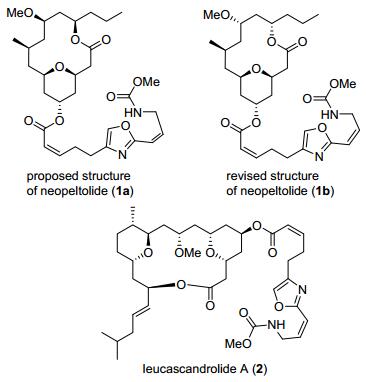
 图1
(+)-Neopeltolide和eucascandrolide A的结构
Figure1.
Structures of (+)-neopeltolide and leucascandrolide A
图1
(+)-Neopeltolide和eucascandrolide A的结构
Figure1.
Structures of (+)-neopeltolide and leucascandrolide A
引用本文:
于江帆, 冯若昆, 杨震. 具有优良抗癌活性的天然产物Neopeltolide的合成研究进展[J]. 有机化学,
2017, 37(10): 2526-2543.
doi:
10.6023/cjoc201703017

Citation: Yu Jiangfan, Feng Ruokun, Yang Zhen. Synthetic Studies toward Neopeltolide:A Potent Anti-cancer Natural Product[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2526-2543. doi: 10.6023/cjoc201703017
Citation: Yu Jiangfan, Feng Ruokun, Yang Zhen. Synthetic Studies toward Neopeltolide:A Potent Anti-cancer Natural Product[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2526-2543. doi: 10.6023/cjoc201703017
具有优良抗癌活性的天然产物Neopeltolide的合成研究进展
English
Synthetic Studies toward Neopeltolide:A Potent Anti-cancer Natural Product
Abstract:
(+)-Neopeltolide was isolated from a deep-water sponge of the family neopeltidae. Due to its attractive novel structure and highly potent anticancer activity, more than twenty total and formal syntheses have been reported in last decade. Herein, the synthetic studies toward the total and formal syntheses of neopeltolide are reviewed according to the synthetic strategies toward the macrolactone core.
-
Key words:
- (+)-neopeltolide
- / macrolactone
- / anti-cancer
- / macrolactonization
- / prins cyclization
- / olefin metathesis
-
大环内酯是一类极为重要的天然产物, 据统计, 在1981~2006年期间发现的新药中, 约有50%的新药都来自天然产物及其衍生物.自1927年发现第一个大环内酯exaltolide以来, 该类化合物逐渐被人们所认识和熟悉.研究发现, 大环内酯类天然产物具有良好的抗肿瘤、抗真菌、抗感染以及抗疟疾等生理活性, 到目前为止已经有多种大环内酯类天然产物或相关衍生物被广泛应用于临床, 比如克拉霉素、阿奇霉素等.
2007年, Wright小组[1]从海洋的neopeltidae类深水海绵中分离得到了大环内酯化合物(+)-neopeltolide, 确定它的结构2a(图 1). Panek研究小组在2007年率先完成了neopeltolide的全合成工作, 并确定了它的绝对构型为化合物1b. (+)-Neopeltolide的结构和leucascan-drolide A (2)[2]拥有很多的相似性(图 1), 拥有环内2, 6-cis-四氢吡喃结构的十四元大环内酯, 在四氢吡喃环4位连接含杂环的长碳羧基侧链, 分子内总共拥有6个手性中心.生理活性研究表明, 该化合物具有优异的抗癌活性, 尤其对人类肺腺癌的A-549细胞系(IC50=1.2 nmol/L)、卵巢癌肉瘤的NCI-ADR-RES细胞系
图1
(+)-Neopeltolide和eucascandrolide A的结构
Figure1.
Structures of (+)-neopeltolide and leucascandrolide A
(IC50=5.1 nmol/L)以及鼠类白血病的P388细胞系(IC50=0.56 nmol/L)等都有很好的细胞毒素作用, 此外, 还能很好地抑制胰腺癌PANC-1和直肠癌DLD-1等细胞系.构效关系研究发现, 大环内酯骨架和噁唑侧链两个结构单元对该分子的生理活性都有重要的影响[3].由于该化合物具有优异的生理活性以及新颖的结构特点, 引起了合成化学家的极大关注, 到目前为止, 已经有许多关于neopeltolide的合成报道, 包括全合成[4]和大环内酯骨架的合成[5]. Cossy[6a], Dai[6b]和Fuwa[6c]等都对neopeltolide的合成工作进行过综述. Neopeltolide的合成难点主要在于如何高效构筑大环内酯骨架和四氢吡喃结构, 它们的合成策略的高效性直接影响了全合成的效率.本文对neopeltolide全合成、neopeltolide大环骨架合成、neopeltolide类似物合成及活性研究进行综述.
1 Neopeltolide全合成
由于Neopeltolide结构的新颖性和优良的生理活性, 它的全合成工作受到广泛关注, Panek和Scheidt两个小组几乎同时报道了首次全合成工作.本小节内容根据构建大环内酯环和四氢吡喃环的方法的不同, 对neopeltolide的全合成工作进行概括和总结.
1.1 利用大环内酯化反应构筑大环结构
1.1.1 以碳正离子环化反应构筑四氢吡喃结构
Panek全合成: 2007年, 波士顿大学的Panek等[4a]率先完成了(+)-neopeltolide的首次不对称合成, 并确定了它的绝对构型.他们利用[4+2]环化反应构建四氢吡喃环, 用Yamaguchi大环化反应完成大环骨架的建立.整个合成从羧酸化合物3开始, 经过选择性还原羧基、羟基保护、酯基还原成醛和羰基的硫代缩醛化等转化得到化合物4.噻烷化合物4对手性环氧5进行开环反应后脱除噻烷得到β-羟基酮6. β-羟基酮经过改良的的Evans-Tishchenko[7]还原可以得到1, 3-anti构型手性羟基, 将新产生的手性羟基进行甲基化后将末端硅基保护基脱除, 利用Swern氧化[8]将伯醇氧化成醛7.硅烷化合物8和醛7在酸催化下发生[4+2]环化反应[2h], 以10:1的非对映选择性构筑了二氢吡喃9.然后发生氰根离子对磺酸酯的亲核取代反应、酰基脱除和氰基的还原等反应可以得到醛10, 将醛转化成羧酸后发生Yamaguchi大环酯化反应得到大环骨架11.最后将四氢吡喃环中的双键通过羟汞化-硼氢化钠还原反应[2h]转化为手性羟基, 并通过酯化反应和Still-Gennari成烯反应[2a]引入侧链即完成了目标分子的全合成.
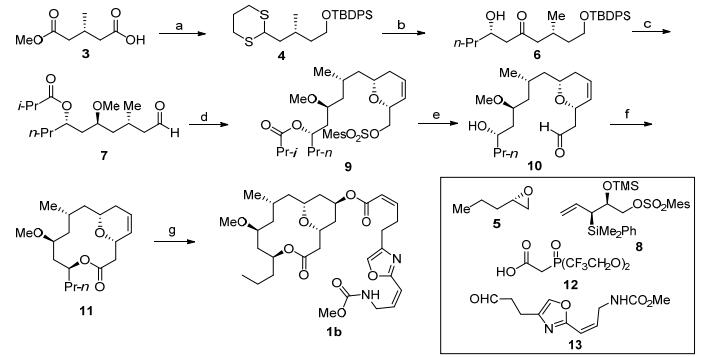 图式 1
Panek的全合成
Scheme1.
Panek's total synthesis
图式 1
Panek的全合成
Scheme1.
Panek's total synthesis
Kozmin消旋体全合成: 2008年, Kozmin等[3a]利用去对称化的Prins环化反应完成了neopeltolide消旋体的合成(Scheme 2).具有对称结构的化合物14发生Prins环化反应构筑了四氢吡喃结构, 对四氢吡喃的5位羟基进行苄基保护后发生Wacker氧化将末端双键转化为甲基酮15.甲基酮和醛16经过羟醛缩合反应[9]和Wittig反应后得到长链化合物17, 再经历羰基的反式还原[10]、酯基的水解后, 进行Yamaguchi大环化反应, 就构筑了大环骨架18.随后对骨架进行官能团的转化, 依次发生双键加氢还原、C(11)位置羟基的手性翻转、羟基的甲基化反应以及苄基的脱除反应, 得到大环内酯消旋体核心骨架19.核心骨架19和侧链20通过Mitsunobu反应连接就完成消旋体的合成[2b].
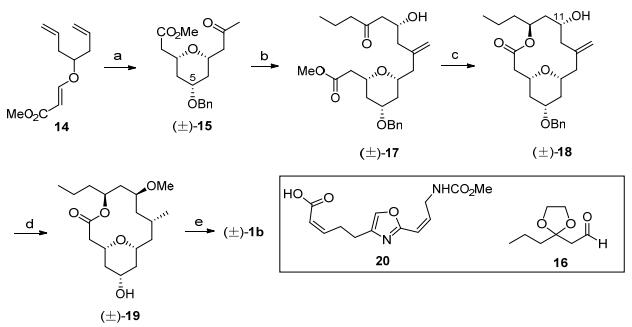 图式 2
Kozmin消旋体全合成
Scheme2.
Kozmin's total synthesis of (±)-neopeltolide
图式 2
Kozmin消旋体全合成
Scheme2.
Kozmin's total synthesis of (±)-neopeltolide
Maier全合成: Maier等[3b, 5a]报道了neopeltolide的大环内酯核心骨架的合成, 合成中利用Prins环化构筑四氢吡喃结构(Scheme 3).
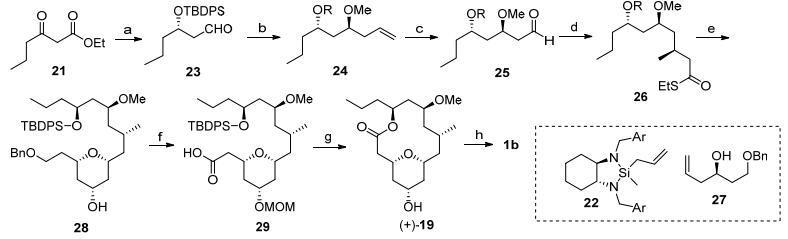 图式 3
Maier全合成
Scheme3.
Maier's total synthesis
图式 3
Maier全合成
Scheme3.
Maier's total synthesis
β-羰基酯21经过Noyori不对称氢化反应[11]、硅基保护和酯基DIBAL-H还原[12]后生成醛23, 然后通过不对称烯丙基化反应得到1, 3-二羟基结构, 并对羟基进行甲基化得到24.将24中的双键转化为醛后进行wittig反应[13], 得到的硫酯发生不对称迈克反应建立手性甲基[14].硫酯26发生还原生成醛, 和高烯丙基醇27发生Prins环化反应构筑四氢吡喃环.随后将28中的活性羟基先进行MOM保护, 再发生加氢脱苄基反应、Dess-martin氧化反应和次氯酸钠氧化反应[15], 将末端苄氧基转化为羧酸, 得到化合物29.脱除化合物29中的硅基保护基后发生Yamaguchi大环内酯化反应[16], 酸性条件下水解甲氧基甲基醚, 和侧链20发生Mitsunobu反应, 就完成完成天然产物的全合成.
Floreancig全合成: Floreancig等[3e, 5c]利用大环内酯化和碳正离子环化为关键反应完成了neopeltolide的全合成(Scheme 4).碘代物30先和三氯乙腈反应后作为保护基对β-羟基酯31中的活性羟基进行保护得到32, 随后与炔33发生Sonogashira偶联得到烯炔化合物34, 在铂催化下[17]将共轭炔键转化为酮的同时脱除所有硅基保护得到β-羟基酮35.通过Evans-Tishchenko反应得到1, 3-anti构型二羟基结构, 将其中的活性羟基甲基化生成烯炔化合物36, 将两个酯基同时水解后发生Yamaguchi大环内酯化反应[16], 得到大环内酯结构.在钌催化下将末端炔基转化成乙酸烯醇酯38[18], 接着在DDQ氧化下发生碳正离子环化反应, 得到四氢吡喃酮39, 最后将大环结构中的双键还原完成了大环内酯骨架40的合成.最后通过硼氢化钠还原羰基以及Mitsunobu反应完成天然产物的全合成.
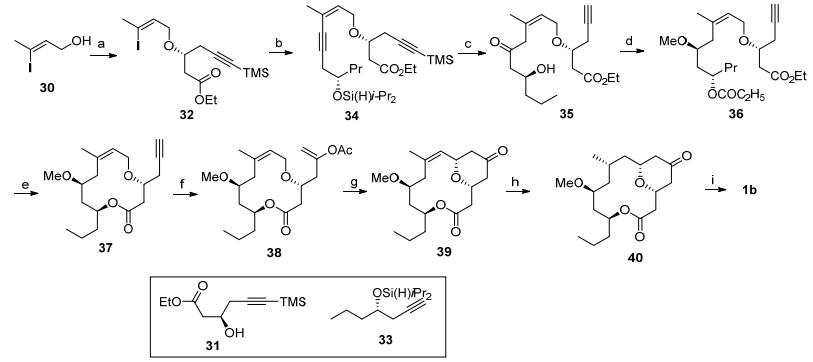 图式 4
Floreancig的全合成
Scheme4.
Floreancig's total synthesis
图式 4
Floreancig的全合成
Scheme4.
Floreancig's total synthesis
1.1.2 利用钌催化反应来完成四氢吡喃环结构
Fuwa和Sasaki全合成: Fuwa和Sasaki等[3d, 4e]以大环内酯化反应和烯烃复分解反应完成了大环内酯环和四氢吡喃环的构建(Scheme 5).底物41经过不对称烯丙基化反应[19]和羟基保护后转化为42, 接着发生的烯烃复分解反应[20]和酯基还原得到43.将烯丙醇43转化为手性烯丙基醇44需要经过Sharpless不对称环氧化反应和末端羟基的碘代-消除开环反应.将44中的仲羟基进行苄氧甲氧基保护, DDQ氧化脱除MPM保护后进行乙酰化, 再转化为乙烯基磷酸酯得到化合物46.乙烯基磷酸酯和碘化物47在Suzuki-Miyaura偶联反应条件下生成的烯醇化合物, 经过烯烃复分解反应构筑二氢吡喃结构, 再经过加氢还原反应得到四氢吡喃化合物48.将48中末端羟基脱除硅基保护后转化为羧酸甲酯50.脱除50中的MPM后将酯基皂化水解, 接着经过Yamaguchi大环内酯化[16]得到51, 完成母核骨架构筑.四氢吡喃环上的羟基在脱除BOM保护基后与侧链羧酸20发生Mitsunobu反应完成了目标产物的合成.
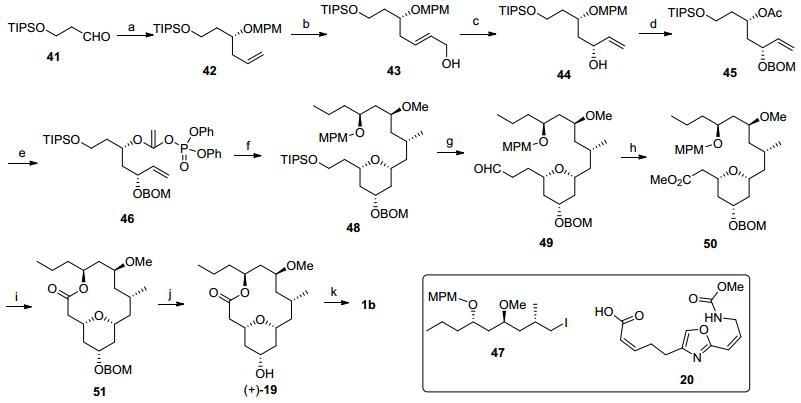 图式 5
Fuwa和Sasaki全合成
Scheme5.
Fuwa and Sasaki's total synthesis
图式 5
Fuwa和Sasaki全合成
Scheme5.
Fuwa and Sasaki's total synthesis
Roulland全合成: 2009年, Roulland等[4f]利用钌催化的烯-炔偶联反应构筑了四氢吡喃环, 并完成了neopeltolide的全合成工作(Scheme 6).从原料21经过钌催化的不对称氢化反应建立C(13)手性羟基[21], 酰胺化后得到的52和2-甲基烯丙基氯化镁反应生成β-羟基酮, 通过SmI2促进的1, 3-还原反应建立C(11)的羟基手性[7a]. 53中的活性羟基和丙烯酰氯反应后接着发生分子内烯烃复分解反应得到54.钯催化下的加氢反应建立甲基手性, 同时内酯环发生酯交换反应得到羧酸异丙基酯, 得到的C(11)位活性羟基进行甲基化, 接着将异丙基酯酯基还原为醛得到中间体55.在InBr3催化下与连二烯发生不对称炔丙基化反应[22], 得到炔丙基醇57.炔丙基醇57和3-丁烯醛发生钌催化的烯-炔偶联反应[23]构筑了四氢吡喃结构58.通过四氧化锇和高碘酸钠切断四氢吡喃的环外双键, 醛基经过Pinnick氧化反应转化为羧基, Yamaguchi大环内酯化反应以及羰基的还原等反应可以将58转化为核心骨架19, 最后通过Mitsunobu反应完成目标产物(+)-Neopeltolide (1b)的合成.
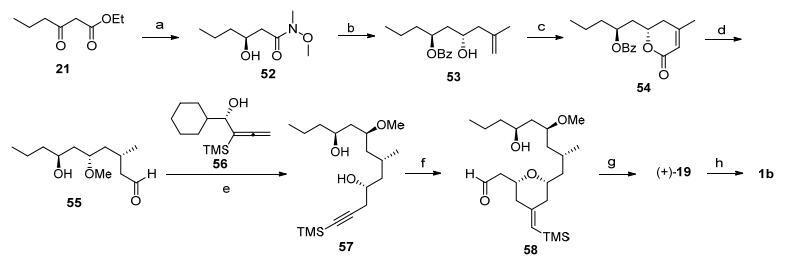 图式 6
Roulland全合成
Scheme6.
Roulland's total synthesis
图式 6
Roulland全合成
Scheme6.
Roulland's total synthesis
1.1.3 利用氧杂Diels-Alder (D-A)反应构筑四氢吡喃结构
Paterson全合成: 2008年, Paterson研究团队[4d]利用D-A反应实现了neopeltolide的全合成(Scheme 7).该小组也利用21的不对称加氢反应建立C(13)手性羟基, 再经过基团保护和酯基还原得到醛59, 随后发生Brown不对称烯丙基化反应[24]构筑C(11)手性中心, 同时将活性羟基甲基化生成60.将60中的双键经过臭氧化切断后进行HWE反应得到61.在氧杂Diels-Alder反应前体63的合成中, 首先是将中间体61的不饱和酯转化为不饱和醛, 随后通过手性配体催化下的汉斯酯参与的双键还原[25]建立C(9)的手性甲基得到63.醛63和双烯体64在手性催化剂65作用下发生Jacobsen不对称D-A反应[26]得到四氢吡喃酮化合物66.将66中PMB保护基脱除并将羟基转化为羧基, 脱除C(13)位TBS保护基就可以得到环化反应前驱体67.在Yamaguchi大环内酯化条件下发生环化, 并对酮羰基进行还原就可以得到核心骨架(+)-19, 最后通过Mitsunobu反应连接侧链20, 完成天然产物的全合成.
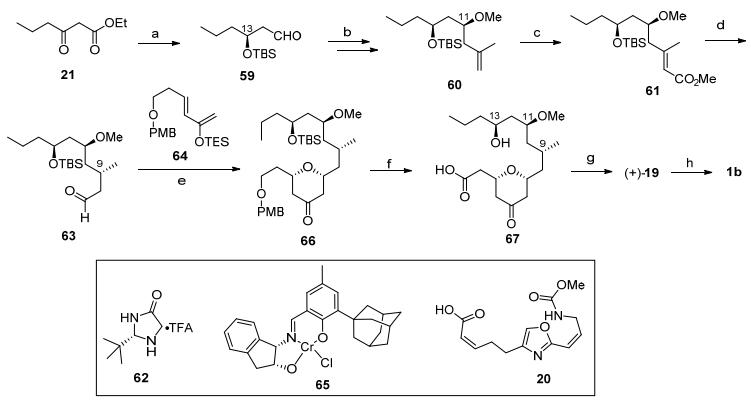 图式 7
Paterson全合成
Scheme7.
Paterson's total synthesis
图式 7
Paterson全合成
Scheme7.
Paterson's total synthesis
Ghosh全合成: Ghosh等[4g]利用大环内酯化反应和氧杂D-A反应为关键步骤完成了neopeltolide的全合成工作(Scheme 8). 3-甲基戊二酸酐在酶催化下发生去对称化得到戊二酸单丙基酯[27], 硼烷选择性还原羧基得到69, 将羟基氧化后进行缩醛保护、酯基转化为醛并发生不对称烯丙基化反应得到手性醇71[28].羟基进行甲基化后双键发生Lemieux-Johnson氧化成醛[29], 再次发生不对称烯丙基化形成手性醇72.缩醛72水解后发生HWE生成α, β-不饱和甲基酮[30], 随后将羰基转化为烯醇硅醚得到二烯体73.在手性催化剂75的催化下, 二烯体73和醛74发生Jacobsen不对称D-A反应构筑四氢吡喃酮, 接着将羰基转化为缩酮后发生氰基对磺酸酯的取代反应得到氰化物77.氰基的水解反应和大环内酯化反应生成大环内酯骨架79[31].接着发生两次还原反应即可完成核心骨架19的合成, 最后和侧链20通过Mitsunobu反应连接完成天然产物的合成.
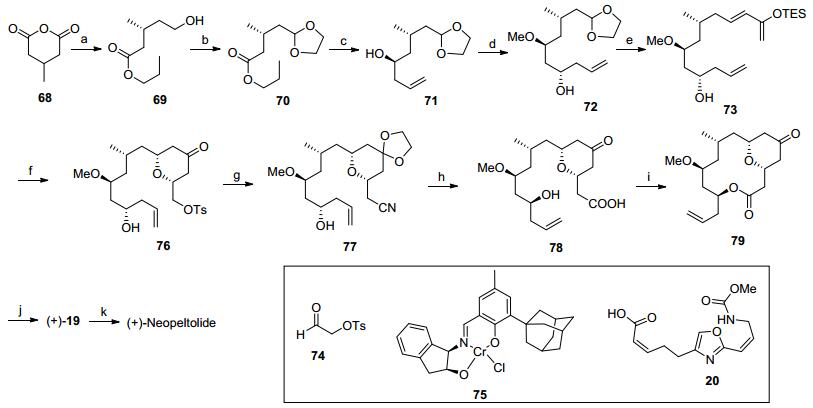 图式 8
Ghosh全合成
Scheme8.
Ghosh's total synthesis
图式 8
Ghosh全合成
Scheme8.
Ghosh's total synthesis
1.2 以碳正离子环化反应构筑大环以及四氢吡喃结构
1.2.1 Scheidt全合成
2007年, Scheidt等[4b]利用酯化反应和Prins成环反应完成了neopeltolide的首次全合成工作(Scheme 9).手性原料80经过酰胺化反应和羟基PMB保护得到Weinreb酰胺81[32].碘代物82[33]转化为锂试剂后对Weinreb酰胺的加成, 进而脱除甲氧基苄基保护得到β-羟基酮83, 羟基酮发生SmI2引发的Evans-Tischenko还原得到1, 3-二羟基构型, 其中的活性羟基被甲基保护, 接着利用碳酸钾脱除苯甲酰基顺利转化为酯化前驱体84.醇84和羧酸85在Yamaguchi条件下发生酯化反应, 在弱酸下脱除两个硅基保护后, 用四甲基吡啶氮氧化物(TEMPO)和醋酸碘苯中将一级羟基选择性氧化[34], 得到Prins环化关键前驱体醛86.在路易斯酸三氟甲磺酸钪作用下发生的Prins成环反应顺利关上大环骨架, 接着发生脱酸反应完成大环内酯骨架40的合成.最后发生C(5)羰基的还原以及Mitsunobu反应就完成了neopeltolide的全合成.
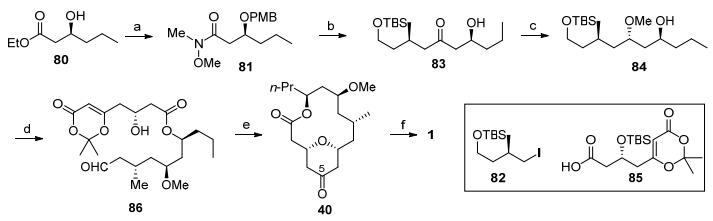 图式 9
Scheidt全合成
Scheme9.
Scheidt's total synthesis
图式 9
Scheidt全合成
Scheme9.
Scheidt's total synthesis
1.2.2 Lee全合成
2008年, Lee教授[4c]带领研究团队利用Prins成环反应作为关键步骤, 同时构筑了大环内酯骨架及四氢吡喃环结构, 完成了neopeltolide的全合成(Scheme 10).丁醛经过不对称加成反应[35]、苄基保护和臭氧化双键切断反应得到醛88, 接着再一次发生底物诱导的烯丙基化反应、羟基酰基化反应得到化合物90.底物诱导的铑催化的氢甲酰化反应将双键转化为醛, 其中C(9)位置甲基的非对映选择性为5:1[36], 接着发生缩醛反应得到缩醛91, 脱除苄基后和羧酸92发生酯化反应, 脱除PMB的保护可以顺利得到Prins环化前驱体93.后者在路易斯酸催化下发生Prins环化反应, 构筑了大环骨架及四氢吡喃结构, 再经过乙酰基的脱除得到核心骨架19, 最后与侧链20发生Mitsunobu反应可以完成Neopeltolide的全合成.
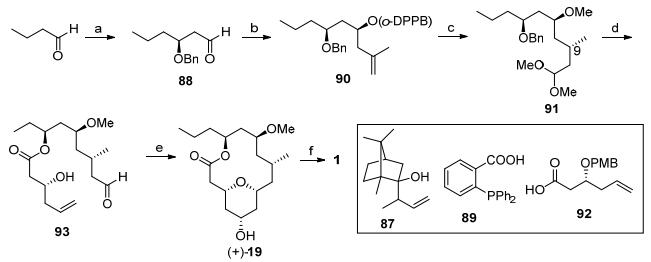 图式 10
Lee全合成
Scheme10.
Lee's total synthesis
图式 10
Lee全合成
Scheme10.
Lee's total synthesis
1.3 利用烯烃复分解反应构筑大环结构
1.3.1 Fuwa全合成
烯烃复分解反应也是构筑大环结构的常用办法[37]. Fuwa小组就以烯烃复分解环化反应(RCM)反应为关键步骤完成了neopelto-lide的全合成(Scheme 11)[3g, 4e].手性化合物94经过硅基保护和双键的切断[38]得到醛95, 不对称烯丙基化反应和烯烃复分解反应可以将95转化为不饱和酯96, 羟基的苄氧甲氧基保护和硅基的脱除, 得到的醇97先后经过分子内氧杂迈克反应、三甲基硅醇钾(TMSOK)下发生酯基水解[39]和分子间酯化反应就得到大环化反应前驱体99.在Grubbs二代催化剂作用下发生分子内烯烃复分解反应[40], 再通过加氢同时发生BOM的脱除和双键的还原反应, 完成大环内酯核心骨架19的合成.最后和侧链20发生Mitsunobu反应可以完成Neopeltolide的全合成.
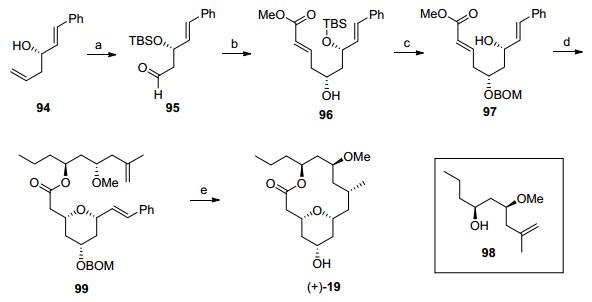 图式 11
Fuwa全合成
Scheme11.
Fuwa's total synthesis
图式 11
Fuwa全合成
Scheme11.
Fuwa's total synthesis
1.3.2 Hoveyda全合成
Hoveyda小组[4h]长期以来从事烯烃复分解反应, 2015年, 他们利用钼和钌催化的烯烃复分解反应非常高效地完成了neopeltolide的全合成工作(Scheme 12).合成工作从不饱和酰胺100开始, 经过手性N-杂卡宾催化的氧化反应[41], 得到β-羟基酰胺化合物52, 接着经过格氏反应、Evans-Tischenko还原、羟基甲基化和苯甲酰基脱除反应, 顺利转化为酯化前驱体98.羧酸合成砌块由手性原料102转化得到, 首先102和正丁基乙烯基醚在配体103作用下发生钼催化的烯烃复分解反应, 高效得到四氢吡喃环104, 接着烯醇醚在酸性下发生水解得到的醛, 经过氧化得到羧酸化合物105.羧酸105和醇98经过酯
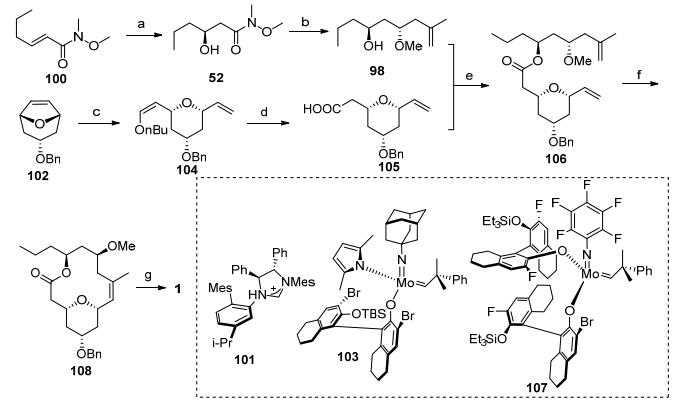 图式 12
Hoveyda全合成
Scheme12.
Hoveyda's total synthesis
图式 12
Hoveyda全合成
Scheme12.
Hoveyda's total synthesis
缩合反应得到的产物106在手性钼配体107催化下发生烯烃复分解反应[42], 以89%的产率得到大环内酯骨架108.最后经过加氢还原和Mitsunobu反应就完成了天然产物neopeltolide的全合成.
其中侧链20的合成主要是通过钼和钌的催化构筑两个顺式双键(Scheme 13).烯丙基胺化合物109和硼酸酯在钼催化剂110作用下得到Z构型的硼酸酯111, 接着和碘代物112发生偶联反应得到醇113, 随后发生溴代反应, 接着和烯丙基格氏试剂发生反应得到二烯烃114.端位烯烃和顺丁烯二醇在钌催化剂115作用下发生烯烃复分解反应得到伯醇116, 接着通过氧化反应得到侧链20.本工作的亮点在于在侧链20和大环骨架的合成中都使用了钼催化的烯烃复分解反应, 使得合成的效率大大提高.
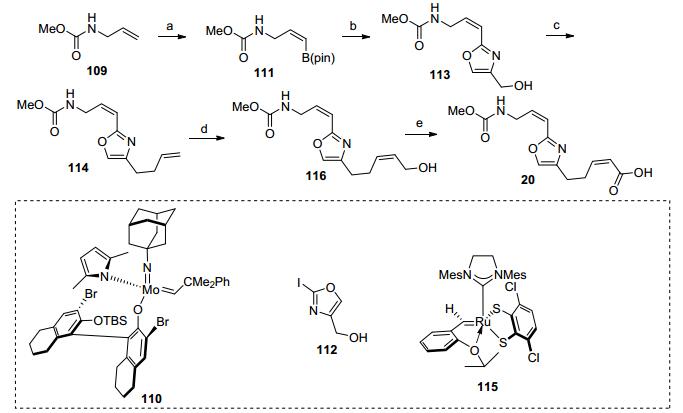 图式 13
Hoveyda侧链合成
Scheme13.
Hoveyda's synthesis of the side chain
图式 13
Hoveyda侧链合成
Scheme13.
Hoveyda's synthesis of the side chain
2 Neopeltolide大环内酯骨架合成
Neopeltolide的结构具有较大的复杂性, 对它的全合成需要大量的工作.构效关系和活性研究表明, 大环内酯母核是不可缺少的结构[3], 因此大环内酯骨架的合成工作也有重要的意义, 很多研究小组对它进行了合成研究.
2.1 利用大环内酯化反应构筑大环骨架
2.1.1 以迈克加成反应建立四氢吡喃环
Hong大环内酯骨架合成: 2009年, Hong教授[5d]利用烯丙位氧化串联氧杂迈克反应构筑四氢吡喃环, 并通过大环内酯化反应完成neopeltolide核心骨架合成(Scheme 14). 1, 3-二噻烷对碘化物进行亲核取代后开环氧反应[43], 得到的氯代物原位形成环氧化合物118, 接着进行乙基格氏试剂开环氧, 并脱除噻烷, 得到的羟基酮119发生Evans-Tischenko还原反应构筑1, 3-二羟基化合物, 再对活性羟基进行甲基化得到120.将末端双键通过不对称双羟基化[44]和环氧化反应形成化合物121[45], 再发生噻烷122对环氧121的开环反应, 得到的产物123在二氧化锰条件下发生烯丙位羟基氧化串联氧杂迈克反应, 同时醛继续氧化为羧酸, 得到大环内酯化前驱体124, 在2-甲基-6-硝基苯甲酸酐催化下发生大环内酯化反应[46], 接着进行噻烷的脱除和羰基的还原就完成了大环内酯骨架19的合成.
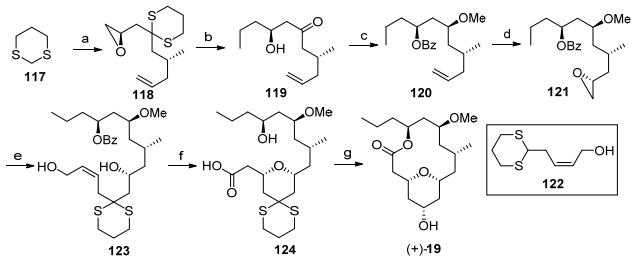 图式 14
Hong大环内酯骨架合成
Scheme14.
Hong's synthesis of macrolactone core
图式 14
Hong大环内酯骨架合成
Scheme14.
Hong's synthesis of macrolactone core
Subhash Ghosh大环内酯骨架合成: Ghosh小组[5k]利用连续利用手性拆分原理构筑C(11)和C(13)羟基手性中心, 最终完成了大环骨架的合成(Scheme 15).从已知的手性原料125出发, 经过两次手性拆分后得到环氧化合物127A[47], 接着发生乙基格氏试剂开环反应、羟基的苄基保护和二苯基叔丁基硅基[48]脱除得到伯醇128, 将末端羟基转化为氰基是增加一个碳原子的常用方法.氰基经过还原得到的醛129和片段130通过HWE反应[49]连接, 就可以得到不饱和酮131.在弱酸条件下脱除硅基保护后, 发生钯催化的氧杂迈克反应就完成了四氢吡喃酮结构的构筑.将132加氢脱除苄基, 先后经过TEMPO氧化和Pinnick氧化转化为羧酸[50], 最后通过Yamaguchi大环化反应完成骨架的合成.
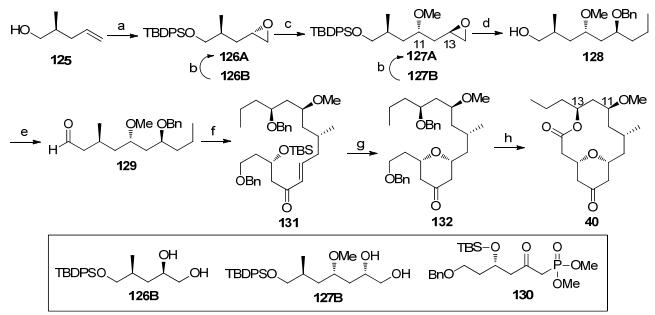 图式 15
Subhash Ghosh大环内酯骨架合成
Scheme15.
Subhash Ghosh's synthesis of macrolactone core
图式 15
Subhash Ghosh大环内酯骨架合成
Scheme15.
Subhash Ghosh's synthesis of macrolactone core
Taylor大环内酯骨架合成: 2008年, Taylor小组[51]通过自由基氧杂迈克加成和自由基加成反应构筑四氢吡喃结构, 通过大环内酯化反应合成了neopeltolide核心骨架(Scheme 16)[5b].手性原料3先后经过羧酸还原、酰胺化反应[52]和Dess-Martin氧化[53]反应转化为醛133, 利用Soderquist手性硼烯丙基化试剂134[54]对醛进行不对称烯丙基化反应后用苄氧甲氧基保护得到135. β-羟基硫醚136在形成双锂试剂对Weinreb酰胺进行亲核加成得到β-羟基酮137, 羟基酮发生Evans-Tischenko还原反应得到1, 3-二羟基构型, 其中的活性羟基被甲基保护后顺利转化为化合物138.碘氯参与的醚转移反应可以高选择性建立C(5)手性羟基(139)[51].碘代物139在三丁基膦作用下与丙炔酸乙酯发生氧杂迈克反应[55], 生成的不饱和酯再次发生自由基迈克加成反应得到四氢吡喃化合物140[56].将两个酯基同时水解并发生Yamaguchi大环内酯化, 在加氢条件下脱除苄基保护就完成大环内酯核心骨架141的合成.
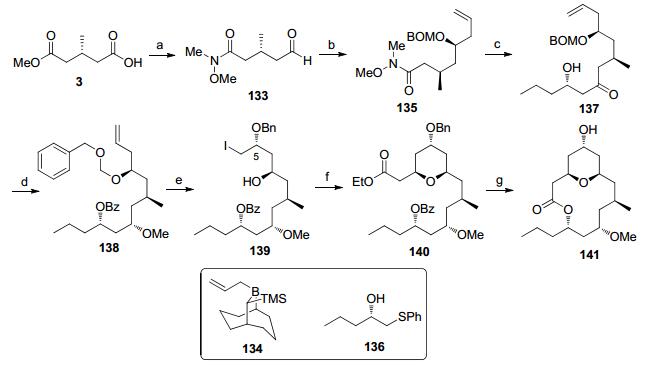 图式 16
Taylor大环内酯骨架合成
Scheme16.
Taylor's synthesis of macrolactone core
图式 16
Taylor大环内酯骨架合成
Scheme16.
Taylor's synthesis of macrolactone core
2.1.2 利用D-A反应和[3, 3]-重排反应构筑四氢吡喃环(Raghavan大环内酯骨架合成)
2012年, Raghavan教授[5i]完成了neopeltolide的大环骨架合成, 该工作利用氧杂D-A反应和[3, 3]-重排反应完成四氢吡喃环的构建(Scheme 17).不饱和醛142在MacMillan条件下先进行双键的不对称还原形成手性甲基[25], 然后与双烯体144发生不对称D-A反应[57]形成二氢吡喃酮145, 对酮羰基进行Luche还原[58]和乙酰基保护得到146, 随后发生Ireland-Claisen重排[59]生成2, 6-cis式的二氢吡喃147, 将酯基还原为醇后进行TBS保护.在LDBB作用下, 发生苯硫醚对醛148的加成反应得到149, 得到的化合物经过羟基氧化反应和硅基脱除得到二羟基酮150.锆催化的Evans-Tishchenko反应[7]还原羰基, 产生的新的手性羟基经过甲基化得到产物151.将酯基水解, TEMPO和醋酸碘苯选择性氧化伯醇得到羧酸, 然后发生Yamaguchi大环内酯化得到11.在合成的最后, 二氢吡喃中的双键经过氧化-汞化反应转化为C(5)手性羟基, 就完成大环内酯核心骨架141的合成.
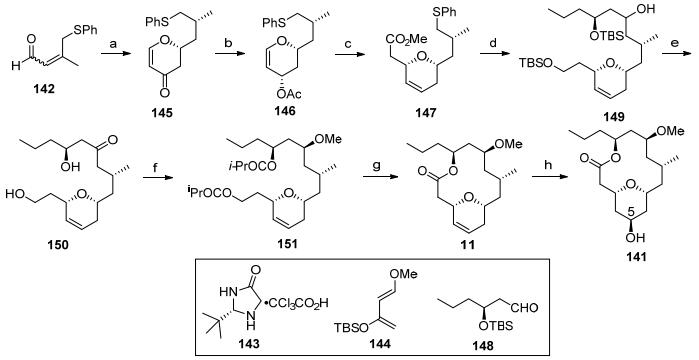 图式 17
Raghavan大环内酯骨架合成
Scheme17.
Raghavan's synthesis of macrolactone core
图式 17
Raghavan大环内酯骨架合成
Scheme17.
Raghavan's synthesis of macrolactone core
2.2 利用碳正离子环化反应构筑大环结构及四氢吡喃环
2.2.1 Yadav大环内酯骨架合成
Yadav教授[5e]也利用Prins环化为合环方法完成了neopeltolide大环内酯骨架的合成(Scheme 18). (S)-香茅醇经过苄基保护后发生双键的氧化-消除反应得到烯丙基醇152, 臭氧化切断双键成醛153[60], 随后与(2S)-4-烯-1, 2-戊二醇发生Prins环化反应得到二醇154[61], 第一次Prins环化反应建立C(11)和C(13)手性羟基, 将155中的一级羟基转化为碘代物后发生消除反应得到中间体156.羟基的甲基化反应、加氢还原双键同时脱除苄基、氧化一级醇为醛, 将醛进行缩醛化同时伴随叔丁基二苯基硅基的脱除, 顺利得到酯化反应的前驱体157.作者将得到的缩醛134按照Lee小组的合成方法完成大环内酯核心骨架的合成.
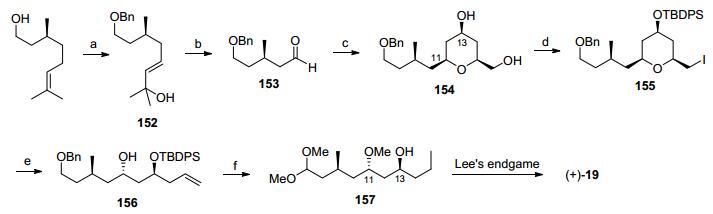 图式 18
大环内酯骨架合成
Scheme18.
Yadav's synthesis of macrolactone coreYadav
图式 18
大环内酯骨架合成
Scheme18.
Yadav's synthesis of macrolactone coreYadav
2.3 利用烯烃复分解反应构筑大环结构
2.3.1 She大环内酯骨架合成
2011年, 厍学功教授[5h]利用钯催化的烯烃的烷氧化羰基化反应和分子内烯烃复分解反应分别完成四氢吡喃结构和neopeltolide大环结构的合成(Scheme 19).利用Krische小组[62]发展的铱催化的双不对称烯丙基化反应合成具有C-2对称性的二醇158, 接着发生钯催化的烷氧化羰基化反应[63]得到四氢吡喃化合物159, 经过苄基保护后, 在Grubbs催化剂下发生末端双键的迁移反应[64], 得到酯化反应前体160.酯基水解后与醇98在2-甲基-6-硝基苯甲酸酐[65]条件下发生酯化反应可以顺利得到烯烃复分解前体161.最后通过分子内烯烃复分解反应构筑大环骨架, 在钯碳加氢条件下同时发生苄基脱除和双键还原反应, 完成大环骨架19的合成.
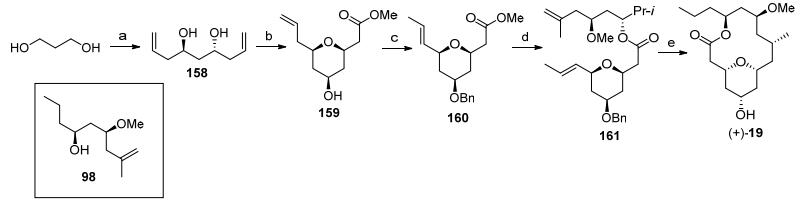 图式 19
She大环内酯骨架合成
Scheme19.
She's synthesis of macrolactone core
图式 19
She大环内酯骨架合成
Scheme19.
She's synthesis of macrolactone core
2.3.2 Sharma大环内酯骨架合成
2012年, Sharma小组[5j]以分子内烯烃复分解反应和汞环化反应等关键反应完成了neopeltolide大环骨架的合成.首先利用已知化合物162经过格氏反应、氧化还原反应以及硅基保护得到163, 接着将丙叉水解脱除后1, 2-二醇转化为环氧化合物, 丙醇化合物对环氧的开环反应可以顺利得到醇166.炔丙基醇在红铝下还原为反式双键[66], 再发生不对称环氧化反应和甲基选择性开环氧得到二醇化合物167.在碘参与下将邻二醇转化为双键[67], 脱除硅基保护就完成醇片段169的合成(Scheme 20).
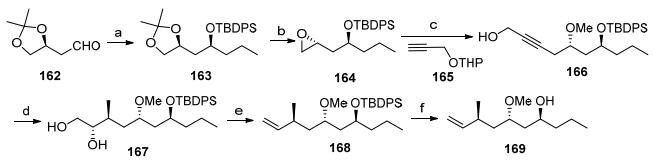 图式 20
Sharma醇片段169的合成
Scheme20.
Sharma's synthesis of alcohol fragment 169
图式 20
Sharma醇片段169的合成
Scheme20.
Sharma's synthesis of alcohol fragment 169
羧酸片段的合成从已知手性化合物170的不对称环氧化开始(Scheme 21), 再经过红铝还原得到1, 3-二醇171, 经过官能团转化后生成172, 随后脱除丙叉并将邻二醇转化为环氧化合物173.乙烯基格氏试剂开环氧和甲氧基甲基保护将环氧化合物173转化为174, 末端保护的羟基经脱硅基后经氧化转化为羧酸175.羧酸片段和醇169酯化后在Grubbs催化下发生大环化反应得到十四元大环内酯骨架[68].脱除甲氧基苄基的保护, 接着发生在汞环化反应构筑四氢吡喃结构177[69].自由基脱除汞并对甲氧基甲基进行水解完成大环内酯核心骨架141的合成.
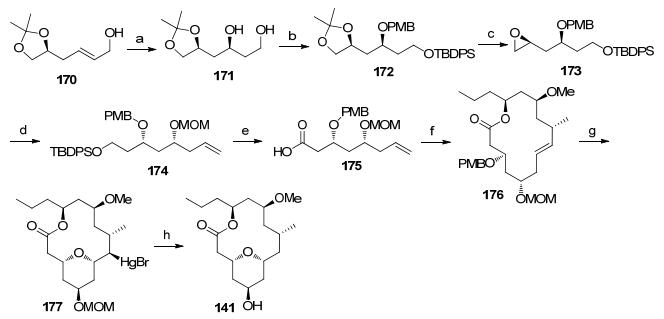 图式 21
Sharma大环内酯骨架合成
Scheme21.
Sharma's synthesis of macrolactone core
图式 21
Sharma大环内酯骨架合成
Scheme21.
Sharma's synthesis of macrolactone core
3 Neopeltolide类似物合成及构效关系研究
由于其广泛而出色的抗癌活性, 关于neopeltolide的合成工作有大量的报道.同时, 很多的研究小组一直致力于它的类似物合成和构效关系研究[3], 希望能够发现具有更好生物活性的化合物.尤其是Fuwa小组, 合成了大量的neopeltolide类似物, 并进行了活性测试.
3.1 Dai类似物合成
Dai研究小组[70]发展了一种钯催化的分子内双键烷氧化羰基化反应, 同时构建了四氢吡喃环和大环内酯环, 具有非常高的合成效率(Scheme 22).利用底物178在醋酸钯作用下发生环化反应得到大环结构179, 发生缩酮的水解和羰基的还原反应得到9-去甲基大环核心骨架180, 最后连接侧链后完成了9-demethylneopelto-lide (181)合成.
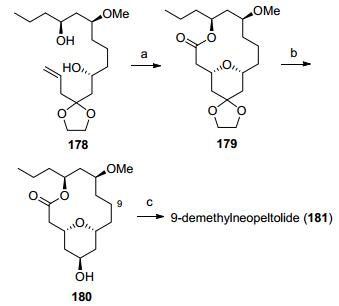 图式 22
Dai’s neopeltolide类似物合成
Scheme22.
Dai's synthesis of neopeltolide analogues
图式 22
Dai’s neopeltolide类似物合成
Scheme22.
Dai's synthesis of neopeltolide analogues
3.2 Maier类似物合成及活性研究
2008年, Maier等[3b]在完成了neopeltolide的全合成工作的基础上, 通过手性中心和双键的立体异构变化, 成功合成了一些结构类似物182~186, 并做了一系列生物活性测试.其中侧链中含有共轭型双键[Z (C2'-C3'), E (C4'-C5')]的化合物184具有很好的生理活性, 对小鼠成纤维细胞(L929)、人肺癌细胞(A549)、还原型辅酶I (NADH)系列细胞系的IC50分别为0.16, 0.22, 12.9 nmol/L, 和天然产物的neopeltolide的活性非常接近, 分别为0.25, 0.16, 10.2 nmol/L (图 2).
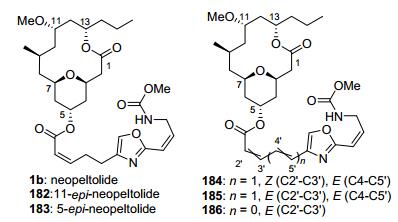 图2
Maier合成的neopeltolide类似物结构
Figure2.
Maier's synthesis of neopeltolide analogues
图2
Maier合成的neopeltolide类似物结构
Figure2.
Maier's synthesis of neopeltolide analogues
3.3 Fuwa类似物合成及活性研究
Fuwa小组[4e, 5f]利用已有的合成路线合成了多种类似物结构(图 3).他们首先合成了9-demethylneopeltolide (181)和11-demethoxyneopeltolide (187), 针对P388细胞系的活性研究表明, 化合物181具有优良的生理活性, 其IC50为0.813 nmol/L, 是neopeltolide的1.11倍(IC50=0.899 nmol/L).接着Fuwa小组根据烯烃复分解反应, 在保留C(3)和C(7)位置的手性始终为cis-构型的情况下, 合成了16种8, 9-dehydroneopeltolide衍生物188a~188p.研究发现, 和天然产物保持一致手性的类似物188a具有很好的生理活性, 其对细胞系A549、人乳腺癌细胞系(MCF-7)、人纤维肉瘤中国仓鼠卵巢细胞(HT-1080)、小鼠白血病细胞(P388)的IC50分别为0.5, 33, 0.51, 0.72 nmol/L, 在人类肺癌细胞系A549的表现中甚至优于天然产物neopeltolide (IC50=1.2 nmol/L).
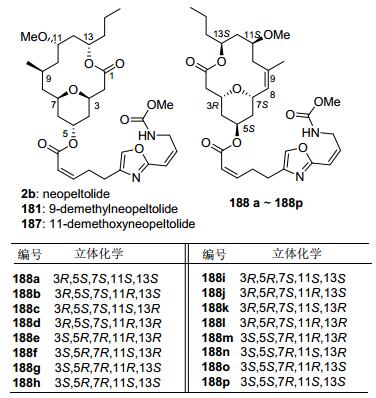 图3
Fuwa合成的neopeltolide类似物结构
Figure3.
Fuwa's synthesis of neopeltolide analogues
图3
Fuwa合成的neopeltolide类似物结构
Figure3.
Fuwa's synthesis of neopeltolide analogues
3.4 Floreancig类似物合成及活性研究
Floreancig等[3e, 3f]利用已有合成路线[5c]合成了多种类似物结构(图 4).他们首先合成了8, 9-dehydroneo-peltolide (188a), 接着利用双键的氧化还原合成了9-epi-neopeltolide (189), 8, 9-epoxyneopeltolide (190), dihydroxyneopeltolide (191), (8R)-hydroxyneopeltolide (192)和(8S)-hydroxy-9-epi-neopeltolide(193).侧链和5-氨基母核进行酰胺化得到194, 以苯环、吡啶环对侧链的噁唑环进行替换分别得到196和197, 并对上述化合物的生理活性进行了活性测试.侧链中C(4')—C(5')为反式双键结构, 同时以呋喃环替代噁唑环, 得到类似物198.活性测试发现, 化合物188a和189在人结肠癌细胞系HCT116中GI50分别为5.41, 6.62 nmol/L, 最接近天然产物的数值(GI50=0.77 nmol/L), 189在人乳腺癌细胞系MCF-7的GI50(1180 nmol/L)要优于neopeltolide (6960 nmol/L).在HCT116-p53KO细胞系的表现中, (8R)-hydroxy neopeltolide (192)的GI50 (13 nmol/L)最接近neopeltolide的数值(9.9 nmol/L).
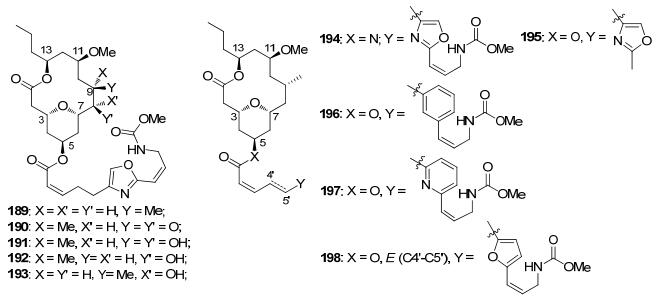 图4
Floreancig合成的neopeltolide类似物结构
Figure4.
Floreancig's synthesis of neopeltolide analogues
图4
Floreancig合成的neopeltolide类似物结构
Figure4.
Floreancig's synthesis of neopeltolide analogues
4 结论与研究前景
抗生素药物自青霉素发现以来对医药领域的发展作出了很大的贡献, 当下社会抗生素滥用现象以及抗生素耐药性的致命缺陷, 使得抗生素新药的研发迫在眉睫.大环内酯作为一类很早应用于临床的抗生素化合物, 由于结构复杂, 合成难度较大, 在很长的一段时间内都没有太大的研究进展.
Neopeltolide作为一个对癌细胞具有广谱的高度抗增殖活性的大环内酯, 自发现以来, 就得到了很大的关注, 到目前为止有20多篇的合成相关研究论文.在neopeltolide的后续研究中, 有两个方面需要关注.其一, 将大环内酯改造成大环内酰胺化合物200, 也可以着重对侧链部分进行衍生化, 包括对侧链杂环结构的改变和侧链末端的碳酸酯的改变, 研究其对活性的改善可能产生的影响(图 5).其二, 自然界存量过少是该化合物难以进一步进行临床试验的限制, 因此如何切实提高合成效率, 量化合成neopeltolide也成为该化合物进一步研究的关键.
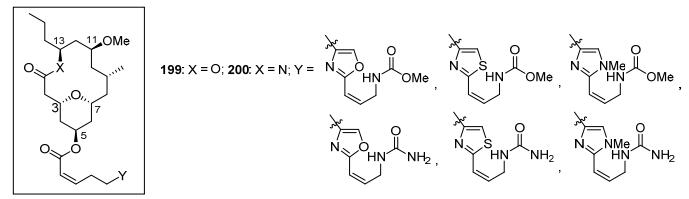 图5
Neopeltolide可衍生化的类似物结构
Figure5.
Desgined structures of neopeltolide analogues
图5
Neopeltolide可衍生化的类似物结构
Figure5.
Desgined structures of neopeltolide analogues
-
-
[1]
Wright, A. E.; Botelho, J. C.; Guzman, E.; Harmody, D.; Linley, P.; McCarthy, P. J.; Pitts, T. P.; Pomponi, S. A.; Reed, J. K. J. Nat. Prod. 2007, 70, 412. doi: 10.1021/np060597h
-
[2]
For total syntheses of leucascandrolide A, see:
(a) Hornberger, K. R.; Hamblett, C. L.; Leighton, J. L. J. Am. Chem. Soc. 2000, 122, 12894.
(b) Wang, Y.; Janjic, J. S.; Kozmin, A. J. Am. Chem. Soc. 2002, 124, 13670.
(c) Fettes, A.; Carreira, E. M. Angew. Chem., Int. Ed. 2002, 41, 4098.
(d) Paterson, I.; Tudge, M. Angew. Chem., Int. Ed. 2003, 42, 343.
(e) Fettes, A.; Carreira, E. M. J. Org. Chem. 2003, 68, 9274.
(f) Paterson, I.; Tudge, M. Tetrahedron 2003, 59, 6833.
(g) Wang, Y.; Janjic, J. S.; Kozmin, A. Pure Appl. Chem. 2005, 77, 1161.
(h) Su, Q.; Dakin, L. A.; Panek, J. S. J. Org. Chem. 2007, 72, 2.
(ⅰ) Van Orden, L. J.; Patterson, B. D.; Rychnovsky, S. D. J. Org. Chem. 2007, 72, 5784. For syntheses of the leucascandrolide A macrolide, see:
(j) Kopecky, D. J.; Rychnovsky, S. D. J. Am. Chem. Soc. 2001, 123, 8420.
(k) Wipf, P.; Reeves, J. T. Chem. Commun. 2002, 2066.
(l) Williams, D. R.; Plummer, S.; Patnaik, V. S. Angew. Chem., Int. Ed. 2003, 42, 3934.
(m) Williams, D. R.; Patnaik, V. S.; Plummer, S. Org. Lett. 2003, 5, 5035.
(n) Crimmins, M. T.; Siliphaivanh, P. Org. Lett. 2003, 5, 4641.
(o) FerriM, L.; Reymond, S.; Capdevielle, P.; Cossy, J. Org. Lett. 2007, 9, 2461. Other reports of leucascandrolide A fragments include:
(p) Crimmins, M. T.; Carroll, C. A.; King, B. W. Org. Lett. 2000, 2, 597.
(q) Kozmin, S. A. Org. Lett. 2001, 3, 755.
(r) Dakin, L. A.; Langille, N. F.; Panek, J. S. J. Org. Chem. 2002, 67, 6812.
(s) Wipf, P.; Graham, T. H. J. Org. Chem. 2001, 66, 3242. -
[3]
(a) Ulanovskaya, O. A.; Janjic, J.; Suzuki, M.; Sabharwal, S. S.; Schumacker, P. T.; Kron, S. J.; Kozmin, S. A. Nat. Chem. Biol. 2008, 4, 418.
(b) Vintonyak, V. V.; Kunze, F. S.; Maier, M. E. Chem.-Eur. J. 2008, 14, 11132.
(c) Custar, D. W.; Zabawa, T. P.; Hines, J.; Crews, C. M.; Scheidt, K. A. J. Am. Chem. Soc. 2009, 131, 12406.
(d) Fuwa, H.; Saito, A.; Naito, S.; Konoki, K.; Yotsu-Yamashita, M.; Sasaki, M. Chem.-Eur. J. 2009, 15, 12807.
(e) Cui, Y.; Tu, W.; Floreancig, P. E. Tetrahedron 2010, 66, 4867.
(f) Cui, Y.; Balachandran, R.; Day, B. W.; Floreancig, P. E. J. Org. Chem. 2012, 77, 2225.
(g) Fuwa, H.; KawakamiA, K.; Noto, K.; Muto, T.; Suga, Y.; Konoki, K.; Yotsu-Yamashita, M.; Sasaki, M. Chem.-Eur. J. 2013, 19, 8100.
(h) D'Ambrosio, M.; Guerriero, A.; Debitus, C.; Pietra, F. Helv. Chim. Acta 1996, 79, 51.
(ⅰ) Fuwa, H.; Sato, M.; Sasaki, M. Mar. Drugs 2014, 12, 5576. -
[4]
(a) Youngsaye, W.; Lowe, J. T.; Pohlki, F.; Ralifo, P.; Panek, J. S. Angew. Chem., Int. Ed. 2007, 46, 9211.
(b) Custar, D. W.; Zabawa, T. P.; Scheidt, K. A. J. Am. Chem. Soc. 2008, 130, 804.
(c) Woo, S. K.; Kwon, M. S.; Lee, E. Angew. Chem., Int. Ed. 2008, 47, 3242.
(d) Paterson, I.; Miller, N. A. Chem. Commun. 2008, 4708.
(e) Fuwa, H.; Naito, S.; Goto, T.; Sasaki, M. Angew. Chem., Int. Ed. 2008, 47, 4737.
(f) Guinchard, X.; Roulland, E. Org. Lett. 2009, 11, 4700.
(g) Ghosh, A. K.; Shurrush, K. A.; Dawson, Z. L. Org. Biomol. Chem. 2013, 11, 7768.
(h) Yu, M.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2015, 54, 215. -
[5]
(a) Vintonyak, V. V.; Maier, M. E.Org. Lett. 2008, 10, 1239.
(b) Kartika, R.; Gruffi, T. R.; Taylor, R. E. Org. Lett. 2008, 10, 5047.
(c) Tu, W.; Floreancig, P. E. Angew. Chem., Int. Ed. 2009, 48, 4567.
(d) Kim, H.; Park, Y.; Hong, J. Angew. Chem., Int. Ed. 2009, 48, 7577.
(e) Yadav, J. S.; Krishana, G. G.; Kumar, S. N. Tetrahedron 2010, 66, 480.
(f) Fuwa, H.; Saito, A.; Sasaki, M. Angew. Chem., Int. Ed. 2010, 49, 3041.
(g) Martinez-Solorio, D.; Jennings, M. P. J. Org. Chem. 2010, 75, 4095.
(h) Yang, Z.; Zhang, B.; Zhao, G.; Yang, J.; Xie, X.; She, X. Org. Lett. 2011, 13, 5916.
(ⅰ) Raghavan, S.; Samanta, P. K. Org. Lett. 2012, 14, 2346.
(j) Sharma, G. V. M.; Reddy, S. V.; Ramakrishna, K. V. S. Org. Biomol. Chem. 2012, 10, 3689.
(k) Athe, S.; Chandrasekhar, B.; Roy, S.; Pradhan, T. K.; Ghosh, S. J. Org. Chem. 2012, 77, 9840. -
[6]
(a) Gallon, J.; Reymond, S.; Cossy, J. C. R. Chim. 2008, 1463.
(b) Bai, Y.; Dai, M. J. Curr. Org. Chem. 2015, 19, 871.
(c) Fuwa, H. Mar. Drugs, 2016, 14, 65. -
[7]
Evans, D. A.; Hoveyda, A. H. J. Am. Chem. Soc. 1990, 112, 6447. doi: 10.1021/ja00173a071
-
[8]
(a) Omura, K.; Swern, D. Tetrahedron 1978, 34, 1651.
(b) Huang, S. L.; Omura, K.; Swern, D.; Further, D. Synthesis 1978, 297. -
[9]
(a) Paterson, I.; Gibson, K. R.; Oballa, R. M. Tetrahedron Lett. 1996, 37, 8585.
(b) Evans, D. A.; Coleman, P. J. J. Org. Chem. 1997, 62, 788. -
[10]
Chen, K. M.; Hardtmann, G. E.; Prasad, K.; Repic, O.; Shapiro, M. J. Tetrahedron Lett. 1987, 28, 155. doi: 10.1016/S0040-4039(00)95673-9
-
[11]
Noyori, R. Angew. Chem., Int. Ed. 2002, 41, 2008. doi: 10.1002/1521-3773(20020617)41:12<2008::AID-ANIE2008>3.0.CO;2-4
-
[12]
Zakharkin, L. I.; Khorlina, I. M. Tetrahedron Lett. 1962, 3, 619. doi: 10.1016/S0040-4039(00)70918-X
-
[13]
Keck, G. E.; Boden, E. P.; Mabury, S. A. J. Org. Chem. 1985, 50, 709. doi: 10.1021/jo00205a036
-
[14]
Arrayás, R. G.; Adrio, J.; Carretero, J. C. Angew. Chem., Int. Ed. 2006, 45, 7674. doi: 10.1002/(ISSN)1521-3773
-
[15]
(a) Kraus, G. A.; Taschner, M. J. J. Org. Chem. 1980, 45, 1175.
(b) Bal, B. S.; Childers, W. E.; Pinnick, H. W. Tetrahedron 1981, 37, 2091. -
[16]
(a) Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn. 1979, 52, 1989.
(b) Yeung, K.-S.; Paterson, I. Chem. Rev. 2005, 105, 4237.
(c) Parenty, A.; Moreau, X.; Campagne, J. M. Chem. Rev. 2006, 106, 911. -
[17]
Tamao, K.; Maeda, K.; Tanaka, T.; Ito, Y. Tetrahedron Lett. 1988, 29, 6955. doi: 10.1016/S0040-4039(00)88485-3
-
[18]
(a) Goossen, L. J.; Paetzold, J.; Koley, D. Chem. Commun. 2003, 706.
(b) Neveux, M.; Bruneau, C.; Dixneuf, P. H. J. Chem. Soc., Perkin Trans. 11991, 1197. -
[19]
Brown, H. C.; Jadhav, P. K. J. Am. Chem. Soc. 1981, 105, 2092.
-
[20]
Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900. doi: 10.1002/anie.200200556
-
[21]
Duprat de Paule, S.; Jeulin, S.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Champion, N.; Dellis, P. Eur. J. Org. Chem. 2003, 1931.
-
[22]
Lee, K.-C.; Lin, M.-J.; Loh, T.-P. Chem. Commun. 2004, 2456.
-
[23]
Vugts, D. J.; Veum, L.; al-Mafraji, K.; Lemmens, R.; Schmitz, R. F.; de Kanter, F. J. J.; Groen, M. B.; Hanefeld, U.; Orru, R. V. A. Eur. J. Org. Chem. 2006, 1672.
-
[24]
Brown, H. C.; Jadhav, P. K.; Perumal, P. T. Tetrahedron Lett. 1984, 25, 5111. doi: 10.1016/S0040-4039(01)81537-9
-
[25]
Ouellet, S. G.; Tuttle, J. B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 32. doi: 10.1021/ja043834g
-
[26]
Dossetter, A. G.; Jamison, T. F.; Jacobsen, E. N. Angew. Chem., Int. Ed. 1999, 38, 2398. doi: 10.1002/(ISSN)1521-3773
-
[27]
Ito, H.; Inoue, T.; Iguchi, K. Org. Lett. 2008, 10, 3873. doi: 10.1021/ol801395q
-
[28]
Jadhav, P. K.; Bhat, K. S.; Perumal, P. T.; Brown, H. C. J. Org. Chem. 1986, 51, 432. doi: 10.1021/jo00354a003
-
[29]
Yu, W.; Mei, Y.; Kang, Y.; Hua, Z.; Jin, Z. Org. Lett. 2004, 6, 3217. doi: 10.1021/ol0400342
-
[30]
Masamune, S.; Roush, W. R.; Blanchette, M. A.; Choy, W.; Davis, J. T.; Essenfeld, A. P.; Sakai, T. Tetrahedron Lett. 1984, 25, 2183. doi: 10.1016/S0040-4039(01)80205-7
-
[31]
Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn. 1990, 55, 7.
-
[32]
Nahm, S.; Weinreb, S. M. Tetrahedron Lett. 1981, 22, 3815. doi: 10.1016/S0040-4039(01)91316-4
-
[33]
Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. J. Am. Chem. Soc. 1997, 119, 6496. doi: 10.1021/ja970402f
-
[34]
Demico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. J. Org. Chem. 1997, 62, 6974. doi: 10.1021/jo971046m
-
[35]
Lee, C.-L. K.; Lee, C.-H. A.; Tan, K.-T.; Loh, T. -P. Org. Lett. 2004, 6, 1281. doi: 10.1021/ol049633z
-
[36]
(a) Breit, B. Chem. Commun. 1997, 591.
(b) Breit, B. Eur. J. Org. Chem. 1998, 63, 1123. -
[37]
For reviews, see:
(a) Hoveyda, A. H.; Zhugralin, A. R. Nature 2007, 450, 243.
(b) Gradillas, A.; Perez-Castells, J. Angew. Chem., Int. Ed. 2006, 45, 6086.
(c) Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4490.
(d) Deiters, A.; Martin, S. F. Chem. Rev. 2004, 104, 2199.
(f) Rstner, A. F. Angew. Chem., Int. Ed. 2000, 39, 3012, and references therein. -
[38]
Yu, W.; Mei, Y.; Kang, Y.; Hua, Z.; Jin, Z. Org. Lett. 2004, 6, 3217. doi: 10.1021/ol0400342
-
[39]
Laganis, E. D.; Chenard, B. L. Tetrahedron Lett. 1984, 25, 5831. doi: 10.1016/S0040-4039(01)81697-X
-
[40]
Hong, S. H.; Sanders, H. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160. doi: 10.1021/ja052939w
-
[41]
(a) Wu, H.; Radomkit, S.; O'Brien, J. M.; Hoveyda, A. H. J. Am. Chem. Soc. 2012, 134, 8277.
(b) Radomkit, S.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2014, 53, 3387. -
[42]
Wang, C.; Haeffner, F.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2013, 52, 1939. doi: 10.1002/anie.v52.7
-
[43]
Smith Ⅲ, A. B.; Adams, C. M.; Kozmin, S. A.; Paone, D. V. J. Am. Chem. Soc. 2001, 123, 5925. doi: 10.1021/ja0106164
-
[44]
Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483. doi: 10.1021/cr00032a009
-
[45]
Hicks, D. R.; Fraser-Reid, B. Synthesis 1974, 203.
-
[46]
Shiina, I.; Fukui, H.; Sasaki, A. Nat. Protoc. 2007, 2, 2312. doi: 10.1038/nprot.2007.316
-
[47]
(a) Tokunaga, M.; Larrow, J. F.; Kakiuchi, F.; Jacobsen, E. N. Science 1997, 277, 936.
(b) Schaus, S. E.; Brandes, B. D.; Larrow, J. F.; Tokunaga, M.; Hansen, K. B.; Gould, A. E.; Furrow, M. E.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 1307. -
[48]
Chakraborty, T. K.; Chattopadhyay, A. K. J. Org. Chem. 2008, 73, 3578. doi: 10.1021/jo800181n
-
[49]
Blanchette, M. A.; Choy, W.; Davis, J. T.; Essenfeld, A. P.; Masamune, S.; Roush, W. R.; Sakai, T. Tetrahedron Lett. 1984, 25, 2183. doi: 10.1016/S0040-4039(01)80205-7
-
[50]
Bal, B. S.; Childers, W. E.; Pinnick, H. W. Tetrahedron 1981, 37, 2091. doi: 10.1016/S0040-4020(01)97963-3
-
[51]
Liu, K.; Taylor, R. E.; Kartika, R. Org. Lett. 2006, 8, 5393. doi: 10.1021/ol0623318
-
[52]
Nahm, S.; Weinreb, S. M. Tetrahedron Lett. 1981, 22, 3815. doi: 10.1016/S0040-4039(01)91316-4
-
[53]
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155. doi: 10.1021/jo00170a070
-
[54]
Burgos, C. H.; Canales, E.; Matos, K.; Soderquist, J. A. J. Am. Chem. Soc. 2005, 127, 8044. doi: 10.1021/ja043612i
-
[55]
Inanaga, J.; Baba, Y.; Hanamoto, T. Chem. Lett. 1993, 241.
-
[56]
Lee, E. Pure Appl. Chem. 2005, 77, 2073.
-
[57]
Schaus, S. E.; Branalt, J.; Jacobsen, E. N. J. Org. Chem. 1998, 63, 403. doi: 10.1021/jo971758c
-
[58]
Luche, J. L. J. Am. Chem. Soc. 1978, 100, 2226. doi: 10.1021/ja00475a040
-
[59]
Ireland, R. E.; Wipf, P.; Armstrong, J. D., Ⅲ J. Org. Chem. 1991, 56, 650. doi: 10.1021/jo00002a030
-
[60]
Mori, G.; Kuwahar, S. Tetrahedron 1982, 38, 521. doi: 10.1016/0040-4020(82)80096-3
-
[61]
(a) Trost, B. M.; Kondo, Y. Tetrahedron Lett. 1991, 32, 1613.
(b) Walsh, T. F.; Toupence, R. B.; Ujjainwalla, F.; Young, J. R.; Goulet, M. T. Tetrahedron 2001, 57, 5233. -
[62]
(a) Lu, Y.; Kim, I. S.; Hassan, A.; Del Valle, D. J.; Krische, M. J. Angew. Chem., Int. Ed. 2009, 48, 5018.
(b) Han, S. B.; Hassan, A.; Kim, I. S.; Krische, M. J. J. Am. Chem. Soc. 2010, 132, 15559. -
[63]
(a) Yang, Z.; Xie, X. G.; Jing, P.; Zhao, G. Y.; Zheng, J. Y.; Zhao, C. G.; She, X. G. Org. Biomol. Chem. 2011, 9, 984.
(b) White, J. D.; Hong, J.; Robarge, L. A. Tetrahedron Lett. 1999, 40, 1463.
(c) Hornberger, K. R.; Hamblett, C. L.; Leighton, J. L. J. Am. Chem. Soc. 2000, 122, 12894. -
[64]
(a) Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953.
(b) Hanessian, S.; Giroux, S.; Larsson, A. Org. Lett. 2006, 8, 5481.
(c) Seden, P. T.; Charmant, J. P. H.; Willis, C. L. Org. Lett. 2008, 10, 1637. -
[65]
Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. J. Org. Chem. 2004, 69, 1822. doi: 10.1021/jo030367x
-
[66]
Denmark, S. E.; Fujimori, S. J. Am. Chem. Soc. 2005, 127, 8971. doi: 10.1021/ja052226d
-
[67]
(a) Garegg, P. G.; Samuclson, B. Synthesis 1979, 813.
(b) Garegg, P. J. Pure Appl. Chem. 1984, 56, 845. -
[68]
Jang, K. P.; Choi, S. Y.; Chung, Y. K.; Lee, E. Org. Lett. 2011, 13, 2476. doi: 10.1021/ol2007296
-
[69]
(a) Evans, D. A.; Bender, S. L.; Morris, J. J. Am. Chem. Soc. 1988, 110, 2506.
(b) Blanchette, M. A.; Malamas, M. S.; Nantz, M. H.; Roberts, J. C.; Somfai, P.; Whritenour, D. C.; Masamune, S.; Kageyama, M.; Tamura, T. J. Org. Chem. 1989, 54, 2817.
(c) de Koning, C. B.; Green, I. R.; Michael, J. P.; Oliveira, J. R. Tetrahedron 2001, 57, 9623.
(d) Liu, B.; Zhou, W.-S. Tetrahedron Lett. 2003, 44, 4933. -
[70]
Bai, Y.; Davis, D. C.; Dai, M. Angew. Chem., Int. Ed. 2014, 53, 6519. doi: 10.1002/anie.201403006
-
[1]
-

图 1 (+)-Neopeltolide和eucascandrolide A的结构
Figure 1 Structures of (+)-neopeltolide and leucascandrolide A

图式 1 Panek的全合成
Scheme 1 Panek's total synthesis
Reagents and conditions: (a) (ⅰ) BH3•SMe2, THF; (ⅱ) TBDPSCl, DMF; (ⅲ) DIBAL-H, Et2O, -78oC; (ⅳ) 1, 3-propanedithiol, I2, CHCl3, 68% (over 4 steps). (b) (ⅰ) t-BuLi, THF, 5, -78oC; (ⅱ) CaCO3, MeI, MeCN/H2O, 50% (over 2 steps). (c) (ⅰ) Zr(Ot-Bu)4, i-PrCHO, -78oC. (ⅱ) Me3OBF4, Proton Sponge, CH2Cl2, 90%. (ⅲ) 49% HF in H2O, MeCN, r.t., 91%. (ⅳ) (COCl)2, DMSO, CH2Cl2, Et3N, -78oC to r.t., 73% (over 4 steps). (d) 8, TfOH, CH2Cl2, benzene, -78oC, 75% (dr 10:1). (e) (ⅰ) NaCN, DMF, 60oC, 84%; (ⅱ) DIBAL-H, Et2O, -78 oC, 96%; (ⅲ) DIBAL-H, CH2Cl2, -78oC, 60%; (f) (ⅰ) NaClO2, NaH2PO4•H2O, t-BuOH, H2O, 85%. (ⅱ) 2, 4, 6-trichlorobenzoyl chloride, toluene, DMAP, Et3N, 44%; (g) (ⅰ) Hg(O2CCF3)2, then NaBH4, 63% (d.r. > 20:1); (ⅱ) 12, EDCI, HOBT•H2O, CH2Cl2, 99%; (ⅲ) 18-crown-6, KHMDS, -78oC, then 13, 85oC, 62%

图式 2 Kozmin消旋体全合成
Scheme 2 Kozmin's total synthesis of (±)-neopeltolide
Reagents and conditions: (a) (ⅰ) CF3CO2H, 0 ℃, aq. NH4OH; (ⅱ) BnOC(NH)CCl3, TfOH (cat.); (ⅲ) PdCl2, CuCl2, DMF, H2O, O2 (64% for 3 steps). (b) (ⅰ) 16, Cy2BCl, Et3N; (ⅱ) PPh3MeBr, KHMDS, then HCl quench (75% for 2 steps). (c) (ⅰ) Et2BOMe, NaBH4; (ⅱ) TMSOK, Et2O; (ⅲ) 2, 4, 6-trichlorobenzoyl chloride, toluene, DMAP, Et3N (55% for 3 steps). (d) (ⅰ) H2, Pd/C; (ⅱ) p-NO2C6H4CO2H, PPh3, DEAD; (ⅲ) K2CO3, MeOH; (ⅳ) Me3OBF4; (ⅴ) H2, Pd(OH)2 (41% for 5 steps). (e) 20, PPh3, DIAD, 83%

图式 3 Maier全合成
Scheme 3 Maier's total synthesis
Reagents and conditions: (a) (ⅰ) (S)-BINAP, Ru(Ⅱ), H2; (ⅱ) TBDPSCl, Imidazole; (ⅲ) DIBAL-H, -78 oC (90% for 3 steps). (b) (ⅰ) 22, CH2Cl2; (ⅱ) Me3OBF4; (c) (ⅰ) OsO4, NMO; (ⅱ) NaIO4, MeOH (70% for 4 steps). (d) Ph3P=CHCOSEt, CH2Cl2; (ⅱ) MeMgBr, CuBr•SMe2, (S, R)-Josiphos (86% for 2 steps). (e) (ⅰ) Et3SiH, Pd/C; (ⅱ) 27, TFA, -5oC; (ⅲ) K2CO3, MeOH (62% for 3 steps). (f) (ⅰ) MOMCl, DIPEA; (ⅱ) H2, Pd/C; (ⅲ) DMP, NaHCO3; (ⅳ) NaClO2, NaH2PO4(86% for 4 steps); (g) (ⅰ) TBAF, THF; (ⅱ) Yamaguchi lactonization; (ⅲ) HCl (80% for 3 steps); (h) 20, PPh3, DIAD, 80%

图式 4 Floreancig的全合成
Scheme 4 Floreancig's total synthesis
Reagents and conditions: (a) (ⅰ) NaH, Cl3CCN, Et2O, 0 ℃, 100%; (ⅱ) 31, TfOH, cyclohexane, 77%; (b) 33, CuI, i-Pr2NH, [(Ph3P)2PdCl2], 89%; (c) [Pt(DVDS)], THF, then H2O2, KF, Bu4NF, KHCO3, DMF, 40 ℃, 57% (67% based on starting-material purity); (d) (ⅰ) EtCHO, SmI2, THF, -10℃, 77%; (ⅱ) Me3OBF4, proton sponge, CH2Cl2, 0 ℃, 93%; (e) (ⅰ) LiOH, H2O, MeOH, 45 ℃; (ⅱ) Et3N, 2, 4, 6-Cl3BzCl, THF, then DMAP, toluene, 65 ℃, 72% (two steps). (f) HOAc, Na2CO3, [{Ru(p-cymene)Cl2}2], 1-decyne, (2-furyl)3P, toluene, 80 ℃, 82%. (g) DDQ, 2, 6-Cl2Py, LiClO4, DCE, 65%. (h) H2, Pd/C, EtOH, 74%. (ⅰ) NaBH4, MeOH, 95%. (ⅱ) 20, PPh3, DIAD, 93%

图式 5 Fuwa和Sasaki全合成
Scheme 5 Fuwa and Sasaki's total synthesis
Reagents and conditions: (a) (ⅰ) (+)-Ipc2BOMe, allylMgBr; (ⅱ) MPMOC(=NH)CCl3, La(OTf)3. (b) (ⅰ) Grubbs' Ⅱ, methyl acrylate; (ⅱ) DIBALH, -78 ℃ (45% for 4 steps). (c) (ⅰ) (-)-DET, Ti(Oi-Pr)4, t-BuO2H; (ⅱ) PPh3, I2; (ⅲ) Zn, AcOH (73% for 3 steps). (d) (ⅰ) BOMCl, DIPEA; (ⅱ) DDQ, pH 7 buffer (72% for 2 steps). (e) (ⅰ) Ac2O, Et3N, DMAP; (ⅱ) KHDMS, (PhO)2P(O)Cl, -78 ℃; (f) (ⅰ) B-MeO-9-BBN, t-BuLi, then 47, Pd(PPh3)4; (ⅱ) Grubbs Ⅱ, 70 ℃; (ⅲ) H2, Pd/C, EtOAc (63% for 3 steps); (g) (ⅰ) TBAF, THF; (ⅱ) SO3•Pyr., Et3N, DMSO; (h) (ⅰ) NaClO2, NaH2PO4, t-BuOH; (ⅱ) TMSCHN2, MeOH (86% for 4 steps). (ⅰ) (ⅰ) DDQ, pH=7 buffer; (ⅱ) TMSOK, Et2O; (ⅲ) Yamaguchi lactonization (92% for 3 steps). (j) H2, Pd(OH)2/C; (k) 20, PPh3, DIAD, 61%

图式 6 Roulland全合成
Scheme 6 Roulland's total synthesis
Reagents and conditions: (a) (ⅰ) (R)-(+)-Synphos-Ru(Ⅱ), H2; (ⅱ) MeNH(OMe)•HCl, AlMe3(87% for 2 steps). (b) (ⅰ) (2-methylallyl)magnesium chloride; (ⅱ) PhCHO, SmI2 (68% for 2 steps). (c) (ⅰ) acryloyl chloride, DIPEA; (ⅱ) Grubbs Ⅱ, reflux (83% for 2 steps). (d) H2, Pd/C, then, PPTS, n-PrOH; (ⅱ) MeO3BF4; (ⅲ) DIBALH, -78 ℃ (69% for 3 steps). (e) InBr3, 56%, 77%. (f) [CpRu(MeCN)3]PF6, but-3-enal, AcOH, 28%~58%. (g) (ⅰ) OsO4, NaIO4; (ⅱ) NaClO2, KH2PO4; (ⅲ) Yamaguchi lactonization; (ⅳ) NaBH4, MeOH (73% for 4 steps). (h) 20, PPh3, DIAD, 75%

图式 7 Paterson全合成
Scheme 7 Paterson's total synthesis
Reagents and conditions: (a) (ⅰ) (S)-BINAP-Ru(Ⅱ), H2; (ⅱ) TBSCl, DMF; (ⅲ) DIBALH, -78 ℃ (76% for 3 steps). (b) (ⅰ) (-)-Ipc2B(2-methylallyl); (ⅱ) NaH, MeI (77% for 2 steps). (c) (ⅰ) O3, DMS; (ⅱ) (OMe)2POCH2CO2Me, NaH (68% for 2 steps). (d) (ⅰ) DIBALH, -78 ℃; (ⅱ) DMP, NaHCO3; (ⅲ) 62, Hantzch ester, dr=3:1 (86% for 3 steps). (e) 65, 4 Å MS, 8 d, CHCl3, 78%. (f) (ⅰ) DDQ, pH 7 buffer; (ⅱ) DMP, NaHCO3; (ⅲ) NaClO2, NaH2PO4; (ⅳ) TBAF, THF (88% for 4 steps). (g) (ⅰ) Yamaguchi lactonization; (ⅱ) NaBH4, MeOH (79% for 2 steps). (h) 20, PPh3, DIAD, 53%

图式 8 Ghosh全合成
Scheme 8 Ghosh's total synthesis
Reagents and conditions: (a) (ⅰ) Lipase PS-30, n-PrOH; (ⅱ) BH3•Me2S, THF (98% for 2 steps). (b) (ⅰ) Swern oxidation; (ⅱ) (CH2OH)2, PPTS (89% for 2 steps). (c) (ⅰ) LiAlH4, Et2O; (ⅱ) OsO4, NaIO4; (ⅲ) (+)-Ipc2BOMe, allylMgBr (58% for 3 steps). (d) (ⅰ) NaH, MeI; (ⅱ) OsO4, NaIO4; (ⅲ) (-)-Ipc2BOMe AllylMgBr (64% for 3 steps). (e) (ⅰ) PPTS, AcOH; (ⅱ) LiCl, DIPEA; MeCOCH2P(O)(OMe)2; (ⅲ) TESOTf, Et3N (81% for 3 steps). (f) 74, TFA, 4 Å MS, 75, 83%; (g) (ⅰ) (CH2OH)2, PTSA; (ⅱ) NaCN, DMF (48% for 2 steps). (h) (ⅰ) 10% NaOH, EtOH; (ⅱ) PTSA, acetone (74% for 2 steps). (ⅰ) Yamaguchi esterification, 40%; (j) (ⅰ) Pd/C, H2; (ⅱ) NaBH4, EtOH (71% for 2 steps). (k) 20, PPh3, DIAD, 78%

图式 9 Scheidt全合成
Scheme 9 Scheidt's total synthesis
Reagents and conditions: (a) (ⅰ) (OMe)MeNH2Cl, THF, i-PrMgBr; (ⅱ) PMB-OC(NH)CCl3, PPTS (68% for 2 steps). (b) (ⅰ) 82, t-BuLi; (ⅱ) DDQ, buffer. (c) (ⅰ) SmI2, PhCHO; (ⅱ) MeOTf, DTBMP; (ⅲ) K2CO3, MeOH (29% from 81). (d) (ⅰ) 85, Yamaguchi condition; (ⅱ) HF•pyridine; (ⅲ) TEMPO, H5C6I(OAc)2 (76% for 3 steps). (e) (ⅰ) Sc(OTf)3, CaSO4; (ⅱ) DMSO, H2O (21% for 2 steps). (f) (ⅰ) NaBH4, MeOH; (ⅱ) 20, DIAD, Ph3P (76% for 2 steps).

图式 10 Lee全合成
Scheme 10 Lee's total synthesis
Reagents and conditions: (a) (ⅰ) 87, CSA, CH2Cl2; (ⅱ) NaH, BnBr, TBAI, THF/DMF (5:1); (ⅲ) O3, CH2Cl2, -78 ℃, Ph3P (49% for 3 steps). (b) (ⅰ) CH2C(CH3)CH2TMS, TiCl4, -78 ℃; (ⅱ) 89, DCC, DMAP, CH2Cl2 (78% for 2 steps). (c) (ⅰ) Rh(CO)2(acac), H2/CO; (ⅱ) H2SO4, HC(OMe)3; (ⅲ) KOH, EtOH; (ⅳ) NaH, MeI (40% for 4 steps). (d) (ⅰ) H2, Pd/C, MeOH; (ⅱ) 92, DCC, DMAP; (ⅲ) DDQ, pH 7 buffer (81% for 3 steps). (e) (ⅰ) TESOTf, TMSOAc; (ⅱ) K2CO3, MeOH (68% for 2 steps). (f) 20, DIAD, Ph3P, 79%

图式 11 Fuwa全合成
Scheme 11 Fuwa's total synthesis
Reagents and conditions: (a) (ⅰ) TBSCl, imidazole; (ⅱ) OsO4, NaIO4 (58% for 2 steps). (b) (ⅰ) (+)-Ipc2BOMe, allylMgBr; (ⅱ) Grubbs' Ⅱ, methyl acrylate (78% for 2 steps). (c) (ⅰ) BOMCl, DIPEA; (ⅱ) TBAF, THF (90% for 2 steps). (d) (ⅰ) DBU, toluene; (ⅱ) TMSOK, Et2O; (ⅲ)98, Yamaguchi esterification (38% for 3 steps). (e) (ⅰ) Grubbs' Ⅱ, toluene, 1, 4-benzoquinone; (ⅱ) H2, Pd/C, Pd(OH)2/C (79% for 2 steps)

图式 12 Hoveyda全合成
Scheme 12 Hoveyda's total synthesis
Reagents and conditions: (a) 101 (7.5 mol%), B2(pin)2, NaBO3, MeOH, THF, 86% (er=95:5). (b) (ⅰ) (2-methylallyl)magnesium chloride, PPTS, CH2Cl2; (ⅱ) SmI2, PhCHO; (ⅱ) Me3OBF4, proton sponge; (ⅲ) KOH, MeOH (53% from 52). (c) 1-(vinyloxy)butane, 103 (0.6 mol%), benzene, 88%, (er=99:1). (d) (ⅰ) 1.0 mol/L HCl, THF; (ⅱ) NaClO2, NaH2PO4 (86% for 2 steps). (e) EDC, DMAP, Et3N, 88%. (f) (ⅰ) 107 (8.0 mol%), toluene, 89%, (Z/E > 98:2). (g) (ⅰ) Pd/C, H2, EtOH; (ⅱ) 20, DIAD, Ph3P (68% for 2 steps)

图式 13 Hoveyda侧链合成
Scheme 13 Hoveyda's synthesis of the side chain
Reagents and conditions: (a) 110 (3.0 mol%), vinyl-B(pin), 86% (Z:E > 98:2). (b) Pd(dppf)Cl2 (10 mol%), 112, 82%. (c) (ⅰ) PPh3, CBr4, 2, 6-lutidine, 81%; (ⅱ) CuCN (50 mol%), allylMgBr, 70%. (d) Z-2-butene-1, 4-diol, 115 (10 mol%), 70%. (e) (ⅰ) Dess-Martin, NaHCO3; (ⅱ) NaClO2, NaH2PO4•H2O, t-BuOH, H2O, 85% (75% for 2 steps)

图式 14 Hong大环内酯骨架合成
Scheme 14 Hong's synthesis of macrolactone core
Reagents and conditions: (a) (ⅰ) n-BuLi, THF, 1 h, then (R)-5-iodo-4-methylpenten; (ⅱ) n-BuLi, THF, then (R)-epichlorohydrin (66% for 2 steps). (b) (ⅰ) EtMgBr, CuI; (ⅱ) MeI, CaCO3 (84% for 2 steps). (c) (ⅰ) PhCHO, SmI2; (ⅱ) MeO3BF4 (81% for 2 steps). (d) (ⅰ) AD mix-β; (ⅱ) NaH, N-p-toluene sulfonylimidazole (85% for 2 steps). (e) (ⅰ) n-BuLi, HMPA, THF, 122, 75%; (f) MnO2, 3 h, then dimethyltriazolium iodide, MnO2, DBU, MeOH; (ⅱ) 0.1 mol/L LiOH, MeOH/THF (V:V=1:3) (77% for 2 steps). (g) (ⅰ) MNBA, CH2Cl2; (ⅱ) MeI, CaCO3; (ⅲ) NaBH4, MeOH (57% for 3 steps)

图式 15 Subhash Ghosh大环内酯骨架合成
Scheme 15 Subhash Ghosh's synthesis of macrolactone core
Reagents and conditions: (a) (ⅰ) TBDPSCl, DMF, imidazole; (ⅱ) mCPBA, CH2Cl2; (ⅲ) (S, S)-(Salen)Co(Ⅲ)(OAc), H2O; (b) (ⅰ) AcCl, -78 ℃; (ⅱ) MsCl, Et3N; (ⅲ) K2CO3, MeOH (71% from 125); (c) (ⅰ) vinylmagnesium bromide, CuI, THF, -40~0 ℃; (ⅱ) KH, MeI (81% for 2 steps); (ⅲ) (R, R)-(Salen)Co(Ⅲ)(OAc), H2O (60% from 126A). (d) (ⅰ) EtMgBr, CuI; (ⅱ) BnO(NH)CCl3, CF3SO3H; (ⅲ) TBAF, THF (77% for 3 steps). (e) (ⅰ) TsCl, Et3N; (ⅱ) NaCN, NaI; (ⅲ) DIBAL-H, -78 ℃ (75% for 3 steps). (f) 130, LiCl, DIPEA, 77%. (g) (ⅰ) HF-Py, CH3CN; (ⅱ) Pd(CH3CN)4BF4, CH2Cl2, r.t. (38% for 2 steps). (h) (ⅰ) Pd/C, H2; (ⅱ) TEMPO, BAIB; (ⅲ) NaClO2, KH2PO4; (ⅳ) Yamaguchi lactonization (61% for 4 steps)

图式 16 Taylor大环内酯骨架合成
Scheme 16 Taylor's synthesis of macrolactone core
Reagents and conditions: (a) (ⅰ) BH3•SMe2, THF; (ⅱ) (OMe)MeNH2Cl, i-PrMgCl; (ⅲ) DMP, NaHCO3 (86% for 3 steps). (b) (ⅰ) 134, BF3•Et2O; (ⅱ) BOMCl, DIPEA (55% for 2 steps). (c) 136, n-BuLi, LiDBB, 92%. (d) (ⅰ) SmI2, PhCHO; (ⅱ) Me3OBF4, Proton sponge (79% for 2 steps). (e) ICl, Na2S2O3, 71%. (f) (ⅰ) ethyl propiolate, Bu3P; (ⅱ) AIBN, n-Bu3SnH (93% for 2 steps). (g) (ⅰ) KOH, MeOH; (ⅱ) TCBCl, Et3N, DMAP; (ⅲ) H2, Pd/C (83% for 3 steps)

图式 17 Raghavan大环内酯骨架合成
Scheme 17 Raghavan's synthesis of macrolactone core
Reagents and conditions: (a) (ⅰ) 143, Hantzch ester; (ⅱ) TBME, TFA, 144, (S, S)-(Salen) (56% for 2 steps). (b) (ⅰ) NaBH4, CeCl3; (ⅱ) Ac2O, TEA (91% for 2 steps). (c) (ⅰ) LDA, TMSCl; (ⅱ) etheral CH2N2 (90% for 2 steps). (d) (ⅰ) LAH, THF; (ⅱ) TBSCl; (ⅲ) LDBB, THF, -78 ℃, then 148 (51% for 3 steps). (e) (ⅰ) IBX, DMSO; (ⅱ) TBAF, THF (81% for 2 steps). (f) (ⅰ) isobutanal, -50 ℃, Zr(Oi-Bu)4; (ⅱ) NaH, MeI (75% for 2 steps). (g) (ⅰ) K2CO3, MeOH; (ⅱ) PhI(OAc)2, TEMPO; (ⅲ) NaClO2, NaH2PO4; (ⅳ) Yamaguchi lactonization (39% for 4 steps). (h) Hg(OTFA)2, THF, 24 h, then NaBH4, aq. NaOH, 24 h, 60%

图式 18 大环内酯骨架合成
Scheme 18 Yadav's synthesis of macrolactone coreYadav
Reagents and conditions: (a) (ⅰ) NaH, BnBr, TBAI; (ⅱ) H2O2, (PhSe)2, t-BuO2H, CH2Cl2 (74% for 2 steps). (b) O3, -78 ℃, Me2S, 88%. (c) (S)-pent-4-ene-1, 2-diol, TFA, CH2Cl2then K2CO3, CH3OH, 56%. (d) (ⅰ) TsCl, Et3N, CH2Cl2; (ⅱ) TBDPSCl, DMAP, imidazole; (ⅲ) NaI, reflux (84% for 3 steps). (e) Zn, EtOH, reflux, 96%. (f) (ⅰ) Me3OBF4, CH2Cl2; (ⅱ) H2, Raney nickel, EtOH; (ⅲ) DMP, NaHCO3, CH2Cl2; (ⅳ) PTSA, CH(OMe)3, CH3OH (56% for 5 steps)

图式 19 She大环内酯骨架合成
Scheme 19 She's synthesis of macrolactone core
Reagents and conditions: (a) [Ir(cod)Cl]2, (R)-Cl, OMe-BIPHEP, allyl acetate, 70%. (b) PdCl2, CuCl2, CO, CH3CN, MeOH, r.t., 4 h, 83%. (c) (ⅰ) BnO(NH=C)CCl3, CF3SO3H; (ⅱ) Grubbs' Ⅱ, MeOH, 60 ℃ (73% for 2 steps). (d) (ⅰ) LiOH, MeOH; (ⅱ) 98, MNBA, DMAP (87% for 2 steps). (e) (ⅰ) Hoveyda-Grubbs' Ⅱ, toluene, 80 ℃; (ⅱ) Pd/C, H2, EtOH (53% for 2 steps)

图式 20 Sharma醇片段169的合成
Scheme 20 Sharma's synthesis of alcohol fragment 169
Reagents and conditions: (a) (ⅰ) n-propyl bromide, Mg, -40 ℃; (ⅱ) (COCl)2, DMSO, Et3N, -78 ℃; (ⅲ) LiAlH4, LiI; (ⅳ) TBDPSCl, imidazole (46% for 4 steps). (b) (ⅰ) CuCl2, CH3CN; (ⅱ) BzCl, Bu2SnO; (ⅲ) TsCl, Et3N; (ⅳ) K2CO3, MeOH (58% for 4 steps). (c) (ⅰ) n-BuLi, BF3•Et2O; (ⅱ) MeI, NaH; (ⅲ) PPTS, MeOH (41% for 3 steps). (d) (ⅰ) Red-Al, Et2O; (ⅱ) (-)DIPT, Ti(Oi-Pr)4; (ⅲ) Me3Al, hexane (60% for 3 steps). (e) PPh3, imidazole, I2, 70%. (f) TBAF, THF, 84%

图式 21 Sharma大环内酯骨架合成
Scheme 21 Sharma's synthesis of macrolactone core
Reagents and conditions: (a) (ⅰ) (-)-DIPT, Ti(OiPr)4, C6H5C(CH3)2OOH, 4 Å MS; (ⅱ) Red-Al, THF, 0 ℃~r.t. (68% for 2 steps). (b) (ⅰ) p-anisaldehyde dimethyl acetal, PPTS, CH2Cl2; (ⅱ) DIBALH, CH2Cl2; (ⅲ) TBDPSCl, imidazole, CH2Cl2 (63% for 3 steps). (c) (ⅰ) CuCl2·2H2O, CH3CN; (ⅱ) p-TsCl, Bu2SnO, Et3N, CH2Cl2; (ⅲ) K2CO3, MeOH (62% for 3 steps). (d) (ⅰ) vinyl magnesium bromide, CuI, THF, -20 ℃; (ⅱ) MOMCl, DIPEA, DMAP (75% for 2 steps). (e) (ⅰ) TBAF, THF; (ⅱ) TEMPO, BAIB (60% for 2 steps). (f) (ⅰ) 152, DCC, DMAP, CH2Cl2; (ⅱ) Grubb's Ⅱ, CH2Cl2 (44% for 2 steps). (g) (ⅰ) DDQ, CH2Cl2; (ⅱ) Hg(CF3COO)2, CH2Cl2, KBr (76% for 2 steps). (h) (ⅰ) n-Bu3SnH, AIBN, toluene, reflux; (ⅱ) conc. HCl, MeOH (80% for 2 steps)

图式 22 Dai’s neopeltolide类似物合成
Scheme 22 Dai's synthesis of neopeltolide analogues
Reagents and conditions: (a) Pd(OAc)2, CuCl2, CO, 4 Å MS, DCE, 58%; (b) (ⅰ) HCl; (ⅱ) NaBH4 (72% for two steps); (c) 20, DIAD, Ph3P, 56%

图 4 Floreancig合成的neopeltolide类似物结构
Figure 4 Floreancig's synthesis of neopeltolide analogues
-


 下载:
下载:


























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 21
- 文章访问数: 4662
- HTML全文浏览量: 760
 下载:
下载:
