 图式1
烯烃的双官能化反应
Scheme1.
Difunctionalization of alkenes
图式1
烯烃的双官能化反应
Scheme1.
Difunctionalization of alkenes
引用本文:
吴正兴, 张万斌. 金属催化的共轭二烯的1, 2-双官能化反应的研究进展[J]. 有机化学,
2017, 37(9): 2250-2262.
doi:
10.6023/cjoc201704031

Citation: Wu Zhengxing, Zhang Wanbin. Recent Advances in Metal-Catalyzed 1, 2-Difunctionalization of Conjugated Dienes[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2250-2262. doi: 10.6023/cjoc201704031
Citation: Wu Zhengxing, Zhang Wanbin. Recent Advances in Metal-Catalyzed 1, 2-Difunctionalization of Conjugated Dienes[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2250-2262. doi: 10.6023/cjoc201704031
金属催化的共轭二烯的1, 2-双官能化反应的研究进展
摘要:
共轭二烯的1,2-双官能化反应是重要的均相催化反应之一.反应所得的双官能化产物广泛存在于天然产物和生物活性的化合物中,也是很多重要的有机中间体的来源;而且双官能化产物中还保留有一个可以继续转化的双键,可以更灵活地构建我们所需的目标结构,或继续官能化,从而实现简单二烯的多官能化反应,得到邻近的多官能化产物.此领域中主要的难点在于反应中区域选择性、化学选择性和立体选择性等复杂选择性的控制.近年来,随着金属有机化学的不断发展,已陆续实现了金属钯、铜、铁或银催化的共轭二烯的1,2-双官能化反应,而且有些报道中通过引入手性配体成功地实现了共轭二烯的不对称1,2-双官能化反应.本综述将对近年来金属催化的共轭二烯的1,2-双官能化反应的研究进展进行重点介绍.
English
Recent Advances in Metal-Catalyzed 1, 2-Difunctionalization of Conjugated Dienes
Abstract:
The 1, 2-difunctionalization of conjugated dienes is an important homogeneous catalytic reaction. The obtained products through 1, 2-difunctionalization are widely existed in natural products and bioactive compounds, and are also sources of important organic intermediates, in addition, the preserved double bond in the difunctionalized product can be further transformed to give the desired structures or be functionalized sequentially to achieve multi-functionalization. The main difficulties focus on the encountered complex selectivities, including the regioselectivity, chemoselectivity and stereoselectivity in reactions. In recent years, with the development of organometallic chemistry, metal palladium, copper, iron or silver catalyzed 1, 2-difunctionalizations of conjugated dienes have been reported in succession. In some cases the enantioselective 1, 2-difunctionalizations of conjugated dienes were achieved via the introduction of chiral ligands. This review mainly focus on the recent metal catalyzed 1, 2-difunctionalizations of conjugated dienes.
-
Key words:
- metal-catalyzed
- / conjugated diene
- / 1, 2-difunctionalization
-
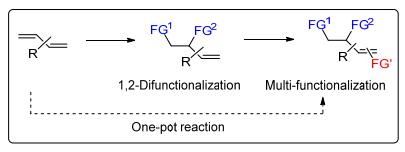
烯烃的1, 2-双官能化反应是有机化学中一类重要的反应.此类反应可以在一步反应中引入两个官能团, 是有机合成中一种高效的转化方法[1]; 反应所得的产物在很多天然产物和生物活性的化合物的合成中具有广泛的应用, 而且也是很多重要的有机中间体的来源, 比如邻二胺化合物、邻氨基醇、邻二醇、氮杂环丙烷化合物和环氧乙烷化合物等(Scheme 1)[2].
图式1
烯烃的双官能化反应
Scheme1.
Difunctionalization of alkenes
烯烃的1, 2-双官能化反应中, 孤立烯烃的双官能化研究较早.早在20世纪初, 通过加入过氧酸实现的烯烃的环氧化反应就有报道[3].在20世纪30年代, 有报道烯烃的双卤化反应可以通过与氟气、氯气和液溴反应实现[4].与此同时, 当量金属锇促进的双羟化反应也被报道[5]. 20世纪60年代, 在当量铜促进下, 通过烯烃与叠氮化物反应获得了烯烃的氮杂环丙烷产物[6]. 20世纪70年代, 当量的金属锇[7]或钯[8]促进的烯烃的1, 2-羟基胺基化和1, 2-双胺化反应被相继报道.早期的烯烃的1, 2-双官能化反应主要是当量的金属促进的反应, 而后随着金属有机化学的发展, 以Sharpless小组[9]开发的金属锇催化的1, 2-羟基胺基化及其不对称合成、不对称1, 2-双羟化及金属钛催化的不对称环氧化反应为代表, 一系列的金属催化的烯烃的1, 2-双官能化反应被陆续报道[1].
共轭二烯的双官能化反应相对于孤立烯烃报道较少, 共轭二烯中由于多出的共轭的双键, 其1, 2-双官能化反应的机理往往与单烯烃的不同, 所以难以直接用单烯烃反应中的条件实现共轭二烯的双官能化反应.但共轭二烯的1, 2-双官能化反应也有重要的意义, 因为其双官能化产物中还保留有一个双键可以继续转化, 可以更灵活地构建我们所需的目标结构, 或继续官能化从而实现简单二烯的多官能化反应, 得到邻近的多官能化产物(Scheme 2).这些在单烯烃1, 2-双官能化反应中较难实现.
 图式2
共轭二烯的1, 2-双官能化反应的优势
Scheme2.
Advantage of 1, 2-difunctionalization of conjugated dienes
图式2
共轭二烯的1, 2-双官能化反应的优势
Scheme2.
Advantage of 1, 2-difunctionalization of conjugated dienes
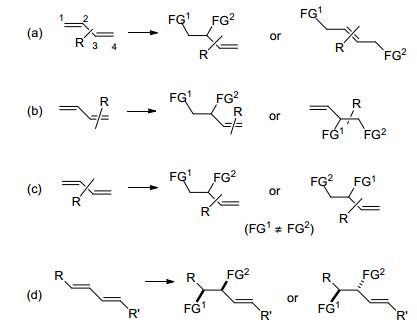
关于共轭二烯的双官能化反应的研究, 早期集中于1, 4-双官能化反应[10], B ckvall小组[10a~10f]在此领域做了系统的工作.共轭二烯的1, 2-双官能化反应早期研究较少, 最近十几年来有了较大的突破和进展[11~37].共轭二烯的1, 2-双官能化反应中主要的挑战在于复杂的选择性的控制, 具体如下(Scheme 3): (1) 在共轭二烯中由于共轭效应的影响, 会存在1, 2-双官能化和1, 4-双官能化反应的区域选择性问题; (2) 当共轭二烯为非对称性的二烯时, 会存在不同双键官能化的化学选择性问题; (3) 当两个亲核试剂不同时, 会存在官能化顺序的问题, 能否选择性地调控亲核试剂的进攻顺序也给我们提出了更高的挑战; (4) 当共轭二烯的两端都有取代基时, 还存在非对映选择性的控制问题.因此, 针对此领域中存在的问题, 本文将概述近年来金属催化的共轭二烯的1, 2-双官能化反应, 以及共轭二烯的不对称1, 2-双官能化反应的研究进展.
 图式3
共轭二烯的1, 2-双官能化反应中复杂的选择性
Scheme3.
Complex selectivities in 1, 2-difunctionalization of conjugated dienes
图式3
共轭二烯的1, 2-双官能化反应中复杂的选择性
Scheme3.
Complex selectivities in 1, 2-difunctionalization of conjugated dienes
1 钯催化的共轭二烯的1, 2-双官能化反应
钯催化共轭二烯的1, 2-双官能化反应有数例报道, 可以分为钯(0) 催化的反应和钯(Ⅱ)催化的反应[12~28].其中, 钯催化的共轭二烯的不对称1, 2-双官能化反应也有报道[21~28].
1.1 钯(0) 催化的共轭二烯的1, 2-双官能化反应
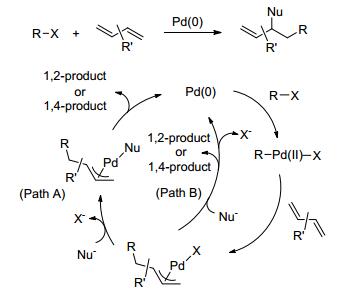
钯(0) 催化的共轭二烯的双官能化反应机理大致如下(Scheme 4):首先钯(0) 和预先活化的底物RX通过氧化加成得到钯(Ⅱ)物种, 然后此物种通过烯烃的亲核钯化过程得到π-烯丙基钯物种, 最后此π-烯丙基钯物种经过另一亲核试剂官能化得到钯(0) 和最终的双官能化产物(π-烯丙基钯物种经过亲核试剂顺式官能化(Path A)或反式官能化(Path B)).钯(0) 再进行下一轮的催化循环.
 图式4
钯(0) 催化的共轭二烯的双官能化反应机理
Scheme4.
Pd(0)-catalyzed catalytic cycle concerning difunctionalization of conjugated dienes
图式4
钯(0) 催化的共轭二烯的双官能化反应机理
Scheme4.
Pd(0)-catalyzed catalytic cycle concerning difunctionalization of conjugated dienes
早在1982年, Dieck小组[12]就以邻碘苯胺为底物, 在原位形成的Pd(0) 的催化下, 以中等的收率实现了共轭二烯的1, 2-双官能化, 构建了吲哚啉类化合物.随后Larock小组[13]使用一系列含官能团的芳基碘化物为碳源、氮源或氧源, 在Pd(0) 的催化下实现了一系列的共轭二烯1, 2-双官能化反应.
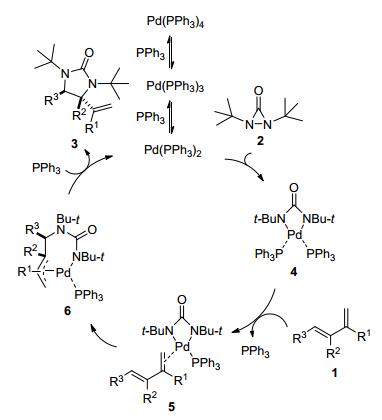
2007年史一安小组[14a]用二叔丁基取代的二氮环甲烷酮2作为氮源, Pd(PPh3)4作为催化剂, 首次实现了Pd(0) 催化的共轭二烯1的区域选择性、化学选择性和非对映选择性的1, 2-双胺化反应(Eq. 1).对各种类型的共轭二烯, 反应都能取得不错的产率和选择性.通过缓慢加入氮源, 可以将Pd(0) 催化剂的量从10%降到1~2 mol%.但是顺式的共轭二烯在目前的条件下还不能取得理想效果.
可能的双胺化反应的机理概括如下(Scheme 5)[14]:首先Pd(0) 与氮源2通过氧化加成得到四元环的Pd(Ⅱ)物种4; 然后Pd(Ⅱ)物种通过配体交换得到烯烃络合物5; 经过烯烃络合物5中双键对氮钯键的插入得到π-烯丙基钯络合物6; 最后π-烯丙基钯络合物经过还原消除得到产物3和Pd(0); Pd(0) 继续参与后续的催化循环.
 图式5
钯(0) 催化的共轭二烯的双胺化反应机理
Scheme5.
Mechanism of Pd(0)-catalyzed diamination of conjugated dienes
图式5
钯(0) 催化的共轭二烯的双胺化反应机理
Scheme5.
Mechanism of Pd(0)-catalyzed diamination of conjugated dienes
2011年Sigman小组[15]实现了共轭二烯分子间的1, 2-烯基芳基化反应(Eq. 2).此反应的关键在于稳定的π-烯丙基钯中间体11的形成(Scheme 6).该中间体是通过Heck反应过程实现双键对Pd(Ⅱ)物种9中碳钯键的插入, 此过程快于Suzuki偶联反应.得到的π-烯丙基钯中间体11通过转金属化和还原消除得到最终产物8.此反应条件简单, 试剂简单易得, 且不需要大过量的试剂.反应条件对官能团具有较好的兼容性, 而且通过此方法得到的产物应用其它的方法不易合成.
 图式6
钯(0) 催化的1, 2-烯基芳基化反应机理
Scheme6.
Mechanism of Pd(0)-Catalyzed 1, 2-alkenylation/ arylation
图式6
钯(0) 催化的1, 2-烯基芳基化反应机理
Scheme6.
Mechanism of Pd(0)-Catalyzed 1, 2-alkenylation/ arylation
1.2 钯(Ⅱ)催化的共轭二烯的1, 2-双官能化反应
钯(Ⅱ)催化的反应过程与钯(0) 的类似, 反应由钯(Ⅱ)启动, 经过一次催化循环得到的钯(0) 通过外加氧化剂再氧化为钯(Ⅱ)继续参与反应循环(Scheme 7).
 图式7
钯(Ⅱ)催化的共轭二烯的双官能化反应机理
Scheme7.
Pd(Ⅱ)-catalyzed catalytic cycle concerning difunctionalization of conjugated dienes
图式7
钯(Ⅱ)催化的共轭二烯的双官能化反应机理
Scheme7.
Pd(Ⅱ)-catalyzed catalytic cycle concerning difunctionalization of conjugated dienes
早在1984年, Larock小组[16]报道了一例使用含官能团的有机汞试剂为底物, 在当量的LiPdCl3的作用下实现了共轭二烯的1, 2-双官能化反应.
2005年Lloyd-Jones和Booker-Milburn小组[17]使用烷基保护的尿素13作为氮源, 用苯醌作为氧化剂, 在Pd(Ⅱ)的催化下, 实现了共轭烯烃的区域选择性的1, 2-双胺化反应, 普遍得到了不错的收率(Eq. 3).该小组同应条件对各种不同类型的二烯具有不错的兼容性, 且反应条件相对温和(60 ℃, DME, 5 mol% Pd), 不需要大过量的二烯.
2008年Lloyd-Jones和Booker-Milburn小组[18]还发展了一种新的Pd(Ⅱ)催化的共轭二烯的1, 2-芳基胺基化反应(Eq. 4).此方法可以从N-苯基保护的尿素类化合物15出发, 高产率地合成官能化的二氢吲哚结构16.反应的关键是使用Pd(OAc)2作为预催化剂, 通过反应中加入TsOH原位生成具有更高亲电活性的对甲苯磺酰氧基钯Pd(OTs)2, 在相对温和的条件下实现了C—H活化的过程, 然后烯烃插入到碳钯键形成π-烯丙基钯物种18, 最后氮进攻π-烯丙基钯物种18得到最终产物16, 同时产生Pd(0). Pd(0) 在苯醌氧化剂氧化下变为Pd(Ⅱ)进行后续的催化循环(Scheme 8).
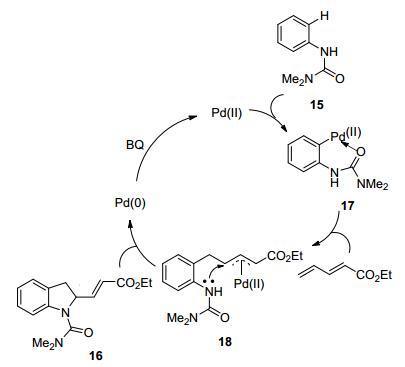 图式8
钯(Ⅱ)催化氧化1, 2-碳胺化反应机理图
Scheme8.
Mechanism of Pd(Ⅱ)-catalyzed oxidative 1, 2-carbo-amination
图式8
钯(Ⅱ)催化氧化1, 2-碳胺化反应机理图
Scheme8.
Mechanism of Pd(Ⅱ)-catalyzed oxidative 1, 2-carbo-amination
2013年杨丹小组[19]报道了一例Pd(Ⅱ)催化分子内的共轭二烯的1, 2-胺基烷基化反应.反应以较好的收率和出色的非对映选择性得到一系列双吡咯烷类化合物20 (Eq. 5).
2015年Booker-Milburn小组[20]还报道了一例Pd(Ⅱ)催化的分子内的共轭烯烃的双官能化构建稠杂环化合物的反应(Eq. 6).该反应选用含有吲哚呋喃结构的化合物21为底物, 通过碳氢活化/串联反应的过程完成.该小组目前正致力于提高形成七元环产物的产率以及将目前的方法应用到生物碱等化合物的合成中.
1.3 钯催化的共轭二烯的不对称1, 2-双官能化反应
20世纪90年代, Shibasaki小组[21]报道了一系列Pd(0) 催化共轭二烯的分子内不对称1, 2-烯基乙酰氧基化、烯基胺基化及烯基烷基化反应(Eq. 7).并且该小组[21c]将此方法应用于三并五元环倍半萜[(-)-∆9(12)-Capnellene]化合物的全合成中.
2007年, 史一安小组[22]报道了首例Pd(0) 催化的共轭烯烃的不对称双胺化反应(Eq. 8).通过一系列手性亚磷酰胺配体的筛选, 配体L1显示出最高的反应活性和最好的对映选择性.通过进一步二烯底物的拓展, 一系列的共轭二烯都可以选择性地得到双胺化在二烯的内双键上的产物3, 并获得不错的收率(60%~95%)和对映选择性(87%~95% ee).这是金属催化烯烃的不对称双胺化领域的重大突破.
磺酰胺类三元环底物25作为氮源也可以应用到共轭烯烃的不对称双胺化反应中(Eq. 9)[23], 史一安小组应用手性亚磷酰胺配体L2, 以66%~98%的收率和90%~93%的对映选择性得到一系列五元环的磺酰胺类产物26.
2014年Sigman小组[24]报道了一例Pd(0) 催化共轭二烯的分子间的不对称1, 2-双芳基化反应.反应实现了不错的区域选择性, 得到了最高为82%的对映选择性.
2015年龚流柱小组[25]以芳基碘化物27、丙二酸酯的钠盐28和共轭二烯1为原料, 用手性钯络合物以高区域选择性和高对映选择性实现了三组分反应, 即共轭二烯的不对称1, 2-双官能化反应(Eq. 10).亚磷酰胺配体L3显示出最佳的效果, 不仅展现出很高的催化反应活性, 而且实现了高区域选择性和高对映选择性的控制.反应通过钯催化串联的芳基化/不对称烯丙基烷基化过程实现, 对一系列二烯底物1和芳基碘化物底物27等具有很好的兼容性.
2016年黄汉民小组[26]开发了一种新型的钯催化的共轭烯烃的不对称1, 2-胺甲基胺化反应(Eq. 11).加入的Pd(Ⅱ)原位生成的Pd(0) 对底物缩酰胺30氧化加成, 通过碳氮键的活化变为Pd(Ⅱ)物种继续参与反应.经过配体筛选, 由联萘酚骨架衍生来的BINAPO配体L4显示出良好的催化效果, 高区域选择性和高对映选择性地得到一系列手性1, 3-二胺产物31.这是首例通过碳氮键的活化实现共轭二烯的不对称双官能化反应的报道.
2016年韩志勇小组[12, 13]在Dieck小组和Larock小组工作的基础上, 以亚磷酰胺L5为配体, 报道了Pd(0) 催化共轭二烯的不对称1, 2-芳基胺基化和1, 2-芳基氧基化反应(Eq. 12).反应得到了中等至较高的收率, 而且得到最高为87%的对映选择性[27].
2017韩志勇小组[28]在Lloyd-Jones和Booker-Mil-burn小组[18]工作的基础上, 使用手性的亚砜-噁唑啉配体L6, 报道了首例Pd(Ⅱ)催化氧化的共轭二烯的不对称1, 2-芳基胺基化反应(Eq. 13).反应普遍得到了不错的对映选择性, 但反应的活性依然是此类Pd(Ⅱ)催化氧化二烯的双官能化反应中需要进一步解决的问题.
2 铜催化的共轭二烯的1, 2-双官能化反应
近年来铜催化共轭二烯的1, 2-双官能化反应也有报道, 史一安小组[29~31, 33, 34]在此领域做了重要工作.此外, 该小组[33, 34]在铜催化的共轭二烯的不对称1, 2-双官能化反应领域也进行了研究.
2.1 铜催化的共轭二烯的1, 2-双官能化反应研究进展
2007年史一安小组[29]报道了首例铜催化的共轭二烯的双胺化反应(Eq. 14).在CuCl/P(OPh)3的催化作用下, 以二氮环甲烷酮2作为氮源, 高收率和高选择性的实现了双胺化反应, 并且对共轭三烯底物反应也能进行.同Pd(0) 催化的反应相比, 此反应主要双胺化在共轭二烯端位的双键上得到产物34.该小组推测此反应很可能通过自由基反应的机理路径, 详细机理见下文.
2010年史一安小组[30]报道当使用CuBr作为催化剂时, 双胺化反应也顺利进行.但与该小组前期工作不同, 该反应发生在共轭二烯的内双键上(Eq. 15).一系列共轭二烯底物都能以高收率(81%~99%)和高选择性能实现双胺化反应.
基于以上结果, 史一安小组[31]对不同的铜盐体系导致选择性反转的现象进行了具体研究.研究显示反应通过两个不同的相互竞争的机理路径(Scheme 9).首先Cu(Ⅰ)与含氮氮键的氮源2作用得到含氮自由基的Cu(Ⅱ)物种35和四元环Cu(Ⅲ)物种36.
 图式9
铜催化共轭烯烃的1, 2-双胺化反应机理
Scheme9.
Mechanism of Cu-catalyzed 1, 2-diamination of conjugated dienes
图式9
铜催化共轭烯烃的1, 2-双胺化反应机理
Scheme9.
Mechanism of Cu-catalyzed 1, 2-diamination of conjugated dienes
两物种之间很可能存在平衡, 氮自由基物种35在加入配体(如PCy3)的作用下占优(Scheme 9, left).氮自由基物种35与共轭二烯反应得到含烯丙基自由基的Cu(Ⅱ)物种37, 继而最终得到双胺化在二烯端位双键的产物34, 同时Cu(Ⅰ)催化剂获得再生.位阻效应和形成的烯丙基自由基物种相对稳定很可能是双胺化发生在二烯端位的原因.
双胺化在二烯的内双键上的机理很可能与Pd(0) 催化的双胺化反应机理类似, 通过四元环Cu(Ⅲ)物种36实现(Scheme 9, right).反应体系中不引入配体促进了四元环Cu(Ⅲ)物种36的形成, 此物种与二烯配位形成二烯的络合物39, 然后经过双键对氮铜键的插入得到π-烯丙基物种40, 最后经过还原消除得到双胺化产物3, 同时再生出Cu(Ⅰ)催化剂.
2008年Yoon小组[32]报道了一例铜(Ⅱ)催化共轭二烯的1, 2-氧基胺基化反应(Eq. 16).此反应以氧氮环丙烷类化合物41作为氮源和氧源, 成功地构建了五元杂环产物42, 得到了不错的收率以及很高的区域选择性和化学选择性.产物经过进一步衍生, 可以方便地构建1, 2-氨基醇类化合物.
2.2 铜催化的共轭二烯的不对称1, 2-双官能化反应
史一安小组[33, 34]对铜催化的共轭二烯的不对称1, 2-双胺化反应进行了研究探索. 2008年该小组[33]报道了在CuCl催化下, (R)-DTBM-SEGPHOS作为配体可以实现不对称1, 2-双官能化反应, 最高达74%ee (Eq. 17), 普遍得到中等的ee值.该反应经过自由基的机理过程, 对实现反应对映选择性的控制提出了很大的挑战.
2009年, 史一安小组[34]报道了手性磷酸铜盐物种43作为催化剂也可以实现不对称1, 2-双官能化反应, 得到中等的ee值(Eq. 18).更高效的不对称催化体系还需要进一步研究.
3 铁催化的共轭二烯的1, 2-双官能化反应
2010年Yoon小组[35]以氧氮环丙烷类化合物44作为氮源和氧源, 报道了一例铁(Ⅲ)催化的共轭二烯的胺基氧基化反应(Eq. 19).反应对于端位取代的共轭二烯底物普遍得到了不错的收率和非对映选择性.不同于此小组之前报道的Eq. 16中的铜(Ⅱ)催化的共轭二烯的反应, 铁催化的反应实现了氮源和氧源的化学选择性的双官能化得到杂环化合物45, 得到的产物对于铜(Ⅱ)催化的反应产物是有效的补充.
2012年Yoon小组[36]以氧氮环丙烷类化合物44作为氮源和氧源, 报道了一例铁(Ⅱ)催化的共轭二烯的不对称胺基氧基化反应(Eq. 20).反应经过配体的筛选, 双噁唑啉配体L8显示出最好的效果.对于端位取代的共轭二烯底物, 可以得到不错的收率和较高的对映选择性.
4 银催化的共轭二烯的1, 2-双官能化反应
2010年Pérez小组[37]报道了银催化的含端位羟基的共轭二烯的氮杂环丙烷化反应(Eq. 21).反应以PhINTs为氮烯的来源, 通过形成氮烯中间体的机理路径实现. [Tp*, BrAg]催化体系显示出很高的反应活性, 以及很高的化学选择性和立体选择性, 氮杂环丙烷化反应主要发生在临近羟基的双键上, 得到产物46.此反应体现出银催化的共轭二烯的氮杂环丙烷化反应在有机合成中是很有前景的.
5 结论与展望
近十几年来, 金属催化的共轭二烯的1, 2-双官能化反应取得了显著的进展, 各种新的双官能化反应被逐步开发, 并且实现了较高的反应活性或较好的选择性.共轭二烯的不对称1, 2-双官能化反应也有报道, 且取得了较好的对映选择性, 尤其是Pd(0) 作为催化剂在不对称催化反应方面显示出明显的优势.整体来说, 目前实现的1, 2-双官能化反应较好地解决了催化反应中复杂的选择性问题.虽然如此, 相比于其它发展较为成熟的烯烃的官能化反应, 此领域还存在很大的发展的空间.首先, 尽管目前实现了一些共轭二烯的1, 2-双官能化反应, 但目前反应的种类还比较有限, 从Scheme 1中可以看出, 很多新的双官能化反应有待开发.其次, 在不同价态金属催化的反应, 如Pd(0) 与Pd(Ⅱ)催化的1, 2-双官能化反应中, Pd(Ⅱ)催化的反应体系通过加入氧化剂, 避免了Pd(0) 催化的反应中预先官能化底物的使用, 显示出反应策略上的优势, 但运用Pd(Ⅱ)作为催化剂开发新反应的难度较大.能否用此策略开发更多的双官能化反应是很有意义的工作, 而且能否将目前的反应体系中使用的氧化剂替换为经济绿色的氧气也是很大的挑战.最后, 关于共轭二烯的不对称1, 2-双官能化反应, Pd(0) 催化的反应普适性较强.但目前Pd(0) 催化的反应中仅史一安小组报道的双胺化反应可以实现不同类型的共轭二烯的不对称双官能化, 其它的不对称催化的报道都集中于端位芳基取代的共轭二烯底物. Pd(Ⅱ)催化氧化的不对称反应也有一例报道, 但反应的活性依然是此类氧化双官能化反应, 中需要进一步解决的问题.所以, 能否通过新的不对称催化体系的开发, 实现更多新的不对称双官能化反应, 以及对不同结构的共轭二烯具有广泛兼容性的反应体系, 是很有价值的研究方向.
-
-
[1]
(a) Kolb, H. C. ; VanNieuwenhze, M. S. ; Sharpless, K. B. Chem. Rev. 1994, 94, 2483.
(b) Bäckvall, J. -E. Modern Oxidation Methods, Wiley-VCH, Weinheim, 2004.
(c) Kotov, V. ; Scarborough, C. C. ; Stahl, S. S. Inorg. Chem. 2007, 46, 1910.
(d) Minatti, A. ; Muñiz, K. Chem. Soc. Rev. 2007, 36, 1142.
(e) Chemler, S. R. ; Fuller, P. H. Chem. Soc. Rev. 2007, 36, 1153.
(f) Jensen, K. H. ; Sigman, M. S. Org. Biomol. Chem. 2008, 6, 4083.
(g) McDonald, R. I. ; Liu, G. ; Stahl, S. S. Chem. Rev. 2011, 111, 2981.
(h) Niu, F. ; Nie, C. ; Chen, Y. ; Sun, X. Prog. Chem. 2014, 26, 1942(in Chinese).
(牛凡凡, 聂昌军, 陈勇, 孙小玲, 化学进展, 2014, 26, 1942. )
(i) He, T. ; Zeng, X. Chin. J. Org. Chem. 2017, 37, 798(in Chinese).
(何天雄, 曾祥华, 有机化学, 2017, 37, 798. ) -
[2]
(a) Smith, J. G. Synthesis 1984, 629.
(b) Tanner, D. Angew. Chem. Int. Ed. 1994, 33, 599.
(c) Kolb, H. C. ; VanNieuwenhze, M. S. ; Sharpless, K. B. Chem. Rev. 1994, 94, 2483.
(d) Ager, D. J. ; Prakash, I. ; Schaad, D. R. Chem. Rev. 1996, 96, 835.
(e) Bennani, Y. L. ; Hanessian, S. Chem. Rev. 1997, 97, 3161.
(f) Lucet, D. ; Gall, T. L. ; Mioskowski, C. Angew. Chem. Int. Ed. 1998, 37, 2580.
(g) Gribble, G. W. Acc. Chem. Res. , 1998, 31, 141.
(h) Bergmeier, S. C. Tetrahedron 2000, 56, 2561.
(i) Lauret, C. Tetrahedron: Asymmetry 2001, 12, 2359.
(j) Schneider, C. Synthesis 2006, 3919.
(k) Singh, G. S. ; D'hooghe, M. ; Kimpe, N. D. Chem. Rev. 2007, 107, 2080.
(l) Gao, Z. ; Xiao, L. ; Chen, J. ; Xia, C. Chin. J. Catal. 2008, 29, 831(in Chinese).
(高志文, 肖林飞, 陈静, 夏春谷, 催化学报, 2008, 29, 831. )
(m) Bataille, C. J. R. ; Donohoe, T. J. Chem. Soc. Rev. 2011, 40, 114.
(n) Callebaut, G. ; Meiresonne, T. ; Kimpe, N. D. ; Mangelinckx, S. Chem. Rev. 2014, 114, 7954.
(o) Wang, Q. ; Chang, H. ; Wei, W. ; Liu, Q. ; Gao, W. ; Li, Y. ; Li, X. Chin. J. Org. Chem. 2016, 36, 939(in Chinese).
(王清宇, 常宏宏, 魏文珑, 刘强, 高文超, 李彦威, 李兴, 有机化学, 2016, 36, 939. ) -
[3]
Prileschajew, N. Eur. J. Inorg. Chem. 1909, 42, 4811.
-
[4]
Gilman, H. Organic Chemistry:An Advanced Treatise, Vol. 1, Wiley, New York, 1938, p. 36.
-
[5]
(a) Criegee, R. Justus Liebigs Ann. Chem. 1936, 522, 75.
(b) Criegee, R. Angew. Chem. 1937, 50, 153. -
[6]
Kwart, H.; Kahn, A. A. J. Am. Chem. Soc. 1967, 89, 1950. doi: 10.1021/ja00984a034
-
[7]
(a) Sharpless, K. B.; Patrick, D. W.; Truesdale, L. K.; Biller, S. A. J. Am. Chem. Soc. 1975, 97, 2305.
(b) Chong, A. O.; Oshima, K.; Sharpless, K. B. J. Am. Chem. Soc. 1977, 99, 3420. -
[8]
(a) Bäckvall, J.-E. Tetrahedron Lett. 1978, 19, 163.
(b) Bäckvall, J.-E.; Björkman, E. E. J. Org. Chem. 1980, 45, 2893. -
[9]
(a) Sharpless, K. B.; Chong, A. O.; Oshima, K. J. Org. Chem. 1976, 41, 177.
(b) Hentges, S. G.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 4263.
(c) Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974.
(d) Jacobsen, E. N.; Marko, I.; Mungall, W. S.; Schroeder, G.; Sharpless, K. B. J. Am. Chem. Soc. 1988, 110, 1968. -
[10]
(a) Aranyos, A.; Szabó, K. J.; Bäckvall, J.-E. J. Org. Chem. 1998, 63, 2523.
(b) Itami, K.; Palmgren, A.; Thorarensen, A.; Bäckvall, J.-E. J. Org. Chem. 1998, 63, 6466.
(c) Palmgren, A.; Larsson, A. L. E.; Bäckvall, J.-E. J. Org. Chem. 1999, 64, 836.
(d) Löfstedt, J.; Närhi, K.; Dorange, I.; Bäckvall, J.-E. J. Org. Chem. 2003, 68, 7243.
(e) Verboom, R. C.; Persson, B. A.; Bäckvall, J.-E. J. Org. Chem. 2004, 69, 3102.
(f) Piera, J.; Persson, A.; Caldentey, X.; Bäckvall, J.-E. J. Am. Chem. Soc. 2007, 129, 14120.
(g) Burks, H. E.; Kliman, L. T.; Morken, J. P. J. Am. Chem. Soc. 2009, 131, 9134.
(h) Schuster, C. H.; Li, B.; Morken, J. P. Angew. Chem. Int. Ed. 2011, 50, 7906. -
[11]
Xu, D.; Crispino, G. A.; Sharpless, K. B. J. Am. Chem. Soc. 1992, 114, 7571. doi: 10.1021/ja00045a044
-
[12]
O'Connor, J. M.; Stallman, B. J.; Clark, W. G.; Shu, A. Y. L.; Spada, R. E.; Stevenson, T. M.; Dieck, H. A. J. Org. Chem. 1983, 48, 807. doi: 10.1021/jo00154a010
-
[13]
(a) Larock, R. C.; Fried, C. A. J. Am. Chem. Soc. 1990, 112, 5882.
(b) Larock, R. C.; Berrios-Pena, N. G.; Narayanan, K. J. Org. Chem. 1990, 55, 3447. -
[14]
(a) Du, H.; Zhao, B.; Shi, Y. J. Am. Chem. Soc. 2007, 129, 762.
(b) Zhao, B.; Du, H.; Cui, S.; Shi, Y. J. Am. Chem. Soc. 2010, 132, 3523. -
[15]
Liao, L.; Jana, R.; Urkalan, K. B.; Sigman, M. S. J. Am. Chem. Soc. 2011, 133, 5784. doi: 10.1021/ja201358b
-
[16]
Larock, R. C.; Harrison, L. W.; Hsu, M. H. J. Org. Chem. 1984, 49, 3662. doi: 10.1021/jo00193a047
-
[17]
Bar, G. L. J.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. J. Am. Chem. Soc. 2005, 127, 7308. doi: 10.1021/ja051181d
-
[18]
Houlden, C. E.; Bailey, C. D.; Ford, J. G.; Gagné, M. R.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. J. Am. Chem. Soc. 2008, 130, 10066. doi: 10.1021/ja803397y
-
[19]
Xing, D.; Yang, D. Org. Lett. 2013, 15, 4370. doi: 10.1021/ol401901h
-
[20]
Cooper, S. P.; Booker-Milburn, K. I. Angew. Chem. Int. Ed. 2015, 54, 6496. doi: 10.1002/anie.201501037
-
[21]
(a) Kagechika, K.; Shibasaki, M. J. Org. Chem. 1991, 56, 4093.
(b) Kagechika, K.; Ohshima, T.; Shibasaki, M. Tetrahedron 1993, 49, 1773.
(c) Ohshima, T.; Kagechika, K.; Adachi, A.; Sodeoka, M.; Shibasaki, M. J. Am. Chem. Soc. 1996, 118, 7108. -
[22]
Du, H.; Yuan, W.; Zhao, B.; Shi, Y. J. Am. Chem. Soc. 2007, 129, 11688. doi: 10.1021/ja074698t
-
[23]
Cornwall, R. G.; Zhao, B.; Shi, Y. Org. Lett. 2013, 15, 796. doi: 10.1021/ol303469a
-
[24]
Stokes, B. J.; Liao, L.; de Andrade, A. M.; Wang, Q.; Sigman, M. S. Org. Lett. 2014, 16, 4666. doi: 10.1021/ol502279u
-
[25]
Wu, X.; Lin, H.-C.; Li, M.-L.; Li, L. L.; Han, Z.-Y.; Gong, L.-Z. J. Am. Chem. Soc. 2015, 137, 13476. doi: 10.1021/jacs.5b08734
-
[26]
Liu, Y.; Xie, Y.; Wang, H.; Huang, H. J. Am. Chem. Soc. 2016, 138, 4314. doi: 10.1021/jacs.6b00976
-
[27]
Chen, S.-S.; Meng, J.; Li, Y.-H.; Han, Z.-Y. J. Org. Chem., 2016, 81, 9402. doi: 10.1021/acs.joc.6b01611
-
[28]
Chen, S.-S.; Wu, M.-S.; Han, Z.-Y. Angew. Chem., Int. Ed. 2017, 56, 6641. doi: 10.1002/anie.201702745
-
[29]
Yuan, W.; Du, H.; Zhao, B.; Shi, Y. Org. Lett. 2007, 9, 2589. doi: 10.1021/ol071105a
-
[30]
Zhao, B.; Peng, X.; Cui, S.; Shi, Y. J. Am. Chem. Soc. 2010, 132, 11009. doi: 10.1021/ja103838d
-
[31]
Zhao, B.; Peng, X.; Zhu, Y.; Ramirez, T. A.; Cornwall, R. G.; Shi, Y. J. Am. Chem. Soc. 2011, 133, 20890. doi: 10.1021/ja207691a
-
[32]
Michaelis, D. J.; Ischay, M. A.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 6610. doi: 10.1021/ja800495r
-
[33]
Du, H.; Zhao, B.; Yuan, W.; Shi, Y. Org. Lett. 2008, 10, 4231. doi: 10.1021/ol801605w
-
[34]
Zhao, B.; Du, H.; Shi, Y. J. Org. Chem. 2009, 74, 8392. doi: 10.1021/jo901685c
-
[35]
Williamson, K. S.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 4570. doi: 10.1021/ja1013536
-
[36]
Williamson, K. S.; Yoon, T. P. J. Am. Chem. Soc. 2012, 134, 12370. doi: 10.1021/ja3046684
-
[37]
(a) Llaveria, J.; Beltrán, Á.; Díaz-Requejo, M. M.; Matheu, M. I.; Castillón, S.; Pérez, P. J. Angew. Chem. Int. Ed. 2010, 49, 7092.
(b) Llaveria, J.; Beltrán, Á.; Sameera, W. M. C.; Locati, A.; Díaz-Requejo, M. M.; Matheu, M. I.; Castillón, S.; Maseras, F.; Pérez, P. J. J. Am. Chem. Soc. 2014, 136, 5342.
-
[1]
-

图式2 共轭二烯的1, 2-双官能化反应的优势
Scheme 2 Advantage of 1, 2-difunctionalization of conjugated dienes

图式3 共轭二烯的1, 2-双官能化反应中复杂的选择性
Scheme 3 Complex selectivities in 1, 2-difunctionalization of conjugated dienes

图式4 钯(0) 催化的共轭二烯的双官能化反应机理
Scheme 4 Pd(0)-catalyzed catalytic cycle concerning difunctionalization of conjugated dienes

图式5 钯(0) 催化的共轭二烯的双胺化反应机理
Scheme 5 Mechanism of Pd(0)-catalyzed diamination of conjugated dienes

图式6 钯(0) 催化的1, 2-烯基芳基化反应机理
Scheme 6 Mechanism of Pd(0)-Catalyzed 1, 2-alkenylation/ arylation

图式7 钯(Ⅱ)催化的共轭二烯的双官能化反应机理
Scheme 7 Pd(Ⅱ)-catalyzed catalytic cycle concerning difunctionalization of conjugated dienes

图式8 钯(Ⅱ)催化氧化1, 2-碳胺化反应机理图
Scheme 8 Mechanism of Pd(Ⅱ)-catalyzed oxidative 1, 2-carbo-amination
-


 下载:
下载:








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 62
- 文章访问数: 3140
- HTML全文浏览量: 657
 下载:
下载:
