 图1
Bifunctional catalysis of enzyme, Lewis base-transi-tion metal cooperation, and Lewis acid-transition metal cooperation in H(E)-H(E) activation
Figure1.
Bifunctional catalysis of enzyme, Lewis base-transi-tion metal cooperation, and Lewis acid-transition metal cooperation in H(E)-H(E) activation
图1
Bifunctional catalysis of enzyme, Lewis base-transi-tion metal cooperation, and Lewis acid-transition metal cooperation in H(E)-H(E) activation
Figure1.
Bifunctional catalysis of enzyme, Lewis base-transi-tion metal cooperation, and Lewis acid-transition metal cooperation in H(E)-H(E) activation
Citation:
Li Yinwu, Zhang Jianyu, Shu Siwei, Shao Youxiang, Liu Yan, Ke Zhuofeng. Boron-Based Lewis Acid Transition Metal Complexes as Potential Bifunctional Catalysts[J]. Chinese Journal of Organic Chemistry,
2017, 37(9): 2187-2202.
doi:
10.6023/cjoc201703002

硼基路易斯酸-过渡金属配合物:新型双功能催化剂
摘要:
路易斯酸-过渡金属(LA-TM)配合物作为一种新型的双功能催化剂崭露头角.有别于传统的过渡金属双功能催化剂,LA-TM催化剂另辟蹊跷,用路易斯酸功能位点作为电子受体,过渡金属作为电子给体,进行新型的双功能催化.近期,LA-TM配合物迅速发展.依据其键合特点,对硼基LA-TM配合物进行综述,包括sp3杂化、sp2杂化和sp杂化的硼基LA-TM配合物,并归纳了其在LA-TM催化方面的潜在应用,包括H-H/E-H/E-E键活化、氢化、硅氢化和转移脱氢化等等.本综述有望将为开拓LA-TM双功能催化的新领域提供有价值的信息.
English
Boron-Based Lewis Acid Transition Metal Complexes as Potential Bifunctional Catalysts
Abstract:
Lewis acid-transition metal (LA-TM) complexes, with the LA site functions as an electron acceptor and the TM center functions as an electron donor, have been emerging as a new type of bifunctional catalysts lately, different from traditional transition metal bifunctional catalysts. Due to their rapid developments recently, the boron-based LA-TM complexes, which are divided into three major types, the sp3, sp2, and sp boron-based complexes, are reviewed in this paper according to their binding features. Reactions promoted by this new type of LA-TM bifunctional catalysts have been surveyed, including migration reactions, activation of H-H/E-H/E-E bonds, hydrogenations, hydrosilylations, and transfer dehydrogenation reactions etc. This overview of boron-based LA-TM complexes could provide valuable information to explore the new horizon in LA-TM bifunctional catalysis.
-
Key words:
- Lewis acid
- / transition metal
- / bifunctional catalysis
- / boron
- / activation
- / hydrogenation
- / dehydrogenation
-
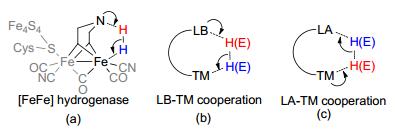
Transition metal (TM) complexes play an important role in catalytic chemistry, owning to their unique d-orbitals.[1] TM catalysts are widely applied in various reactions, including oxidative addition, reductive elimination, insertion, elimination, metathesis, etc.[2] To improve the efficiency, selectivity, and scope of TM catalysis, the concept of bifunctional catalysis via metal ligand cooperation has been introduced and applied as a very successful strategy.[3] If traced back to the long history of life on the earth, which began about 3.8 billion years ago, much longer than the age of science of human being, enzymes have mastered the bifunctional catalysis via metal ligand cooperation long before.[4] For example, the [FeFe] hydrogenase was proved to be equipped with a pendant amine close to the distal Fe center in the active pocket.[5] The pendant amine is expected to act as a Lewis base to interact with the proton, while the vacant coordination site of iron acts as a Lewis acid to interact with the hydride, promoting a reversible heterolytic H2 activation or production via bifunctional metal ligand cooperation manner (Figure 1a).
图1
Bifunctional catalysis of enzyme, Lewis base-transi-tion metal cooperation, and Lewis acid-transition metal cooperation in H(E)-H(E) activation
Figure1.
Bifunctional catalysis of enzyme, Lewis base-transi-tion metal cooperation, and Lewis acid-transition metal cooperation in H(E)-H(E) activation
With this useful concept that mimics the enzymatic cooperation feature, chemists have developed numerous types of ligands that can function as a Lewis base, together with transition metals, to construct the Lewis base-transition metal (LB-TM) systems for bifunctional catalysis (Figure 1b). In the traditional LB-TM systems, ligands with a heteroatom lone pair or a delocalized moiety are usually used as an LB.[6] Since 1980s, after the pioneering works of Noyori, [7] the traditional LB-TM bifunctional approach has attracted considerable interest. Up today, it has been successfully applied in a wide range of catalysis.[1b, 3a, 8]
Significantly, a novel Lewis acid-transition metal (LA-TM) bifunctional concept can be conceived, if the LB-TM bifunctional system is extended to an alternative, which is composed of TM and a Lewis acid, rather than Lewis base, as a functional site (Figure 1c).[9] In this kind of LA-TM systems, the Lewis acid site functions as an electron acceptor and the transition metal as an electron donor to facilitate the molecular transformation via LA-TM cooperation, which is interestingly different from the traditional transition metal bifunctional catalysts.[10] The LA-TM complexes only emerged after 2000s, when chemists expanded the utilization of Lewis acids from acting as external activators, co-catalysts, or additives to becoming one important part of the ligands in TM complexes.[9e, 10a]
Although the LA-TM cooperative catalysis is still in its infant stage, it has attracted incremental interests recently. Some interesting LA-TM complexes appeared in the literature that could promote significant transformations via LA-TM cooperation. The general reaction modes for LA-TM catalyzed H2 activation were proposed by Ke and co-workers.[10b] As shown in Figure 2, the H2 can undergo (1) a fac (cis) homolytic cleavage, where the reaction plane is perpendicular to the LA-TM bond; (2) a mer (trans) homolytic cleavage, where the reaction plane is parallel to the LA-TM bond; (3) a synergetic heterolytic cleavage via the cooperation of LA-TM bond, or (4) a dissociative heterolytic cleavage assisted by separated TM center and LA center. There are also some other good mechanistic studies.[11] However, the reaction mechanisms for different LA-TM cooperative catalysis remain a challenge to be fully understood yet. As a new strategy for metal ligand cooperation, the LA-TM complex has shown significant potential in bifunctional catalysis. For example, the nickel-borane complex, ferraboratrane complex, and cobalt-boryl complex had been achieved in H2 activation and hydrogenation under 101 kPa pressure and room temperature.[12] LA-TM complexes promoted migration reactions, activation of H—H/E—H/E—E bonds, hydrogenations, hydrosilylations, and transfer dehydrogenation reactions have also been reported in the literature. Until now, the Lewis acid atoms utilized in ligands of LA-TM complexes have extended from the commonly boron, to aluminum, [13] gallium, [14] indium, [14] zinc, [15] antimony, [16] etc. A series of Lewis acid ligands with various coordination numbers, including monodentate, bidentate, tridentate, and tetradentate, have also been developed. Among them, the boron-based ligands are particularly interesting due to its variation in binding. This review aims at providing a brief overview on the structures of boron-based LA-TM complexes according to their binding features, and their achieved reactions via LA-TM type bifunctional cooperation. This review is expected to provide valuable information for future exploration of the new horizon of LA-TM bifunctional catalysis.
 图2
General H2 activation modes for LA-TM catalysts.
Figure2.
General H2 activation modes for LA-TM catalysts.
图2
General H2 activation modes for LA-TM catalysts.
Figure2.
General H2 activation modes for LA-TM catalysts.
1 Boron-based LA-TM complexes with different binding features
According to the binding features of the LA center, the boron-based LA-TM complexes can be divided into three major types: the sp3 type, the sp2 type, and the sp type boron-based LA-TM complexes, as shown in Figure 3. (1) In the sp3 boron-based LA-TM complexes, Type A is the most typical one, in which the boron atom with three covalent B—X bonds accepts the electron pair of TM (TM→LA). The sp3 boron-based LA-TM complex can also adopt type B or type C, upon introducing one or two L-type groups onto the boron center. The polarity of the LA-TM bond in type B complex depends on the electronegativity difference between TM and boron centers. Type C complex exhibits an LA→TM bonding feature, where there is an umpolung boron center acting as an L-type ligand to the TM center. (2) The sp2 boron-based LA-TM complexes feature a planar structure with a Y shape boron center, which can be distinguished by type D, type E, and type F. In type D, boron center is expected to be a potential LA site for LA-TM cooperation with a Fischer type TM=B double bond. In type E, an L-type ligand datively binds to the sp2 boron enter. Therefore, either the empty p orbital or the sp2 orbital (after L dissociation) of boron center can participate in the LA-TM cooperation. Type F complex should be a rare example bearing a Schrock type TM=B double bond. The more common cases are sp2 boron-based LA-TM complexes with delocalization features, like types G and H in Figure 3, where the empty p orbital of boron may serve as a potential LA site for LA-TM cooperation. (3) The sp boron-based LA-TM complexes usually adopt linear structures. Type Ⅰ and type J both involve an allene like moiety. Type K and type L complexes contain a TM≡B triple bond. The strength (as well the reactivity) of the triple TM≡B bond in type K complexes should highly depends on the TM center. Type L is a Lewis acid and Lewis base adduct between TM≡B and L-type ligand. Type M is a borane complex with B≡X triple bond, where the empty sp orbital of the boron center accepts the TM electron pair. Type N is a boryl complex with a B≡X triple bond, where either the TM-B or the B≡X bond can be a potential moiety for bifunctional LA-TM catalysis.
 图3
Boron-based LA-TM complexes with different binding features
Figure3.
Boron-based LA-TM complexes with different binding features
图3
Boron-based LA-TM complexes with different binding features
Figure3.
Boron-based LA-TM complexes with different binding features
2 sp3 boron-based LA-TM complexes
Type A complex is the largest family in the sp3 boron-based LA-TM complexes. An early example of transition metal-borane complex was reported in 1999 by Hill (1, as shown in Figure 4).[17] In this interesting structure, tetradentate Reglinski ligand precursor, which consists of an sp3 boron as the Lewis acid center, was used as a typical electron deficient Z-type ligand to coordinate with TM center. A d8 ruthenium was used as the TM center, where the filled dz2 orbital is stabilized by LA center via σ(dz2-p) interaction. Later, using d8 Rh(Ⅰ) as the TM center, B(azaindolyl)3 as the LA ligand, Owen et al.[18] reported the structure of complex 2. With Ir(Ⅰ) as the TM center, an [Ir(η1-cod){κ3N, N, B-B(azaindolyl)3}(CO)2 complex was confirmed by NMR in Owen's study.[11a] Since then, a series of complexes in analogous structures were synthesized. This kind of LA-TM complexes usually utilizes N-heterocycle as the tethered skeleton to construct tetradentate borane ligands, which can be divided into nitrogen donor ligands and sulfur donor ligands (Figure 5). The sulfur donor ligands containing different tethered skeletons like pyridazine-thione, pyridine-thione, imidazole-thione, triazole-thione, and thiazole-thiones, were reported in various LA-TM systems.[19] The nitrogen donor ligands with pyrrolo-pyridine and pyrazole as tethered skeletons were also studied.[20] Tetradentate N-heterocyclic ligands with mixed sulfur and nitrogen donors were also reported by Connelly to form type A LA-TM complexes with iridium (4 in Figure 4).[21] Sulfur group was suggested to be a stronger donor than nitrogen group. Along with ongoing efforts, researchers extended the N-heterocyclic ligands from tetradentate to tridentate, and synthesized many complexes with various transition metals, including manganese, [19c] iron, [22] cobalt, [23] rhodium, [24] nickel, [19c] platinum, [19e] etc.
 图4
Tetradentate and tridentate boron-based LA-TM complexes bearing N-heterocyclic ligands
Figure4.
Tetradentate and tridentate boron-based LA-TM complexes bearing N-heterocyclic ligands
图4
Tetradentate and tridentate boron-based LA-TM complexes bearing N-heterocyclic ligands
Figure4.
Tetradentate and tridentate boron-based LA-TM complexes bearing N-heterocyclic ligands
 图5
Structures of the N-heterocyclic nitrogen donor ligands and sulfur donor ligands
Figure5.
Structures of the N-heterocyclic nitrogen donor ligands and sulfur donor ligands
图5
Structures of the N-heterocyclic nitrogen donor ligands and sulfur donor ligands
Figure5.
Structures of the N-heterocyclic nitrogen donor ligands and sulfur donor ligands
Later, the phosphorus donor ligands (TPB and DBP in Figure 6) were also developed to construct Type A boron-based LA-TM complexes.[25] This kind of ligands with benzene as the tethered skeleton soon became one of the most prevalent ligands in LA-TM complexes. Various LA-TM complexes bearing this kind of ligands have been synthesized with different steric hindrance groups and transition metals, including iron, cobalt, rhodium, nickel, palladium, platinum, copper, silver, and gold, [26] leading to interesting variation in their coordination structures. As for the TPB ligands, the trigonal bipyramidal LA-TM complex with k4-L3B coordination structure is the majority. Peters and coworkers reported a paramagnetic k4-L3B TPB complex using Fe(0) as the TM center and N2 as the apical ligand, and a diamagnetic analogous with CO as the apical ligand (8, in Figure 6).[27] When changing the apical ligands from N2 to Br and imido, complexes d6 and d7 iron centers were also able to obtain. Due to the strong σ(dz2-p) stabilization, an interesting negative charged analogous was also successfully synthesized.[27] A series of k4-L3B TPB complexes using cobalt as the TM center was also systematic studied with similar TPB ligands.[26d] With H2 as the apical ligand, the TBP cobalt complex was proposed to be doublet, showing typical σ(dz2-p) LA-TM interaction, sharply different from the diamagnetic TSiP cobalt complex.
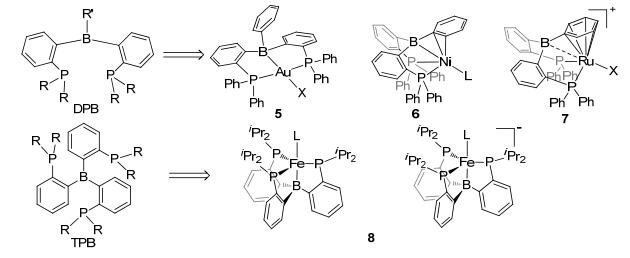 图6
Type A boron-based LA-TM complexes with DPB and TPB ligands
Figure6.
Type A boron-based LA-TM complexes with DPB and TPB ligands
图6
Type A boron-based LA-TM complexes with DPB and TPB ligands
Figure6.
Type A boron-based LA-TM complexes with DPB and TPB ligands
The DPB ligands formed different coordination structures from TPB ligands in LA-TM complexes. An early example of DBP boron-based LA-TM complex was reported by Bourissou.[28] As shown in 5 of Figure 6, the square-planar k3-PPB-DPB-Au(Ⅰ) complex was suggested to exhibit a typical TM→LA interaction, where the TM center remained at a low oxidation state without being further oxidized. An interesting kind of DPB LA-TM complexes using Ni(0) as TM center was reported by Peters (6 in Figure 6).[12a]The DPB ligand formed a pseudo tetrahedron nickel complex with a ƞ2 C-N B-phenyl group and a THF as solvent ligand. While the ligand bearing B-mesityl group formed a pseudo three-coordinated LA-TM complex with a vacant site on the nickel center, which was found to be efficient for hydrogen activation. Using earlier transition metal, Ru(Ⅱ), as TM center, Stephen and coworkers synthesized an interesting DPB LA-TM complex, [(TPB(η6-Ph))RuCl][B(C6F5)4], which was suggested to behave a C-based Lewis reactivity as shown in 7, Figure 6.[29] Besides, DPB-TM complexes with Fe, Co, Rh, Ir, Pd, Cu, etc. as the TM center, have been reported in recent years.[30] Apart from the above type A complexes with boron tethered chelate ligands, complexes with more flexible ligands that bear terminal boron as Lewis acid, had also attracted many interests. An early example of a terminal LA ligand complex was reported by Bourissou and co-workers with Au(Ⅰ) as TM center in 2006.[31] The ambiphilic bidentate monophosphine-borane ligand readily displaced the labile dimethyl sulfide ligand from (Me2S)AuCl to get complex 9 (Figure 7). In complex 9, the d10 Au(Ⅰ) and ambiphilic bidentate P-B ligand were proposed to exhibit a typical TMLA interaction.[31] Derived from the monophosphine-borane complex, a platinum complex 10 [Fe(ƞ5-C5H4PPh2)(ƞ5-C5H4PtBu{C6H4(BPh2)-ortho})] (FcPPB), was reported by Emslie, [32] with bis(phosphine)-borane as an ambiphilic ligand, in which the borane atom occupied a terminal position. This Pt(0)(FcPPB) complex has a wide bite-angle for terminal PPB ligand, leading to a different situation for TM→LA interaction. Similar to terminal phosphine-borane complexes, terminal nitrogen-borane complexes had been reported as well. Figueroa et al.[33] synthesized a T-shape platinum complex 11 with a (boryl)iminomethane (BIM) and an isocyanide as ligands. Different from other monodentate borane ligands, the small bite angle enforced by the BIM ligand was suggested to promote a significant reversible TM→LA σ-interaction.
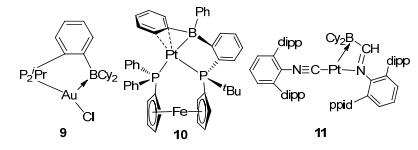 图7
LA-TM complexes with terminal borane ligands
Figure7.
LA-TM complexes with terminal borane ligands
图7
LA-TM complexes with terminal borane ligands
Figure7.
LA-TM complexes with terminal borane ligands
Compared with type A complexes, there are only a few examples for type B complexes. In 2004, through 4-methylpyridine and 3, 5-dimethylpyridine coordinated with (dichloroboryl)iron complex, Braunschweig et al.[34] reported the type B complexes 12a and 12b (Figure 8). In complexes 12a and 12b, boron forms three covalent bonds with the iron center and two chlorine atoms, respectively. The nitrogen in pyridine derivative donates a pair of electrons to the empty sp3 orbital of boron. Typical electron donor group PMe3 could also be used to form analogous complex 12c.[35] Although only monodentate boryl complexes were found until now, type B complexes with multidentate chelate ligands should also be able to be designed and synthesized by using the same strategy in the future. After the dissociation of the additional electron donor ligand, type B complexes may be expected to exhibit unique LA-TM catalytic feature.
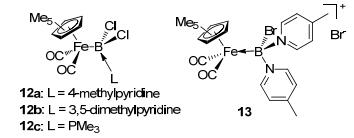 图8
Type B and type C sp3 boron-based complexes
Figure8.
Type B and type C sp3 boron-based complexes
图8
Type B and type C sp3 boron-based complexes
Figure8.
Type B and type C sp3 boron-based complexes
With two 4-methylpyridine as additional donor groups, complex 13 was reported as an early example for type C complexes.[34b] It should be noted that, in complex 13, boron donates a pair of electrons to iron, acting as a Lewis base instead of a traditional Lewis acid. The umpolung in LA-TM interaction (TM→LA to LA→TM) should be of great interest to enrich the diversity in LA-TM catalysis. Since the synthesis of complex 13, rare type C complex was reported. Very recently, in 2016, Langer et al.[36] reported a boron-based pincer complex 15. With excess NaBH4, FeCl2 reacted with two equivalents of dppa, resulting in phosphine-borane complex 14 (Figure 9).[37] By releasing H2, complex 14 could transfer to the phosphine-stabilized borylene complex 15. In this interesting complex, each phosphine donates one electron pair to the boron atom, resulting in a borylene structure that the boron center acts as a Lewis base to the metal center. This work further proved the possibility that the Lewis acid boron atom could change into an L-type ligand for the TM center in sp3 boron-based pincer complexes, which is intriguing for the catalysis design with LA-TM systems.
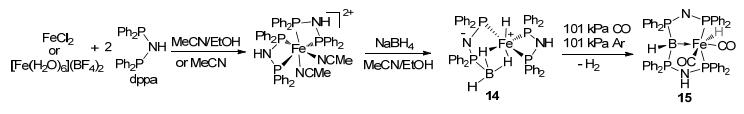 图9
Synthesis of type C sp3 boron-based pincer iron complex.
Figure9.
Synthesis of type C sp3 boron-based pincer iron complex.
图9
Synthesis of type C sp3 boron-based pincer iron complex.
Figure9.
Synthesis of type C sp3 boron-based pincer iron complex.
3 Reactions for the sp3 boron-based LA-TM complexes
Hydrogen activation and hydrogenation are important catalytic reactions catalyzed by the sp3 boron-based LA-TM complexes, which have great potentials in the fields of hydrogen energy and synthesis. Owen and co-workers[18] firstly reported the H2 activation using the rhodium-borane complex 16 as LA-TM catalyst (Figure 10). This borohydride complex 16, utilizing tri(azaindolyl) as a ligand skeleton, [20a] possesses a Rh-H-B bridge bond. The migration of hydride into the norbornadiene ligand transferred complex 16 to rhodium-borane complex 2, which is a typical sp3 boron-based LA-TM complex. Through addition of trialkylphosphines to complex 2, one azaindole arm loosed coordination and was replaced by the PMe3 ligand to form 17. The rhodium-borane complex 2 reacted with two equivalents of H2 in the presence of PtBu3 to yield borohydride complex 18. Further hydrogenation of olefins was observed by reacting with styrene and cyclooctene at H2 atmosphere (250 kPa, 85 ℃). This is the first time that LA-TM complex with nitrogen donor ligand was found to show capacity in the activation of H2 and the hydrogenation of unsaturated bonds.
 图10
Activation of H2 by rhodium-borane complex
Figure10.
Activation of H2 by rhodium-borane complex
图10
Activation of H2 by rhodium-borane complex
Figure10.
Activation of H2 by rhodium-borane complex
Soon after Owen's research, in 2012, Peter et al.[12a] achieved the H2 activation and hydrogenation reaction by using nickel-borane complex 19 as the LA-TM catalyst (Figure 11). Complex 19 is composed of the DPB ligand and a low valent Ni(0) TM center, in which the B—Ni bond, serving as the LA-TM bifunctional site, could rapidly and reversibly activate H2 to borohydride intermediate 20 under mild conditions (101 kPa, room temperature). Moreover, olefins hydrogenation was observed with intermediate 20 by reacting with styrene under mild reaction conditions as well. The catalytic cycle of hydrogenation was successfully achieved by the combination of these two reactions. This is one of the few examples that bifunctional catalyst is able to catalyze the nonpolar H—H bond activation under 101 kPa pressure and room temperature. Moreover, further studies were carried out to extend the substrates from nonpolar H—H bond to E—H bonds. With nickel-borane complex 19, SiH3Ph and SiH2Ph2 were found to be efficiently activated to form oxidized adduct 21.[38] Significantly, hydrosilylation reaction had also been achieved by reacting benzaldehyde with 20 as the LA-TM catalyst.[38] It is still an exciting challenge to further extend the substrate to other E—H bonds, especially more popular bonds, with LA-TM complexes as potential bifunctional catalysts.
 图11
Activation of H—H/Si—H bond and olefin hydrogenation by DPB-Ni complex
Figure11.
Activation of H—H/Si—H bond and olefin hydrogenation by DPB-Ni complex
图11
Activation of H—H/Si—H bond and olefin hydrogenation by DPB-Ni complex
Figure11.
Activation of H—H/Si—H bond and olefin hydrogenation by DPB-Ni complex
H2 activation was also observed with [(TPB(η6-Ph))RuCl][B(C6F5)4] (22), which reacted with H2 in the presence of PCy3, to form product 23a and 23b, reversibly (Figure 12).[29, 39] Different from the H2 activation to form M—H and M-H-B bridge bond in nickel-borane complex, the C-site of phenyl acted as a Lewis acid site to accept the hydride in this ruthenium-borane complex. Complex 22 could be used for catalytic hydrogenation of aldimines at room temperature under high H2 pressure (10.3 MPa).
 图12
H2 activation and aldimines hydrogenation by DPB-Ru complex
Figure12.
H2 activation and aldimines hydrogenation by DPB-Ru complex
图12
H2 activation and aldimines hydrogenation by DPB-Ru complex
Figure12.
H2 activation and aldimines hydrogenation by DPB-Ru complex
Progress of the E—H activations, including O—H, S—H, N—H, C—H, and Si—H bonds, were also made using similar DPB-M complexes. Complexes DPB-Fe(N2) (24) and DPB-Co(N2) (25), as two analogous LA-TM catalysts, show potential to activate the E—H bonds. As showed in Figures 13 and 14, phenol, thiophenol, benzo[h]quinolone, 8-aminoquinoline, and diphenylsilane were activated, respectively, by these two complexes to corresponding borohydride species.[30b] After the E—H activation by 24 and 25, borohydride adducts with typical M-H-B bridge bonds were generally observed. The activation of SiH2Ph2 was reported with DPB-Co complex 25. Si—H activation had been found in DPB-Fe complex 26 as well, in which Fe(Ⅰ) was the TM center (Figure 15).[40] With the reduction by Na/Hg, 26 reacted with H3SiPh in the present N2 to form complex 27. These progresses of the E—H activation imply the potential of the LA-TM complexes to react with versatile reactants.
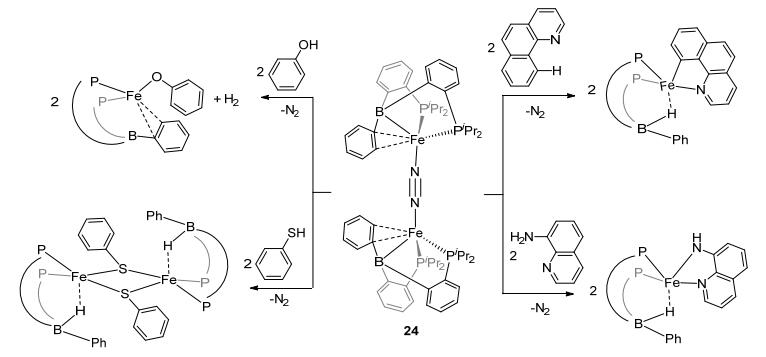 图13
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Fe complex
Figure13.
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Fe complex
图13
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Fe complex
Figure13.
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Fe complex
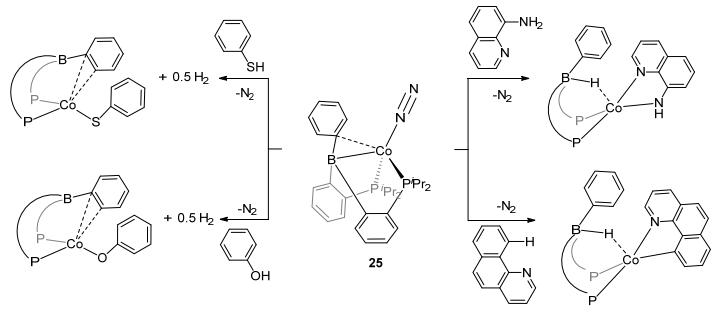 图14
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Co complex
Figure14.
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Co complex
图14
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Co complex
Figure14.
Activation of O—H, S—H, C—H, and N—H bonds by DPB-Co complex
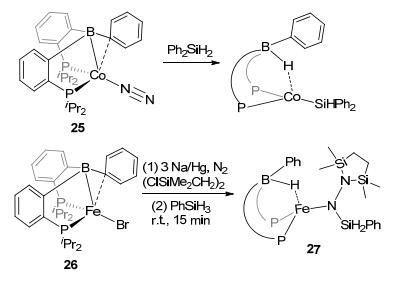 图15
Activation of Si—H bond by DPB-Fe/Co complexes
Figure15.
Activation of Si—H bond by DPB-Fe/Co complexes
图15
Activation of Si—H bond by DPB-Fe/Co complexes
Figure15.
Activation of Si—H bond by DPB-Fe/Co complexes
Besides activation of E—H bonds, activation of E—E bonds was studied by the LA-TM complexes as well. Tauchert et al.[30a] reported the C—O bond activation by the palladium borane complex 28 (Figure 16), by reacting with the allyl substrates. After the C—O cleavage, the OR group interacted with borane atom, and the alkyl group combined with the palladium center. More interesting discovery was reported for C—C coupling by DPB-Fe(CO)2 (29).[30c] In the present of excess K, ligand exchange was observed in DPB-Fe(CO)2 with two equivalents of TMSOTf to form 30. Subsequently, with addition of H2, C—C coupling was observed under mild conditions (Figure 17). The C—C coupling reaction was also observed by the air-stable DPB-Au complex 31 (Figure 18).[41] This sp3 boron-based LA-TM complex was suggested to have several interesting characteristics.[41] First, the LA ligand can increase the Lewis acidity of the TM atom due to electron withdrawing effect. Second, reductive elimination might be promoted because of the electron density decrease on the TM center. Third, LA and TM might work as multipoint recognition systems. Complex 31 was found to react with enynes to form five-, six-, and seven-membered rings by cycloisomerization. These investigations greatly expanded the catalytic scope of the LA-TM complexes from the activation of H—H(E) bonds to the activation of E—E bonds, and also exhibited their capability in coupling reactions.
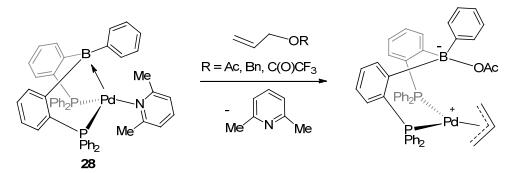 图16
Activation of C—O bond by DPB-Pd complex
Figure16.
Activation of C—O bond by DPB-Pd complex
图16
Activation of C—O bond by DPB-Pd complex
Figure16.
Activation of C—O bond by DPB-Pd complex
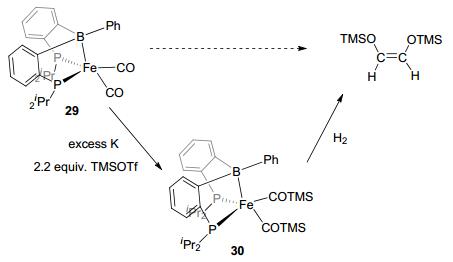 图17
C—C coupling of CO by DPB-Fe complex
Figure17.
C—C coupling of CO by DPB-Fe complex
图17
C—C coupling of CO by DPB-Fe complex
Figure17.
C—C coupling of CO by DPB-Fe complex
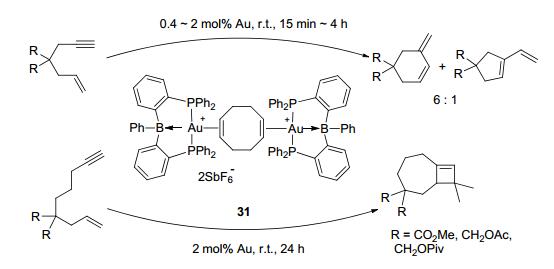 图18
Catalytic cyclization reactions by DPB-Au complex
Figure18.
Catalytic cyclization reactions by DPB-Au complex
图18
Catalytic cyclization reactions by DPB-Au complex
Figure18.
Catalytic cyclization reactions by DPB-Au complex
Apart from the DPB ligands, the sp3 boron-based LA-TM complexes with TPB ligands also showed potentials in catalysis. Complex TPB-Fe(N2) 32 was reported to be able to activate H2 to complexes 33 and 34 (Figure 19).[27, 12b] By reacting with methanol and alkyne, C—H bond activation was also observed with this complex to form complexes 35 and 36. It is worth noting that N2 fixation reaction could be achieved by TPB-FeBr complex 37 (Figure 20).[42] Furthermore, with (ClSiMe2CH2)2, the salinization hydrosilylation product 38 could be furnished from complex 37 during N2 activation. Besides, a direct hydrogenation of N2 was reported as well. Using HBArF4 as the hydrogen source, in the present of KC8, complex 37 activated N2 to 39 in the N2 atmosphere. These reactions suggested the potential of LA-TM catalysts in N2 fixation, because Lewis acid functional site was suggested to be able to reduce the electron density within the N2 molecule.
A series of activation reactions were observed for the sp3 boron-based LA-TM complexes with ligands bearing terminal Lewis acid. Complex 10, Pt-(FcPPB), which reported by Emslie et al., was observed to activate the H—H bond (Figure 21), [10a, 32] leading to product 40. In complex 40, the terminal hydride and the Pt-H-B bridge hydride were in cis positions. Complex 10 also reacted with phenylacetylene to form complex 41. In this reaction, after the heterolytic cleavage of the C—H bond, hydride was attracted by boron to form the Pt-H-B bridge bond, while the alkynyl group attached to TM center cis to hydride.
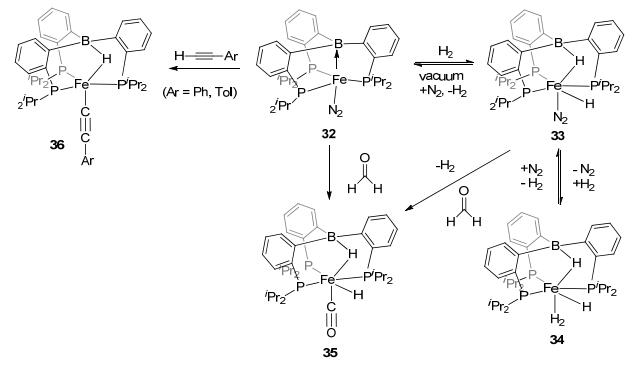 图19
Activation of H—H and C—H bonds by TBP-Fe complex
Figure19.
Activation of H—H and C—H bonds by TBP-Fe complex
图19
Activation of H—H and C—H bonds by TBP-Fe complex
Figure19.
Activation of H—H and C—H bonds by TBP-Fe complex
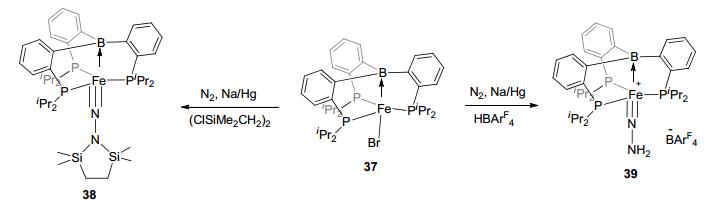 图20
Fixation of N2 by TPB-Fe complex
Figure20.
Fixation of N2 by TPB-Fe complex
图20
Fixation of N2 by TPB-Fe complex
Figure20.
Fixation of N2 by TPB-Fe complex
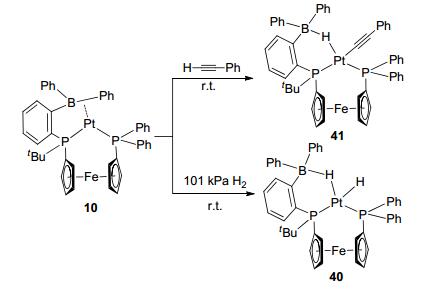 图21
Activation of H2 and phenylacetylene by terminal borane complex Pt-(FcPPB)
Figure21.
Activation of H2 and phenylacetylene by terminal borane complex Pt-(FcPPB)
图21
Activation of H2 and phenylacetylene by terminal borane complex Pt-(FcPPB)
Figure21.
Activation of H2 and phenylacetylene by terminal borane complex Pt-(FcPPB)
Another interesting LA-TM complex bearing terminal Lewis acid ligand, the Pt-(BIM), showed significant activation ability in H—H and E—H bonds activation. By reacting with H2, this T-shape complex 11 formed product 42, in which the terminal hydride and the Pt-H-B bridge hydride were in trans positions (Figure 22).[33] This result was different from that of the Pt-(FcPPB) complex. More differences were reflected in the activation of E—H bonds. By reacting with phenylamine, methyl alcohol, and phenylacetylene, the hydride would remain at terminal position rather than forming a Pt-H-B bridge bond after E—H activations. It should be noted that these reactions were found to be irreversible.
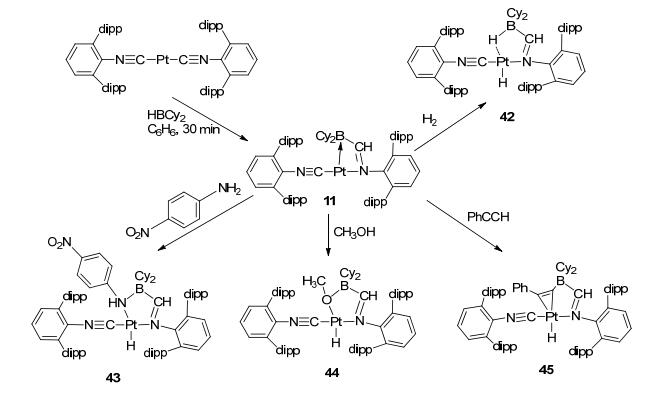 图22
Activation of H—H, N—H, and O—H bonds by terminal borane complex Pt-(BIM)
Figure22.
Activation of H—H, N—H, and O—H bonds by terminal borane complex Pt-(BIM)
图22
Activation of H—H, N—H, and O—H bonds by terminal borane complex Pt-(BIM)
Figure22.
Activation of H—H, N—H, and O—H bonds by terminal borane complex Pt-(BIM)
As shown above, as a new type of bifunctional catalysts, a series of sp3 boron-based LA-TM complexes have been applied in H—H, E—H, and E—E activations. The complexes with phosphorus donor ligands have been explored in reactivity more than other analogous, for example complexes with nitrogen donor ligands. The LA-TM complexes with DPB and TBP ligands show more potentials in catalytic activity at present stage. Extension of the ligand types other than phosphines would be highly desired. And it should be note that, although several sp3 boron-based LA-TM complexes were utilized in E—H bond activation, many of them are limited in stoichiometric reactions, resulting in irreversible intermediates. Therefore, future developments are desired to explore more efficient catalytic transformations via activation of E—H bonds.
4 sp2 boron-based LA-TM complexes
The TM=B double bond involving a d-p interaction between TM and boron is usually not strong enough. Therefore, type D sp2 boron-based LA-TM complexes are not so common in the literature. The vacant p orbital of the sp2 hybridized boron generally requires resonance stabilization from adjacent heteroatom(s) to construct type G or type H complexes. In 1990, an early beautiful example of type D LA-TM complex was reported by Baker et al.[43] (46 in Figure 23), in which a typical TM=B double bond was formed by donating the electron pair of iridium to boron's empty pz orbital. Later, Hartwig[44] and Aldridge et al.[45] reported the synthesis and structure of type D biarylboryl complexes CpFe(CO)2B(Ar)2 (47) (Figure 23). A rare example of type D dialkylboryl tungsten complex 48 with β-H agostic interaction was reported by Pritzkow et al.[46] As for type E sp2 boron-based LA-TM complexes, only very few examples have been found until now. An alkoxylborylene osmium complex 49 (Figure 24) was reported by Wright et al., [47] with pyridine as an L-group donating an electron pair to the boron atom. With an additional pyridine derivative as the L-donor, another two examples are aminoborylene iron complex 50[48] and aminoborylene platinum complex 51[49]. To the best of our knowledge, type F complexes remain a challenge and have not been reported so far.
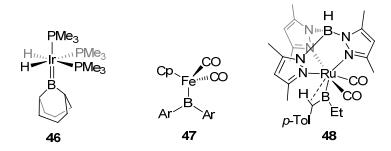 图23
Type D sp2 boron-based LA-TM complexes
Figure23.
Type D sp2 boron-based LA-TM complexes
图23
Type D sp2 boron-based LA-TM complexes
Figure23.
Type D sp2 boron-based LA-TM complexes
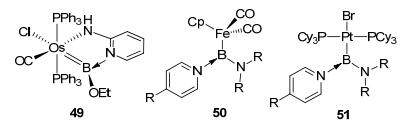 图24
Type E sp2 boron-based LA-TM osmium complexes
Figure24.
Type E sp2 boron-based LA-TM osmium complexes
图24
Type E sp2 boron-based LA-TM osmium complexes
Figure24.
Type E sp2 boron-based LA-TM osmium complexes
Type G sp2 boron-based LA-TM complexes are shown in Figure 25. The bidentate boryl osmium complex 52 was synthesized by Rickard et al.[47]Complexes 53 and 54 were derived from mono-imine-boron ligand.[50] This kind of aminochroloboryl complexes have the sp2 nitrogen that could donate electrons to boron. With a similar structure to 52, the aminochroloboryl complex 55 was reported as a type G complex (Figure 25).[47]
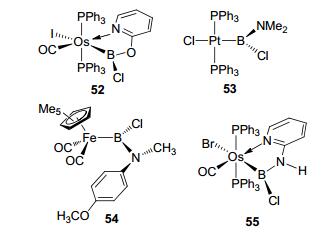 图25
Type G sp2 boron-based LA-TM complexes
Figure25.
Type G sp2 boron-based LA-TM complexes
图25
Type G sp2 boron-based LA-TM complexes
Figure25.
Type G sp2 boron-based LA-TM complexes
Apart from the type D~G complexes mentioned above, type H complexes are the majority in the sp2 boron-based LA-TM complexes, which were widely studied. Type H complexes with BN-heterocyclic boryl ligands are a big family in the literature.[51] These BN-heterocyclic boryl ligands are structural similar to classical NHC. Since the early report of type H complex by Tanaka et al., [52] a number of type H complexes bearing various TM centers and mono-dentate BN-heterocyclic ligands composed of unsaturated and saturated backbones and different steric hindrance have been synthesized (56~59 in Figure 26), [51a, 53] including Os, Cu, Ag, Au, Ti, Hf, etc. The BO-heterocyclic sp2 boron-based LA-TM complexes were studied earlier. In 1990, 1, 2-diphenylenedioxyboryl complex 61 was reported by Merola et al.[43b] Since then, lots of BO-heterocyclic sp2 boron-based complexes such as 60 were studied.[54] As a kind of ligand usually derived from catecholborane or pinacolborane, mono-dentate BO-heterocyclic ligand is generally regarded as a stable moiety in complexes. Therefore it will only be briefly discussed in this review. It is worth noting that, a novel B-heterocyclic sp2 boron-based LA-TM complex [(Cy3P)2Pt(Cl)(BC4Ph4)] (62, Figure 26) derived from borole was synthesized and characterized by Braunschweig et al.[55] Another ferroborirene complex 63, derived from borirene, was also reported.[56] In B-heterocyclic sp2 boron-based LA-TM complexes, the empty p orbital of boron is delocalized with the π electrons of C=C double bond(s) and d electrons of TM center.
图26
Type H sp2 boron-based LA-TM complexes with mono-dentate heterocyclic ligands
Figure26.
Type H sp2 boron-based LA-TM complexes with mono-dentate heterocyclic ligands
BN-heterocyclic boryl pincer ligands have attracted many interests recently. In 2009, by reacting diphosphino-boryl (PBP) pincer ligand with [Ir(cod)Cl]2, Yamashita and Nozaki et al.[57] reported the synthesis of PBP-Ir(H)Cl complex 64, which was further reduced to PBP-Ir(C2H4) complex 65 by LiTMP (Figure 27). The skeleton of this PBP-Ir complex is similar to classical PNP and PCP complexes, but the property of the boron is unique from the N or C atoms in other pincer ligands. In this PBP complex, boron atom binds to TM center as a typical X-type ligand, obtaining additional conjugation interactions with adjacent nitrogen atoms and the Ir center with its empty pz orbital. On the one hand, the empty pz orbital of boron can act as a Lewis acid. On the other hand, the M—B bond can act as a Lewis base to accept the nucleophile. This functional variation in one structure might provide interesting catalytic transformations for various substrates.
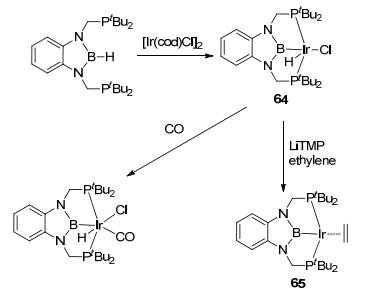 图27
Synthesis and structures of PBP-Ir complexes
Figure27.
Synthesis and structures of PBP-Ir complexes
图27
Synthesis and structures of PBP-Ir complexes
Figure27.
Synthesis and structures of PBP-Ir complexes
Soon after, a series of analogous complexes had been synthesized with PBP ligands. Peter et al.[12c] reported the synthesis of complex PBP-Co(N2) (66, Figure 28). Complex 66 adopts a four coordination square planar structure, in which N2 coordinates with the cobalt center trans to the boron atom. By reacting with RuH(OAc)(PPh3)2, Yamashita et al.[58] further synthesized complex PBP-(μ-H)2Ru(OAc-κ2O). Interestingly, this PBP-(μ-H)2Ru(OAc-κ2O) (67, Figure 28) possesses two Ru-H-B bridge bonds. With a long-tethered PBP ligand, complex 68 was synthesized, in which the plane of the PBP ligand was bended due to the large 7-membered ring.[59]
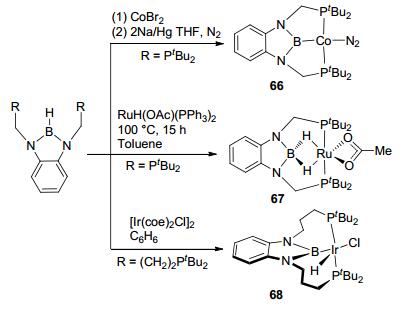 图28
Synthesis and structures of PBP-Co, PBP-Ru, and long-tethered PBP-Ir complexes
Figure28.
Synthesis and structures of PBP-Co, PBP-Ru, and long-tethered PBP-Ir complexes
图28
Synthesis and structures of PBP-Co, PBP-Ru, and long-tethered PBP-Ir complexes
Figure28.
Synthesis and structures of PBP-Co, PBP-Ru, and long-tethered PBP-Ir complexes
5 Reactions for the sp2 boron-based LA-TM complexes
Attempt on applying the sp2 boron-based LA-TM complex in catalysis was reported by Nozaki et al. with PBP-Rh complex 69 (Figure 29).[60] By ligand exchange with AgOTf and reductive elimination with LiCH2SiMe3, complex 70 was synthesized as a catalyst precursor, which could activate a series of O—H bonds. However, no M-H-B bridge bond was observed in the product, suggesting that boron possibly wasn't involved in the reaction. E—H addition was observed in the sp2 LA-TM complexes as well. The PBP-PtCl complex 71 could activate HSiEt3 to further addition of alkene (Figure 30). Complex 72, however, showed low activity in the same reaction.[61]
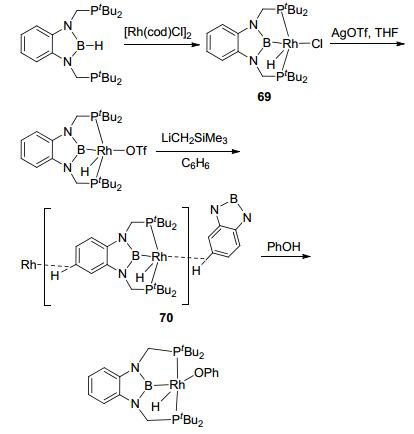 图29
Synthesis and structures of PBP-Rh complexes
Figure29.
Synthesis and structures of PBP-Rh complexes
图29
Synthesis and structures of PBP-Rh complexes
Figure29.
Synthesis and structures of PBP-Rh complexes
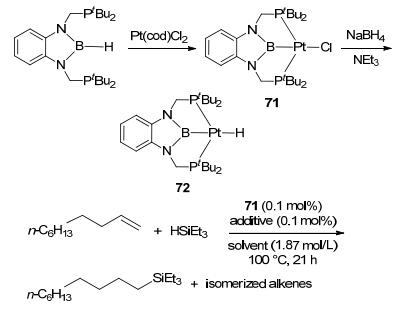 图30
Synthesis, structures and Si—H addition by PBP-Pt complex
Figure30.
Synthesis, structures and Si—H addition by PBP-Pt complex
图30
Synthesis, structures and Si—H addition by PBP-Pt complex
Figure30.
Synthesis, structures and Si—H addition by PBP-Pt complex
Further progress was made by Peter et al.[12c] with complex 66, which was reported to react with two equivalents of H2 to form product 73 with two Co-H-B bridge bonds (Figure 31). Hydrogenation reaction was achieved by reacting with styrene under 101 kPa H2. NH3-BH3 was applied as another hydrogen source to perform the reaction as well. An analogous complex, PBP-Ni, was reported to be able to hydrogenate olefins.[62] The catalytic hydrogenation reactions could also be promoted by mono-hydrogen addition complex. [PBP](μ-H)2Ru(OAc-κ2O) (67) was found to be the catalyst for the hydrogenation of 1-hexanal under 6 MPa H2 with low catalyst loadings (0.01 mol%).[58]
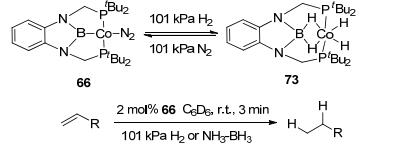 图31
Activation of H2 and olefin hydrogenation by PBP-Co complex
Figure31.
Activation of H2 and olefin hydrogenation by PBP-Co complex
图31
Activation of H2 and olefin hydrogenation by PBP-Co complex
Figure31.
Activation of H2 and olefin hydrogenation by PBP-Co complex
The catalytic transfer dehydrogenation reactions were also been achieved with the sp2 boron-based LA-TM catalysts (Figure 32).[63] By using PBP-Ir complex 65, cyclooctane can be catalytic transfer dehydrogenated to tert-butylethylene. Similar transfer dehydrogenation reaction was found in long-tethered PBP-Ir complex 68.[59] Higher catalytic efficiency was observed with 68 as catalyst for the same reaction. The catalytic transfer dehydrogenation reaction is a potential transformation for small molecule conversion, enriching the chemistry of LA-TM catalysts.
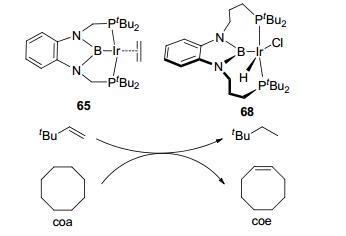 图32
Transfer dehydrogenation by PBP-Ir complex
Figure32.
Transfer dehydrogenation by PBP-Ir complex
图32
Transfer dehydrogenation by PBP-Ir complex
Figure32.
Transfer dehydrogenation by PBP-Ir complex
Compared with the sp3 boron-based LA-TM complexes, the reactivity explorations of the sp2 boron-based LA-TM complexes are still less at present. The reported examples are mainly related to type H complexes. The BN-hetero-cyclic boryl pincer ligands complexes (PBP type) is one of the most potential catalysts among the sp2 type LA-TM complexes. These PBP complexes have been achieved in hydrogenation and transfer dehydrogenation reactions. The vacant p orbital of boron can be involved in tautomerization/aromatization, which may competes with its function as a Lewis acid site to accept nucleophiles. It may be expected that tuning the balance between this competition would be able to overcome the irreversibility in E—H bond activation, providing an opportunity to develop efficient catalytic transformation via H—E(H) activations.
6 sp boron-based LA-TM complexes
In 1998, as an early example of the sp boron-based LA-TM complex, (OC)5W{BN(SiMe3)2} (74a), was reported by Cowley et al (Figure 33).[64] The iminoborylene complex 74a and analogous complexes, in which imino nitrogen donated a pair of electrons to borylene boron, were typical type J LA-TM complexes. So far, type J boron-based LA-TM complexes such as 74, 75, 76, and 77 had been synthesized with different TM centers and ligands including Cr, [65] V, [66] Mo, [67] Fe, [48a, 68] Ru, [48c] Ir, [69] Pt[70] etc. It should be noted that, although it is quite similar to type J complex, type Ⅰ complexes are however very rare in the literature, which may be expected if a linear boryl ligand is introduced to the TM center.
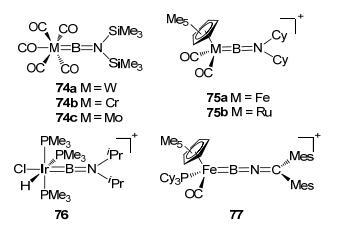 图33
Type J sp boron-based LA-TM complexes
Figure33.
Type J sp boron-based LA-TM complexes
图33
Type J sp boron-based LA-TM complexes
Figure33.
Type J sp boron-based LA-TM complexes
Type K sp boron-based LA-TM complex 78 was reported by Aldridge and co-workers in 2003.[71] Analogous linear terminal alkylborylene complex 79 was obtained by phosphine-induced cleavage of a homodinuclear manganese precursor.[72] In these type K complexes, the TM center tends to donate a pair of electrons to the boron to form a relatively weak TM≡B triple bond, featured with linear structure (Figure 34). This weak M≡B triple bond was also generally regarded in the literature as a TM=B double bond for borylene type ligands. Similar method was utilized to synthesize complex 80.[73] By reacting with Na2[Cr(CO)5] and Cl2BSi(SiMe3)3, analogous complex 81 with M≡B—Si structure was reported as well.[74] However, to the best of our knowledge, type L complexes have not been reported so far, which might require an L-group strong enough in eletron donating.
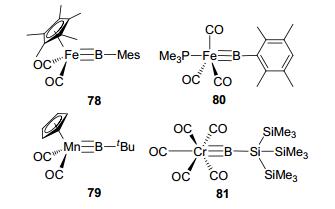 图34
Typical type K sp boron-based LA-TM complexes
Figure34.
Typical type K sp boron-based LA-TM complexes
图34
Typical type K sp boron-based LA-TM complexes
Figure34.
Typical type K sp boron-based LA-TM complexes
In 2006, a type N complex bearing terminally iminoborylene was reported by Braunschweig et al. using platinum(Ⅰ) as the TM center (complex 82, Figure 35).[75] A few type N complexes were reported with rhodium[75] and palladium[76] as the TM centers. Oxylboryl complexes 83 and 84 were also reported as type N examples[77] in which oxygen donated a pair of electrons to the boron to form B≡O bond. While in complex 84, BAr3F interacts with oxyl oxygen to stabilize its lone pair.
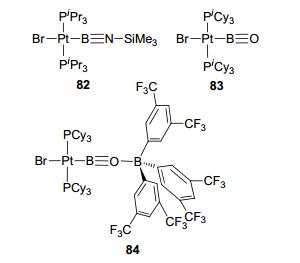 图35
Type N sp boron-based LA-TM complexes
Figure35.
Type N sp boron-based LA-TM complexes
图35
Type N sp boron-based LA-TM complexes
Figure35.
Type N sp boron-based LA-TM complexes
7 Reactions for the sp boron-based LA-TM complexes
Comparing with the sp3 and sp2 boron-based LA-TM catalysts, the catalytic reactions for the sp boron-based LA-TM complexes are very rare. In 2008, by reacting RuHCl(H2)(PCy3)2 with 1.5 equiv. of mesitylborane, type K complex 85 was synthesized by Sabo-Etienne (Figure 36).[78] Complex 85 could react with H2 (3 bar) at room temperature, leading to complex 86 bearing two Ru-H-B bridge bonds. Olefin hydrogenation was observed with the hydrogenated derivatives 87 by Aldrudge et al. (Figure 37).[79] Reaction between complex 87 and 10 equiv. of 3, 3-dimethylbutene yielded 2, 2-dimethylbutane and type Ⅰ complex 88 at room temperature. The reactivity exploration of the sp boron-based LA-TM complexes has till yet to begin. However, it should be exciting to see the future development of stoichiometric/catalytic reactions utilizing the sp boron-based LA-TM complexes, considering that it has various resonance forms that can participate in bifunctional reactivity.
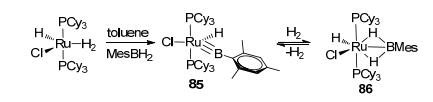 图36
Activation of H2 by type K ruthenium complex
Figure36.
Activation of H2 by type K ruthenium complex
图36
Activation of H2 by type K ruthenium complex
Figure36.
Activation of H2 by type K ruthenium complex
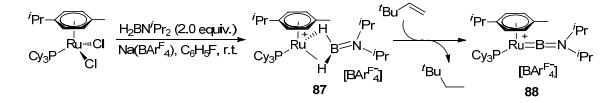 图37
Olefin hydrogenation by type Ⅰ ruthenium hydrogenated derivatives
Figure37.
Olefin hydrogenation by type Ⅰ ruthenium hydrogenated derivatives
图37
Olefin hydrogenation by type Ⅰ ruthenium hydrogenated derivatives
Figure37.
Olefin hydrogenation by type Ⅰ ruthenium hydrogenated derivatives
8 Conclusion and perspective
The LA-TM cooperation has been merging as a novel bifunctional strategy for catalysis, which is a fundamental alternative to the traditional LB-TM bifunctional catalysis and frustrated Lewis pair catalysis. Perspectively, as a new strategy for metal ligand cooperation, the LA-TM catalysis has several advantages to enrich the chemistry of bifunctional catalysis. (1) LA-TM cooperation is a novel bifunctional mode different from traditional LB-TM cooperation. In the LA-TM system, the LA part accepts electron pair and the TM donates electron(s) cooperatively, which is expected to bring new types of transformations for bifunctional catalysis. (2) The LA site of the ligand can stabilize low valent metal center by a dative bond from the metal to the LA ligand, bringing unique feature to this type of catalysts. (3) Both the LA part and the TM center can stabilize the reactive anion(s), for example, hydride(s), in the key intermediates during transformations. Therefore, LA-TM complexes can be expected to act as potential catalysts for the activation of H2 or other unreactive E—H(E) bonds, providing an alternative way to achieve green bifunctional catalysis under mild conditions with low catalyst loading. (4) Unlike LB-TM catalysis, the LA-TM catalysis is not necessary to experience a proton or cation species during reaction, which could be alternative way to expand the scope of reactions to substrates that dislike proton or cation. (5) Especially for hydrogen transformation, the obtained intermediates with multiple hydrides can serve as unique and more robust species for hydrogenation. (6) Furthermore, the idea of LA-TM cooperation will expand the options to develop multi-nuclear catalyst systems, or to develop LA-TM cooperation sites on heterogeneous catalysts.
There have been remarkable developments of LA-TM complexes in recent years. This review has summarized most of boron-based LA-TM complexes according to their binding features. The boron-based ligands are generally divided by borane, boryl, and borylene. However, It can be seen that their structures and binding properties are more complicated (Types A~N), which bring fantastic variations not only to the structure, but also to the reactivity and the catalytic transformation diversity. These variations also bring a big challenge to understand the mechanism of LA-TM catalysis for both theorists and experimentalists. Up today, although many types of LA-TM complexes have been synthesized and studied, some of them are still rare, or even remain a challenge to be explored.
Important reactions promoted by boron-based LA-TM complexes have been summarized. Until now, although significant progress has been made for LA-TM bifunctional catalysis, the applications of LA-TM complexes in catalysis are still limited. The sp3 boron-based LA-TM complexes were explored in reactivity relatively more than the sp2 and sp analogous. The LA-TM complexes with DPB and TBP ligands have been approved as capable catalysts for H2 activation and hydrogenation. However, the activation of E—H bonds is still mainly limited in stoichiometric reactions, in which the irreversible intermediates could not take further transformations. The reactivity explorations of the sp2 and sp boron-based LA-TM complexes are still very few at present. However, the diversity in resonance structure could provide opportunities to develop efficient catalytic transformation by electronic and geometric tuning for the sp2 and sp boron-based LA-TM complexes. Therefore, future developments should focus on exploring more efficient catalytic transformations via activation of H—H, E—H and E—E bonds. Expansion of reaction types for LA-TM catalysts, for example the cross coupling and asymmetric reactions etc., is also highly desired. These developments in the future should bring more impacts of LA-TM catalysts on synthesis and hydrogen energy. The combination of diverse LA and TM, the cooperation of LA and TM in different manners, the umpolung in LA-TM interaction, the bridge bonds, and the multi-nuclear complexes, etc., are all unique characteristics and powerful tools to design and develop novel LA-TM catalysis in the future.
-
-
[1]
(a) Hu, X. L. Chem. Sci. 2011, 2, 1867.
(b) Khusnutdinova, J. R. ; Milstein, D. Angew. Chem. , Int. Ed. 2015, 54, 12236. -
[2]
(a) Kubas, G. J. Chem. Rev. 2007, 107, 4152. 10, 701.
-
[3]
(a) Ikariya, T. ; Blacker, A. J. Acc. Chem. Res. 2007, 40, 1300.
(b) Kuwata, S. ; Ikariya, T. Chem. Commun. 2014, 50, 14290. -
[4]
(a) Szostak, J. W. Medicina-Buenos Aires 2016, 76, 199.
(b) Szostak, J. W. Nature 2009, 459, 171.
(c) Szostak, J. W. ; Bartel, D. P. ; Luisi, P. L. Nature 2001, 409, 387.
(d) Mansy, S. S. ; Szostak, J. W. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13351. -
[5]
(a) Fontecilla-Camps, J. C. ; Volbeda, A. ; Cavazza, C. ; Nicolet, Y. Chem. Rev. 2007, 107, 4273.
(b) Silakov, A. ; Wenk, B. ; Reijerse, E. ; Lubitz, W. Phys. Chem. Chem. Phys. 2009, 11, 6592.
(c) Barton, B. E. ; Olsen, M. T. ; Rauchfuss, T. B. J. Am. Chem. Soc. 2008, 130, 16834.
(d) Gordon, J. C. ; Kubas, G. J. Organometallics 2010, 29, 4682. -
[6]
(a) Hou, C. ; Jiang, J. X. ; Li, Y. W. ; Zhao, C. Y. ; Ke, Z. F. ACS Catal. 2017, 7, 786.
(b) Hou, C. ; Zhang, Z. H. ; Zhao, C. Y. ; Ke, Z. F. Inorg. Chem. 2016, 55, 6539. -
[7]
Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97. doi: 10.1021/ar9502341
-
[8]
(a) Morris, R. H. Chem. Rec. 2016, 16, 2644.
(b) Annibale, V. T. ; Song, D. T. RSC Adv. 2013, 3, 11432.
(c) Zhao, B. G. ; Han, Z. B. ; Ding, K. L. Angew. Chem. , Int. Ed. 2013, 52, 4744.
(d) DuBois, D. L. Inorg. Chem. 2014, 53, 3935.
(e) Crabtree, R. H. New J. Chem. 2011, 35, 18.
(f) Appel, A. M. ; Bercaw, J. E. ; Bocarsly, A. B. ; Dobbek, H. ; DuBois, D. L. ; Dupuis, M. ; Ferry, J. G. ; Fujita, E. ; Hille, R. ; Kenis, P. J. A. ; Kerfeld, C. A. ; Morris, R. H. ; Peden, C. H. F. ; Portis, A. R. ; Ragsdale, S. W. ; Rauchfuss, T. B. ; Reek, J. N. H. ; Seefeldt, L. C. ; Thauer, R. K. ; Waldrop, G. L. Chem. Rev. 2013, 113, 6621.
(g) Fujita, E. ; Muckerman, J. T. ; Himeda, Y. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1031. -
[9]
(a) Owen, G. R. Chem. Commun. 2016, 52, 10712.
(b) Bouhadir, G. ; Bourissou, D. Chem. Soc. Rev. 2016, 45, 1065.
(c) Dang, L. ; Lin, Z. ; Marder, T. B. Chem. Commun. 2009, 3987.
(d) Braunschweig, H. ; Dewhurst, R. D. ; Schneider, A. Chem. Rev. 2010, 110, 3924.
(e) Braunschweig, H. ; Colling, M. Coord. Chem. Rev. 2001, 223, 1. -
[10]
(a) Devillard, M. ; Bouhadir, G. ; Bourissou, D. Angew. Chem. , Int. Ed. 2015, 54, 730.
(b) Li, Y. W. ; Hou, C. ; Jiang, J. X. ; Zhang, Z. H. ; Zhao, C. Y. ; Page, A. J. ; Ke, Z. F. ACS Catal. 2016, 6, 1655. -
[11]
(a) Tsoureas, N. ; Hamilton, A. ; Haddow, M. F. ; Harvey, J. N. ; Orpen, A. G. ; Owen, G. R. Organometallics 2013, 32, 2840.
(b) Harman, W. H. ; Lin, T. P. ; Peters, J. C. Angew. Chem. , Int. Ed. 2014, 53, 1081.
(c) Zeng, G. ; Sakaki, S. Inorg. Chem. 2013, 52, 2844. -
[12]
(a) Harman, W. H. ; Peters, J. C. J. Am. Chem. Soc. 2012, 134, 5080.
(b) Fong, H. ; Moret, M. E. ; Lee, Y. ; Peters, J. C. Organometallics 2013, 32, 305.
(c) Lin, T. P. ; Peters, J. C. J. Am. Chem. Soc. 2013, 135, 15310. -
[13]
Rudd, P. A.; Liu, S. S.; Gagliardi, L.; Young, V. G.; Lu, C. C. J. Am. Chem. Soc. 2011, 133, 20724. doi: 10.1021/ja2099744
-
[14]
Cammarota, R. C.; Lu, C. C. J. Am. Chem. Soc. 2015, 137, 12486. doi: 10.1021/jacs.5b08313
-
[15]
Riddlestone, I. M.; Rajabi, N. A.; Lowe, J. P.; Mahon, M. F.; Macgregor, S. A.; Whittlesey, M. K. J. Am. Chem. Soc. 2016, 138, 11081. doi: 10.1021/jacs.6b05243
-
[16]
Yang, H. F.; Gabbai, F. P. J. Am. Chem. Soc. 2015, 137, 13425. doi: 10.1021/jacs.5b07998
-
[17]
Hill, A. F.; Owen, G. R.; White, A. J. P.; Williams, D. J. Angew. Chem., Int. Ed. 1999, 38, 2759. doi: 10.1002/(ISSN)1521-3773
-
[18]
Tsoureas, N.; Kuo, Y. Y.; Haddow, M. F.; Owen, G. R. Chem. Commun. 2011, 47, 484. doi: 10.1039/C0CC02245D
-
[19]
(a) Blagg, R. J. ; Charmant, J. P. H. ; Connelly, N. G. ; Haddow, M. F. ; Orpen, A. G. Chem. Commun. 2006, 2350.
(b) Landry, V. K. ; Melnick, J. G. ; Buccella, D. ; Pang, K. L. ; Ulichny, J. C. ; Parkin, G. Inorg. Chem. 2006, 45, 2588.
(c) Senda, S. ; Ohki, Y. ; Hirayama, T. ; Toda, D. ; Chen, J. L. ; Matsumoto, T. ; Kawaguchi, H. ; Tatsumi, K. Inorg. Chem. 2006, 45, 9914.
(d) Crossley, I. R. ; Foreman, M. R. S. J. ; Hill, A. F. ; Owen, G. R. ; White, A. J. P. ; Williams, D. J. ; Willis, A. C. Organometallics 2008, 27, 381.
(e) Crossley, I. R. ; Hill, A. F. Dalton Trans. 2008, 201.
(f) Crossley, I. R. ; Hill, A. F. ; Willis, A. C. Organometallics 2008, 27, 312.
(g) Pang, K. L. ; Tanski, J. M. ; Parkin, G. Chem. Commun. 2008, 1008.
(h) Blagg, R. J. ; Adams, C. J. ; Charmant, J. P. H. ; Connelly, N. G. ; Haddow, M. F. ; Hamilton, A. ; Knight, J. ; Orpen, A. G. ; Ridgway, B. M. Dalton Trans. 2009, 8724.
(i) Rudolf, G. C. ; Hamilton, A. ; Orpen, A. G. ; Owen, G. R. Chem. Commun. 2009, 553.
(j) Crossley, I. R. ; Hill, A. F. ; Willis, A. C. Organometallics 2010, 29, 326.
(k) Lopez-Gomez, M. J. ; Connelly, N. G. ; Haddow, M. F. ; Hamilton, A. ; Orpen, A. G. Dalton Trans. 2010, 39, 5221. -
[20]
(a) Tsoureas, N. ; Bevis, T. ; Butts, C. P. ; Hamilton, A. ; Owen, G. R. Organometallics 2009, 28, 5222.
(b) Tsoureas, N. ; Haddow, M. F. ; Hamilton, A. ; Owen, G. R. Chem. Commun. 2009, 2538.
(c) Blagg, R. J. ; Connelly, N. G. ; Haddow, M. F. ; Hamilton, A. ; Lusi, M. ; Orpen, A. G. ; Ridgway, B. M. Dalton Trans. 2010, 39, 11616. -
[21]
Lopez-Gomez, M. J.; Connelly, N. G.; Haddow, M. F.; Hamilton, A.; Lusi, M.; Baisch, U.; Orpen, A. G. Dalton Trans. 2011, 40, 4647. doi: 10.1039/c0dt01718c
-
[22]
Figueroa, J. S.; Melnick, J. G.; Parkin, G. Inorg. Chem. 2006, 45, 7056. doi: 10.1021/ic061353n
-
[23]
Mihalcik, D. J.; White, J. L.; Tanski, J. M.; Zakharov, L. N.; Yap, G. P. A.; Incarvito, C. D.; Rheingold, A. L.; Rabinovich, D. Dalton Trans. 2004, 1626.
-
[24]
Crossley, I. R.; Foreman, M. R. S.; Hill, A. F.; White, A. J. P.; Williams, D. J. Chem. Commun. 2005, 221.
-
[25]
Bontemps, S.; Bouhadir, G.; Dyer, P. W.; Miqueu, K.; Bourissou, D. Inorg. Chem. 2007, 46, 5149. doi: 10.1021/ic7006556
-
[26]
(a) Bontemps, S. ; Bouhadir, G. ; Gu, W. ; Mercy, M. ; Chen, C. H. ; Foxman, B. M. ; Maron, L. ; Ozerov, O. V. ; Bourissou, D. Angew. Chem. , Int. Ed. 2008, 47, 1481.
(b) Sircoglou, M. ; Bontemps, S. ; Bouhadir, G. ; Saffon, N. ; Miqueu, K. ; Gu, W. X. ; Mercy, M. ; Chen, C. H. ; Foxman, B. M. ; Maron, L. ; Ozerov, O. V. ; Bourissou, D. J. Am. Chem. Soc. 2008, 130, 16729.
(c) Kameo, H. ; Hashimoto, Y. ; Nakazawa, H. Organometallics 2012, 31, 4251.
(d) Suess, D. L. M. ; Tsay, C. ; Peters, J. C. J. Am. Chem. Soc. 2012, 134, 14158. -
[27]
Moret, M. E.; Peters, J. C. Angew. Chem., Int. Ed. 2011, 50, 2063. doi: 10.1002/anie.v50.9
-
[28]
Sircoglou, M.; Bontemps, S.; Mercy, M.; Saffon, N.; Takahashi, M.; Bouhadir, G.; Maron, L.; Bourissou, D. Angew. Chem., Int. Ed. 2007, 46, 8583. doi: 10.1002/anie.v46:45
-
[29]
Boone, M. P.; Stephan, D. W. J. Am. Chem. Soc. 2013, 135, 8508. doi: 10.1021/ja403912n
-
[30]
(a) Schindler, T. ; Lux, M. ; Peters, M. ; Scharf, L. T. ; Osseili, H. ; Maron, L. ; Tauchert, M. E. Organometallics 2015, 34, 1978.
(b) Nesbit, M. A. ; Suess, D. L. M. ; Peters, J. C. Organometallics 2015, 34, 4741.
(c) Suess, D. L. M. ; Peters, J. C. J. Am. Chem. Soc. 2013, 135, 12580.
(d) Sircoglou, M. ; Bontemps, S. ; Mercy, M. ; Miqueu, K. ; Ladeira, S. ; Saffon, N. ; Maron, L. ; Bouhadir, G. ; Bourissou, D. Inorg. Chem. 2010, 49, 3983.
(e) Kameo, H. ; Nakazawa, H. Organometallics 2012, 31, 7476.
(f) Conifer, C. M. ; Law, D. J. ; Sunley, G. J. ; White, A. J. P. ; Britovsek, G. J. P. Organometallics 2011, 30, 4060. -
[31]
Bontemps, S.; Bouhadir, G.; Miqueu, K.; Bourissou, D. J. Am. Chem. Soc. 2006, 128, 12056. doi: 10.1021/ja0637494
-
[32]
Cowie, B. E.; Emslie, D. J. H. Chem.-Eur. J. 2014, 20, 16899. doi: 10.1002/chem.201404846
-
[33]
Barnett, B. R.; Moore, C. E.; Rheingold, A. L.; Figueroa, J. S. J. Am. Chem. Soc. 2014, 136, 10262. doi: 10.1021/ja505843g
-
[34]
(a) Braunschweig, H. ; Radacki, K. ; Seeler, F. ; Whittell, G. R. Organometallics 2004, 23, 4178.
(b) Braunschweig, H. ; Radacki, K. ; Seeler, F. ; Whittell, G. R. , Organometallics 2006, 25, 4605. -
[35]
Bissinger, P.; Braunschweig, H.; Damme, A.; Dewhurst, R. D.; Kraft, K.; Kramer, T.; Radacki, K. Chem.-Eur. J. 2013, 19, 13402. doi: 10.1002/chem.201302263
-
[36]
Vondung, L.; Frank, N.; Fritz, M.; Alig, L.; Langer, R. Angew. Chem., Int. Ed. 2016, 55, 14448.
-
[37]
Frank, N.; Hanau, K.; Flosdorf, K.; Langer, R., Dalton Trans. 2013, 42, 11252. doi: 10.1039/c3dt51254a
-
[38]
MacMillan, S. N.; Harman, W. H.; Peters, J. C. Chem. Sci. 2014, 5, 590. doi: 10.1039/C3SC52626G
-
[39]
Boone, M. P.; Stephan, D. W. Chem.-Eur. J. 2014, 20, 3333. doi: 10.1002/chem.v20.12
-
[40]
Suess, D. L. M.; Peters, J. C. J. Am. Chem. Soc. 2013, 135, 4938. doi: 10.1021/ja400836u
-
[41]
Inagaki, F.; Matsumoto, C.; Okada, Y.; Maruyama, N.; Mukai, C., Angew. Chem., Int. Ed. 2015, 54, 818. doi: 10.1002/anie.201408037
-
[42]
(a) Anderson, J. S. ; Rittle, J. ; Peters, J. C. Nature 2013, 501, 84.
(b) Anderson, J. S. ; Moret, M. E. ; Peters, J. C. J. Am. Chem. Soc. 2013, 135, 534.
(c) Moret, M. E. ; Peters, J. C. J. Am. Chem. Soc. 2011, 133, 18118. -
[43]
(a) Baker, R. T. ; Ovenall, D. W. ; Calabrese, J. C. ; Westcott, S. A. ; Taylor, N. J. ; Williams, I. D. ; Marder, T. B. J. Am. Chem. Soc. 1990, 112, 9399.
(b) Knorr, J. R. ; Merola, J. S. Organometallics 1990, 9, 3008. -
[44]
Hartwig, J. F.; Huber, S. J. Am. Chem. Soc. 1993, 115, 4908. doi: 10.1021/ja00064a069
-
[45]
Aldridge, S.; Al-Fawaz, A.; Calder, R. J.; Dickinson, A. A.; Willock, D. J.; Light, M. E.; Hursthouse, M. B. Chem. Commun. 2001, 1846.
-
[46]
Wadepohl, H.; Arnold, U.; Pritzkow, H. Angew. Chem., Int. Ed. 1997, 36, 974. doi: 10.1002/(ISSN)1521-3773
-
[47]
Rickard, C. E. F.; Roper, W. R.; Williamson, A.; Wright, L. J. Organometallics 2002, 21, 4862. doi: 10.1021/om0203431
-
[48]
(a) Aldridge, S. ; Jones, C. ; Gans-Eichler, T. ; Stasch, A. ; Kays, D. L. ; Coombs, N. D. ; Willock, D. J. Angew. Chem. , Int. Ed. 2006, 45, 6118.
(b) De, S. ; Pierce, G. A. ; Vidovic, D. ; Kays, D. L. ; Coombs, N. D. ; Jemmis, E. D. ; Aldridge, S. Organometallics 2009, 28, 2961.
(c) Pierce, G. A. ; Vidovic, D. ; Kays, D. L. ; Coombs, N. D. ; Thompson, A. L. ; Jemmis, E. D. ; De, S. ; Aldridge, S. Organometallics 2009, 28, 2947. -
[49]
Braunschweig, H.; Radacki, K.; Uttinger, K. Chem.-Eur. J. 2008, 14, 7858. doi: 10.1002/chem.v14:26
-
[50]
(a) Curtis, D. ; Lesley, M. J. G. ; Norman, N. C. ; Orpen, A. G. ; Starbuck, J. J. Chem. Soc. , Dalton Trans. 1999, 1687.
(b) Addy, D. A. ; Phillips, N. ; Pierce, G. A. ; Vidovic, D. ; Kramer, T. ; Mallick, D. ; Jemmis, E. D. ; Reid, G. ; Aldridge, S. Organometallics 2012, 31, 1092. -
[51]
(a) Segawa, Y. ; Yamashita, M. ; Nozaki, K. Angew. Chem. , Int. Ed. 2007, 46, 6710.
(b) Asay, M. ; Jones, C. ; Driess, M. Chem. Rev. 2011, 111, 354.
(c) Frank, R. ; Howell, J. ; Campos, J. ; Tirfoin, R. ; Phillips, N. ; Zahn, S. ; Mingos, D. M. P. ; Aldridge, S. Angew. Chem. , Int. Ed. 2015, 54, 9586.
(d) Lu, W. ; Hu, H. T. ; Li, Y. X. ; Ganguly, R. ; Kinjo, R. J. Am. Chem. Soc. 2016, 138, 6650. -
[52]
Onozawa, S.-y.; Hatanaka, Y.; Sakakura, T.; Shimada, S.; Tanaka, M. Organometallics 1996, 15, 5450. doi: 10.1021/om9607199
-
[53]
(a) Borner, C. ; Brandhorst, K. ; Kleeberg, C. Dalton Trans. 2015, 44, 8600.
(b) Clark, G. R. ; Irvine, G. J. ; Roper, W. R. ; Wright, L. J. J. Organomet. Chem. 2003, 680, 81.
(c) Habereder, T. ; Noth, H. Appl. Organomet. Chem. 2003, 17, 525.
(d) Onozawa, S. ; Tanaka, M. Organometallics 2001, 20, 2956.
(e) Okuno, Y. ; Yamashita, M. ; Nozaki, K. Angew. Chem. , Int. Ed. 2011, 50, 920;
(f) Kajiwara, T. ; Terabayashi, T. ; Yamashita, M. ; Nozaki, K. Angew. Chem. , Int. Ed. 2008, 47, 6606.
(g) Terabayashi, T. ; Kajiwara, T. ; Yamashita, M. ; Nozaki, K. J. Am. Chem. Soc. 2009, 131, 14162. -
[54]
(a) Kawamura, K. ; Hartwig, J. F. J. Am. Chem. Soc. 2001, 123, 8422.
(b) Aldridge, S. ; Calder, R. J. ; Baghurst, R. E. ; Light, M. E. ; Hursthouse, M. B. J. Organomet. Chem. 2002, 649, 9.
(c) Braunschweig, H. ; Bertermann, R. ; Brenner, P. ; Burzler, M. ; Dewhurst, R. D. ; Radacki, K. ; Seeler, F. Chem. -Eur. J. 2011, 17, 11828.
(d) Esteruelas, M. A. ; Lopez, A. M. ; Mora, M. ; Onate, E. Chem. Commun. 2013, 49, 7543. -
[55]
Braunschweig, H.; Chiu, C. W.; Radacki, K.; Brenner, P. Chem. Commun. 2010, 46, 916. doi: 10.1039/b923652j
-
[56]
Braunschweig, H.; Fernandez, I.; Frenking, G.; Radacki, K.; Seeler, F. Angew. Chem., Int. Ed. 2007, 46, 5215. doi: 10.1002/(ISSN)1521-3773
-
[57]
(a) Segawa, Y. ; Yamashita, M. ; Nozaki, K. J. Am. Chem. Soc. 2009, 131, 9201.
(b) Segawa, Y. ; Yamashita, M. ; Nozaki, K. Organometallics 2009, 28, 6234. -
[58]
Miyada, T.; Kwan, E. H.; Yamashita, M. Organometallics 2014, 33, 6760. doi: 10.1021/om500585j
-
[59]
Kwan, E. H.; Kawai, Y. J.; Kamakura, S.; Yamashita, M. Dalton Trans. 2016, 45, 15931. doi: 10.1039/C6DT02075E
-
[60]
Hasegawa, M.; Segawa, Y.; Yamashita, M.; Nozaki, K. Angew. Chem., Int. Ed. 2012, 51, 6956. doi: 10.1002/anie.201201916
-
[61]
Ogawa, H.; Yamashita, M. Dalton Trans. 2013, 42, 625. doi: 10.1039/C2DT31892J
-
[62]
Lin, T. P.; Peters, J. C. J. Am. Chem. Soc. 2014, 136, 13672. doi: 10.1021/ja504667f
-
[63]
Tanoue, K.; Yamashita, M. Organometallics 2015, 34, 4011. doi: 10.1021/acs.organomet.5b00376
-
[64]
Braunschweig, H.; Kollann, C.; Englert, U. Angew. Chem., Int. Ed. 1998, 37, 3179. doi: 10.1002/(ISSN)1521-3773
-
[65]
Braunschweig, H.; Colling, M.; Kollann, C.; Stammler, H. G.; Neumann, B. Angew. Chem., Int. Ed. 2001, 40, 2298. doi: 10.1002/1521-3773(20010618)40:12<2298::AID-ANIE2298>3.0.CO;2-E
-
[66]
Braunschweig, H.; Colling, M.; Hu, C. H.; Radacki, K. Angew. Chem., Int. Ed. 2003, 42, 205. doi: 10.1002/anie.200390079
-
[67]
Blank, B.; Colling-Hendelkens, M.; Kollann, C.; Radacki, K.; Rais, D.; Uttinger, K.; Whittell, G. R.; Braunschweig, H. Chem.-Eur. J. 2007, 13, 4770. doi: 10.1002/(ISSN)1521-3765
-
[68]
Niemeyer, J.; Addy, D. A.; Riddlestone, I.; Kelly, M.; Thompson, A. L.; Vidovic, D.; Aldridge, S. Angew. Chem., Int. Ed. 2011, 50, 8908. doi: 10.1002/anie.v50.38
-
[69]
O'Neill, M.; Addy, D. A.; Riddlestone, I.; Kelly, M.; Phillips, N.; Aldridge, S. J. Am. Chem. Soc. 2011, 133, 11500. doi: 10.1021/ja2050748
-
[70]
Braunschweig, H.; Radacki, K.; Rais, D.; Schneider, A.; Seeler, F. J. Am. Chem. Soc. 2007, 129, 10350. doi: 10.1021/ja0742636
-
[71]
Coombs, D. L.; Aldridge, S.; Jones, C.; Willock, D. J. J. Am. Chem. Soc. 2003, 125, 6356. doi: 10.1021/ja0346936
-
[72]
Braunschweig, H.; Burzler, M.; Kupfer, T.; Radacki, K.; Seeler, F. Angew. Chem., Int. Ed. 2007, 46, 7785. doi: 10.1002/(ISSN)1521-3773
-
[73]
Braunschweig, H.; Ye, Q.; Radacki, K. Chem. Commun. 2012, 48, 2701. doi: 10.1039/c2cc17445f
-
[74]
Braunschweig, H.; Colling, M.; Kollann, C.; Merz, K.; Radacki, K. Angew. Chem., Int. Ed. 2001, 40, 4198. doi: 10.1002/1521-3773(20011119)40:22<>1.0.CO;2-D
-
[75]
Braunschweig, H.; Kupfer, T.; Radacki, K.; Schneider, A.; Seeler, F.; Uttinger, K.; Wu, H. J. Am. Chem. Soc. 2008, 130, 7974. doi: 10.1021/ja8016889
-
[76]
Braunschweig, H.; Radacki, K.; Rais, D.; Uttinger, K. Angew. Chem., Int. Ed. 2006, 45, 162. doi: 10.1002/(ISSN)1521-3773
-
[77]
(a) Braunschweig, H. ; Radacki, K. ; Schneider, A. Science 2010, 328, 345.
(b) Braunschweig, H. ; Radacki, K. ; Schneider, A. Chem. Commun. 2010, 46, 6473. -
[78]
Alcaraz, G.; Helmstedt, U.; Clot, E.; Vendier, L.; Sabo-Etienne, S. J. Am. Chem. Soc. 2008, 130, 12878. doi: 10.1021/ja805164n
-
[79]
Addy, D. A.; Bates, J. I.; Kelly, M. J.; Riddlestone, I. M.; Aldridge, S. Organometallics 2013, 32, 1583. doi: 10.1021/om400040q
-
[1]
-

Figure 1 Bifunctional catalysis of enzyme, Lewis base-transi-tion metal cooperation, and Lewis acid-transition metal cooperation in H(E)-H(E) activation

Figure 4 Tetradentate and tridentate boron-based LA-TM complexes bearing N-heterocyclic ligands

Figure 5 Structures of the N-heterocyclic nitrogen donor ligands and sulfur donor ligands

Figure 26 Type H sp2 boron-based LA-TM complexes with mono-dentate heterocyclic ligands

Figure 28 Synthesis and structures of PBP-Co, PBP-Ru, and long-tethered PBP-Ir complexes
-


 下载:
下载:



































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 43
- 文章访问数: 2433
- HTML全文浏览量: 519
 下载:
下载:
