010年, 赵晓明课题组[87]也使用127b作为催化剂, 研究了γ-取代烯丙基碳酸甲酯6与苯硫酚钠254发生烯丙基取代反应(Eq. 69).研究表明:添加物的种类对于反应产物的产率和ee值均有显著的影响.他们分别研究了不同的添加物Cs2CO3, CsF, CsCl, AgBr, LiCl和四丁基氟化铵(TBAF)对反应的影响, 研究发现:只有加入CsF能够使主要产物255的产率和ee值有明显的提高(产率由25%上升为82%, ee值由90%上升为97%).
2011年, 赵晓明课题组[88]继续报道了使用127b作为催化剂, 催化γ-取代烯丙基碳酸甲酯6与Na2S•9H2O (257)发生烯丙基取代反应(Eq. 70).研究表明:少量水与CsF结合有利于反应的进行.在最优反应条件下, 他们对底物6的结构进行了分析, 发现带有给电子基γ-芳基取代烯丙基碳酸甲酯6发生取代反应, 能够获得更好的结果, 主要的二烯丙基硫化物258的产率高达99%, 对映体过量值>99% ee.同年该课题组[89]还研究了以NaSSi(i-Pr)3作为亲核试剂的铱催化烯丙基取代反应.
2013年, 赵晓明课题组[90]报道了以硫代乙酸钾KSAc (260)作为亲核试剂, 铱催化剂127b催化的烯丙基取代反应(Eq. 71).研究表明: 6与KSAc (260)的比例不同将会影响反应产物的产率和区域选择性, 当6与KSAc的比例为1:1时, 产物261的产率仅为20%, 产物261与262的产率比值为55:45.而当6与KSAc的比例增加至4:1时, 产物261的产率和区域选择性都有显著的提高, 产物261产率为72%, 产物261与262的产率比值为89:11.
5 有机氟类亲核试剂
有机氟化合物表现出独特的物理、化学和生物活性等性质.因此在有机合成、药物化学和材料化学等领域具有广泛的应用.
2011年, Nguyen等[91]报道了[Ir(COD)Cl]2作为催化剂, 催化三氯乙亚氨酸酯衍生物263的烯丙基氟化反应.研究表明:当使用Et3N•3HF (264)作为亲核试剂时, 反应1~2 h, 生成一系列支链烯丙基氟化物265, 产率高达91% (Eq. 72).而使用[19F]KF•Kryptofix (266)作为亲核试剂时, 仅反应20 min就能生成烯丙基氟化物265a, 产率达82% (Eq. 73).
2015年, 该课题组[92]继续研究了三氯乙亚氨酸酯衍生物263与Et3N•3HF (264)的不对称烯丙基氟化反应(Eq. 81).他们使用[IrCl(267)]2作为催化剂, 实现了α-取代烯丙基氟化物265b的合成, 产率达90%, ee值达94%.
6 结果与展望
铱催化烯丙基酯的不对称烯丙基取代反应提供了一种很有效的合成手性烯丙基胺、烯丙基醚(醇)、烯丙基硫化物和含氟烯丙基化合物的方法, 因此该类反应的研究, 具有一定的科学意义和广阔的应用前景.由于底物烯丙基酯反应活性相对烯丙基卤温和, 没有催化剂则不能反应, 所以被广泛地应用在烯丙基类型的手性中心的构建.对于新的手性药物结构分子的设计与合成, 具有一定的科学意义和潜在的应用价值.目前, 铱催化烯丙基酯的不对称烯丙基取代反应的研究已经取得了很大的进展.近几年来对于这类反应所研究的配体主要是N—P配体, 而对其它类型的配体研究较少.手性配体与催化剂的设计与合成是不对称催化研究中永恒的主题, 尽管有成百上千的优秀手性配体已经合成出来, 但没有任何一种配体或催化剂是通用的.因此, 发展催化效率高、立体化学选择性好、底物范围广、结构简单、合成方便的铱催化剂仍然是将来需要研究的重点课题之一.
-
-
[1]
(a) Liu, Z.-Q.; Du, H.-F. Org. Lett. 2010, 12, 3054.
(b) Zhang, P.; Le, H.; Kyne, R. E.; Morken, J. P. J. Am. Chem. Soc. 2011, 133, 9716.
(c) Suetsugu, S.; Nishiguchi, H.; Tsukano, C.; Takemoto, Y. Org. Lett. 2014, 16, 996.
(d) Katcher, M. H.; Norrby, P. O.; Doyle, A. G. Organometallics 2014, 33, 2121. -
[2]
(a) Hughes, D. L.; Lloyd-Jones, G. C.; Krska, S. W.; Gouriou, L.; Bonnet, V. D.; Jack, K.; Sun, Y.-K.; David, J. M.; Reamer, R. A. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5379.
(b) Litto, R. D.; Benessere, V.; Ruffo, F.; Moberg, C. Eur. J. Org. Chem. 2009, 1352. -
[3]
Moberg, C. Top Organomet. Chem. 2012, 38, 209.
-
[4]
(a) Jegelka, M.; Plietker, B. Org. Lett. 2009, 11, 3462.
(b) Jegelka, M.; Plietker, B. Chem. Eur. J. 2011, 17, 10417. -
[5]
(a) Trost, B. M.; Rao, M.; Dieskau, A. P. J. Am. Chem. Soc. 2013, 135, 18697.
(b) Kawatsura, M.; Uchida, K.; Terasaki, S.; Tsuji, H.; Minakawa, M.; Itoh, T. Org. Lett. 2014, 16, 1470. -
[6]
Tan, Z.-Z.; Wan, X.-L.; Zang, Z.-H.; Qian, Q.; Deng, W.; Gong, H.-G. Chem. Commun. 2014, 50, 3827. doi: 10.1039/C3CC49859J
-
[7]
(a) Vrieze, D. C.; Hoge, G. S.; Hoerter, P. Z.; Van Haitsma, J. T.; Samas, B. M. Org. Lett. 2009, 11, 3140.
(b) Arnold, J. S.; Nguyen, H. M. J. Am. Chem. Soc. 2012, 134, 8380. -
[8]
(a) Yang, S.-C.; Feng, W.-H.; Gan, K.-H. Tetrahedron 2006, 62, 3752.
(b) Zhang, M.; Watanabe, K. J.; Tsukamoto, M.; Shibuya, R.; Morimoto, H.; Ohshima, T. Chem.-Eur. J. 2015, 21, 1. -
[9]
Guduguntla, S.; Hornillos, V.; Tessier, R.; Fañanás-Mastral, M.; Feringa, B. L. Org. Lett. 2016, 18, 252. doi: 10.1021/acs.orglett.5b03396
-
[10]
Tosatti, P.; Nelson, A.; Marsden, S. P. Org. Biomol. Chem. 2012, 10, 3147. doi: 10.1039/c2ob07086c
-
[11]
Zhuo, C.-X.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2014, 47, 2558. doi: 10.1021/ar500167f
-
[12]
Hethcox, J. C.; Shockley, S. E.; Stoltz, B. M. ACS Catal. 2016, 6, 6207. doi: 10.1021/acscatal.6b01886
-
[13]
Takeuchi, R.; Kashio, M. Angew. Chem., Int. Ed. 1997, 36, 263. doi: 10.1002/(ISSN)1521-3773
-
[14]
Janssen, J. P.; Helmchen, G. Tetrahedron Lett. 1997, 38, 8025. doi: 10.1016/S0040-4039(97)10220-9
-
[15]
Butt, N. A.; Zhang, W.-B. Chem. Soc. Rev. 2015, 44, 7929. doi: 10.1039/C5CS00144G
-
[16]
Helmchen, G.; Dahnz, A.; Dubon, P.; Schelwies, M.; Weihofen, R. Chem. Commun. 2007, 675. http://www.ncbi.nlm.nih.gov/pubmed/17392951
-
[17]
吴钰娟, 龙玉华, 杨定乔, 有机化学, 2010, 29, 1522. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu200910005&dbname=CJFD&dbcode=CJFQWu, Y.-J.; Long, Y.-H.; Yang, D.-Q. Chin. J. Org. Chem. 2009, 29, 1522(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu200910005&dbname=CJFD&dbcode=CJFQ
-
[18]
Giacomina, F.; Riat, D.; Alexakis, A. Org. Lett. 2010, 12, 1156. doi: 10.1021/ol100162y
-
[19]
Stanley, L. M.; Bai, C.; Ueda, M.; Hartwig, J. F. J. Am. Chem. Soc. 2010, 132, 8918. doi: 10.1021/ja103779e
-
[20]
Zhao, Z.-L.; Gu, Q.; Wu, X.-Y.; You, S.-L. Chin. Chem. Lett. 2016, 27, 619. doi: 10.1016/j.cclet.2016.02.017
-
[21]
Bondzic, B. P.; Farwick, A.; Liebich, J.; Eilbracht, P. Org. Biomol. Chem. 2008, 6, 3723. doi: 10.1039/b809143a
-
[22]
Liu, W.-B.; Zheng, S.-C.; He, H.; Zhao, X.-M.; Dai, L.-X.; You, S.-L. Chem. Commun. 2009, 6604. http://europepmc.org/abstract/MED/19865664
-
[23]
Zhang, H.-B.; Chen, J.-T.; Zhao, X.-M. Org. Biomol. Chem. 2016, 14, 7183. doi: 10.1039/C6OB01246A
-
[24]
Liu, W.-B.; Zheng, C.; Zhuo, C.-X.; Dai, L.-X.; You, S.-L. J. Am. Chem. Soc. 2012, 134, 4812. doi: 10.1021/ja210923k
-
[25]
Xu, Q.-L.; Dai, L.-X.; You, S.-L. Adv. Synth. Catal. 2012, 354, 2275. doi: 10.1002/adsc.v354.11/12
-
[26]
Zhan, M.; Li, R.-Z.; Mou, Z.-D.; Cao, C.-G.; Liu, J.; Chen, Y.-W.; Niu, D.-W. ACS Catal. 2016, 6, 3381. doi: 10.1021/acscatal.6b00719
-
[27]
(a) Trost, B. M.; Jiang, C. H. Synthesis 2006, 369.
(b) Behenna, D. C.; Stoltz, B. M. Top Organomet. Chem. 2013, 44, 281. -
[28]
Chen, W.-Y.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 2068. doi: 10.1021/ja311363a
-
[29]
Chen, W.-Y.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 377. doi: 10.1021/ja410650e
-
[30]
Liu, W.-B.; Reeves, C. M.; Virgil, S. C.; Stoltz, B. M. J. Am. Chem. Soc. 2013, 135, 10626. doi: 10.1021/ja4052075
-
[31]
Liu, W.-B.; Reeves, C. M.; Stoltz, B. M. J. Am. Chem. Soc. 2013, 135, 17298. doi: 10.1021/ja4097829
-
[32]
Liu, W.-B.; Okamoto, N.; Alexy, E. J.; Hong, A. Y.; Tran, K.; Stoltz, B. M. J. Am. Chem. Soc. 2016, 138, 5234. doi: 10.1021/jacs.6b02153
-
[33]
Liu, J.; Cao, C.-G.; Sun, H.-B.; Zhang, X.; Niu, D.-W. J. Am. Chem. Soc. 2016, 138, 13103. doi: 10.1021/jacs.6b05288
-
[34]
(a) Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020.
(b) Sandmeier, T.; Krautwald, S.; Zipfel, H. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 14363. -
[35]
(a) Yao, K.; Liu, D.-L.; Yuan, Q.-J.; Imamoto, T.; Liu, Y.-G.; Zhang, W.-B. Org. Lett. 2016, 18, 6296.
(b) Huo, X.-H.; Yang, G.-Q.; Liu, D.-L.; Liu, Y.-G.; Gridnev, I. D.; Zhang, W.-B. Angew. Chem., Int. Ed. 2014, 53, 6776. -
[36]
Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065. doi: 10.1126/science.1237068
-
[37]
Wei, X.; Liu, D.-L.; An, Q.-J.; Zhang, W.-B. Org. Lett. 2015, 17, 5768. doi: 10.1021/acs.orglett.5b02868
-
[38]
Liu, W.-B.; He, H.; Dai, L.-X.; You, S.-L. Org. Lett. 2008, 10, 1815. doi: 10.1021/ol800409d
-
[39]
Zhou, C.-X.; Wu, Q.-F.; Zhou, Q.; Xu, Q.-L.; You, S.-L. J. Am. Chem. Soc. 2013, 135, 8169. doi: 10.1021/ja403535a
-
[40]
Zhuo, C.-X.; Cheng, Q.; Liu, W.-B.; Zhao, Q.; You, S.-L. Angew. Chem., Int. Ed. 2015, 54, 8475. doi: 10.1002/anie.201502259
-
[41]
Wu, Q.-F.; Liu, W.-B.; Zhuo, C.-X.; Rong, Z.-Q.; Ye, K.-Y.; You, S.-L. Angew. Chem., Int. Ed. 2011, 50, 4455. doi: 10.1002/anie.201100206
-
[42]
Xu, Q.-L.; Dai, L.-X.; You, S.-L. Org. Lett. 2012, 14, 2579. doi: 10.1021/ol3008793
-
[43]
Cheng, Q.; Wang, Y.; You, S.-L. Angew. Chem., Int. Ed. 2016, 55, 3496. doi: 10.1002/anie.201511519
-
[44]
Chen, W.-Y.; Hartwig, J. F. J. Am. Chem. Soc. 2012, 134, 15249. doi: 10.1021/ja306850b
-
[45]
Chen, W.-Y.; Chen, M.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 15825. doi: 10.1021/ja506500u
-
[46]
Chen, M.; Hartwig, J. F. Angew. Chem., Int. Ed. 2014, 53, 8691. doi: 10.1002/anie.201403844
-
[47]
Chen, M.; Hartwig, J. F. J. Am. Chem. Soc. 2015, 137, 13972. doi: 10.1021/jacs.5b09980
-
[48]
Chen, M.:Hartwig, J. F. Angew. Chem., Int. Ed. 2014, 53, 12172. doi: 10.1002/anie.201406778
-
[49]
Chen, M.; Hartwig, J. F. Angew. Chem., Int. Ed. 2016, 55, 11651. doi: 10.1002/anie.201607053
-
[50]
Jiang, X.-Y.; Chen, W.-Y.; Hartwig, J. F. Angew. Chem., Int. Ed. 2016, 55, 5819. doi: 10.1002/anie.201600235
-
[51]
Alexakis, A.; Hajjaji, S. E.; Polet, D.; Rathgeb, X. Org. Lett. 2007, 9, 3393. doi: 10.1021/ol0713842
-
[52]
Polet, D.; Rathgeb, X.; Falciola, C. A.; Langlois, J. B.; Hajjaji, S. E.; Alexakis, A. Chem. Eur. J. 2009, 15, 1205. doi: 10.1002/chem.200801879
-
[53]
Hamilton, J. Y.; Sarlah, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 7644. doi: 10.1002/anie.201501851
-
[54]
Liu, X.-J.; You, S.-L. Angew. Chem., Int. Ed. 2017, 56, 4002. doi: 10.1002/anie.201700433
-
[55]
Breitler, S.; Carreira, E. M. J. Am. Chem. Soc. 2015, 137, 5296. doi: 10.1021/jacs.5b01951
-
[56]
Pouy, M. J.; Leitner, A.; Weix, D. J.; Ueno, S.; Hartwig, J. F. Org. Lett. 2007, 9, 3949. doi: 10.1021/ol701562p
-
[57]
Pouy, M. J.; Stanley, L. M.; Hartwig, J. F. J. Am. Chem. Soc. 2009, 131, 11312. doi: 10.1021/ja905059r
-
[58]
Weihofen, R.; Tverskoy, O.; Helmchen, G. Angew. Chem., Int. Ed. 2006, 45, 5546. doi: 10.1002/(ISSN)1521-3773
-
[59]
Spiess, S.; Berthold, C.; Weihofen, R.; Helmchen, G. Org. Biomol. Chem. 2007, 5, 2357. doi: 10.1039/B708571K
-
[60]
Weix, D. J.; Markovic, D.; Ueda, M.; Hartwig, J. F. Org. Lett. 2009, 11, 2944. doi: 10.1021/ol901151u
-
[61]
Markovic, D.; Hartwig, J. F. J. Am. Chem. Soc. 2007, 129, 11680. doi: 10.1021/ja074584h
-
[62]
Ye, K.-Y.; Dai, L.-X.; You, S.-L. Org. Biomol. Chem. 2012, 10, 5932. doi: 10.1039/c2ob00036a
-
[63]
Ye, K.-Y.; Dai, L.-X.; You, S.-L. Chem. Eur. J. 2014, 20, 3040. doi: 10.1002/chem.201400026
-
[64]
Stanley, L. M.; Hartwig, J. F. J. Am. Chem. Soc. 2009, 131, 8971. doi: 10.1021/ja902243s
-
[65]
Liu, W.-B.; Zhang, X.; Dai, L.-X.; You, S.-L. Angew. Chem., Int. Ed. 2012, 51, 5183. doi: 10.1002/anie.201200649
-
[66]
Yang, Z.-P.; Wu, Q.-F.; You, S.-L. Angew. Chem., Int. Ed. 2014, 53, 6986. doi: 10.1002/anie.201404286
-
[67]
Zhang, X.; Yang, Z.-P.; Huang, L.; You, S.-L. Angew. Chem., Int. Ed. 2015, 54, 1873 doi: 10.1002/anie.201409976
-
[68]
Yang, Z.-P.; Wu, Q.-F.; Shao, W.; You, S.-L. J. Am. Chem. Soc. 2015, 137, 15899. doi: 10.1021/jacs.5b10440
-
[69]
Zhuo, C.-X.; Zhang, X.; You, S.-L. ACS Catal. 2016, 6, 5307. doi: 10.1021/acscatal.6b01585
-
[70]
Ye, K.-Y.; Cheng, Q.; Zhuo, C.-X.; Dai, L.-X.; You, S.-L. Angew. Chem., Int. Ed. 2016, 55, 8113. doi: 10.1002/anie.201603266
-
[71]
Yang, Z.-P.; Zheng, C.; Huang, L.; Qian, C.; You, S.-L. Angew. Chem., Int. Ed. 2017, 56, 1530. doi: 10.1002/anie.201611056
-
[72]
Miyabe, H.; Yoshida, K.; Reddy, V. K.; Takemoto, Y. J. Org. Chem. 2009, 74, 305. doi: 10.1021/jo802271d
-
[73]
Gärtner, M.; Jäkel, M.; Achatz, M.; Sonnenschein, C.; Tverskoy, O.; Helmchen, G. Org. Lett. 2011, 13, 2810. doi: 10.1021/ol200688d
-
[74]
Lee, J.-H.; Lee, S.-G. Chem. Sci. 2013, 4, 2922. doi: 10.1039/c3sc50901j
-
[75]
Satyanarayana, G.; Helmchen, G. Eur. J. Org. Chem. 2014, 2242.
-
[76]
Grange, R. L.; Clizbe, E. A.; Counsell, E. J.; Evans, A. P. Chem. Sci. 2015, 6, 777. doi: 10.1039/C4SC01317D
-
[77]
Kimura, M.; Uozumi, Y. J. Org. Chem. 2007, 72, 707. doi: 10.1021/jo0615403
-
[78]
He, H.; Ye, K.-Y.; Wu, Q.-F.; Dai, L.-X.; You, S.-L. Adv. Synth. Catal. 2012, 354, 1084. doi: 10.1002/adsc.201100809
-
[79]
Qu, J.-P.; Roβberg, L.; Helmchen, G. J. Am. Chem. Soc. 2014, 136, 1272. doi: 10.1021/ja411869r
-
[80]
Zhao, D.-P.; Martin, F. M.; Chang, M.-C.; Otten, E.; Feringa, B. L. Chem. Sci. 2014, 5, 4216. doi: 10.1039/C4SC01940G
-
[81]
Zheng, S.-C.; Zhang, M.; Zhao. X.-M. Chem. Eur. J. 2014, 20, 1. doi: 10.1002/chem.201390210
-
[82]
Zhang, M.; Zheng, S.-C.; Zhao, X.-M. Chem. Commun. 2014, 50, 4455. doi: 10.1039/c4cc00413b
-
[83]
Gärtner, M.; Mader, S.; Seehafer, K.; Helmchen, G. J. Am. Chem. Soc. 2011, 133, 2072. doi: 10.1021/ja109953v
-
[84]
Xu, Q.-L.; Dai, L.-X.; You, S.-L. Org. Lett. 2010, 12, 800. doi: 10.1021/ol902873q
-
[85]
Ueda, M.; Hartwing, J. F. Org. Lett. 2010, 12, 92. doi: 10.1021/ol9023248
-
[86]
Liu, W.; Zhao, X.-M.; Zhang, H.-B.; Zhang, L.; Zhao, M.-Z. Chem. Eur. J. 2014, 20, 16873. doi: 10.1002/chem.201405058
-
[87]
Zheng, S.-C.; Gao, N.; Liu, W.; Liu, D.-G.; Zhao, X.-M.; Cohen, T. Org. Lett. 2010, 12, 4454. doi: 10.1021/ol101915b
-
[88]
Zheng, S.-C.; Huang, W.-Q.; Gao, N.; Cui, R.-M.; Zhang, M.; Zhao, X.-M. Chem. Commum. 2011, 47, 6969. doi: 10.1039/c1cc11930c
-
[89]
Huang, W.-Q.; Zheng, S.-C.; Tang, J.-L.; Zhao, X.-M. Org. Biomol. Chem. 2011, 9, 7897. doi: 10.1039/c1ob06332d
-
[90]
Gao, N.; Zhao, X.-M. Eur. J. Org. Chem. 2013, 2708.
-
[91]
Topczewski, J. J.; Tewson, T. J.; Nguyen, H. M. J. Am. Chem. Soc. 2011, 133, 19318. doi: 10.1021/ja2087213
-
[92]
Zhang, Q.; Stockdale, D. P.; Mixdorf, J. C.; Topczewski, J. J.; Nguyen, H. M. J. Am. Chem. Soc. 2015, 137, 11912. doi: 10.1021/jacs.5b07492
-
[1]
-

图式 3 铱催化α, β不饱和丙二酸酯49和酮酸酯52的不对称烯丙基烷基化反应
Scheme 3 Ir-catalyzed asymmetric allylic alkylation of α, β-unsaturated malonates 49 and ketoesters 52

图式 4 铱催化亚胺衍生物56的烯丙基取代反应
Scheme 4 Iridium-catalyzed allylic substitution reaction of imine derivatives 56

图式 5 3-吲哚烯丙基碳酸酯68分子内取代反应可能的反应机理
Scheme 5 Plausible mechanism of 3-indolyl allyl carbonates 68 intramolecular substitution reaction

图式 6 铱催化α-烷氧基酮108的烯丙基取代反应
Scheme 6 Iridium-catalyzed allylic substituted reaction of α-alkoxy ketones 108

图式 7 “一锅法”合成手性N-烯丙基酰胺衍生物129
Scheme 7 One-pot synthesis of chiral N-allylamide derivatives 129

图式 8 铱催化21与苯胺143的烯丙基氨基化反应可能的反应机理
Scheme 8 Plausible mechanism of the iridium-catalyzed allylic amination of 21 with aniline 143

图式 9 分子内取代反应可能的反应机理
Scheme 9 Plausible mechanism of intramolecular substitution reaction

图式 10 铱催化分子内烯丙基氨基化反应
Scheme 10 Iridium-catalyzed intramolecular allylic amination reaction

图式 11 铱催化胍类衍生物的烯丙基取代反应
Scheme 11 Iridium-catalyzed allylic substitution reactions of guanidine derivatives

图式 13 铱催化6与脂肪胺233的烯丙基取代反应可能的反应机理
Scheme 13 Plausible mechanism of the iridium-catalyzed allylic substitution of 6 with aliphatic amine 233
-


 下载:
下载:













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 36
- 文章访问数: 4930
- HTML全文浏览量: 528
 下载:
下载:
