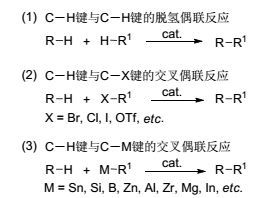
 图式1
C—H键参与的交叉偶联反应
Scheme1.
Cross-coupling reactions of C—H bonds
图式1
C—H键参与的交叉偶联反应
Scheme1.
Cross-coupling reactions of C—H bonds
引用本文:
李桦, 任相伟, 赵温涛, 唐向阳, 王光伟. 官能团导向的C-H键与有机金属试剂构建C-C键的偶联反应[J]. 有机化学,
2017, 37(9): 2287-2302.
doi:
10.6023/cjoc201703036

Citation: Li Hua, Ren Xiangwei, Zhao Wentao, Tang Xiangyang, Wang Guangwei. Cross-Coupling of Directed C-H and Organometallic Reagents for C-C Bond Formation[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2287-2302. doi: 10.6023/cjoc201703036
Citation: Li Hua, Ren Xiangwei, Zhao Wentao, Tang Xiangyang, Wang Guangwei. Cross-Coupling of Directed C-H and Organometallic Reagents for C-C Bond Formation[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2287-2302. doi: 10.6023/cjoc201703036
官能团导向的C-H键与有机金属试剂构建C-C键的偶联反应
English
Cross-Coupling of Directed C-H and Organometallic Reagents for C-C Bond Formation
Abstract:
Transition metal-catalyzed C-H activation is one of the most important areas in organic synthesis. Directed C-H activation can obviate the prefunctionalization of substrate, therefore providing a highly efficient and concise strategy for C-C formation. The cross-coupling of transition metal-activated C-H bond with organic electrophilic reagents has been proven effective for construction of various C-C bonds. Meanwhile, the oxidative coupling between the corresponding intermediates with organometallic reagents has become the focus for chemists due to their high reactivity, and notable achievements have been made in recent years. Here the oxidative couplings of C-H bond and organometallic reagents have been discussed and summarized according to the hybridization of substrate and organometallic reagents.
-
近年来C—H键活化领域发展迅速, 国内外科研工作者在该领域做了大量的工作, 不断有新的研究成果涌现. C—H键活化-偶联反应, 不但避免了传统化学中繁冗的合成步骤, 更减少了一些有毒试剂的应用.过渡金属催化的C—H键活化-偶联反应的发展, 对于医药、生物、材料等方面的发展也起到了重要的推动作用.有机化合物分子通常含有多个键能相近的C—H键, 因此C—H键的选择性断裂及官能团化就成为了具有挑战性的课题[1].在已有的报道中, 通常需要引入导向基团来实现定位作用, 常用的导向基有羧酸、酰胺、咔唑及氨基喹啉等, 这些基团能够很好地提高反应的选择性, 而且在一定条件下容易脱去, 实现了导向基团的可离去性.
C—H键直接参与C—C键形成的方法可分为三类:两分子化合物的C—H键直接脱氢偶联; C—H键与有机卤化物的交叉偶联; C—H键与有机金属试剂的氧化偶联(Scheme 1).在这三类偶联反应中, 两分子化合物直接脱氢偶联最为简便, 但由于产物选择性较差, 底物应用范围窄, 反应仍有待进一步发展.发展比较成熟的是C—H键与有机卤化物的交叉偶联, 反应体系通常需要加碱、配体且温度较为苛刻, 从一定程度上限制了此类反应的应用.和前两种偶联方法相比, 有机金属试剂种类多, 反应活性高, 过渡金属催化的C—H键与有机金属试剂的偶联反应可以在比较温和的条件下反应.一般而言, 此类反应的机理相似, 现以金属钯催化的反应加以说明[2]. Pd(Ⅱ)活性中间体首先在导向基团的作用下, 与烷基或芳基C—H键发生反应, 生成烷基钯(Ⅱ)或芳基钯(Ⅱ)物种, 然后与金属有机试剂发生金属交换, 再经过还原消除过程得到偶联产物, 生成的Pd(0) 被氧化剂氧化成Pd(Ⅱ), 完成催化循环(Scheme 2).
图式1
C—H键参与的交叉偶联反应
Scheme1.
Cross-coupling reactions of C—H bonds
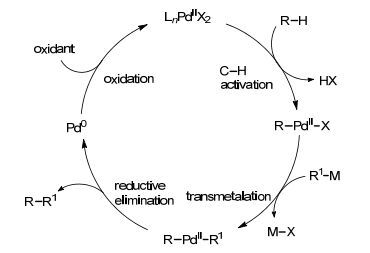 图式2
C—H键与有机金属试剂的偶联反应的机理
Scheme2.
Mechanism for the cross-couplings of C—H bonds with organometallic reagents
图式2
C—H键与有机金属试剂的偶联反应的机理
Scheme2.
Mechanism for the cross-couplings of C—H bonds with organometallic reagents
到目前为止, 在C—H键活化领域已经发表的的综述主要涉及到以下几个方面:过渡金属催化剂Pd[3]、Fe[4]、Co[5]、Ni[6]等催化下的C—H键活化偶联反应; 两分子C—H键的脱氢偶联[7]; C—H键活化用于构建C—O键, C—N键或C—S键等碳杂原子键[8]等.对于在导向基定位条件下的用于构建C—C键的C—H键与有机金属试剂的偶联反应, 尤其是近年来的最新反应进展尚没有系统的总结.本篇综述将以C—H及C—M中的碳原子的杂化类型作为分类标准, 对近十年来不同类型的碳氢键与有机金属试剂的偶联反应进行概括和总结.
对于不同类型的C—H键, 其活化的难易程度有很大差别.由于炔烃的碳碳叁键的碳原子的电负性较大, 使得末端炔烃的碳氢键即C(sp)—H键比较活泼, 容易发生反应, 比如末端炔烃和卤代物之间发生的Sonogashira偶联反应[9]能够在比较温和的条件下实现.又比如末端炔烃与烷基锌试剂在Pd(OAc)2的催化下也可以顺利发生氧化偶联反应形成C(sp)—C(sp3)键[10].鉴于此, 本文将不对C(sp)—H键参与的氧化偶联反应进行详细介绍.与C(sp)—H键相比, C(sp2)—H和C(sp3)—H键更广泛地存在于有机化合物分子中, 而它们的反应活性远低于C(sp)—H键, 因此开展这两类碳氢键的活化研究意义重大.下面我们将着重介绍官能团导向的C(sp2)—H和C(sp3)—H键与有机金属试剂的交叉偶联反应.
1 官能团导向的C(sp2)—H键与有机金属试剂的偶联反应
烯烃和芳香烃是两大类常见的有机化合物分子, C(sp2)—H键的反应活性低于末端炔烃中的C(sp)—H键.由于碳碳双键容易与过渡金属配位, 使得C(sp2)—H键的活化较为容易.近年来, 芳香族化合物的C(sp2)—H键活化取得了很好的进展, 相关的研究成果不断涌现.与此同时, 烯烃的C(sp2)—H键的活化也取得了一定的进展.为了提高反应的选择性, 常借助一些导向基团来完成, 常用的导向基团有吡啶、8-氨基喹啉、酰胺、酮羰基以及羧酸等.常用的有机金属试剂包括有机硼试剂、有机锡试剂、有机锌试剂、有机铝试剂及有机镁试剂(格氏试剂)等, 这些不同金属试剂的应用, 极大地丰富了C—H键活化偶联反应.下面将根据有机金属试剂的种类, 对官能团导向的C(sp2)—H键与有机金属试剂的氧化偶联反应展开描述.
1.1 C(sp2)—H键与C(sp3)—M的偶联反应
有机硼试剂较稳定、毒性低、易制备, 在C—H键活化偶联反应中应用广泛.常见的烷基硼试剂有烷基硼酸、烷基硼酸酯、烷基氟硼酸盐[11]以及N-甲基亚胺二乙酸(MIDA)硼酸酯[12]等, 其中烷基氟硼酸盐在与C—H键的偶联反应中应用比较广泛.
2007年, Yu课题组[13]利用甲基硼酸为金属试剂, 以羧酸为导向基, 实现了苯甲酸的邻位C(sp2)—H的甲基化反应(Eq. 1), 但反应的应用范围较小.有机三氟硼酸盐是结晶性硼酸类化合物, 在空气中和室温下比较稳定, 且取用方便.为了提高反应的应用范围, 2011年, 该课题组[14]对导向基团作了精心的筛选, 选用氟代芳基酰胺代替羧酸作为导向基团, 利用烷基氟硼酸盐作为硼试剂, 实现了苯甲酸或苯乙酸衍生物C(sp2)—H的烷基化反应(Eq. 2).该反应用Pd(Ⅱ)作催化剂, Li2CO3和Ag2CO3分别作为碱和氧化剂, 在BQ(苯醌)作添加剂的情况下可以顺利进行.但反应选择性较差, 通常得到邻位单取代和双取代混合产物, 反应有一定的局限性.
配体在钯催化的芳烃C(sp2)—H的烷基化反应中可以起到很重要的作用. 2013年, Yu课题组[15]报道了以氨基酸衍生物作为配体的芳烃C(sp2)—H的烷基化反应(Eq. 3).和上述反应相比, 该反应不使用氟代芳基酰胺作导向基团, 而是采用苯甲酸或苯乙酸底物中的羧基作导向基团, 在Pd(OAc)2的催化下, 用N-Boc保护的氨基酸做配体能够促使反应顺利进行, 反应被认为是按自由基机理进行.底物中的电子效应对反应影响不大, 反应只适用于伯烷基氟硼酸钾盐, 对于仲、叔烷基氟硼酸钾盐不适用.
2013年, Sanford课题组[16]报道了以吡啶为导向基的芳基底物与烷基氟硼酸钾的偶联反应, 反应用Pd(OAc)2和MnF3的组合来实现催化循环, 在20~40 ℃的条件下即可发生(Eq. 4).作者认为该反应中MnF3作为氧化剂, 将Pd(Ⅱ)中间体氧化为Pd(IV)中间体.反应被认为是按自由基机理进行, 但具体反应历程还有待一步研究.
由于含有吡啶酮结构的化合物具有一定的生物活性, 因此合成吡啶酮的衍生物有着潜在的应用价值. 2016年, Liu课题组[17]用吡啶作为导向基, 在Rh(Ⅲ)的催化作用下, 完成了吡啶酮N原子α位的烷基化反应(Eq. 5).反应以Ag2O作氧化剂, 其中氧化剂的量对反应十分重要, 产物收率随氧化剂量的降低而减少.反应条件温和, 导向基团容易离去.文章对机理部分也作了探究, 认为反应首先是Rh(Ⅲ)催化剂与底物中的导向基团的N原子螯合形成中间体, 然后和烷基硼试剂进行金属交换, 最后经过还原消除得到目标产物(Scheme 3).
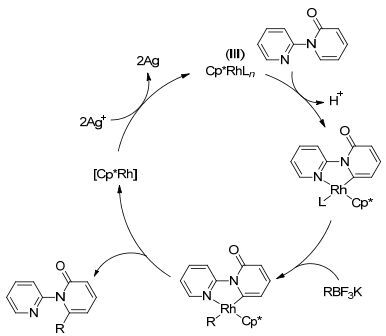 图式3
Rh催化的有机硼试剂和吡啶酮的氧化偶联反应的机理
Scheme3.
Mechanism of rhodium(Ⅲ)-catalyzed oxidative coupling of organoboron reagents with pyridines
图式3
Rh催化的有机硼试剂和吡啶酮的氧化偶联反应的机理
Scheme3.
Mechanism of rhodium(Ⅲ)-catalyzed oxidative coupling of organoboron reagents with pyridines
烷基格氏试剂是常用的活泼金属试剂, 它也可以参与到C(sp2)—H的烷基化反应中, 且该反应可在温和条件下进行. 2011年, Nakamura课题组[18]报道了在Co(acac)2的催化下, 苯甲酰胺衍生物与烷基格氏试剂能够在室温和空气的存在下, 实现芳烃C(sp2)—H的烷基化反应(Eq. 6).其中1, 3-二甲基-3, 4, 5, 6-四氢-2-嘧啶酮(DMPU)作为关键配体, 可以稳定烷基钴中间体, 使反应能够顺利进行.
烷基锌试剂也是一类常用的活泼金属试剂. 2015年, Nakamura课题组[19]又报道了利用烷基锌试剂, 以8-氨基喹啉为导向基团对烯烃或芳烃的C(sp2)—H进行烷基化反应(Eq. 7).反应用Fe(acac)3催化剂, dppen作配体.反应历程经历了Fe(Ⅲ)中间体, 该中间体在双膦配体和导向基团的配位作用下比较稳定, 可以有效抑制烷基金属物种的β-H消除和自身偶联反应. 2015年, Ackermann课题组[20]又实现了甲基锌试剂参与的以三氮唑为导向基的C(sp2)—H键甲基化反应(Eq. 8).在反应中, 甲基锌试剂是通过甲基格氏试剂和氯化锌原位生成的.在相同条件下, 当以氨基喹啉作为导向基团时, 反应收率大大降低, 因此在该催化循环中, 三氮唑作为导向基更具有优势.
烷基铝试剂是一类非常活泼的金属试剂, 在有机合成中有着广泛的应用.其中应用较多的烷基铝试剂是三甲基铝, 比如锆催化的炔烃和烯烃的甲基铝化反应, 可以分别合成含有甲基取代基的三取代烯烃[21]和含α-甲基的手性醇[22].含有甲基取代基的烯烃或芳香烃化合物往往具有独特的生物活性和药理价值, 通过C—H活化在烯烃或芳香烃的特定位置引入甲基对于药物化学具有重要意义.通过甲基铝试剂参与的C—H键活化偶联反应引入甲基的报道比较少. 2015年, Nakamura课题组[23]报道了AlMe3参与的以8-氨基喹啉作导向基团的酰胺类底物的C—H键活化偶联反应(Eq. 9).该反应用Fe(acac)3作催化剂, 顺-1, 2-双(二苯基膦)乙烯(dppen)作配体, 以2, 3-二氯丁烷作氧化剂, 该氧化剂的使用大大减少了催化剂和配体的用量.反应用活泼的金属铝试剂替代了以往的格氏试剂作甲基的供体, 无需添加锌试剂, 就可以实现芳基的直接甲基化反应.反应收率高, 催化剂用量低, 应用性较强.
有机锡试剂毒性较大, 在有机合成中、尤其是在药物分子的合成中的应用受到了一定限制.对于烷基锡试剂参与的C(sp2)—H烷基化反应, 文献报道比较少. Yu课题组[24]在2006年报道了钯催化的C(sp2)—H键与烷基锡试剂的氧化偶联反应(Scheme 4).该方法以Pd(OAc)2作催化剂, 利用噁唑啉作导向基团, 分批加入烷基锡试剂, 以较高的收率和选择性实现C(sp2)—H键的烷基化.
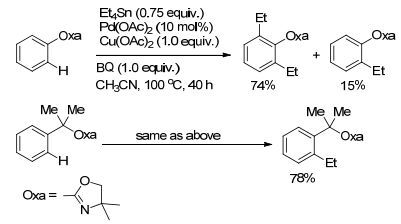 图式4
烷基锡试剂与芳烃C—H键的偶联反应
Scheme4.
C(sp2)—H coupling with alkyltin reagents
图式4
烷基锡试剂与芳烃C—H键的偶联反应
Scheme4.
C(sp2)—H coupling with alkyltin reagents
1.2 C(sp2)—H键与C(sp2)—M的偶联反应
C(sp2)—H键与芳基或烯基金属试剂的偶联反应, 为构建C(sp2)—C(sp2)键提供了一种高效便捷的方法.在这类反应中, 最常用的金属试剂是芳基或烯基硼试剂.虽然芳基或烯基锌试剂、硅试剂以及格氏试剂也可以参与到C(sp2)—H的活化偶联反应中, 但相关的报道比较少.下面将主要根据导向基团的不同对C(sp2)—H键与芳基或烯基硼试剂之间的氧化偶联反应进行描述, 同时也涵盖少量其它芳基金属试剂参与的偶联反应.
含氮官能团中由于氮原子具有良好的配位性能, 经常被用作碳氢键活化中的导向基团. 2007年, Shi课题组[25]利用N, N-二甲基胺基作为有效的导向基团, 成功实现了N, N-二甲基苄胺的邻位烯基化反应, 且二甲基胺基可以在温和的条件下通过一步或两步反应转换成其它基团. 2013年, Zhang课题组[26]同样以N, N-二甲基胺基为导向基, 在Pd作催化剂的条件下, 实现了苄胺邻位的C(sp2)—H与芳基碘代物的交叉偶联反应.基于以上工作, Dixon课题组[27]对导向基团进行修饰, 使用含氮杂环作为导向基团, 实现了苄胺衍生物与芳基硼酸酯的偶联反应(Eq. 10).反应以Pd(OAc)2作催化剂, Ag2CO3作氧化剂, 体系中添加少量的BQ, 有利于还原消除过程的顺利进行.底物范围广, 适用于吡咯、哌啶、吗啉及哌嗪等一系列含氮杂环的C(sp2)—H的芳基化反应.
利用氨基作导向基团, 通过向反应体系中加入手性配体还可以实现手性胺化合物的制备. 2015年, Yu课题组[28]探究了N-对硝基苯磺酰胺基团在芳烃C(sp2)—H活化中的导向作用(Scheme 5), 证明了这一导向基团的可行性.接着, 以二芳基甲胺类化合物为研究对象, 通过对手性氨基酸配体的筛选, 最终以较高的收率和ee值获得了相应的手性芳基化产物. 2016年, 该课题组[29]进一步利用N-对硝基苯磺酰胺基团作为导向基团, 完成了苄胺底物邻位C(sp2)—H键与芳基硼试剂的偶联反应, 并实现了手性拆分(Eq. 11).反应以Pd(OAc)2作催化剂, 引入的手性氨基酸配体对反应起重要作用.在外消旋的底物与芳基硼试剂的偶联过程中, 只有一种构型的底物能与芳基硼酸酯进行偶联反应得到构型单一的产物, 而另一种相反构型的底物则不反应, 从而实现了动力学拆分.该反应是外消旋化合物通过氨基酰化反应来实现手性拆分方法的重要补充[30].反应收率适中, ee值高, 所得的芳基化产物可以通过进一步的转化形成菲啶类化合物.
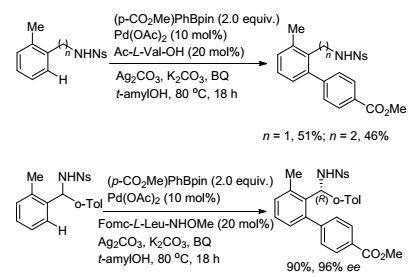 图式5
钯催化二芳基甲胺化合物C(sp2)—H键与有机硼试剂对映选择性氧化偶联
Scheme5.
Pd-catalyzed enantioselective C(sp2)—H oxidative coupling of diarylmethylamines with organoborons
图式5
钯催化二芳基甲胺化合物C(sp2)—H键与有机硼试剂对映选择性氧化偶联
Scheme5.
Pd-catalyzed enantioselective C(sp2)—H oxidative coupling of diarylmethylamines with organoborons
吡啶作为导向基团也经常在C—H键活化反应中用作导向基团. 2008年Studer课题组[31]报道了以2-吡啶或亚胺为导向基的芳烃C(sp2)—H键与芳基硼酸的偶联反应(Eq. 12).反应以2-吡啶杂环为底物, 用[RhCl-(C2H4)2]2作催化剂, 使用便宜易得的四甲基哌啶氧化物(TEMPO)为氧化剂, 无需加碱, 为合成联芳烃提供了一种简单有效的方法. 2009年, Wu课题组[32]同样以2-苯基吡啶为底物, 以Pd(OAc)2为催化剂, 实现了芳基氟硼酸盐与2-苯基吡啶的交叉偶联反应.在芳基硼酸及芳基硼酸酯作为金属试剂参的C—H键活化偶联反应中, 以Pd作催化剂时, 有机硼试剂容易发生自身偶联反应.当用氟硼酸盐时, 在同样反应条件下, 比芳基硼酸参与此反应时的收率大大提高(Eq. 13).
2010年, Nakamura课题组[33]报道了以Fe(acac)3作催化剂, 以吡啶位为导向基的条件下, 实现了硅乙烯基C(sp2)—H与芳基锌试剂或芳基格氏试剂的偶联反应.该工作以1-溴-2-氯乙烷作为氧化剂, 不需添加额外的配体, 反应条件温和, 主要得到反式烯烃.主要反应历程是催化剂在导向基中的氮原子的螯合作用下与烯烃生成五元环状金属中间体, 再通过β-H消除得到比较稳定的反式烯烃(Scheme 6).
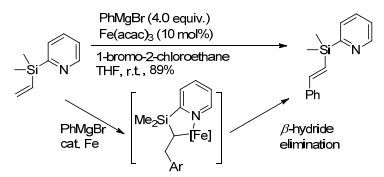 图式6
铁催化烯烃C—H键的选择性官能团化
Scheme6.
Fe-catalyzed selective functionalization of olefinic C—H bonds
图式6
铁催化烯烃C—H键的选择性官能团化
Scheme6.
Fe-catalyzed selective functionalization of olefinic C—H bonds
2011年, Nakamura课题组[34]又报道了以Fe(acac)3作催化剂, 芳基吡啶或芳基亚胺衍生物和芳基格氏试剂之间的氧化偶联反应(Scheme 7a), 反应的关键点在于缓慢滴加格氏试剂, 其次用氯苯作助溶剂, 氯苯可能起着配体的作用, 它可以稳定低价铁的络合物.同年, 该课题组[35]又报道了以吡啶或亚胺为导向基团的烯烃与格氏试剂反应(Scheme 7b), 通过溶剂的选择, 实现了反应产物的顺反构型的调控.
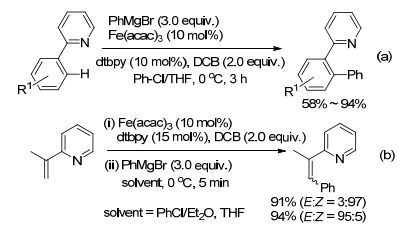 图式7
铁催化的C(sp2)—H的芳基化反应
Scheme7.
Iron-catalyzed arylation of C(sp2)—H
图式7
铁催化的C(sp2)—H的芳基化反应
Scheme7.
Iron-catalyzed arylation of C(sp2)—H
在上述反应中, 吡啶中的氮原子作为螯合原子能够和金属催化剂形成五元环金属化中间体, 使反应顺利进行.但吡啶导向基难离去, 反应底物受限[36]. 2005年, Daugulis课题组[37]引入了双齿导向基团8-氨基喹啉实现C(sp3)—H键的芳基化反应.该导向基团以氮原子作为螯合原子, 容易制备且易脱去, 应用性更强. 2014年, Nakamura课题组[38]报道了芳基硼试剂与以8-氨基喹啉为导向基团的底物的烯基或芳基C(sp2)—H的芳基化反应(Eq. 14).在已经报道的通过C(sp2)—H键活化偶联生成双烯的反应中, 大部分用贵金属催化, 如Pd、Rh、Ru等, 且生成的双烯很容易异构化, 原因是这些金属的4d或5d轨道很容易与产物烯烃相互作用, 这就限制了反应的应用性.由于Fe(Ⅲ)的d轨道与烯烃的π键相互作用弱, 从一定程度上减少了异构化的发生.反应用Fe(acac)3作催化剂, dppen作配体, 体系中加入与硼试剂等当量的正丁基锂试剂, 将硼试剂变成硼酸盐, 使碳硼键更容易断裂, 从而加快反应的进行.反应体系中加入的少量锌试剂, 可以促进硼到铁的金属交换.
2016年, Tan课题组[39]在Cu的催化下实现了芳基酰胺底物邻位的C(sp2)—H键芳基化反应(Eq. 15).反应用8-氨基喹啉为导向基团, 不需要添加额外的氧化剂, 在空气中即可进行反应.该反应能够高选择性地实现邻位单芳基化反应, 对官能团兼容性好.当把8-氨基喹啉换成其它导向基时, 反应不发生.反应同样是经过了五元环金属化过程, 在反应过程中形成了Cu(Ⅲ)中间体, 再经过金属交换和还原消除等过程得到目标产物(Scheme 8).
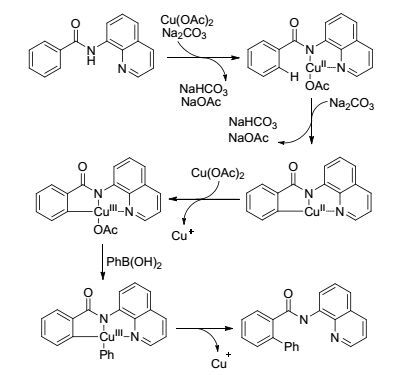 图式8
铜催化的有机硼试剂对C(sp2)—H的官能团化反应的机理
Scheme8.
Mechanism of Cu-catalyzed C(sp2)—H bond functionalization with organoboron compounds
图式8
铜催化的有机硼试剂对C(sp2)—H的官能团化反应的机理
Scheme8.
Mechanism of Cu-catalyzed C(sp2)—H bond functionalization with organoboron compounds
随后, Tan课题组[40]又选用Co(acac)2作催化剂, 用含有8-氨基喹啉的酰胺为底物, 同样也能实现酰胺邻位C(sp2)—H键芳基化反应, 反应收率较高, 对官能团兼容性好(Scheme 9).反应最大的亮点是底物范围不仅局限于芳基酰胺, 丙烯酰胺同样能够反应, 反应收率较高, 芳基化产物中的双键的构型能够很好的保持.
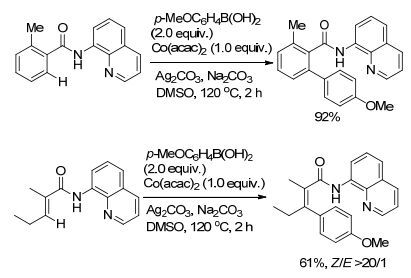 图式9
钴催化的有机硼试剂对C(sp2)—H的官能团化反应
Scheme9.
Cobalt-catalyzed C(sp2)—H bond functionalization with organoboron compounds
图式9
钴催化的有机硼试剂对C(sp2)—H的官能团化反应
Scheme9.
Cobalt-catalyzed C(sp2)—H bond functionalization with organoboron compounds
芳基酰胺衍生物同样可以作为导向基团应用到C(sp2)—H键活化反应中[41, 42]. 2011年, Yu课题组[14]选用氟代芳基酰胺作为导向基团, 实现了苯甲酸或苯乙酸衍生物的C(sp2)—H与芳基和烯基硼试剂的偶联(Scheme 10).该课题组通过对导向基团的筛选, 发现利用氟代酰胺导向基, 可以实现C(sp2)—H或C(sp3)—H键的芳基、烯基、羰基、氨基及氟化等一系列反应[43]. C(sp2)—H与芳基和烯基硼试剂的偶联反应利用Pd(Ⅱ)作催化剂, Ag2CO3作为氧化剂, 体系中添加少量的水可以促进转金属化过程的进行, 使转化率提高15%~20%.少量的DMSO可以稳定Pd(0) 物种, 使反应收率有一定提高. 2016年, Yu课题组[44]对导向基进行了一定修饰, 以全氟芳基酰胺作为导向基, 实现了与芳基硼酸酯的氧化偶联反应(Eq. 16).反应以Rh(Ⅲ)作催化剂, 以Binap作为配体, 该配体对反应的进行起了关键性作用, 底物中的导向基团容易离去, 反应选择性高, 对底物中的官能团容忍性好, 应用性较强.
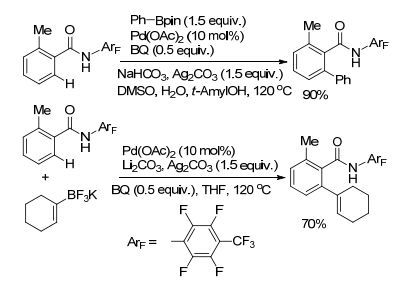 图式10
钯催化的芳基和烯基硼试剂与C(sp2)—H键的交叉偶联反应
Scheme10.
Pd(Ⅱ)-catalyzed cross-coupling of C(sp2)—H bonds and aryl-and vinyl-boron reagents
图式10
钯催化的芳基和烯基硼试剂与C(sp2)—H键的交叉偶联反应
Scheme10.
Pd(Ⅱ)-catalyzed cross-coupling of C(sp2)—H bonds and aryl-and vinyl-boron reagents
利用特定的酰胺类导向基团, 可以实现芳基间位C(sp2)—H的芳基化反应. Yu课题组[45]曾经报道过利用含有氰基的U型远程导向基团, 完成了苯丙酸衍生物与烯烃的脱氢偶联反应.借助此导向基团, 2013年, 该课题组[46]报道了首例利用含有氰基的T型远程导向基团, 在Pd(Ⅱ)催化作用下, 实现了苯丙酸衍生物间位C(sp2)—H键与芳基硼酸酯的偶联反应(Eq. 17).反应突破了以往反应历程中的五元或六元环金属中间体, 通过与远程导向基的螯合作用, 形成了十二元环的环钯中间体.反应中使用的N-保护的氨基酸配体是反应进行的关键, 添加的CsF也起了重要作用, 它促进金属交换过程的进行.
利用简单的酰胺作为导向基团, 也可以实现C(sp2)—H与芳基硼试剂的偶联反应. 2012年, Jeganmohan课题组[47]报道了金属Ru催化的以酰胺为导向基的芳基C(sp2)—H的芳基化反应(Eq. 18).该反应底物范围广, 对芳基底物上取代基的电子效应不敏感, 且有着很高的区域选择性.该反应还可以实现一锅法完成两步C—H键活化反应, 为菲啶酮的合成提供了一种简便方法.当把反应底物由苯甲酰胺变成N-乙酰苯胺时, 反应也能够很好地进行[48](Scheme 11).在这种情况下, 导向基团中的螯合原子由氮原子变成氧原子.在该反应中, 反应底物适用范围广, 芳基上取代基的电子效应对反应影响不大, 其中加入Ag2O作氧化剂起关键性作用, 得到的产物可进一步反应得到菲啶或咔唑.
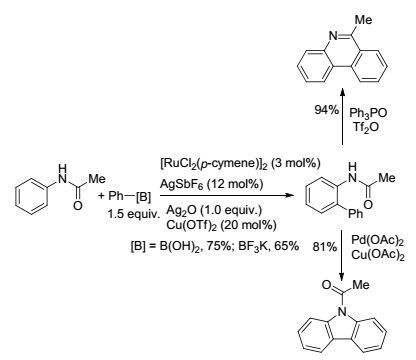 图式11
Ru催化的芳基硼酸对乙酰苯胺的邻位芳香化反应
Scheme11.
Ruthenium-catalyzed ortho-arylation of acetanilides with aromatic boronic acids
图式11
Ru催化的芳基硼酸对乙酰苯胺的邻位芳香化反应
Scheme11.
Ruthenium-catalyzed ortho-arylation of acetanilides with aromatic boronic acids
类似底物也可以在钯催化下进行, Lipshutz课题组[49]用[Pd(MeCN)4](BF4)2作催化剂, BQ作添加剂, 实现了以酰胺为底物的芳基化合物C(sp2)—H与芳基硼酸的偶联反应(Eq. 19).导向基团中的螯合原子同样是酰胺中的氧原子, 与催化剂形成五元环金属化中间体协助反应顺利进行, 当把催化剂换成其它中性催化剂如Pd(OAc)2、PdCl2时, 反应无法顺利进行.这表明在该反应中, 阳离子钯物种的路易斯酸性对反应非常重要, 相比之下, Pd(OAc)2、PdCl2中的阴离子都有配位作用, 钯的路易斯酸性会减弱. BQ的加入, 使反应收率大幅度提高.溶剂的选择对反应也十分重要, 乙酸乙酯最适用于本反应.
酰胺衍生物作为导向基团时, 一些廉价易得的Cu、Ni催化剂也可以用来活化芳基C(sp2)—H键. 2014年Yu课题组[50]实现了铜催化活化芳基C(sp2)—H键与芳基硼酸酯的氧化偶联(Eq. 20).反应以酰胺类化合物为底物, 用酰胺和噁唑啉作为导向基团, 该反应可能历经了Cu(Ⅲ)中间体, 并以此推出了催化循环机理.在利用芳基硅试剂实现的芳烃C(sp2)—H键的芳基化反应已经有所报道[51], 但均使用Pd、Rh等贵金属作为催化剂. 2016年, Shi课题[52]组使用廉价的Ni作催化剂, 实现了底物邻位C—H键的芳基化反应(Eq. 21).该反应利用具有偕二甲基效应的1-(2-吡啶基)-1-甲基乙胺(PIP)作为导向基, 在Ni(Ⅱ)作催化剂的条件下, 用芳基硅试剂实现了芳基底物邻位C(sp2)—H的芳基化反应.该导向基团易离去, 底物范围广, 为联芳烃的合成提供了便捷方法.
酮羰基同样也可作为导向基团, 这类导向基团结构更为简单, 有潜在的应用价值. 2003年, Kakiuchi等[53]报道了钌催化C(sp2)—H键与芳基硼试剂的氧化偶联反应.随后, 作者又发现在反应中添加H—B(OR)2的受体可以提高反应收率.由于频哪酮可以很好地结合H-B(OR)2, 当烯基硼试剂与芳基酮C(sp2)—H键发生氧化偶联时[54], 以频哪酮作为反应溶剂, 有效提高了反应收率(Scheme 12).该反应用RuH2(CO)(PPh3)3作催化剂, 以羰基为导向基团, 用芳基硼酸酯作为偶联试剂, 对芳基酮类化合物实现了邻位C(sp2)—H键的芳基化.作者强调了Ru(0) 的催化作用, 并对反应机理进行了阐述(Scheme 13). Ru(0) 络合物首先对苯环上羰基邻位C—H键进行活化断裂, 生成的中间体中的钌氢物种对酮羰基加成又生成烷氧基钌中间体, 该中间体与芳基硼酸酯进行金属交换生成环钌中间体, 同时生成了三烷氧基硼化合物(经11B NMR和GC-MS证实), 环钌中间体经还原消除得到偶联产物, 同时再生Ru(0) 催化剂.
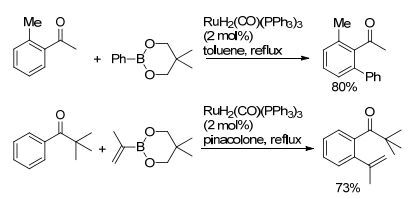 图式12
钌催化有机硼酸酯对芳基酮C—H键的芳基化
Scheme12.
Ru-catalyzed C—H arylation of aromatic ketones with organoboronates
图式12
钌催化有机硼酸酯对芳基酮C—H键的芳基化
Scheme12.
Ru-catalyzed C—H arylation of aromatic ketones with organoboronates
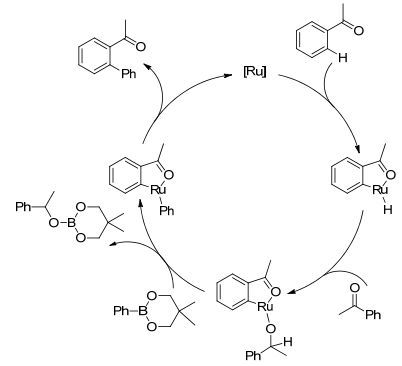 图式13
钌催化苯硼酸酯对芳基酮C—H键的芳基化反应机理
Scheme13.
General mechanism of Ru-catalyzed C—H arylation of aromatic ketones with phenylboronate
图式13
钌催化苯硼酸酯对芳基酮C—H键的芳基化反应机理
Scheme13.
General mechanism of Ru-catalyzed C—H arylation of aromatic ketones with phenylboronate
2015年, Ramana课题组[55]同样以酮羰基作为导向基团, 实现了苯并呋喃氧原子β位C(sp2)—H的芳基化反应, 反应在空气中即可进行, 不需添加额外的氧化剂(Eq. 22).文章通过对芳基硼酸和芳基氟硼酸盐两部分进行实验条件的筛选, 发现两者均能和酮羰基底物发生偶联反应生成目标产物, 扩大了底物的应用范围.文章通过控制实验证明了酮羰基作为导向基的必要性, 在没有酮羰基存在的条件下反应是不进行的.
2017年, Kapur课题组[56]实现了在酮羰基作导向基的条件下, 使同一种底物通过不同的催化体系得到N原子邻位或间位的芳基化产物(Scheme 14).在以Ru(Ⅱ)作催化剂时, 与导向基团中的羰基氧原子螯合形成五元环, 然后经过转金属化和还原消除得到邻位芳基化产物.而在Pd(Ⅱ)作为导向基时, 通过亲电性的Pd催化剂对间位C—H键进行活化, 得到间位芳基化反应.
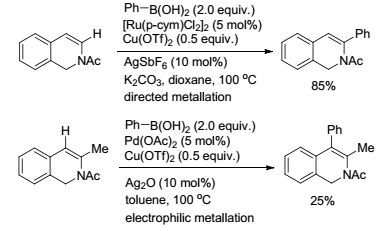 图式14
金属控制的芳基硼试剂对芳基酮的选择性C—H键芳基化反应
Scheme14.
Metal-controlled selective C—H arylation of aromatic ketones with phenyl boron reagents
图式14
金属控制的芳基硼试剂对芳基酮的选择性C—H键芳基化反应
Scheme14.
Metal-controlled selective C—H arylation of aromatic ketones with phenyl boron reagents
除了使用酮羰基作为导向基, 大位阻的膦酰氧基也可作为导向基团. 2016年, Shi课题组[57]以P(O)tBu2作为导向基, 在Pd(OAc)2作催化剂的条件下, 实现了底物C(7) 位置的C(sp2)—H与芳基硼酸的偶联反应(Eq. 23).反应的实现是具有挑战性的, 在吲哚作为反应底物时, N-膦酰氧基可以活化两个反应位点, 即吲哚的C(2) 位和C(7) 位, 分别形成五元环和六元环中间体(Eq. 24).在该反应中使用的导向基位阻较大, 有一定的吸电子性, 从一定程度上降低了C(2) 位置的C—H键的反应活性, 从而使反应发生在C(7) 位置.在催化循环中经历了C(7) 位的C—H键与催化剂形成不稳定的六元环金属中间体.该反应对官能团容忍性好, 选择性高, 且导向基团可离去, 应用范围广.
杂环嘧啶同样可以作为导向基团, 2015年, Peng课题组[58]报道了吡咯衍生物与芳基硼试剂的偶联反应(Eq. 25).反应以嘧啶作导向基, Rh(Ⅲ)作催化剂, 在温和条件下即可进行反应.反应收率高, 操作简单, 并可应用于放大量反应.
在芳基C(sp2)—H与芳基金属试剂的偶联反应中, 羧酸也可以作为导向基团活化芳基C(sp2)—H. Yu课题组[59]于2008年发表了通过使用芳基氟硼酸钾来实现苯甲酸或苯乙酸的邻位芳基化反应(Scheme 15).该反应使用氧气或空气作为氧化剂, 反应中的K+对于C—H键活化过程至关重要.该反应底物适用范围广、收率高.反应还可以适用于3-吡啶氟硼酸钾盐与苯甲酸的活化偶联反应, 实现邻位杂环化.例如当用2-氟-3-吡啶氟硼酸盐与苯甲酸反应时, 不但可以得到邻位吡啶化产物, 还可以继续进行下一步反应, 生成直接环化产物.
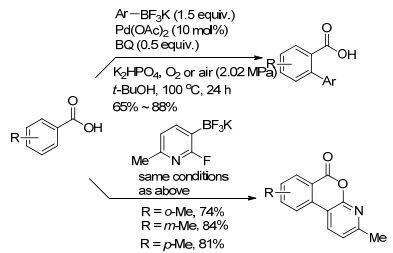 图式15
C—H键活化/芳基-芳基偶联
Scheme15.
C—H activation/aryl-aryl coupling
图式15
C—H键活化/芳基-芳基偶联
Scheme15.
C—H activation/aryl-aryl coupling
2011年, Yu课题组[60]又报道了芳基乙酸与芳基氟硼酸钾在Pd(OAc)2的催化下实现了羧基邻位的芳基化反应, 该反应有以下几点优势:首先反应使用了N-保护的氨基酸作配体, 使反应时间大大缩短, 2 h即可完成反应; 其次, 该反应可用氧气作氧化剂, 并且氧气的压力越大反应速度越快(Eq. 26).文章对反应机理也做了阐述, 反应中羧酸导向基与钯催化剂形成稳定的六元环钯中间体, 再与硼试剂发生金属交换, 进而还原消除得到邻位芳基化产物.钯催化剂被还原为零价, 再通过氧化剂BQ氧化进入下一次催化循环(Scheme 16).
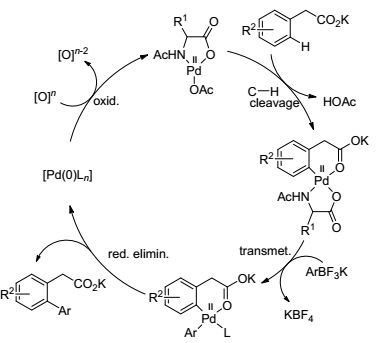 图式16
C(sp2)—H与硼试剂的交叉偶联反应的机理
Scheme16.
Mechanism of cross-coupling of C(sp2)—H bonds with arylboron reagents.
图式16
C(sp2)—H与硼试剂的交叉偶联反应的机理
Scheme16.
Mechanism of cross-coupling of C(sp2)—H bonds with arylboron reagents.
在过渡金属催化的反应中, 硫原子通常被认为是过渡金属催化剂的毒化剂.但近些年来, 在含硫化合物作为反应底物的反应中, 通过反应体系的筛选, 使得过渡金属催化仍然能够顺利进行.在C—H活化反应中, 硫醚[61]也可作为导向基团, 并表现出了良好的应用性. 2012年, Zhang课题组[62]以硫醚为导向基团, 通过采用Pd(OAc)2/Ag2CO3/BQ体系, 实现了芳环邻位C—H键与芳基三氟硼酸钾的氧化偶联(Eq. 27).同时, 通过对硫醚基团的移除或者官能团化, 作者制备了一系列联苯类含硫化合物, 以此证明该方法的应用性.
在C(sp2)—H键活化反应中, 也有一些不需要导向基团直接进行碳氢键活化偶联的报道, 具有代表性的是醌类及萘醌类C(sp2)—H键与芳基硼试剂的偶联反应. 2012年, Yu课题组[63]报道了苯醌C(sp2)—H键与芳基硼试剂的偶联反应, 反应不需要配体, 在FeS与K2S2O8催化体系下, 实现了醌类C(sp2)—H键的芳基化反应.在此基础上, Ding和Wang课题组报道了醌类C(sp2)—H的芳基化反应[64]和磺酰化反应[65, 66], Liu课题组[67]报道了吲哚N-α位C(sp2)—H键芳基化反应.这些反应为C(sp2)—H键直接活化偶联反应做了很好的补充.
1.3 C(sp2)—H键与C(sp)—M的偶联反应
尽管以导向基团定位的芳烃C(sp2)—H键与炔基卤代物的偶联有所报道[68, 69], 但关于芳烃C(sp2)—H键与炔基金属试剂的偶联反应的报道则很少, 在这里对此类反应将不作描述.
2 导向基团定位的C(sp3)—H键与有机金属试剂的偶联反应
通过脂肪族C(sp3)—H键活化偶联反应来构建新的C—C键, 是碳链延长的最直接的方法.但由于C(sp3)—H键的反应活性很低, 且不同C(sp3)—H键之间的键能的差异很小, 选择性很差, 所以普通的C(sp3)—H键的选择性活化成为了碳氢键活化领域的终极目标[70, 71].下面也将根据有机金属试剂的种类, 对C(sp3)—H键活化-偶联反应展开描述.
2.1 C(sp3)—H键与C(sp3)—M的偶联反应
通过氧化偶联反应来构建新的C(sp3)—C(sp3)报道的比较少, 主要是由于C(sp3)—M在反应体系中, 与过渡金属(Mt)发生金属交换后生成C(sp3)—Mt物种容易发生β-H消除反应.目前该副反应通常采用大位阻的配体或使用不含有β-H的底物来避免.在C(sp3)—H键活化反应中常涉及区域选择性和对映选择性问题, 如何实现反应的高区域选择性及高对映选择性成为了化学家进一步研究的对象, 往往需要选择合适的导向基团或外加手性配体来实现此类反应.近年来, Yu课题组在C(sp3)—H键与C(sp3)—M键的偶联反应领域取得了非常重要的成果.
2006年, Yu课题组[72]报道了首例以吡啶为导向基的C(sp3)—H与烷基硼酸的氧化偶联反应(Eq. 28).反应以Pd(OAc)2为催化剂, Ag2O/BQ作为氧化体系, 其中BQ既可作为氧化剂, 又可以作为配体促进还原消除反应的进行, Ag2O的添加起了至关重要的作用, 它能够促进金属交换过程的进行. 2008年, 该课题组[73]以酰胺为导向基, 实现了羰基β位C(sp3)的烷基化反应(Eq. 29), 反应以碳酸钾为碱在相似条件下进行.文中提及以空气中的氧气作为氧化剂代替Ag2O时, 反应同样可以发生, 但需要在反应过程中不断通入高压空气.另外, 溶剂2, 2, 5, 5-四甲基四氢呋喃是反应进行的重要条件, 它可以抑制硼试剂自身偶联产物的产生以及β-H消除反应的发生.该反应对含α-H的酰胺底物不兼容, 所以底物的适用范围有限.
在具有潜手性中心的C(sp3)原子发生C—H键活化反应时, 常常会遇到对映选择性问题.由于很难找到合适的配体骨架从空间上协助C—H键选择性断裂, 因此高对映选择性地合成含有手性中心的化合物具有很大挑战性. 2011年, Yu课题组[74]选用在天然产物及药物中常见的环丙基酰胺作为底物, 通过与有机硼试剂的氧化偶联反应, 实现了含有潜手性中心的C(sp3)—H键的烷基化反应(Eq. 30).反应以4-氰基全氟苯酰胺作为导向基, Pd(OAc)2作为催化剂, Li2CO3作碱, THF作溶剂以利于烷基化反应的进行.反应过程中, 除了底物外, 其他试剂分两次加入到反应体系中, 相比于一次投料法能够获得更好的收率和ee值, 作者猜测是由于催化剂的有效浓度不同导致实验结果的不同.其中配体对对映选择性起关键性作用, 通过对配体的修饰, 发现在选用N-单保护的氨基酸做配体时, 可以以较高的ee值得到反应产物.
2.2 C(sp3)—H键与C(sp2)—M的偶联反应
通过C(sp3)—H键与C(sp2)—M的偶联反应来实现烷烃的芳基化反应报道的比较多.与C(sp3)—M相比, C(sp2)—M参与的反应中, 不易发生β-H消除反应, 其中M可以是硅、硼、镁和锌等金属.在有机硼试剂参与的C—H键活化氧化偶联反应中, Yu课题组做了大量的研究工作.例如2007年, Yu课题组[23]首次实现了芳基硼酸酯与脂肪族羧酸羰基β位的C(sp3)—H键活化偶联反应, 但反应收率低, 底物范围窄(Eq. 31).随后该课题组又通过Weinreb酰胺作为导向基团, 实现了羰基β位C(sp3)—H的芳基化.由于Thorpe-Ingold效应[75], 反应对含有α-H的底物不兼容, 所以底物的适用范围有限. 2014年该课题组[76]实现了环丁烷羧酸衍生物的羰基β位C(sp3)—H的芳基化反应, 反应中添加手性的单N-保护的α-氨基酸做配体(MPAHA), 该配体能够加速反应的进行, 是Pd(Ⅱ)催化的C—H键官能团化反应能够发生的关键, 其次反应中缺电子的酰胺作为弱的辅助导向基, 也对反应至关重要(Eq. 32).
2015年, Glorius课题组[77]报道了在以吡啶为导向基定位条件下, 芳基硼酸酐对C(sp3)—H的芳基化反应.反应以Rh(Ⅲ)作催化剂, 高选择性地实现了吡啶氮原子β位的伯C(sp3)—H的芳基化反应(Scheme 17).在该反应中, 不同硼试剂对反应收率的影响很大, 当用苯基三氟硼酸盐时, 没有目标产物生成, 当采用芳基硼酸酐, 反应收率最高.在底物扩展中, 对于吡啶氮原子α位有取代基的底物收率高于无取代基的底物, 作者认为可能是由于底物α位取代基的空间位阻作用, 更有助于C(sp3)—H与Rh(Ⅲ)形成环金属化中间体.作者通过氘代实验证明了反应并没有β-H消除的产物生成, 猜想是由于吡啶N原子有着较强的螯合作用, 使得金属交换/还原消除过程快于β-H消除过程, 从而避免了β-H消除产物的生成.
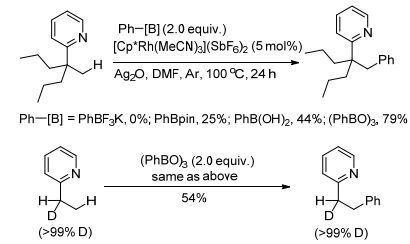 图式17
Rh催化的C(sp3)—H键的芳香化反应
Scheme17.
Rh(Ⅲ)-catalyzed arylation of C(sp3)—H bonds
图式17
Rh催化的C(sp3)—H键的芳香化反应
Scheme17.
Rh(Ⅲ)-catalyzed arylation of C(sp3)—H bonds
除了实现羧酸类化合物中的羰基β位的C—H键活化反应, 对含氮化合物中氮原子的α位及γ位的C(sp3)—H活的研究, 也有了一定进展.早期报道的文章中, 氮的α位C(sp3)—H键直接芳香化反应一般选用Ru(0) 作催化剂, 反应需要较高温度, 很难控制产物的立体选择性[78]; 其次, 通常选用杂环作为导向基, 导向基不易脱去.对于氮的γ位的C(sp3)—H键活化, 在催化循环过程中需要经过不稳定的六元环金属化过程, 因此反应有一定难度. 2015年, Yu课题组[79]报道了以硫代酰胺为导向基团的吡咯、哌啶及N-甲基胺类底物的氮原子的α位C(sp3)—H键活化-芳基化反应(Scheme 18).该反应以Pd(OTf)2作催化剂, 在反应过程中得到了五元环钯中间体, 这为机理的探究提供有力的证据.反应还通过一锅法制备出二取代芳基(芳杂环)化产物(Scheme 18).
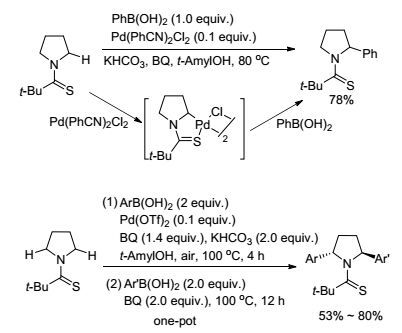 图式18
钯催化的C(sp3)—H键与有机硼试剂的氧化偶联
Scheme18.
Pd-catalyzed oxidative coupling of C(sp3)—H bonds with organoborons
图式18
钯催化的C(sp3)—H键与有机硼试剂的氧化偶联
Scheme18.
Pd-catalyzed oxidative coupling of C(sp3)—H bonds with organoborons
关于胺类化合物中氮原子的γ位C(sp3)—H键活化-偶联反应, Daugulis[37], Chen课题组[80]都做过相关报道, 他们均使用芳基卤代物作为偶联底物, 实现了氮原子的γ位C(sp3)—H的芳基化反应.反应都是通过底物中导向基的强螯合作用与催化剂形成稳定的环钯中间体所实现, 当将芳基卤代物换作硼试剂时氧化偶联反应是不进行的.为了进一步扩大反应的应用性, Yu课题组[81]通过引入单N-保护的氨基酸作配体, 成功实现了氮原子的γ位C(sp3)—H键与芳基硼试剂的氧化偶联.反应以三氟甲磺酰胺基团作为导向基团, 以Pd(OAc)2为催化剂, 在不加配体的情况下反应是不进行的, 当引入氨基酸配体时反应收率最高可达96% (Eq. 33).反应中配体起了关键性作用, 它能够影响催化剂的立体和电子效应, 从而加速反应的进行.该方法同样适用于合成1, 2-和1, 3-氨基醇化合物以及氨基酸类衍生物. 2017年, Zhao课题组[82]报道了吡唑类化合物氮原子β或γ位的C(sp3)—H键的单芳基化反应(Scheme 19).反应以Rh(Ⅲ)作催化剂, 初步研究认为Rh(Ⅲ)作催化剂能够促进无取代的甲基C—H键官能团化反应, 而对亚甲基C—H键不起作用, 这对γ位的无取代的甲基C—H键活化同样适用.反应选择三甲基吡唑作为导向基, 并证明了导向基的电子云密度是影响过渡金属催化C—H键活化的关键, 而经过机理研究初步证明, 该导向基可以协助催化剂形成不稳定的金属化中间体, 再经过金属交换和去质子化过程得到最终产物.反应得到单取代产物, 可用于放大量反应.
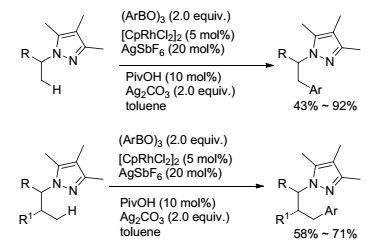 图式19
Rh催化的C(sp3)—H键活化芳基化反应
Scheme19.
Rhodium-catalyzed C(sp3)—H arylation reactions
图式19
Rh催化的C(sp3)—H键活化芳基化反应
Scheme19.
Rhodium-catalyzed C(sp3)—H arylation reactions
除了芳基硼试剂可与C(sp3)—H键发生氧化偶联反应外, 芳基硅试剂也可以发生类似反应. 2015年, Yu课题组[83]以喹啉衍生物作配体, 在单N-保护的酰胺基团作导向基的条件下成功实现了α-氨基酰胺底物的羰基β位C(sp3)—H键与硅试剂的氧化偶联反应(Eq. 34), 其中配体对反应的进行起了重要作用.此外, 作者还以相似的反应体系实现布洛芬羰基β位C(sp3)—H键的芳基化.值得注意的是, 当用芳基卤代物代替芳基硅试剂时, 可以完成芳基C(sp2)—H的芳基化[84], 从而实现了布洛芬的选择性C—H键衍生化(Scheme 20).
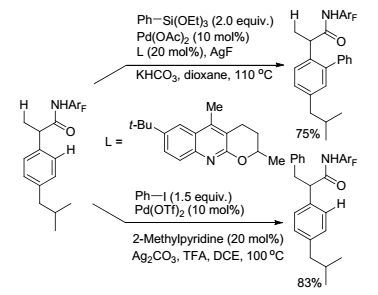 图式20
布洛芬酰胺衍生物C—H键的芳基化
Scheme20.
C—H arylation of buprofen-derived amide
图式20
布洛芬酰胺衍生物C—H键的芳基化
Scheme20.
C—H arylation of buprofen-derived amide
2015年, Gaunt课题组[85]报道了以脂肪族胺上的氮原子作为螯合原子, 在Pd(OAc)2作催化剂条件下, 实现了烷烃C(sp3)—H键与芳基硼试剂的偶联反应, 其中配体的添加对反应起重要作用(Eq. 35).反应不同于以往在催化循环过程中经历的五元环或六元环金属过渡态过程, 而是与Pd催化剂螯合形成少见的四元环金属中间体进行[86].在此之前, 该课题组[87]已报道过仲胺可经过四元环金属化过程实现C—H键活化, 反应成功的关键是仲胺周围取代基的空间位阻效应, 该反应则是借助单N-保护的氨基酸配体的协同作用, 实现环状仲氨的邻位甲基上的C(sp3)—H键芳基化反应, 对于非环状及小位阻取代的仲胺反应是不进行的.该反应对芳基硼试剂中芳环上的电子效应不敏感, 但对于吡啶、噻吩等杂环类的芳基硼试剂, 反应是不进行的.
芳基锌试剂及芳基格氏试剂等活泼金属试剂也可以作为偶联对象, 参与到C(sp3)—H键活化偶联反应中来. 2013, Nakamura课题组[88]报道了以8-氨基喹啉为导向基团的2, 2-二取代的丙酰胺的羰基β位C(sp3)—H芳基化反应(Eq. 36).这是首例Fe催化的C(sp3)—H键活化偶联反应.该反应的进行首先是引入了8-氨基喹啉作导向基团, 该导向基团上的两个N原子可以和Fe原子形成稳定的五元环配合物.其次是加入了1, 2-双(二苯基膦基)苯(dppbz)双膦配体, 该配体具有不可替代性.反应加入7 equiv.的芳基格氏试剂, 3 equiv.的锌试剂, 其中形成3 equiv.的二芳基锌试剂, 剩余1 equiv.格氏试剂可能作为强碱用于拔去酰胺上的氢原子, 详细机理有待进一步研究. 2014年, Ackermann课题组[89]报道了用三唑作导向基团, 以FeCl3作催化剂, dppe作配体, 也实现了芳基格氏试剂对羰基β位C(sp3)—H键芳基化反应(Eq. 37), 收率高达87%.若相同的催化体系用8-氨基喹啉作导向基团, 则收率只有9%, 可见三唑导向基团具有独特的优势与反应活性.以上反应最大的不足是需要使用大量的格氏试剂及锌试剂, 限制了反应的应用.
2.3 C(sp3)—H键与C(sp)—M的偶联反应
对于C(sp3)—C(sp)键的形成, 虽然脂肪族C—H键与炔基卤代物的交叉偶联, 脂肪族C—H键与炔烃的C—H键脱氢偶联, 以及脂肪族卤代物和炔烃的偶联等反应都有所报道[90~92], 但对于以导向基团定位的脂肪族C—H键与炔基金属试剂的偶联反应, 则未看到此类反应的相关报道, 在这里不做阐述.
3 结论与展望
综上所述, 过渡金属催化的碳氢键活化反应已取得很大进展, 可以用来快速构建各种类型的化合物[93].尽管官能团导向的碳氢键活化与其它试剂的偶联反应涵盖了各种底物类型, 但是目前碳氢键活化反应与理想的反应类型仍有很大距离.在现有的碳氢键活化反应中, 虽然不必在反应位点引入活化基团, 但是大多底物需要在适当的位置引入导向基团以实现选择性碳氢键活化, 导向基团的引入和去除同样会导致反应步骤的增加和能源的浪费, 并伴随大量废物的产生, 降低反应的应用价值.对于无导向基团的碳氢键活化虽已实现, 但是底物适用范围很窄, 只有较少的底物和催化体系能够实现.如何从简单的烷烃、烯烃和芳烃这些便宜易得的工业原料出发合成复杂的有机化合物, 仍是合成化学领域尚未解决的难题.自然界生物体中的碳氢键转化反应并不少见, 生物酶催化的化学转化被证实是一种非常高效、温和、高选择选择性的方法, 可以用于对简单烷烃进行修饰, 但是由于生物酶的专一性, 适用的底物范围很窄.因此如何理解生物酶催化的机理, 并对现有的催化体系进行修饰和优化, 保存酶的高活性的同时克服现有酶的底物局限性, 并发展新型碳氢键活化催化体系, 将其广泛应用于各类廉价易得的石油化工原料的直接转化, 这将真正意义上实现化学反应进程的绿色环保.
-
-
[1]
(a) Li, H. ; Shi, Z. -J. Prog. Chem. 2010, 22, 1914(in Chinese).
(李湖, 施章杰, 化学进展, 2010, 22, 1914. )
(b) Zhou, L. -H. ; Lu, W. -J. Acta Chim. Sinica 2015, 73, 1250(in Chinese).
(周励宏, 陆文军, 化学进展, 2015, 73, 1250. )
(c) Yu, J. -Q. ; Ding, K. -L. Acta Chim. Sinica 2015, 73, 1223(in Chinese).
(余金权, 丁奎岭, 化学学报, 2015, 73, 1223. )
(d) Zhu, Q. ; Wang, L. ; Xia, C. -G. ; Liu, C. Chin. J. Org. Chem. 2016, 36, 2813(in Chinese).
(朱庆, 王露, 夏春谷, 刘超, 有机化学, 2016, 36, 2813. ) -
[2]
(a) Liu, C. ; Liu, G. ; Zhao, H. Chin. J. Chem. 2016, 34, 1048.
(b) Ren, X. ; Kong, S. ; Shu, Q. ; Shu, M. Chin. J. Chem. 2016, 34, 373.
(c) Xu, J. -B. ; Chen, P. -H. ; Ye, J. -S. ; Liu, G. -S. Acta Chim. Sinica 2015, 73, 1294(in Chinese).
(徐佳斌, 陈品红, 叶金星, 刘国生, 化学学报, 2015, 73, 1294. ) -
[3]
Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Chem. Commun. 2010, 46, 677. doi: 10.1039/b908581e
-
[4]
李娟华, 刘昆明, 段新方, 刘晋彪, 有机化学, 2017, 37, 314. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345773.shtmlLi, J.; Liu, K.; Duan, X.; Liu, J. Chin. J. Org. Chem. 2017, 37, 314(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345773.shtml
-
[5]
Gao, K.; Yoshikai, N. Acc. Chem. Res. 2014, 47, 1208. doi: 10.1021/ar400270x
-
[6]
Su, B.; Cao, Z.-C.; Shi, Z.-J. Acc. Chem. Res. 2015, 48, 886. doi: 10.1021/ar500345f
-
[7]
Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787. doi: 10.1021/acs.chemrev.6b00567
-
[8]
(a) Dastbaravardeh, N.; Christakakou, M.; Haider, M.; Schnürch, M. Synthesis 2014, 46, 1421.
(b) Shang, M.; Sun, S.-Z.; Wang, H.-L.; Wang, M.-M.; Dai, H.-X. Synthesis 2016, 48, 4381. -
[9]
Negishi, E.; Anastasia, L. Chem. Rev. 2003, 103, 1979. doi: 10.1021/cr020377i
-
[10]
Chen, M.; Zheng, X.; Li, W; He, J.; Lei, A. J. Am. Chem. Soc. 2010, 132, 4101. doi: 10.1021/ja100630p
-
[11]
Molander, G. A.; Ellis, N. Acc. Chem. Res. 2007, 40, 275. doi: 10.1021/ar050199q
-
[12]
Dick, G. R.; Woerly, E. M.; Burke, M. D. Angew. Chem., Int. Ed. 2012, 51, 2667. doi: 10.1002/anie.201108608
-
[13]
Giri, R.; Maugel, N.; Li, J.-J.; Wang, D.-H.; Breazzano, S. P.; Saunders, L. B.; Yu, J.-Q. J. Am. Chem. Soc. 2007, 129, 3510. doi: 10.1021/ja0701614
-
[14]
Wasa, M.; Chan, K. S. L.; Yu, J.-Q. Chem. Lett. 2011, 40, 1004. doi: 10.1246/cl.2011.1004
-
[15]
Thuy-Boun, P. S.; Villa, G.; Dang, D.; Richardson, P.; Su, S.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 17508. doi: 10.1021/ja409014v
-
[16]
Neufeldt, S. R.; Seigerman, C. K.; Sanford, M. S. Org. Lett. 2013, 15, 2302. doi: 10.1021/ol400888r
-
[17]
Peng, P.; Wang, J.; Jiang, H.; Liu, H. Org. Lett. 2016, 18, 5376. doi: 10.1021/acs.orglett.6b02755
-
[18]
Chen, Q.; Ilies, L.; Yoshikai, N.; Nakamura, E. Org. Lett. 2011, 13, 3232. doi: 10.1021/ol2011264
-
[19]
Ilies, L.; Ichikawa, S.; Asako, S.; Matsubara, T.; Nakamura, E. Adv. Synth. Catal. 2015, 357, 2175. doi: 10.1002/adsc.v357.10
-
[20]
Graczyk, K.; Haven, T.; Ackermann, L. Chem.-Eur. J. 2015, 21, 8812. doi: 10.1002/chem.201501134
-
[21]
Negishi, E.; Okukado, N.; King, A. O.; Van Horn, D. E.; Spiegel, B. I. J. Am. Chem. Soc. 1978, 100, 3354. doi: 10.1021/ja00479a018
-
[22]
Xu, S.; Negishi, E. Acc. Chem. Soc. 2016, 49, 2158. doi: 10.1021/acs.accounts.6b00338
-
[23]
Shang, R.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2015, 137, 7660. doi: 10.1021/jacs.5b04818
-
[24]
Chen, X.; Li, J.-J.; Hao, X.-S.; Goodhue, C.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 78. doi: 10.1021/ja0570943
-
[25]
Cai, G.; Fu, Y.; Li, Y.; Wan, X.; Shi, Z. J. Am. Chem. Soc. 2007, 129, 7666. doi: 10.1021/ja070588a
-
[26]
Feng, R.; Yao, J.; Liang, Z.; Liu, Z.; Zhang, Y. J. Org. Chem. 2013, 78, 3688. doi: 10.1021/jo400186p
-
[27]
Tan, P.-W.; Haughey, M.; Dixon, D. J. Chem. Commun. 2015, 51, 4406. doi: 10.1039/C5CC00410A
-
[28]
Laforteza, B. N.; Chan, K. S. L.; Yu, J.-Q. Angew. Chem., Int. Ed. 2015, 54, 11143. doi: 10.1002/anie.201505204
-
[29]
Xiao, K.-J.; Chu, L.; Chen, G.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 7796. doi: 10.1021/jacs.6b04660
-
[30]
Krasnov, V. P.; Gruzdev, D. A.; Levit, G. L. Eur. J. Org. Chem. 2012, 1471.
-
[31]
Vogler, T.; Studer, A. Org. Lett. 2008, 10, 129. doi: 10.1021/ol702659a
-
[32]
Chu, J.-H.; Tsai, S.-L.; Wu, M.-J. Synthesis 2009, 3757.
-
[33]
Ilies, L.; Okabe, J.; Yoshikai, N.; Nakamura, E. Org. Lett. 2010, 12, 2838. doi: 10.1021/ol1009448
-
[34]
Yoshikai, N.; Asako, S.; Yamakawa, T.; Ilies, L.; Nakamura, E. Chem.-Asian J. 2011, 6, 3059. doi: 10.1002/asia.v6.11
-
[35]
Ilies, L.; Asako, S.; Nakamura, E. J. Am. Chem. Soc. 2011, 133, 7672. doi: 10.1021/ja2017202
-
[36]
Shang, M.; Sun, S.-Z.; Wang, H.-L.; Wang, M.-M.; Dai, H.-X. Synthesis 2016, 48, 4381. doi: 10.1055/s-0035-1562795
-
[37]
Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154. doi: 10.1021/ja054549f
-
[38]
Shang, R.; Ilies, L.; Asako, S.; Nakamura, E. J. Am. Chem. Soc. 2014, 136, 14349. doi: 10.1021/ja5070763
-
[39]
Gui, Q.; Chen, X.; Hu, L.; Wang, D.; Liu, J.; Tan, Z. Adv. Synth. Catal. 2016, 358, 509. doi: 10.1002/adsc.201500884
-
[40]
Hu, L.; Gui, Q.; Chen, X.; Tan, Z.; Zhu, G. Org. Biomol. Chem. 2016, 14, 11070. doi: 10.1039/C6OB02224C
-
[41]
Wang, D.; Yu, X.; Xu, X.; Ge, B.; Wang, X.; Zhang, Y. Chem. Eur. J. 2016, 22, 8663. doi: 10.1002/chem.v22.25
-
[42]
Yu, X.; Wang, D.-S.; Xu, Z.; Yang, B.; Wang, D. Org. Chem. Front. 2017, 4, 1011. doi: 10.1039/C6QO00793G
-
[43]
(a) Wasa, M.; Engle, K. M.; Yu, J.-Q. J. Am. Chem. Soc. 2009, 131, 9886.
(b) Wasa, M.; Engle, K. M.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 3680.
(c) Wasa, M.; Worrell, B. T.; Yu, J.-Q. Angew. Chem., Int. Ed. 2010, 49, 1275.
(d) Wasa, M.; Yu, J.-Q. Tetrahedron 2010, 66, 4811.
(e) Yoo, E. J.; Wasa, M.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 17378.
(f) Yoo, E.-J.; Ma, S.; Mei, T.-S. Chan, K. S. L. Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 7652. -
[44]
Wang, H.-W.; Cui, P.; Lu, Y.; Sun, W.-Y.; Yu, J.-Q. J. Org. Chem. 2016, 81, 3416. doi: 10.1021/acs.joc.6b00083
-
[45]
(a) Leow, D.; Li, G.; Mei, T.-S.; Yu, J.-Q. Nature 2012, 486, 518.
(b) Dai, H.-X.; Li, G.; Zhang, X.-G.; Stepan, A. F.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 7567. -
[46]
Wan, L.; Dastbaravardeh, N.; Li, G.; Yu, J-Q. J. Am. Chem. Soc. 2013, 135, 18056. doi: 10.1021/ja410760f
-
[47]
Chinnagolla, R. K.; Jeganmohan, M. Org. Lett. 2012, 14, 5246. doi: 10.1021/ol3024067
-
[48]
(a) Chinnagolla, R. K.; Jeganmohan, M. Chem. Commun. 2014, 50, 2442.
(b) Hubrich, J.; Himmler, T.; Rodefeld, L.; Ackermanna, L. Adv. Synth. Catal. 2015, 357, 474. -
[49]
Nishikata, T.; Abela, A. R.; Huang, S.; Lipshutz, B. H. J. Am. Chem. Soc. 2010, 132, 4978. doi: 10.1021/ja910973a
-
[50]
Shang, M.; Sun, S.-Z.; Dai, H.-X.; Yu, J.-Q. Org. Lett. 2014, 16, 5666. doi: 10.1021/ol5027377
-
[51]
(a) Zhou, H.; Xu, Y.-H.; Chung, W.-J.; Loh, T.-P. Angew. Chem., Int. Ed. 2009, 48, 5355.
(b) Li, W.; Yin, Z.; Jiang, X.; Sun, P. J. Org. Chem. 2011, 76, 8543. -
[52]
Zhao, S.; Liu, B.; Zhan, B.-B.; Zhang, W.-D.; Shi, B.-F. Org. Lett. 2016, 18, 4586. doi: 10.1021/acs.orglett.6b02236
-
[53]
Kakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Murai, S. J. Am. Chem. Soc. 2003, 125, 1698. doi: 10.1021/ja029273f
-
[54]
Ueno, S.; Chatani, N.; Kakiuchi, F. J. Org. Chem. 2007, 72, 3600. doi: 10.1021/jo070182g
-
[55]
Paymode, D. J.; Ramana, C. V. J. Org. Chem. 2015, 80, 11551. doi: 10.1021/acs.joc.5b01932
-
[56]
Tiwari, V. K.; Kamal, N.; Kapur, M. Org. Lett. 2017, 19, 262. doi: 10.1021/acs.orglett.6b03558
-
[57]
Yang, Y.; Qiu, X.; Zhao, Y.; Mu, Y.; Shi, Z. J. Am. Chem. Soc. 2016, 138, 495. doi: 10.1021/jacs.5b11569
-
[58]
Wang, L.; Li, Z.; Qu, X.; Peng, W. Chin. J. Chem. 2015, 33, 1015. doi: 10.1002/cjoc.201500354
-
[59]
Wang, D.-H.; Mei, T.-S.; Yu, J.-Q. J. Am. Chem. Soc. 2008, 130, 17676. doi: 10.1021/ja806681z
-
[60]
Engle, K. M.; Thuy-Boun, P. S.; Dang, M.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 18183. doi: 10.1021/ja203978r
-
[61]
(a) Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2010, 132, 3965.
(b) Samanta, R.; Antonchick, A. P. Angew. Chem., Int. Ed. 2011, 50, 5217.
(c) Yu, M.; Xie, Y.; Xie, C.; Zhang, Y. Org. Lett. 2012, 14, 2164.
(d) Wang, D.; Yu, X.; Yao, W.; Hu, W.; Ge, C.; Shi, X. Chem.-Eur. J. 2016, 22, 5543. -
[62]
Yao, J.; Yu, M.; Zhang, Y. Adv. Synth. Catal. 2012, 354, 3205. doi: 10.1002/adsc.201200447
-
[63]
Wang, J.; Wang, S.; Wang, G.; Zhang, J.; Yu, X.-Q. Chem. Commun. 2012, 48, 11769. doi: 10.1039/c2cc35468c
-
[64]
Wang, D.; Ge, B.; Li, L.; Shan, J.; Ding, Y. J. Org. Chem. 2014, 79, 8607. doi: 10.1021/jo501467v
-
[65]
Ge, B.; Wang, D.; Dong, W.; Ma, P.; Li, Y.; Ding, Y. Tetrahedron Lett. 2014, 55, 5443. doi: 10.1016/j.tetlet.2014.08.023
-
[66]
Wang, D.; Yu, X.; Ge, B.; Miao, H.; Ding, Y. Chin. J. Org. Chem. 2015, 35, 676. doi: 10.6023/cjoc201412047
-
[67]
Liu, C.; Yang, F. Chin. J. Chem. 2016, 34, 1213. doi: 10.1002/cjoc.v34.12
-
[68]
Xie, F.; Qi, Z.; Yu, S.; Li, X. J. Am. Chem. Soc. 2014, 136, 4780. doi: 10.1021/ja501910e
-
[69]
Landge, V. G.; Midya, S. P.; Rana, J.; Shinde, D. R.; Balaraman, E. Org. Lett. 2016, 18, 5252. doi: 10.1021/acs.orglett.6b02549
-
[70]
Arndtsen, B. A.; Bergman, R.; Mobley, T. A.; Peterson, T. Acc. Chem. Res. 1995, 28, 154. doi: 10.1021/ar00051a009
-
[71]
赵金钵, 张前, 化学学报, 2015, 73, 1235. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmZhao, J.-B.; Zhang, Q. Acta Chim. Sinica 2015, 73, 1235(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[72]
Chen, X.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 12634. doi: 10.1021/ja0646747
-
[73]
Wang, D.-H.; Wasa, M.; Giri, R.; Yu, J.-Q. J. Am. Chem. Soc. 2008, 130, 7190. doi: 10.1021/ja801355s
-
[74]
Wasa, M.; Engle, K. M.; Lin, D. W.; Yoo, E. J.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 19598. doi: 10.1021/ja207607s
-
[75]
(a) Bachrach, S. M. J. Org. Chem. 2008, 73, 2466.
(b) Li, S.; Zhu, R.-Y.; Xiao, K.-J.; Yu, J.-Q. Angew. Chem., Int. Ed. 2016, 55, 4317. -
[76]
Xiao, K.-J.; Lin, D. W.; Miura, M.; Zhu, R.-Y.; Gong, W.; Wasa, M.; Yu, J.-Q. J. Am. Chem. Soc. 2014, 136, 8138. doi: 10.1021/ja504196j
-
[77]
Wang, X.; Yu, D.-G.; Glorius, F. Angew. Chem., Int. Ed. 2015, 54, 10280. doi: 10.1002/anie.201503888
-
[78]
(a) Pastine, S. J.; Gribkov, D. V.; Sames, D. J. Am. Chem. Soc. 2006, 128, 14220.
(b) Phani Kumar, N. Y.; Jeyachandran, R.; Ackermann, L. J. Org. Chem. 2013, 78, 4145.
(c) Dastbaravardeh, N.; Schnürch, M.; Mihovilovic, M. D. Org. Lett. 2012, 14, 1930. -
[79]
Spangler, J. E.; Kobayashi, Y.; Verma, P.; Wang, D.-H.; Yu, J.-Q. J.Am. Chem. Soc. 2015, 137, 11876. doi: 10.1021/jacs.5b06740
-
[80]
(a) He, G.; Zhao, Y.; Zhang, S.; Lu, C.-X.; Chen, G. J. Am. Chem. Soc. 2012, 134, 3.
(b) Zhang, S.-Y.; He, G.; Nack, W. A.; Zhao, Y.-S.; Li, Q.; Chen, G. J. Am. Chem. Soc. 2013, 135, 2124. -
[81]
Chan, K. S. L.; Wasa, M.; Chu, L.; Laforteza, B. N.; Miura, M.; Yu, J.-Q. Nat. Chem. 2014, 6, 146. doi: 10.1038/nchem.1836
-
[82]
Yuan, C.; Tu, G.; Zhao, Y. Org. Lett. 2017, 19, 356. doi: 10.1021/acs.orglett.6b03522
-
[83]
He, J.; Takise, R.; Fu, H.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 4618. doi: 10.1021/jacs.5b00890
-
[84]
He, J.; Li, S.; Deng, Y.; Fu, H.; Laforteza, B. N.; Spangler, J. E.; Homs, A.; Yu, J.-Q. Science 2014, 343, 1216. doi: 10.1126/science.1249198
-
[85]
He, C.; Gaunt, M. J. Angew. Chem., Int. Ed. 2015, 54, 15840. doi: 10.1002/anie.201508912
-
[86]
McNally, A.; Haffemayer, B.; Collins, B. S. L.; Gaunt, M. J. Nature 2014, 510, 129. doi: 10.1038/nature13389
-
[87]
Smalley, A. P.; Gaunt, M. J. J. Am. Chem. Soc. 2015, 137, 10632. doi: 10.1021/jacs.5b05529
-
[88]
Shang, R.; Ilies, L.; Matsumoto, A.; Nakamura, E. J. Am. Chem. Soc. 2013, 135, 6030. doi: 10.1021/ja402806f
-
[89]
Gu, Q.; Al Mamari, H. H.; Graczyk, K.; Diers, E.; Ackermann, L. Angew. Chem., Int. Ed. 2014, 53, 3868. doi: 10.1002/anie.201311024
-
[90]
Ano, Y.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2011, 133, 12984. doi: 10.1021/ja206002m
-
[91]
Luo, F-X.; Xu, X.; Wang, D.; Cao, Z.-C.; Zhang, Y.-F.; Shi, Z.-J. Org. Lett. 2016, 18, 2040. doi: 10.1021/acs.orglett.6b00289
-
[92]
Luo, F.-X.; Cao, Z.-C.; Zhao, H.-W.; Wang, D.; Zhang, Y.-F.; Xu, X.; Shi, Z.-J. Organometallics 2017, 36, 18. doi: 10.1021/acs.organomet.6b00529
-
[93]
(a) Zhu, Q. ; Zhu, C. ; Deng, Z. ; He, G. ; Chen, J. ; Zhu, J. ; Xia, H. Chin. J. Chem. 2016, 34, 1.
(b) Yuan, S. -T. ; Wang, Y. -H. ; Qiu, G. -Y. ; Liu, J. -B. Chin. J. Org. Chem. 2017, 37, 566(in Chinese).
(袁斯甜, 王艳华, 邱观音生, 刘晋彪, 有机化学, 2017, 37, 566. )
(c) Wang, M. -M; Wang, Z. -X; Shang, M. ; Dai, H. -X. Chin. J. Org. Chem. 2015, 35, 570(in Chinese).
(王明明, 王子萧, 商明, 戴辉雄, 有机化学, 2015, 35, 570. )
(d) Lu, B. -N. ; Li, X. -Y. ; Lin, Y. -M. Chin. J. Org. Chem. 2015, 35, 2275(in Chinese).
(卢贝丽, 李现艳, 林咏梅, 有机化学, 2015, 35, 2275. )
-
[1]
-

图式2 C—H键与有机金属试剂的偶联反应的机理
Scheme 2 Mechanism for the cross-couplings of C—H bonds with organometallic reagents

图式3 Rh催化的有机硼试剂和吡啶酮的氧化偶联反应的机理
Scheme 3 Mechanism of rhodium(Ⅲ)-catalyzed oxidative coupling of organoboron reagents with pyridines

图式5 钯催化二芳基甲胺化合物C(sp2)—H键与有机硼试剂对映选择性氧化偶联
Scheme 5 Pd-catalyzed enantioselective C(sp2)—H oxidative coupling of diarylmethylamines with organoborons

图式6 铁催化烯烃C—H键的选择性官能团化
Scheme 6 Fe-catalyzed selective functionalization of olefinic C—H bonds

图式8 铜催化的有机硼试剂对C(sp2)—H的官能团化反应的机理
Scheme 8 Mechanism of Cu-catalyzed C(sp2)—H bond functionalization with organoboron compounds

图式9 钴催化的有机硼试剂对C(sp2)—H的官能团化反应
Scheme 9 Cobalt-catalyzed C(sp2)—H bond functionalization with organoboron compounds

图式10 钯催化的芳基和烯基硼试剂与C(sp2)—H键的交叉偶联反应
Scheme 10 Pd(Ⅱ)-catalyzed cross-coupling of C(sp2)—H bonds and aryl-and vinyl-boron reagents

图式11 Ru催化的芳基硼酸对乙酰苯胺的邻位芳香化反应
Scheme 11 Ruthenium-catalyzed ortho-arylation of acetanilides with aromatic boronic acids

图式12 钌催化有机硼酸酯对芳基酮C—H键的芳基化
Scheme 12 Ru-catalyzed C—H arylation of aromatic ketones with organoboronates

图式13 钌催化苯硼酸酯对芳基酮C—H键的芳基化反应机理
Scheme 13 General mechanism of Ru-catalyzed C—H arylation of aromatic ketones with phenylboronate

图式14 金属控制的芳基硼试剂对芳基酮的选择性C—H键芳基化反应
Scheme 14 Metal-controlled selective C—H arylation of aromatic ketones with phenyl boron reagents

图式16 C(sp2)—H与硼试剂的交叉偶联反应的机理
Scheme 16 Mechanism of cross-coupling of C(sp2)—H bonds with arylboron reagents.

图式18 钯催化的C(sp3)—H键与有机硼试剂的氧化偶联
Scheme 18 Pd-catalyzed oxidative coupling of C(sp3)—H bonds with organoborons
-


 下载:
下载:



















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 28
- 文章访问数: 2476
- HTML全文浏览量: 372
 下载:
下载:
