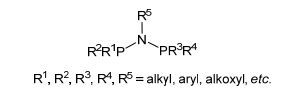
 图1
PNP配体的结构通式
Figure1.
General formula of the PNP ligands
图1
PNP配体的结构通式
Figure1.
General formula of the PNP ligands
引用本文:
刘睿, 钟向宏, 刘振宇, 梁胜彪, 朱红平. N-四氢糠基PNP配体/铬催化体系及其乙烯选择性齐聚性能[J]. 有机化学,
2017, 37(9): 2315-2321.
doi:
10.6023/cjoc201703010

Citation: Liu Rui, Zhong Xianghong, Liu Zhenyu, Liang Shengbiao, Zhu Hongping. Selective Ethylene Oligomerization Catalyzed by the Chromium Complex Bearing N-Tetrahydrofurfuryl PNP Ligand[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2315-2321. doi: 10.6023/cjoc201703010
Citation: Liu Rui, Zhong Xianghong, Liu Zhenyu, Liang Shengbiao, Zhu Hongping. Selective Ethylene Oligomerization Catalyzed by the Chromium Complex Bearing N-Tetrahydrofurfuryl PNP Ligand[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2315-2321. doi: 10.6023/cjoc201703010
N-四氢糠基PNP配体/铬催化体系及其乙烯选择性齐聚性能
摘要:
通过胺基锂盐分离的方法合成了一种未有报道过的N-四氢糠基PNP配体E.E分别与CrCl3(THF)3和Cr(CO)6反应生成化合物[{Ph2PN(CH2OC4H7) PPh2}CrCl2(μ-Cl)]2(1)和[Ph2PN(CH2OC4H7) PPh2]Cr(CO)4(2).这三个化合物通过谱学和元素分析表征,化合物2进一步经过X射线单晶结构确认.在甲基铝氧烷(MAO)或其它助剂作用下,考察了1、2以及E/CrCl3(THF)3、E/Cr(acac)3、E/CrCl2(THF)2催化体系催化乙烯齐聚的性能.这些体系高选择性地催化乙烯四聚,最高活性为15.9 kg(product)/g(Cr)·h,1-辛烯的选择性最高可达63.6%.
English
Selective Ethylene Oligomerization Catalyzed by the Chromium Complex Bearing N-Tetrahydrofurfuryl PNP Ligand
Abstract:
The N-tetrahydrofurfuryl diphoshinoamine (PNP) ligand (E) was synthesized by means of two-step salt elimination reactions where separation of the two kinds of the aminyl lithium salts for the respective reactions is necessary for obtaining a high yield of E. The ligand reacted with CrCl3(THF)3 and Cr(CO)6 to give P, P-chelation complexes[{Ph2PN(CH2OC4H7)-PPh2}CrCl2(μ-Cl)]2 (1) and[Ph2PN(CH2OC4H7)PPh2]Cr(CO)4 (2), respectively. Complexes E, 1 and 2 were characterized by spectroscopy and elemental analysis, of which complex 2 was further confirmed by X-ray crystallography. Upon activation with methylaluminoxane (MAO) or AlEt3, the catalyst systems including 1, 2, E/CrCl3(THF)3, E/Cr(acac)3 and E/CrCl2(THF)2 were investigated. The best catalytic activity was achieved by 15.9 kg (product)/g (Cr)·h in which a selectivity of 63.6% for 1-C8 was obtained.
-
线性α-烯烃中的1-己烯和1-辛烯是重要的有机化工原料和化学中间体, 用以制备增塑剂、表面活性剂、润滑剂、长链羧酸和环氧化合物等, 它们也是制备聚烯烃弹性体(POE)的不可缺少的共聚单体.近年来随着市场对1-己烯和1-辛烯需求量的不断增长, 学术界和工业界对这两种烯烃的制备技术和方法倍加关注[1].传统的方法如蜡裂解、烷烃催化裂解、烷烃脱氢、煤品抽提、萃取分离、脂肪醇脱氢等往往得到多种α-烯烃的混合物, 并含有其它杂质, 最后经纯化分离得到的1-己烯和1-辛烯质量占比很低[1d].乙烯齐聚催化法是多年来发展起来的重要方法, 它包括乙烯选择性齐聚和非选择性齐聚.乙烯非选择性齐聚法往往给出碳数呈Schulz-Flory或Poisson分布的多种α-烯烃产物[2], 相应的1-己烯和1-辛烯质量占比有一定限制, 而乙烯选择性齐聚法则能够高选择性制这两种烯烃, 这是目前该领域的研究重点和热点.
早在1964年, 联碳公司(UCC)的Manyik、Walker和Wilson在申请的聚乙烯专利中陈述, 使用部分氢化的三异丁基铝活化的2-乙基己酸铬催化乙烯聚合生成含丁基侧链的聚合物, 他们推测反应过程中发生了部分乙烯三聚生成1-己烯, 并进一步共聚插入聚乙烯增长链中[3]. 1977年, Manyik等[4]首次报道了一种由2-乙基己酸铬和部分水解的三异丁基铝组成的乙烯选择性三聚催化剂体系. 1999年, Phillips公司[5]改进了乙烯三聚催化剂, 优化形成2-乙基己酸铬/2, 5-二甲基吡咯/二乙基氯化铝/三乙基铝催化剂体系, 反应得到占比93.8%的1-己烯, 并将该技术推向工业化. 2002年, 英国石油(BP)公司Wass等[6]发现三氯化铬/磷氮磷(PNP)配体/甲基铝氧烷(MAO)体系也能高效地催化乙烯三聚, 生成近90%的1-己烯. 2004年, Sasol技术公司的研究人员[7]在英国石油公司的研究基础上, 对磷氮磷配体进行改进, 催化体系催化乙烯三聚联四聚, 生成67.5% 1-辛烯和22.7% 1-己烯, 目前该技术也正推向工业化. 2010年Gambarotta等[8]报道采用Ph2PN(R)(CH2)nN(R)PPh2 (n=2, R=Me; n=3, R=Me, Et, i-Pr)配体/三氯化铬/甲基铝氧烷体系催化乙烯四聚, 1-辛烯选择性高达91.0%, 但是活性不高.
目前的研究表明PNP配体是一种优异的配体.以PNP为骨架, 改变P和N原子上的取代基, 可以影响金属铬中心的电子和立体空间环境, 因而调控催化行为和结果[9]. PNP配体的结构通式如图 1所示, 本研究小组以及其他课题组[1, 10]也对其与铬组成的催化体系进行了综述. P原子上取代基变换的研究已经报道很多, 相比较而言N原子上的取代基变换研究尚有待深入.取代基中含有电子给体的原子, 这些原子易与铬中心发生电子给体-受体(Donor-Acceptor)作用.如图 2所示, 目前报道的N原子上含有烷氧基团的PNP配体A~D由Bercaw课题组研究, 其与CrCl3和MAO组成的催化体系催化乙烯得到C6组分在45%~66%范围内, C8组分在16%~34%内变化.取代基上给电子基团的长度和刚性影响到氧原子和铬中心的作用, 因而影响到催化过程中CrC6和CrC8环中间体的形成以及协同的3, 7-H和3, 9-H迁移, 最后还原消除生成1-己烯和1-辛烯[11].
图1
PNP配体的结构通式
Figure1.
General formula of the PNP ligands
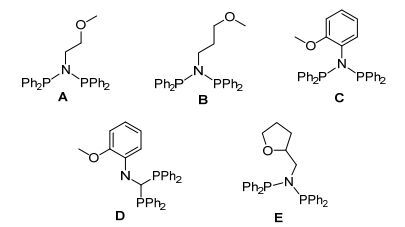 图2
N原子上含有醚基和酚氧烷基的PNP配体A~D和N原子上含四氢糠基的配体E
Figure2.
Diphoshinoamine ligands containing the N-ether or N-phenolic alkyl group for A~D and the N-tetrahydrofurfuryl group for E
图2
N原子上含有醚基和酚氧烷基的PNP配体A~D和N原子上含四氢糠基的配体E
Figure2.
Diphoshinoamine ligands containing the N-ether or N-phenolic alkyl group for A~D and the N-tetrahydrofurfuryl group for E
我们在这里报道一种N原子上连接四氢糠基的PNP配体(如图 2中的E), 这是一种新的PNP配体.以E/CrCl3/MAO组成的体系催化乙烯生成1-辛烯为主要产物(C8组分为65.5%, 其中1-C8占96.1%), 并含有1-己烯(C6组分为20.3%, 其中1-C6占42.2%), 这些结果与Bercaw课题组的结果有很大差异.本文将详细报道目标配体及其铬配合物的合成、表征, 以及催化性能, 并进一步考察N原子上四氢糠基的电子和空间位阻效应的影响.
1 结果与讨论
1.1 PNP配体E的制备
以伯胺为前驱体合成PNP配体主要有两种方法:一种是以路易斯碱如中性有机胺为HCl分子的络合剂, 在反应过程中形成[R3NH]+Cl-盐从有机溶剂中析出, 促进伯胺直接与两分子的二取代膦氯化物反应的动力学转化, 生成目标配体; 另一种则是形成胺基锂与二取代膦氯化物发生盐消除反应, 因为含两个胺基质子, 所以反应需要两次盐消除, 其中中间产物为单膦基取代的胺, 可分离也可以不分离[10].我们以N-四氢糠基伯胺C4H7OCH2NH2为原料, 先采取第一种合成方法, 但是没有成功.很可能是由于四氢糠基的电子特性, 使得胺基质子的反应性受到影响.紧接着尝试第二种方法.最初我们采用一锅进行的方法, 即两次形成的胺基锂盐不从溶液中分离出来, 直接与二苯基膦氯化物Ph2PCl发生盐消除反应, 但是得到的配体产率非常低.于是我们对这种方法进行改进, 即将伯胺与丁基锂n-BuLi反应后生成的胺基锂C4H7OCH2NHLi分离出来, 再与Ph2PCl反应生成C4H7OCH2NH(PPh2).该化合物可以不用分离, 但是其进一步与n-BuLi生成的C4H7OCH2N-(PPh2)Li需要再次分离, 然后与Ph2PCl反应得到配体E, 这样总的产率高达70%.整个反应过程如Scheme 1所示.有文献报道使用该方法可能会导致产物重排, 生成类似Ph2P(Ph)2P=N(CH2OC4H7)的化合物[12], 但是实验证明, 改进后的方法能够成功地获得目标配体E.化合物E的熔点经测试为78 ℃, 说明该化合物具有很好的热稳定性.该化合物也通过1H NMR、13C NMR、13C-DEPT90、13C-HSQCETGP谱以及31P NMR谱图证实, 其C、H、N元素组成经元素分析确认. 31P NMR仅在δ 63.86处出现一个单峰, 说明不存在重排产物.在1H NMR中, 胺氢质子的宽峰消失, 说明其完全被膦基取代.在δ 1.03~1.07 (m, 1H)和1.41~1.44 (m, 1H)的共振吸收峰可以指认为3C位上的两个质子, 在δ 1.20~1.30 (m, 2H)和3.86 (m, 1H)的峰分别指认为2C和4C位上的质子, 在δ 3.33~3.46 (m, 3H)和3.61~3.66 (m, 1H)的峰为1C和5C位上的质子, 相应的这些碳原子的共振吸收峰也可以一一确定.
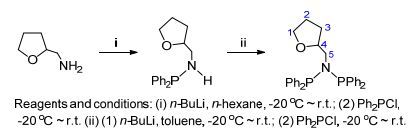 图式1
两步法反应合成配体E
Scheme1.
Synthesis of ligand E by the two-step salt elimination reactions
图式1
两步法反应合成配体E
Scheme1.
Synthesis of ligand E by the two-step salt elimination reactions
1.2 铬配合物的合成
我们采用两种金属铬前驱体CrCl3(THF)3和Cr(CO)6来分别合成N-四氢糠基PNP配体铬化合物.以甲苯作溶剂, CrCl3(THF)3和E的反应需要在加热下(80 ℃)进行.可以观察到反应溶液逐渐变为蓝色, 同时产物1也在反应的过程中从溶液中析出, 反应12 h后收集得到94%产率的蓝色固体. Cr(CO)6和E的反应需要在130 ℃加热下进行.相比于1, 产物2在甲苯中有很好的溶解度, 最后通过移走溶剂并在正己烷的洗涤后得到85%产率的黄色固体, 反应式如Scheme 2所示.熔点测试给出化合物1的熔点为211 ℃, 而化合物2的熔点为217 ℃.这两个化合物在惰性气氛下(N2)都具有很好的热稳定性; 而在空气气氛下, 化合物1在150 ℃以内稳定性良好(TGA谱图见辅助材料图S10), 化合物2在200 ℃以内稳定性良好(TGA谱图见辅助材料图S11). C、H、N元素分析揭示这两个化合物的组成与Scheme 2中的结构相符.配合物1中的铬为三价, 整个化合物具顺磁性, 因而该化合物无法用NMR谱确认, 试图培养其单晶进行X射线结构解析, 但是没有成功.我们推测化合物1可能是二聚体结构, 通过氯离子桥联, 满足铬中心六配位的稳定配位几何.一些文献也报道类似的化合物是二聚体结构[7, 11].但是也不能排除单体结构, 因为四氢糠基上的氧原子有可能与铬中心配位.化合物2中的铬为零价, 不具有顺磁性.该化合物可以进一步通过1H NMR、13C NMR、13C-DEPT90、13C-HSQCETGP谱以及31P NMR谱图证实. 31P NMR显示化学位移位于δ 116.00, 与配体E的相比明显向低场偏移.因此, 可以推测磷原子上的孤对电子向铬中心偏移, 与铬形成配位键. 1H NMR和13C NMR谱清楚地看到N-四氢糠基PNP配体的存在. 13C NMR还显示CO的化学位移在δ 136.24~136.86 (m)处, 同时IR谱证实C≡O键的伸缩振动, 振动频率位于常规的1872、1907、1910和2004 cm-1处.这些数据可以与其它PNP配体配位的铬羰基化合物相类比[12].
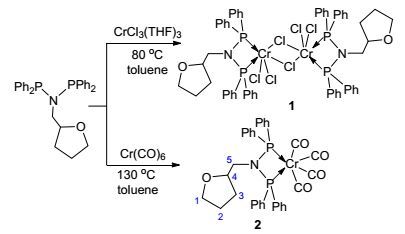 图式2
化合物1和2的合成
Scheme2.
Synthesis of complexes 1 and 2
图式2
化合物1和2的合成
Scheme2.
Synthesis of complexes 1 and 2
通过重结晶, 我们成功地得到了化合物2的单晶, 并进行了X射线的衍射测试.结构分析确认该化合物呈单核状态, PNP配体与铬螯合配位, 同时铬中心与四个CO分子配位, 形成稳定六配位的畸变八面体配位几何.晶体结构数据如表 2所示, 单晶结构见图 3. P(1)— Cr(1)—P(2) 键角为68.29°, 略大于化合物[Ph2PN(iPr)-PPh2Cr(CO)4]中的67.9°[14]和化合物[CrCl2[P, P-κ2-Ph2PN-(CH2CH2OCH3)PPh2](µ2-Cl)]2中的66.8°[11]. P(1)— Cr(1)—P(2) 平面与P(1)—N(1)—P(2) 平面所成二面角仅3.46°, 表明P(1)—Cr(1)—P(2)—N(1) 四元环基本上共面.
 表 2
配合物2的晶体结构数据
Table 2.
Crystallographic data for complex 2
表 2
配合物2的晶体结构数据
Table 2.
Crystallographic data for complex 2
Parameter Value Formula C33H29CrNO5P2 Mr 633.51 Cryst syst. Monoclinic Space group P2(1)/n a/Å 11.3439(3) b/Å 19.3338(5) c/Å 14. 0390(4) α/(°) 90 β/(°) 94.106(2) γ/(°) 90 V/Å3 3071.14(14) Z 4 ρcalcd/(g•cm-3) 1.370 μ/mm-1 0.518 F(000) 1312 Crystal size/mm3 0.40×0.40×0.30 θ range/(°) 3.07~26.00 Index ranges -13≤h≤9 -23≤k≤17 -17≤l≤14 Collected data 13961 Unique data 6015 (Rint=0.0296) Completeness to θ (%) 99.8 Data/restraints/params 6015/422/434 GOF on F2 1.022 Final R indices [I>2(I)] R1=0.0404
wR2=0.0865R indices (all data) R1=0.0525
wR2=0.0909Largest diff peak/hole (e•Å-3) 0.336/-0.355 aAll data were collected at 173(2) K using Mo Kα (λ=0.71073 Å) radiation. R1=∑(||Fo|-|Fc||)/∑|Fo|, Fc2)2/∑w(Fo2)]1/2, GOF=[∑w(Fo2-Fc2)2/(No-Np)]1/2. 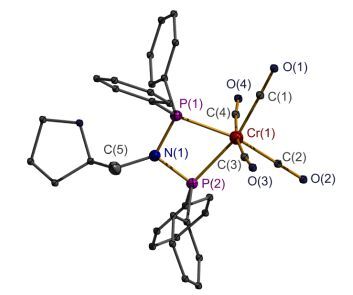 图3
配合物2的晶体结构图及重要的键长和键角参数
Figure3.
X-ray crystal structure of complex 2
图3
配合物2的晶体结构图及重要的键长和键角参数
Figure3.
X-ray crystal structure of complex 2
1.3 乙烯齐聚
采用MAO或其它助催化剂, 我们系统地研究了配合物1、2以及E和金属铬化合物CrCl3(THF)3、Cr(acac)3 (acac为乙酰丙酮配体)、CrCl2(THF)2原位组成的催化体系催化乙烯齐聚的性能.为了阐明配体的作用, 首先进行了三组空白对照实验, 即只用铬源和助催化剂MAO组成的体系, 结果发现在35 ℃、4.0 MPa乙烯压力、甲苯溶剂、750倍MAO中, CrCl3(THF)3、Cr(acac)3和CrCl2(THF)2都只有很低的催化活性[0.7、0.7、0.6 kg (PE)/g (Cr)•h, 表 1, Entries 1~3], 并且只生成聚合物.该结果与我们以前报道的结果类似[15].当加入配体E后, 在相同条件下, 三种铬源的反应活性分别提高至13.0、6.5、2.2 kg (product)/g (Cr)•h (Entries 4~6), 产物以齐聚物为主, C8组分分别占61.7% (1-C8的选择性为96.0%)、65.5% (1-C8的选择性为96.1%)、60.1% (1-C8的选择性为89.7%); C6组分分别占23.9% (1-C6的选择性为45.5%)、20.3% (1-C6的选择性为42.2%)、29.9% (1-C6的选择性为54.5%), 其它是C10+组分.同时, 我们还发现有一定量的聚乙烯(PE)生成, 分别占所有产物的28.8%、46.4%、39.3%. Bercaw课题组[11]报道了N-烷氧基或N-芳基烷氧基PNP配体A~D稳定的铬催化体系, 在300 Al/Cr比、25 ℃、100 kPa乙烯压力下的催化活性只有0.4~0.9 kg (product)/g (Cr)•h, 并且以乙烯三聚为主, C6的选择性为61%~66%.对比配体E与A~D的结构发现, 氮原子上烷氧取代基具环状与非环状的差异.可能是环状取代基减弱了氧原子对铬中心的给电子作用, 因而CrC8中间体的稳定性提高, 有利于C8组分的形成, 协同的3, 9-H迁移导致选择性生成1-C8为主导产物.这些结果表明, PNP配体配位稳定铬中心有利于提高反应的催化活性, 同时也诱导产物发生根本性的改变.
表 1
1/MAO, 2/Ag[Al(OC(CF3)3)4]/TEA和E/Cr/MAO催化乙烯齐聚的结果a
Table 1.
Ethylene oligomerization results catalyzed by 1/MAO, 2/Ag[Al(OC(CF3)3)4]/TEA, 和E/Cr/MAO
Entry Cat. Al:Cr p/MPa T/℃ Run time/min Act.b PEc/wt% Oligomer distributiond/wt% C6 (1-C6) C8 (1-C8) C10+ 1 CrCl3(THF)3 750 4.0 35 30 0.7 100 — — — 2 Cr(acac)3 750 4.0 35 30 0.7 100 — — — 3 CrCl2(THF)2 750 4.0 35 30 0.6 100 — — — 4 E/CrCl3(THF)3 750 4.0 35 30 13.0 28.8 23.9 (45.5) 61.7 (96.0) 14.4 5 E/Cr(acac)3 750 4.0 35 30 6.5 46.4 20.3 (42.2) 65.5 (96.1) 14.2 6 E/CrCl2(THF)2 750 4.0 35 30 2.2 39.3 29.9 (54.5) 60.1 (89.7) 10.0 7 1 750 4.0 35 30 13.5 27.1 22.6 (43.9) 63.3 (95.8) 14.1 8 1 600 4.0 35 30 13.7 16.7 20.5 (55.3) 63.5 (96.3) 16.0 9 1 500 4.0 35 30 12.4 19.3 19.8 (59.2) 65.0 (92.7) 15.2 10 1 400 4.0 35 30 4.4 42.1 19.8 (56.4) 56.7 (94.5) 23.5 11 1 600 4.0 28 30 11.7 16.1 13.9 (52.3) 63.8 (96.9) 22.3 12 1 600 4.0 40 30 10.6 51.4 21.8 (51.2) 61.1 (96.8) 17.1 13 1 600 4.0 45 30 12.9 58.3 23.0 (49.7) 60.2 (97.8) 16.8 14 1 600 4.0 50 30 13.8 65.4 24.6 (66.5) 57.7 (98.6) 17.7 15 1 600 3.0 35 30 10.5 18.4 22.6 (54.3) 60.2 (97.5) 17.2 16 1 600 3.5 35 30 11.9 25.2 22.0 (49.6) 61.0 (97.1) 17.0 17 1 600 4.5 35 30 15.9 16.4 17.1 (60.6) 64.8 (98.1) 18.1 18 1 600 4.0 35 60 12.5 18.5 25.5 (51.2) 63.5 (96.0) 11.0 19 1 600 4.0 35 120 11.8 20.8 27.5 (51.6) 61.3 (97.5) 11.2 20 1 600 4.0 35 240 7.8 32.6 28.2 (53.4) 60.5 (96.7) 11.3 21e 1 600 4.0 35 30 15.5 18.6 20.6 (56.2) 63.2 (96.8) 16.2 22f 1 600 4.0 35 30 13.8 16.8 20.8 (54.5) 62.8 (96.5) 16.4 23g 1 600 4.0 35 30 10.5 32.8 32.4 (56.1) 56.7 (95.6) 10.9 24h 1 600 4.0 35 30 13.1 15.0 29.1 (53.5) 59.1 (95.7) 11.8 25i 2 500 4.0 35 30 3.4 45.5 36.4 (56.1) 54.7 (100) 9.8 aReaction conditions: n(Cr)=5 μmol; solvent, toluene, 80 mL; co-catalyst, MAO; stirring speed, 500 r/min. b kg (product)/g (Cr)•h. c wt% of total product. dwt% of liquid products. en(Cr)=2 μmol.fn(Cr)=10 μmol. g Solvent, chlorobenzene, 80 mL. h Solvent, cyclohexane, 80 mL. i Conditions: 5.0 μmol of complex 2, 10 μmol of Ag[Al(OC(CF3)3)4], 500 equiv of TEA, 80 mL of chlorobenzene. 我们也发现对于同一种配体E, 不同的铬源催化活性以及产物的选择性略有不同. PNP配体对铬中心的配位稳定作用也基本上被认同, 那么不同的铬源意味着在MAO的作用下可能形成相同的活性中心, 但是活化诱导期应该不同.这可能是导致催化结果有所差异的原因[16].此外, 产物中不但包含齐聚物, 也包含聚合物.我们以前探讨过同一种NNP配体稳定的不同价态的铬中心会形成不同的结构, 因而会给出齐聚和聚合两种不同的结果[15].但是本文的PNP配体E也与不同价态的铬源组合[如CrCl3(THF)3与CrCl2(THF)2], 两种结果都出现, 只是数值大小有差异.显然在各自的体系中均会形成不同结构的活性中心, 来引发行为相差很大的齐聚和聚合行为.
我们进一步以配合物1为催化剂, 控制几乎相同的反应组成和条件, 得到的结果与CrCl3(THF)3/E非常接近(Entry 7).这点可以理性地解释E配体的稳定作用, 并且形成相同的反应活性中心.当然应该包含两种, 一种引发乙烯齐聚, 另一种则引发聚合.
选择化合物1, 详细地研究了MAO量、反应温度、乙烯压力、催化反应时间、催化剂浓度和溶剂对催化性能的影响.
首先, 保持温度、乙烯压力和反应时间不变, 当Al/Cr比由750降至600以及500时, 催化活性略有降低, 产物组成变化不大(Entries 8, 9);当进一步降至400时, 反应活性急剧降低(约3倍), 同时C8组分减少8个百分点, C10+组分增加8个百分点, PE量增加22个百分点(Entry 10).众所周知, 助催化剂MAO对反应体系的净化、活性中心的产生以及乙烯聚合过程中的链转移起着决定性的作用.过少的量可能会导致两种催化活性中心量的变化.因此, MAO的最佳使用量在500比例为佳.
其次, 固定Al/Cr比为600、乙烯压力为4.0 MPa、反应时间仍为30 min, 随着温度从35 ℃升高至40、45、50 ℃时, 催化活性变化不大, 在10.6~13.8 kg (product)/g (Cr)•h间变化(表 1, Entries 8, 12~14); C8组分选择性略有降低, 而C6组分选择性略有升高, 但是PE量与Entry 8相比从16.7%增加至51.4%、58.3%、65.4%.当温度降低至28 ℃时, PE的比例略有降低(16.1%).反应温度升高, 分子运动加剧, 反应活性应该明显增加.但是该体系促进了聚合反应.要保持齐聚反应性, 该体系的反应温度应控制在35 ℃.
再次, 保持Al/Cr比为600、温度为35 ℃、反应时间为30 min, 增大乙烯压力至4.5 MPa, 催化活性和C8组分的选择性均有提高, 而C6的选择性降低, PE量几乎不变(Entry 17).当降低压力至3.5、3.0 MPa时, 活性少许降低, PE增加少许, 齐聚物组分选择性变化很小(Entries 15, 16).乙烯压力升高, 乙烯在甲苯中的溶解度逐渐增大, 有利于提高乙烯分子对CrC6中间体的配位插入速率, 从而提高了催化活性和C8组分的选择性.
当选择Al/Cr为600、反应温度为35 ℃、乙烯压力为4.0 MPa时, 反应时间从30 min延长至60、120 min, 催化反应活性略有降低, C6组分增大5%和7%, 其它也变化不大(Entries 18, 19);但是再延长至240 min时, 反应活性降低近一半, 但是PE量增加约16% (Entry 20).在该反应条件下, 控制时间为30 min, 当催化剂量由5 μmol (Entry 8) 降至2 μmol, 表观催化反应活性略有提高, 但C6和C8组分的比例变化很小(Entry 21);而将催化剂量由5 μmol升高至10 μmol, 催化反应活性和各组分选择性基本保持不变(Entry 22).此外, 溶剂对催化反应活性和产物选择性都有一定的影响.相比于甲苯, 以氯苯作溶剂, 催化反应活性明显降低, 产物中C6组分增加了12%, 而C8组分降低了约7%, PE比例明显升高(Entry 23);以环己烷为溶剂, C6组分增大约9%, C8组分降低约4%, 其它变化不大(Entry 24).这些结果表明该催化剂体系可以在一定时间内保持反应活性(如延长至120 min), 但过长会引起活性降低.该体系选择甲苯溶剂为佳, 其它溶剂如氯苯或环己烷会引起活性降低或1-C8组分的减少.
PNP配位的羰基铬化合物的催化活性通常都不高, 该化合物往往需要特殊的处理, 一般需要先加入氧化剂将其氧化升高氧化态, 然后再加入助剂活化[13, 17].其活性中心形成机理也非常复杂.在经过系列考察发现, 配合物2在氧化剂Ag[Al(OC(CF3)3)4]的作用以及进一步三乙基铝的活化下, 催化乙烯齐聚生成C8组分54.7% (1-C8的选择性为100%)、C6组分36.4% (1-C6的选择性为56.1%)、C10+组分9.8%;同时得到PE 45.5%.但是反应活性较低, 为3.4 kg (product)/g (Cr)•h (Entry 25).
2 结论
在本文中, 成功地合成了一种新型的N-四氢糠基PNP配体E, 该化合物需要通过胺基锂盐分离的特殊步骤来合成.进一步制备了两种铬配合物[{Ph2PN(CH2O-C4H7)PPh2}CrCl2(μ-Cl)]2 (1)和[Ph2PN(CH2OC4H7)PPh2]-Cr(CO)4 (2), 并对这两种化合物进行了详细的表征.基于铬六配位的稳定配位几何, 推测化合物1为以氯离子为桥联的二聚体.在助催化剂MAO的作用下, 考察了1、2以及E和金属铬化合物CrCl3(THF)3、CrCl2(THF)2、Cr(acac)3原位组成的催化体系催化乙烯齐聚和聚合的性能.控制Al/Cr比为600、温度35 ℃、乙烯压力4.5 MPa时, 化合物1的催化活性可达15.9 kg (product)/g (Cr)•h, 且以63.6%的选择性生成1-辛烯和10%的选择性生成1-己烯, 聚合物的量为16.4%.
3 实验部分
3.1 仪器与试剂
所有涉及水氧敏感化合物的实验操作均采用Schlenk技术或在充满氩气的手套箱中进行.溶剂四氢呋喃、甲苯和正己烷都是经过钠丝预干燥, 然后利用钠钾合金/二苯甲酮回流, 显蓝紫色后蒸馏使用.氯苯和环己烷是先用氢化钙处理, 然后重蒸使用. C6D6经钠钾合金干燥后使用, CDCl3则是用氢化钙处理后使用.试剂四氢糠胺、二苯基氯化磷、正丁基锂、甲基铝氧烷(MAO)(10%的甲苯溶液)、无水CrCl3、无水CrCl2、Cr(acac)3、Cr(CO)6均是购买于百灵威试剂公司.前驱体CrCl3(THF)3[18]、CrCl2(THF)2[18]、Ag[Al(OC(CF3)3)4][19]均是采用文献方法制得.
1H NMR (500 MHz)、13C NMR (125 MHz)和31P NMR (202 MHz)是在Bruker Avance Ⅱ 500 MHz核磁共振仪上测试, 熔点在Büchi B-540型显微熔点仪上测试; 红外光谱在Nicolet FT-IR330光谱仪上测试; 元素分析使用Vario ELⅢ元素分析仪, 进行CHN含量测试; 热重分析在NETZSCH STA 409 PC/PG仪器上测试.
齐聚产物通过海欣GC-950气相色谱仪进行定性定量分析.色谱柱型号: Agilent Technologies, Inc19091Z-236 HP-1, 60 m×0.25 mm, 中性氧化铝柱.分析条件:柱温采取程序升温, 初始温度35 ℃, 初温时间10 min, 升温速率10 ℃/min, 终止温度280 ℃, 汽化室和检测器温度均为280 ℃.
3.2 实验方法
3.2.1 PNP配体E的制备
准确称取四氢糠胺(2.8 mL, 26 mmol)溶于60 mL正己烷中.当冷却至-20℃时, 缓慢滴加n-BuLi (11.5 mL, 2.4 mol•L-1, 28 mmol).滴加完毕后自然升至室温, 并继续反应6 h.反应结束后过滤收集白色固体四氢糠胺锂盐, 用正己烷(10 mL)洗涤、干燥, 称量为2.72 g.将该锂盐重新置于60 mL正己烷中, 冷却至-20℃时缓慢滴加二苯基氯化膦(4.6 mL, 25.4 mmol), 滴加完毕后自然升至室温继续反应12 h.反应结束后过滤除去白色沉淀, 收集生成N-二苯基膦基四氢糠胺的滤液.再经过上述n-BuLi脱质子反应以及随后与二苯基氯化膦的脱LiCl的盐消除反应, 最后得到Ph2PN(CH2OC4H7)PPh2 (E), 白色固体, 产量为8.50 g, 产率为70%. m.p. 78 ℃; 1H NMR (500 MHz, C6D6, 298 K) δ: 1.03~1.07 (m, 1H, 3CH2), 1.20~1.30 (m, 2H, 2CH2), 1.41~1.44 (m, 1H, 3CH2), 3.33~3.46 (m, 3H) (1CH2, N5CH2), 3.61~3.66 (m, 1H, N5CH2), 3.86 (m, 1H, 4CH), 7.06~7.66 (m, 20H, C6H5); 31P NMR (202 MHz, C6D6, 298 K) δ: 63.86; 13C NMR (125 MHz, C6D6, 298 K) δ: 25.40 (2CH2), 29.09 (3CH2), 57.15 (N5CH2), 67.02 (1CH2), 78.33 (4CHO), 128.54 (d, JP, C=31.7 Hz), 133.17 (dd, JP, C=62.3, 21.2 Hz), 140.19 (dd, JP, C=26.9, 11.1 Hz, C6H5), 222.49, 228.41 (CO). Anal. calcd for C29H29NOP2: C 74.19, H 6.23, N 2.98; found C 74.39, H 6.56, N 3.23.
3.2.2 配合物1和2的合成
准确称取配体E (0.55 g, 1.18 mmol)和CrCl3(THF)3 (0.41 g, 1.10 mmol), 溶于50 mL甲苯中, 加热至80 ℃搅拌12 h.反应结束后冷至室温, 得到蓝色悬浊液, 过滤收集蓝紫色固体, 用正己烷洗涤, 然后干燥、称重, 得[{Ph2PN(CH2OC4H7)PPh2}CrCl2(μ-Cl)]2 (1) 0.65 g, 产率为94%. m.p. 211 ℃. Anal. calcd for C58H58N2O2P4Cl6-Cr2: C 55.48, H 4.66, N 2.23; found C 55.26, H 4.52, N 2.05.
准确称取配体E (0.56 g, 1.18 mmol)和Cr(CO)6 (0.48 g, 2.2 mmol)于100 mL Schlenk瓶中, 加入80 mL甲苯, 加热至130 ℃回流72 h.反应结束后冷至室温, 过滤除去不溶物, 滤液通过减压法除去溶剂, 得到黄色固体, 用正己烷洗涤, 然后真空干燥, 得到[Ph2PN-(CH2OC4H7)PPh2]Cr(CO)4 (2) 0.54 g, 产率为85%. m.p. 217 ℃; 1H NMR (500 MHz, CDCl3, 298 K) δ: 0.87~0.91 (m, 1H, 3CH2), 1.48~1.61 (m, 3H, 2CH2), 2.82~3.00 (m, 2H, N5CH2), 3.37~3.57 (m, 2H, 1CH2), 3.71~3.76 (m, 1H, 4CH), 7.47~7.61 (m, 20H, C6H5); 31P NMR (202 MHz, CDCl3, 298 K) δ: 116.00; 13C NMR (125 MHz, CDCl3, 298 K) δ: 25.02 (2CH2), 29.07 (3CH2), 54. 68 (N5CH2), 67.62 (1CH2), 76.53 (4CH), 128.31~132.13 (m, C6H5), 136.24~136.86 (m, CO); IR (KBr) v: 1872 (C≡O), 1907 (C≡O), 1910 (C≡O), 2004 (C≡O) cm-1. Anal. calcd for C33H29NO5P2Cr: C 62.56, H 4.61, N 2.21; found C 62.43, H 4.69, N 2.35.
3.2.3 单晶的培养与测试
适合于X射线单晶衍射的化合物2, 通过如下操作培养单晶:将2的甲苯溶液上层铺上一层正己烷, 然后置于-20 ℃条件下.正己烷向甲苯溶液中缓慢扩散, 三天后析出黄色晶体.
化合物2的衍射数据在Oxford Gemini S Ultra单晶X-射线衍射仪上收集.在173(2) K温度下, 用石墨单色器单色化的Mo-Kα (λ=0.71073 Å)射线, 采用ω-2θ扫描方式收集衍射数据.分子结构中的非氢原子采用直接法由SHELX-97程序[20]解出, 氢原子由理论计算模型加上去.晶体数据在剑桥数据库存放, 编号为CCDC 1533644.
3.2.4 聚合操作方法
将高压釜安装好, 预热至100℃, 真空干燥1 h, 经氮气置换三次后充入乙烯, 并配置循环水冷却, 降至设定温度.在手套箱中, 称取一定量的催化剂, 溶于溶剂中, 搅拌5 min, 置于注射器中, 密封好, 移出手套箱.然后分别将溶剂、助剂、催化剂溶液依次在乙烯气流下注入反应釜中, 设定反应温度、压力以及搅拌速度和时间, 开始反应.反应结束后, 停止通入乙烯, 使用低温循环水泵将反应釜降温至5 ℃, 缓缓卸压后, 将反应后的产物倾入含质量分数为10%的盐酸酸化的乙醇溶液中, 再加水100 mL.通过萃取收集有机组分, 经无水硫酸钠干燥后称量; 聚合物固体产物经过滤收集, 并于50 ℃下真空干燥至恒重, 称量.有机组分加入内标物正庚烷, 注入GC-FID中分析, 得到色谱图, 通过与标准样品色谱图比较, 给出定量分析结果.
辅助材料(Supporting Information) 配体E和化合物2的核磁谱图, 化合物1和2的热重分析谱图, 以及代表性的齐聚产物气相色谱分析图谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
(a) McGuinness, D. S. Chem. Rev. , 2011, 111, 2321.
(b) Dixon, J. T. ; Green, M. J. ; Hess, F. M. Morgan, D. H. J. Organomet. Chem. 2004, 689, 3641.
(c) Agapie, T. Coord. Chem. Rev. , 2011, 255, 861.
(d) Qian, B. Z. Petrochem. Ind. Technol. 2011, 18, 58(in Chinese).
(钱伯章, 石化技术, 2011, 18, 58. ) -
[2]
(a) Lappin, G. Butene-1 and Other LLDPE Comonmers, Chem SysSystems, Inc, New York, 1986.
(b) Sauser, J. Alpha-olefins Applications Handbook, Marcel Dekker, New York, 1989.
(c) Britovsek, G. J. P.; Bruse, M.; Gibson, V. C.; Kimberley, B. S.; Maddox, P. J.; Mastroianni, S.; McTavish, S. J.; Redshaw, C.; Solan, G. A.; Williams, D. J. J. Am. Chem. Soc. 1999, 121, 8728.
(d) Small, B. L.; Brookhart, M.; Bennett, A. M. A. J. Am. Chem. Soc. 1998, 120, 4049.
(e) Brookhart, B. L.; Small, M. J. Am. Chem. Soc. 1998, 120, 7143. -
[3]
Robert, M. M.; Albans, S. US 3300458, 1967[Chem. Abstr. 1967, 66, 66008].
-
[4]
Manyik, R. M.; Walker, W. E.; Wilson, T. P. J. Catal. 1977, 47, 197. doi: 10.1016/0021-9517(77)90167-1
-
[5]
Freeman, J. W.; Buster, J. L.; Knudsen, R. D. Phillips Petroleum Company, US 5856257, 1999[Chem. Abstr. 1999, 130, 95984].
-
[6]
Carter, A.; Cohen, S. A.; Cooley, N. A.; Murphy, A.; Scutt, J.; Wass, D. F. Chem. Commun. 2002, 858.
-
[7]
Bollmann, A.; Blann, K.; Dixon, J. T.; Hess, F. M.; Killian, E.; Maumela, H.; McGuinness, D. S.; Morgan, D. H.; Neveling, A.; Otto, S.; Overett, M.; Slawin, A. M. Z.; Wasserscheid, P.; Kuhlmann, S. J. Am. Chem. Soc. 2004, 126, 14712. doi: 10.1021/ja045602n
-
[8]
(a) Shaikh, Y.; Albahily, K.; Sutcliffe, M.; Fomitcheva, V.; Gambarotta, S, Korobkov, I.; Duchateau, R. Angew. Chem. Int. Ed. 2012, 51, 1366.
-
[9]
(a) Jiang, T.; Ning, Y. N.; Zhang, B. J.; Li, J. Z.; Wang, G.; Yi, J. J.; Huang, Q. J. Mol. Catal. A:Chem. 2006, 259, 161.
(b) Jiang, T.; Zhang, S.; Jiang, X. L.; Yang, C. F.; Niu, B.; Ning, Y. N. J. Mol. Catal. A:Chem. 2008, 279, 90.
(c) Jiang, T.; Tao, Y. Q.; Gao, X. L.; Mao, G. L.; Chen, H. X.; Chen, C. G..; Ning, Y. N. Chin. Sci. Bull. 2012, 57, 1510.
(d) Zhang, J.; Wang, X.; Zhang, X.; Wu, W.; Zhang, G.; Xu, S.; Shi, M. ACS Catal. 2013, 3, 2311. -
[10]
Liu, R.; Xiao, S.; Zhong, X.; Cao, Y.; Liang, S.; Liu, Z.; Ye, X.; Shen, A.; Zhu, H. Chin. J. Org. Chem. 2015, 35, 1861(in Chinese). doi: 10.6023/cjoc201504009
-
[11]
Elowe, P. R.; McCann, C.; Pringle, P. G.; Spitzmesser, S. K.; Bercaw, J. E. Organometallics 2006, 25, 5255. doi: 10.1021/om0601596
-
[12]
Maumela, M.; Blann, K.; de Bod, H. T.; Dixon, J.; Gabrielli, W.; Williams, D. B. Synthesis 2007, 3863.
-
[13]
Dulai, A.; de Bod, H. T.; Hanton, M. J.; Smith, D. M.; Downing, S.; Mansell, S. M.; Wass, D. F. Organometallics 2009, 28, 4613. doi: 10.1021/om900285e
-
[14]
Rucklidge, A. J.; McGuinness, D. S.; Tooze, R. P.; Slawin, A. M. Z.; Pelletier, J. D. A.; Hanton, M. J.; Webb, P. B. Organometallics 2007, 26, 2782. doi: 10.1021/om0701975
-
[15]
Liu, R.; Zhu, K. T.; Zhong, X. H.; Li, J. C.; Liu, Z. Y.; Chen, S. B.; Zhu, H. P. Dalton Trans. 2016, 45, 17020. doi: 10.1039/C6DT03216H
-
[16]
(a) Blann, K.; Bollmann, A.; Dixon, J. T.; Hess, F. M.; Killian, E.; Maumela, H.; Morgan, D. H.; Neveling, A.; Otto, S.; Overett, M. J. Chem. Commun. 2005, 620.
(b) Blann, K.; Bollmann, A.; Debod, H.; Dixon, J.; Killian, E.; Nongodlwana, P.; Maumela, M.; Maumela, H.; McConnell, A.; Morgan, D. J. Catal. 2007, 249, 244.
(c) Overett, M. J.; Blann, K..; Bollmann, A.; Dixon, J. T.; Hess, F.; Killian, E.; Maumela, H.; Morgan, D. H.; Neveling, A.; Otto, S. Chem. Commun. 2005, 622. -
[17]
Dulai, A.; McMullin, C. L.; Tenza, K.; Wass, D. F. Organometallics 2011, 30, 935. doi: 10.1021/om100912y
-
[18]
Herwig, W.; Zeiss, H. J. Org. Chem. 1958, 23, 1404.
-
[19]
Carter, E.; Cavell, K. J.; Gabrielli, W. F.; Hanton, M. J.; Hallett, A. J.; McDyre, L.; Platts, J. A.; Smith, D. M.; Murphy, D. M. Organometallics 2013, 32, 1924. doi: 10.1021/om400029y
-
[20]
Sheldrick, G. M. SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen:Göttingen, Germany, 1997.
-
[1]
-

图 2 N原子上含有醚基和酚氧烷基的PNP配体A~D和N原子上含四氢糠基的配体E
Figure 2 Diphoshinoamine ligands containing the N-ether or N-phenolic alkyl group for A~D and the N-tetrahydrofurfuryl group for E

图式1 两步法反应合成配体E
Scheme 1 Synthesis of ligand E by the two-step salt elimination reactions

图 3 配合物2的晶体结构图及重要的键长和键角参数
Figure 3 X-ray crystal structure of complex 2
Thermal ellipsoids are at 50% probability and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cr(1)—P(1), 2.3407(6); Cr(1)—P(2), 2.3462(6); P(1)—N(1), 1.7056(17); P(2)—N(1), 1.7071(17); P(1)—Cr(1)—P(2), 68.29(2); P(1)—N(1)—P(2), 100.86(9); N(1)—P(1)—Cr(1), 95.49(6); N(1)—P(2)—Cr(1), 95.25(6)
表 2 配合物2的晶体结构数据
Table 2. Crystallographic data for complex 2
Parameter Value Formula C33H29CrNO5P2 Mr 633.51 Cryst syst. Monoclinic Space group P2(1)/n a/Å 11.3439(3) b/Å 19.3338(5) c/Å 14. 0390(4) α/(°) 90 β/(°) 94.106(2) γ/(°) 90 V/Å3 3071.14(14) Z 4 ρcalcd/(g•cm-3) 1.370 μ/mm-1 0.518 F(000) 1312 Crystal size/mm3 0.40×0.40×0.30 θ range/(°) 3.07~26.00 Index ranges -13≤h≤9 -23≤k≤17 -17≤l≤14 Collected data 13961 Unique data 6015 (Rint=0.0296) Completeness to θ (%) 99.8 Data/restraints/params 6015/422/434 GOF on F2 1.022 Final R indices [I>2(I)] R1=0.0404
wR2=0.0865R indices (all data) R1=0.0525
wR2=0.0909Largest diff peak/hole (e•Å-3) 0.336/-0.355 aAll data were collected at 173(2) K using Mo Kα (λ=0.71073 Å) radiation. R1=∑(||Fo|-|Fc||)/∑|Fo|, Fc2)2/∑w(Fo2)]1/2, GOF=[∑w(Fo2-Fc2)2/(No-Np)]1/2.  下载: 导出CSV
下载: 导出CSV
表 1 1/MAO, 2/Ag[Al(OC(CF3)3)4]/TEA和E/Cr/MAO催化乙烯齐聚的结果a
Table 1. Ethylene oligomerization results catalyzed by 1/MAO, 2/Ag[Al(OC(CF3)3)4]/TEA, 和E/Cr/MAO
Entry Cat. Al:Cr p/MPa T/℃ Run time/min Act.b PEc/wt% Oligomer distributiond/wt% C6 (1-C6) C8 (1-C8) C10+ 1 CrCl3(THF)3 750 4.0 35 30 0.7 100 — — — 2 Cr(acac)3 750 4.0 35 30 0.7 100 — — — 3 CrCl2(THF)2 750 4.0 35 30 0.6 100 — — — 4 E/CrCl3(THF)3 750 4.0 35 30 13.0 28.8 23.9 (45.5) 61.7 (96.0) 14.4 5 E/Cr(acac)3 750 4.0 35 30 6.5 46.4 20.3 (42.2) 65.5 (96.1) 14.2 6 E/CrCl2(THF)2 750 4.0 35 30 2.2 39.3 29.9 (54.5) 60.1 (89.7) 10.0 7 1 750 4.0 35 30 13.5 27.1 22.6 (43.9) 63.3 (95.8) 14.1 8 1 600 4.0 35 30 13.7 16.7 20.5 (55.3) 63.5 (96.3) 16.0 9 1 500 4.0 35 30 12.4 19.3 19.8 (59.2) 65.0 (92.7) 15.2 10 1 400 4.0 35 30 4.4 42.1 19.8 (56.4) 56.7 (94.5) 23.5 11 1 600 4.0 28 30 11.7 16.1 13.9 (52.3) 63.8 (96.9) 22.3 12 1 600 4.0 40 30 10.6 51.4 21.8 (51.2) 61.1 (96.8) 17.1 13 1 600 4.0 45 30 12.9 58.3 23.0 (49.7) 60.2 (97.8) 16.8 14 1 600 4.0 50 30 13.8 65.4 24.6 (66.5) 57.7 (98.6) 17.7 15 1 600 3.0 35 30 10.5 18.4 22.6 (54.3) 60.2 (97.5) 17.2 16 1 600 3.5 35 30 11.9 25.2 22.0 (49.6) 61.0 (97.1) 17.0 17 1 600 4.5 35 30 15.9 16.4 17.1 (60.6) 64.8 (98.1) 18.1 18 1 600 4.0 35 60 12.5 18.5 25.5 (51.2) 63.5 (96.0) 11.0 19 1 600 4.0 35 120 11.8 20.8 27.5 (51.6) 61.3 (97.5) 11.2 20 1 600 4.0 35 240 7.8 32.6 28.2 (53.4) 60.5 (96.7) 11.3 21e 1 600 4.0 35 30 15.5 18.6 20.6 (56.2) 63.2 (96.8) 16.2 22f 1 600 4.0 35 30 13.8 16.8 20.8 (54.5) 62.8 (96.5) 16.4 23g 1 600 4.0 35 30 10.5 32.8 32.4 (56.1) 56.7 (95.6) 10.9 24h 1 600 4.0 35 30 13.1 15.0 29.1 (53.5) 59.1 (95.7) 11.8 25i 2 500 4.0 35 30 3.4 45.5 36.4 (56.1) 54.7 (100) 9.8 aReaction conditions: n(Cr)=5 μmol; solvent, toluene, 80 mL; co-catalyst, MAO; stirring speed, 500 r/min. b kg (product)/g (Cr)•h. c wt% of total product. dwt% of liquid products. en(Cr)=2 μmol.fn(Cr)=10 μmol. g Solvent, chlorobenzene, 80 mL. h Solvent, cyclohexane, 80 mL. i Conditions: 5.0 μmol of complex 2, 10 μmol of Ag[Al(OC(CF3)3)4], 500 equiv of TEA, 80 mL of chlorobenzene.
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 3251
- HTML全文浏览量: 474
 下载:
下载:
