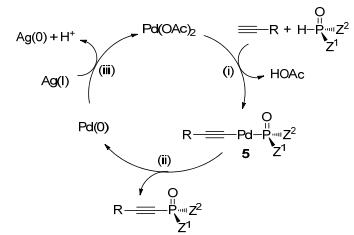
 图 图式1
钯催化C—H/P—H交叉偶联可能的反应机理
Figure 图式1.
roposed mechanism of the palladium-catalyzed C— H/P—H cross coupling
图 图式1
钯催化C—H/P—H交叉偶联可能的反应机理
Figure 图式1.
roposed mechanism of the palladium-catalyzed C— H/P—H cross coupling
引用本文:
杨佳, 肖晶, 周永波, 陈铁桥, 尹双凤, 韩立彪. 简单易得试剂与磷-氢化合物交叉偶联合成有机磷化合物[J]. 有机化学,
2017, 37(5): 1055-1068.
doi:
10.6023/cjoc201702050

Citation: Yang Jia, Xiao Jing, Zhou Yongbo, Chen Tieqiao, Yin Shuangfeng, Han Libiao. Recent Advances in the Synthesis of Organophosphorus Compounds via Cross Coupling between Readily Available Materials and P-H Compounds[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1055-1068. doi: 10.6023/cjoc201702050
Citation: Yang Jia, Xiao Jing, Zhou Yongbo, Chen Tieqiao, Yin Shuangfeng, Han Libiao. Recent Advances in the Synthesis of Organophosphorus Compounds via Cross Coupling between Readily Available Materials and P-H Compounds[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1055-1068. doi: 10.6023/cjoc201702050
简单易得试剂与磷-氢化合物交叉偶联合成有机磷化合物
English
Recent Advances in the Synthesis of Organophosphorus Compounds via Cross Coupling between Readily Available Materials and P-H Compounds
Abstract:
This mini-review focuses on the recent advances in the synthesis of organophosphorus compounds via cross coupling of P-H compounds with readily available starting materials, mainly including the reactions of terminal alkynes and heteroatom compounds (oxygen, sulfur or nitrogen-contained compounds) with P-H compounds forming sp-C-P, sp2-C-P, sp3-C-P, and P-Z bonds. Related reaction mechanisms are also discussed.
-
Key words:
- P-H compounds
- / cross coupling
- / organophosphorus compounds
-
有机磷化合物在有机合成、医药、农药、材料以及生命科学中都有着广泛的应用[1].传统的制备有机磷化合物的方法主要是通过磷卤试剂的亲核取代反应[2], 由于该方法中使用的反应底物大都对空气敏感, 使得反应条件比较苛刻, 底物局限性较大, 且反应产生当量有机卤代副产物, 不符合绿色化学发展要求.与磷卤试剂相比, 磷-氢化合物更为绿色稳定.利用磷-氢化合物与其他偶联试剂反应直接构建有机磷化合物, 其反应的原子经济性较高, 更为绿色环保.近年来, 通过过渡金属催化磷-氢化合物交叉偶联构建有机磷化合物是有机磷化学中的研究热点之一[3].该方法主要有以下几点优势: (1) 避免对水氧敏感的磷卤试剂的使用, 反应更简单绿色; (2) 反应条件温和, 底物适用范围更广; (3) 原子经济性和步骤经济性高, 符合绿色化学发展需求.
简单易得试剂来源广泛, 通常具有廉价、稳定、易储存等特点, 因此利用该类化合物作为反应底物进行交叉偶联反应具有较高的研究价值, 且符合绿色化学发展要求.但部分底物存在惰性强, 难以活化, 选择性较差等问题, 是有机合成化学研究的一个难点.近几年来, 我们课题组主要致力于以简单易得的偶联试剂与磷-氢化合物为底物合成有机磷化合物的新反应研究, 发展了一系列简单、高效、绿色地构建有机磷化合物的新方法.本文将主要围绕我们课题组近期的研究工作, 对近年来基于简单易得偶联试剂与磷-氢化合物交叉偶联反应合成有机磷化合物的反应进行综述, 并对部分反应的机理作扼要总结.分别从简单易得的末端炔烃、含氧有机化合物、含硫有机化合物以及含氮有机化合物作为偶联试剂进行归纳总结.
1 末端炔烃与磷-氢化合物绿色合成炔基膦化合物
炔基膦化合物是一类重要的有机合成中间体[4], 传统的构建该类化合物的方法主要是通过有机金属试剂与磷卤试剂反应[5], 反应底物均较活泼, 反应条件相对苛刻, 底物兼容性较差.通过对简单底物的C—H键活化[6]来构建C—C、C—Z键, 具有原子经济性高, 步骤经济性高以及对环境友好等优点.因此, 直接从简单易得的末端炔烃出发, 与磷-氢化合物通过C—H/P—H交叉偶联反应构建炔基膦化合物具有重要的研究价值.
2009年, 我们与赵玉芬课题组[7]合作, 报道了首例铜催化磷-氢化合物与末端炔烃需氧氧化交叉偶联合成炔基膦酸酯的新方法(Eq. 1).该方法的反应条件非常简单温和, 在干燥空气气氛下, 以二甲基亚砜(DMSO)为溶剂, 10 mol%的CuI作催化剂, 催化量的K2CO3作碱, 末端炔烃与磷-氢化合物55 ℃反应过夜, 即可得到相应的氧化偶联产物.该反应底物官能团耐受性好, 一系列官能团, 如羟基、烷氧基、羧基、羰基、氰基、氯、氨基、氨甲酰基和磺酰胺基都能在该反应体系中兼容.特别是含有端炔基团的具有特殊生物活性的大分子核苷酸类似物也可作为反应底物, 高选择性地与磷-氢化合物交叉偶联, 表明该方法能够合成高度官能团化的有机化合物.此外, 不同种类的亚磷酸酯类化合物均在该体系中取得很好的反应效果.遗憾的是, 在相似的反应条件下, 二苯基膦氧化物[Ph2P(O)H]在该反应体系中易被氧化成二苯基膦酸[Ph2P(O)OH]而不能得到相应的偶联产物.
针对上述问题, 我们利用钯催化实现了末端炔烃与二级膦氧化物的脱氢偶联反应(Eq. 2), 并对其反应机理进行了研究[8].在5 mol% Pd(OAc)2和2 equiv. AgBF4作用下, 以四氢呋喃(THF)为溶剂, 等量的苯乙炔与二苯基膦氧化物在60 ℃下反应可得到几乎定量的炔基膦氧类化合物, 且在反应中没有检测到二苯基膦氧化物与苯乙炔的加成副产物.此外, 该反应的底物适用范围较广, 芳香族和脂肪族的末端炔烃均可在该体系中取得较好的反应效果.一系列官能团, 如氟、氯、三氟甲基、酯基、羰基、羧基等均能在该体系中兼容.且含有较活泼碳-溴键的4-溴苯乙炔也能在该体系中发生交叉脱氢偶联反应得到溴原子保留的目标产物.另外, 通过向体系中加入适量三乙胺并升高反应温度, 亚磷(膦)酸酯类化合物也能与苯乙炔顺利发生交叉脱氢偶联反应.
接着, 我们对该反应的机理进行了探索, 研究了反应过程的立体化学, 并分离得到反应的活性中间体.计量反应结果表明, 磷-氢化合物与Pd(OAc)2在三乙基膦作配体时, 在室温下即可迅速地发生配体交换反应得到活性中间体4 (Eq. 3).且不同种类的磷-氢化合物, 如具有RP构型的[(-)MenO]BnP(O)H、亚磷酸二乙酯、环状亚磷酸酯、苯基膦酸乙酯以及二苯基膦氧化物均能与醋酸钯发生配体交换反应得到几乎定量的活性中间体4; 另外, 活性中间体4可与苯乙炔在室温下进行第二次配体交换反应得到中间体5 (Eq. 4).通过磷谱监测反应过程以及对中间体4a和5a的单晶结构解析确认, 这两步配体交换反应中磷原子的绝对构型均是保持的.最后, 分离得到的中间体5a可在80 ℃发生还原消除反应得到磷原子绝对构型保持的偶联产物3d (Eq. 5).
在计量反应的基础上, 我们继续考察了不同种类的手性磷-氢化合物在催化反应中的反应活性(Eq. 6).除了苄基手性磷-氢化合物外, 苯基以及大位阻的mesityl取代的手性磷-氢化合物均能与末端炔烃在钯催化下发生脱氢偶联得到中等收率的目标产物.
综上, 提出了如下反应机理: (1) Pd(OAc)2与磷-氢化合物、苯乙炔发生配体交换反应得到中间体5; (2) 中间体5经还原消除反应得到目标产物并释放零价钯; (3) 银盐再将零价钯氧化成二价钯进入下一个催化循环(Scheme 1).此催化过程中, 磷原子的绝对构型始终是保持的.同时, 还通过对照实验排除了银媒介的反应历程.
图 图式1
钯催化C—H/P—H交叉偶联可能的反应机理
Figure 图式1.
roposed mechanism of the palladium-catalyzed C— H/P—H cross coupling
同年, 雷爱文课题组[9]报道了银促进末端炔烃与磷-氢化合物的氧化脱氢偶联构建炔基二苯基膦氧类化合物, 为炔基膦氧化合物的合成提供了一条新途径.在DMSO中, 2 equiv. Ag2CO3的作用下, 1.5 equiv.末端炔烃与二苯基膦氧化物在120 ℃下反应可得到产率最高为70%的交叉偶联产物(Eq. 7).该反应体系的底物适用范围较广, 芳环上不论是拉电子、给电子基团还是卤素原子取代的末端炔烃都能在体系中耐受.此外, 杂环以及脂肪族的末端炔烃也能与二苯基膦氧化物进行氧化交叉偶联反应得到中等收率的目标产物.芳环上带有氟、氯或甲基等官能团的二苯基膦氧化物以及苯基膦酸乙酯也都能在该体系中兼容.遗憾的是, 亚磷酸酯在该体系中不反应.
对照实验结果表明, 该反应可能是经过如下反应历程:首先, 苯乙炔(1a)与二苯基膦氧(2c)分别与银盐形成相应的炔基银6和膦银盐7, 6和7在过量的银盐的作用下形成中间体8, 中间体8再分解得到交叉偶联产物3并生成零价银(Scheme 2).
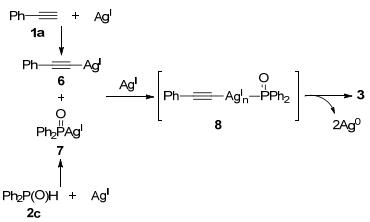 图 图式2
银媒介C—H/P—H交叉偶联可能的反应机理
Figure 图式2.
Proposed mechanism of the silver-mediated C—H/P—H cross coupling
图 图式2
银媒介C—H/P—H交叉偶联可能的反应机理
Figure 图式2.
Proposed mechanism of the silver-mediated C—H/P—H cross coupling
2 含氧有机化合物与磷-氢化合物交叉偶联构建sp2/sp3-C—P键或P—O键
2.1 含氧有机化合物与磷-氢化合物交叉偶联构建sp2/sp3-C—P键
传统地构建sp2/sp3-C—P键的方法[10]主要包含以下几种: (1) 有机金属试剂与磷卤试剂反应; (2) Michaelis-Arbuzov重排反应; (3) 过渡金属催化有机卤代物与磷-氢化合物直接交叉偶联反应(Scheme 3).以上三类反应均需使用有机卤代物作为起始原料.
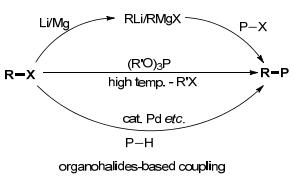 图 图式3
传统的构建sp2/sp3-C—P键的方法
Figure 图式3.
Traditional method for the construction of sp2/sp3-C—P bond
图 图式3
传统的构建sp2/sp3-C—P键的方法
Figure 图式3.
Traditional method for the construction of sp2/sp3-C—P bond
与有机卤代物相比, 含氧有机化合物如醇、酚及其衍生物等广泛存在于自然界中, 相对廉价易得, 绿色环保.近年来, 以含氧有机化合物为新型亲电偶联试剂, 通过过渡金属催化C—O键活化构建C—C及C—Z键[11]是研究热点之一.因此, 发展以含氧有机化合物作为有机卤代物的替代物与磷-氢化合物交叉偶联构建C—P键具有重要意义.
2012年, 韩福社课题组[12]报道了镍催化酚类化合物与磷-氢化合物交叉偶联构建C—P键的反应.该反应分为两步进行:首先以乙腈为溶剂, 碱存在下使用PyBroP对酚类化合物进行活化, 在100 ℃反应3 h后再原位加入镍催化剂和1.5 equiv.磷-氢化合物, 在100 ℃反应18 h可高产率地得到芳基膦氧化合物(Eq. 8).该反应体系中, 氰基、酯基或乙酰基取代的芳基酚和含氮杂环酚类化合物均可在该体系中取得较好的反应效果.不同种类的磷-氢化合物, 如二级膦氧化物以及亚磷酸酯均能在该体系中与酚类化合物交叉偶联得到中等至高产率的芳基膦氧化合物.该方法通过一锅两步实现高效磷-芳基化反应, 无需分离活化的酚类中间体, 在一定程度上提高了反应效率, 且使用廉价稳定的二价镍作催化剂, 展现出较好的应用前景, 为C—P键构建提供了新思路.
同年, 张万斌课题组[13]以易得的甲磺酸酯及对甲苯磺酸酯类化合物作为反应底物, 通过镍催化实现磷酰化反应, 拓宽了过渡金属催化磷-芳基化反应的底物范围.该反应体系以10 mol%的NiCl2(dppf)作为催化剂, 外加20 mol% dppf, 在当量的锌粉和N, N-二异丙基乙胺(DIPEA)的作用下, 磺酸酯类化合物与磷-氢化合物在N, N-二甲基甲酰胺(DMF)中反应得到中等至良好收率的芳基膦氧类化合物(Eq. 9).多种芳环及含氧杂环的磺酸酯类化合物都能与二苯基膦氧化物反应, 得到中等收率的偶联产物.此外, 亚磷酸酯及亚膦酸酯类化合物都能在该体系中兼容.
2015年, Kwong课题组[14]通过钯催化实现了甲磺酸酯及对甲苯磺酸酯与亚磷(膦)酸酯的交叉偶联反应.该方法使用Pd(OAc)2/L1催化体系, 在3 equiv. DIPEA作用下, 磺酸酯类化合物在叔丁醇和异丙醇(或乙醇)的体积比为1:1混合溶剂中与亚磷酸酯反应合成相应的芳基膦酸酯(Eq. 10), 其催化剂用量最低可降至0.25 mol%.该钯催化体系的化学选择性非常高, 如游离氨基酸、酮基、酯基、氨基以及杂环取代的磺酸酯类化合物都可以在该体系中耐受.值得注意的是, 具有生物活性的酚类衍生物也能在该体系中顺利反应.另外, 该体系还可一锅两步进行磷酰化-氨化反应, 为合成含磷药物提供了更加直接便利的途径.
2015年, 我们课题组[15]报道了首例镍催化酚类及醇类衍生物惰性C—O键活化与磷-氢化合物交叉偶联构建sp2/sp3-C—P键的新方法(Eq. 11).在Ni(cod)2的催化下, 以dcype为配体, 酯类化合物与磷-氢化合物在碱的作用下可进行C—O/P—H交叉偶联得到相应的有机磷化合物(Eq. 11).该反应简单高效, 底物兼容性好.不同种类的酯类化合物如叔戊酸酯、碳酸酯以及氨基甲酸酯类化合物均能与二苯基膦氧化物反应得到高产率的偶联产物.芳基、烯基、苄基和烯丙基取代的叔戊酸酯类化合物都可在该体系中耐受.特别是醛基这类容易与磷-氢化合物发生加成反应的官能团, 也能在该体系中兼容.多种磷-氢化合物, 包含二芳基膦氧、二烷基膦氧、芳基烷基膦氧、亚膦酸酯以及亚磷酸酯类化合物均可作为交叉偶联的底物.当将温度升高至120 ℃时, 具有高配位性的三价磷化合物, 二苯基膦也能与叔戊酸酯类化合物发生C—O/P—H交叉偶联直接得到相应的三价有机膦配体产物.
根据文献报道和对照实验结果, 我们提出了可能的反应机理:首先零价镍对叔戊酸酯类化合物进行氧化加成生成中间体B, 磷-氢化合物在碱的作用下与B发生配体交换得到中间体C, C再经过还原消除得到相应的产物10并释放零价镍进入下一个催化循环(Scheme 4).
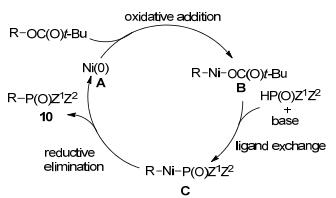 图 图式4
镍催化C—O/P—H交叉偶联可能的反应机理
Figure 图式4.
Proposed mechanism of the nickel-catalyzed C—O/P—H cross coupling
图 图式4
镍催化C—O/P—H交叉偶联可能的反应机理
Figure 图式4.
Proposed mechanism of the nickel-catalyzed C—O/P—H cross coupling
随后, 我们又将该体系的底物范围拓展至结构更简单的苯酚酯类化合物(Eq. 12)[16].在Ni(cod)2/dcype催化下, 不同官能团取代的苯酚叔戊酸酯和多种磷-氢化合物在Cs2CO3作用下甲苯中于100 ℃反应得到高产率的交叉偶联产物.然而, 不足的是, 二苯基膦氧化物不能在该体系中兼容.
为弥补上述方法的缺陷, 我们课题组报道了以相对活泼的三氟甲烷磺酸酯类化合物为底物, 在Ni(cod)2/dppf催化下合成三芳基膦氧类化合物的方法(Eq. 13)[17].各种类型的磷-氢化合物均能在该体系中兼容.
镍催化剂在C—O键活化反应中表现出高活性, 常见的廉价铁催化剂也可催化C—O键断裂. 2016年, 我们课题组[18]实现了廉价铁催化简单的缩醛类化合物与磷-氢化合物P—H/C—O交叉偶联构建sp3-C—P键的新方法(Eq. 14).在催化量的FeCl3作用下, 磷-氢化合物与缩醛发生P—H/C—O交叉偶联可高效地制备相应的α-烷氧基膦氧化合物.该反应非常简单高效, 无需外加配体和添加剂, 绿色高效地实现了磷-氢化合物与缩醛的交叉偶联反应, 为合成α-烷氧基膦氧化合物提供了一种新的途径.该体系的底物适用范围非常广泛, 特别是对卤素基团如氯、溴、碘均具有极强的容忍性, 可通过对芳基碳-卤键的后续官能团化得到结构更复杂的α-烷氧基膦氧化合物.另外, 含硝基这类强拉电子基团的底物也能在该体系中得到中等收率的产物.除了芳基缩醛外, 脂肪族的缩醛也能与二级膦氧化物反应以良好的收率得到相应的α-烷氧基膦氧化合物.不同种类的磷-氢化合物如二级膦氧化物、亚磷酸酯、亚膦酸酯以及三价的二苯基膦都可被用于与缩醛反应.
2.2 醇或酚类化合物与磷-氢化合物交叉偶联构建磷酸酯类化合物
含P—O键的化合物在医药化学、农业化学以及材料化学中都有着广泛的应用[1, 19].传统的构建这类有机磷化合物的方法[20]大都是通过醇类或酚类化合物与磷卤试剂发生取代反应制备.因此, 发展更为绿色稳定的磷-氢化合物替代磷卤试剂, 与醇或酚类化合物直接氧化脱氢偶联构建P—O键, 具有重要研究意义.
Prabhu课题组[21]在2013年报道了一例碘促进醇类化合物与亚磷酸二乙酯交叉偶联构建P—O键的反应.在室温下, 10 mol%的碘单质和1 equiv.双氧水作用, 简单的碳链醇以及炔基、烯基或苯基取代的脂肪醇均能在该体系中与亚磷酸酯氧化偶联得到高产率的磷酸酯类化合物(Eq. 15).环状的二级醇如薄荷醇和胆固醇也可在该体系中分别得到产率为32%和23%的偶联产物.
2015年, 我们课题组[22]报道了一例基于氯仿的Atherton-Todd型反应, 在叔丁醇锂的作用下, 酚类或醇类化合物在室温下氯仿中与二级膦氧化物反应0.5 h即可发生高效偶联(Eq. 16).反应条件温和, 利用氯仿替代了高毒的四氯化碳作为卤化试剂和溶剂, 为膦酸酯的合成提供了新途径.
2016年, 我们进一步报道了铁催化醇类化合物与磷-氢化合物直接脱氢偶联构建P—O键的新方法(Eq. 17)[23].该反应无需氧化剂, 无卤参与, 反应释放氢气作为副产物, 是一类直接、高效、绿色的合成有机磷化合物的方法.此外, 该反应具有高官能团容忍性, 不论是含拉电子基或给电子基取代的2-苯基乙醇, 简单的一级醇如甲醇、乙醇等, 或二级醇如异丙醇均能在该体系中耐受.除了二苯基膦氧化物外, 二正丁基膦氧化物以及五元环状亚磷酸酯都能在该体系中兼容.当将反应放大至1 mmol时, 只需在1 mol% Fe(acac)2/1 mol% 1, 10-菲啰啉, 10 mol%碳酸钾作用下, 二苯基膦氧化物即可与苯乙醇反应高产率地得到膦酸酯.
3 含硫有机化合物与磷-氢化合物交叉偶联构建sp2-C—P键或P—S键
3.1 含硫有机化合物C—S键活化构建sp2C—P键
有机硫化合物来源丰富, 稳定易得, 但因其容易对金属催化剂产生毒化作用, 也为过渡金属催化该类化合物的反应带来一定的难度.目前, 通过过渡金属催化C—S键断裂构建C—C键的研究已经有了较多的报道, 而在构建C—Z键方面研究较少[24].因此, 若能使用有机硫化合物与同样易于对金属进行配位的磷-氢化合物作为反应底物, 在过渡金属催化下来构建C—P键, 则具有重要的研究价值.
2014年, 王磊课题组[25]发展了钯催化芳基亚磺酸钠与亚磷酸酯通过C—S/P—H交叉偶联构建sp2-C—P键的新方法.在5 mol% Pd(PPh3) Cl2、Ag2CO3和四丁基氯化铵(TBAC)存在下, 苯基亚磺酸钠与亚磷酸酯在DMSO中80 ℃下反应, 可得到相应的芳基膦酸酯类化合物(Eq. 18).芳环上含不同官能团(如甲基、甲氧基、叔丁基、酰胺基或卤原子等)取代的亚磺酸钠化合物均能在该体系中耐受.同年, 肖君华课题组[26]又报道了一例高效的微波促进的钯催化芳基亚磺酸钠与亚磷酸酯的交叉偶联反应(Eq. 19).
2016年, 我们课题组[27]采用以简单的硫醚或砜类化合物为偶联试剂, 通过镍催化C—S/P—H交叉偶联合成芳基膦氧类化合物的新方法.以Ni(cod)2作为催化剂, 在叔丁醇钠的作用下, 芳基硫醚或砜类化合物与磷-氢化合物在1, 4-二氧六环中100 ℃下进行交叉偶联反应(Eq. 20).该反应体系无需外加配体即可实现磷-氢化合物与苯甲硫醚及砜类化合物的高效偶联.放大反应至克级规模时只需0.1 mol%的镍催化剂即可高产率地得到偶联产物.通过该方法还可对药物硫利达嗪进行相应的磷酰基化修饰.此外, 一系列的芳基甲基硫醚类化合物都能在该反应体系中取得较好的反应效果, 吡啶或喹啉取代的甲基硫醚也在该反应体系中兼容.同时, 苯硫醚在该体系中也能顺利发生反应.
除了芳基硫醚类化合物外, 各种芳基砜类化合物也可与磷-氢化合物发生交叉偶联反应, 且部分相对活泼的底物如氰基、嘧啶基取代的苯甲砜等无需镍催化剂即可得到相应的偶联产物(Eq. 21).
最后, 我们提出了可能的反应机理, 该反应的机理与C—O键活化类似.不同的是, 在叔丁醇钠的作用下, 磷氧氢类化合物会先与其成磷盐E, E再与氧化加成中间体D进行配体交换得到C, 最后还原消除得到目标产物10并释放出零价镍(Scheme 5).
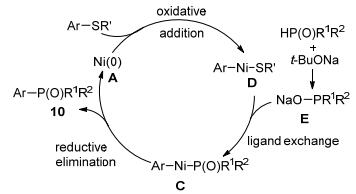 图 图式5
镍催化C—S/P—H交叉偶联可能的反应机理
Figure 图式5.
Proposed mechanism of the nickel-catalyzed C—S/ P—H cross coupling
图 图式5
镍催化C—S/P—H交叉偶联可能的反应机理
Figure 图式5.
Proposed mechanism of the nickel-catalyzed C—S/ P—H cross coupling
3.2 含硫有机化合物S—H/P—H交叉偶联构建P—S键
含P—S键的硫代磷酸酯化合物在生物化学分子中较为常见, 在医药和农药中都有着广泛的应用[28].与构建P—O键类似, 传统的构建P—S键的方法[29]也是基于对水氧敏感的磷卤试剂与有机硫化合物的亲核取代反应.发展简单易得含硫有机化合物与磷-氢化合物交叉偶联构建P—S键的反应尤为重要.
2015年, 潘远江课题组[30]报道了一例简单高效的过苯甲酸特丁酯(TBPB)促进硫醇或硫酚类化合物与磷-氢化合物S—H/P—H交叉偶联构建P—S键的新方法.在室温下, 2 equiv. TBPB, 20 mol% KI作用下, 硫醇或硫酚与1.5 equiv.磷-氢化合物在DMSO中进行交叉偶联反应(Eq. 22).该反应官能团容忍性非常高, 羧基、巯基以及苄基取代的硫醇类化合物均能在该体系中兼容.卤原子和酰胺取代的苯硫酚类化合物也能顺利与磷-氢化合物进行氧化脱氢偶联反应.除了二芳基膦氧化物外, 芳基苄基膦氧化物和亚膦酸酯类化合物也能在该体系中取得较好的反应效果.作者通过对照实验推测, 该反应可能是经过自由基历程.
同年, 我们课题组[22]报道了的氯仿促进的Atherton-Todd类型反应, 实现硫酚或硫醇类化合物与磷-氢化合物的交叉偶联合成硫代膦酸酯.在2 equiv.叔丁醇锂的作用下, 以氯仿为溶剂, 多种芳基硫酚在室温下与二级膦氧化物迅速反应, 得到高产率的交叉偶联产物(Eq. 23).简单的脂肪族硫醇也能在该体系中兼容.
2016年, 肖君华课题组[31]又报道了一例二氯二甲基海因(DCDMH)促进的亚磷酸酯与硫酚或硫醇化合物交叉偶联构建硫代磷酸酯的新反应(Eq. 24).该反应无需过渡金属催化剂及添加剂的参与, 以CH2Cl2为溶剂, 反应底物在空气中室温下搅拌10 min即可反应完全, 条件温和, 简单高效, 底物兼容性较好.多种硫酚或硫醇均能与亚磷酸酯类化合物反应, 特别是含有强拉电子的硝基取代的苯硫酚也能在该体系中耐受, 得到中等收率的目标产物.
除了上述无过渡金属参与的交叉偶联反应外, 同年, 我们课题组[32]发展了钯催化磷-氢化合物与硫酚或硫醇类化合物直接脱氢偶联构建P—S键的反应(Eq. 25).该方法利用Pd2(dba)3为催化剂, dppf为配体, 使用苯乙烯作为氢气捕获试剂, 在100 ℃下可实现磷-氢化合物与硫酚或硫醇的直接脱氢偶联反应构建相应的硫代磷酸酯类化合物.该反应非常高效通用, 且底物范围广.三种磷-氢化合物包含亚磷酸酯、亚膦酸酯以及二级膦氧化物都能与芳基或脂肪族硫醇反应.该方法打破了传统方法的底物限制, 如具有磷手性的硫代磷酸酯以及羟基或氨基取代的硫代磷酸酯, 均能通过该方法合成.
进而我们对该反应的机理进行了探索.研究表明, 该反应历程是经过零价钯到二价钯再到零价钯的催化过程.我们通过五元环状亚磷酸酯与四三乙基磷钯反应得到活性中间体钯-氢复合物21a, 中间体21a再与硫酚发生配体交换反应, 释放出氢气生成磷-钯-硫中间体22a (Scheme 6), 并通过对22a的单晶结构解析确定金属复合物的结构.
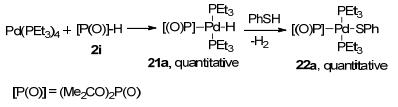 图 图式6
钯催化脱氢偶联的化学计量反应
Figure 图式6.
Stoichiometric reactions of the dehydrogenative coupling catalyzed by Pd
图 图式6
钯催化脱氢偶联的化学计量反应
Figure 图式6.
Stoichiometric reactions of the dehydrogenative coupling catalyzed by Pd
结合催化反应及计量反应结果, 我们提出了可能的反应机理:首先零价钯与磷-氢化合物发生氧化加成反应得到钯-氢化合物21, 21继续与硫酚或硫醇进行配体交换反应得到中间体22, 22再经还原消除过程即可得到交叉偶联产物20, 并释放出零价钯进入到下一个催化循环.
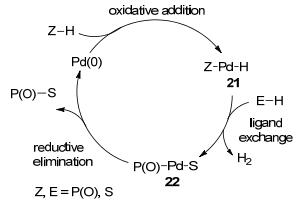 图 图式7
钯催化S—H/P—H交叉偶联反应的可能机理
Figure 图式7.
Proposed mechanism of the palladium-catalyzed S—H/P—H cross coupling
图 图式7
钯催化S—H/P—H交叉偶联反应的可能机理
Figure 图式7.
Proposed mechanism of the palladium-catalyzed S—H/P—H cross coupling
此外, 光催化反应也可用于合成硫代磷酸酯类化合物.张波和李萍课题组[33]报道了一例通过有机染料作为光催化剂在可见光下催化硫醇或硫酚类化合物与磷-氢化合物脱氢偶联构建硫代磷酸酯类化合物的新方法(Eq. 26).该方法使用空气作为绿色氧化剂, 反应条件温和, 官能团容忍性高, 并且可进行放大, 操作简单, 具有非常强的实用性.该反应体系利用5 mol%的虎红钠盐作为光催化剂, 硫醇或硫酚类化合物与磷-氢化合物在10 W蓝光照射下空气气氛中室温反应12 h即可得到中等至良好收率的硫代磷酸酯类化合物.氨基、羟基这类活泼基团均能在该体系中耐受.长链、大位阻以及苄基取代的硫醇也可与二苯基膦氧化物反应得到中等收率的目标产物.多种二级膦氧化物都能在该体系中兼容, 而亚膦酸酯在该反应中活性相对较低, 需延长反应至36 h可得到产率为30%的偶联产物.
通过大量对照实验, 作者提出了该反应可能的机理:首先, 虎红钠盐在可见光照射下产生激发态的虎红钠盐, 再与氧气反应得到1O2; 硫酚或硫醇和磷-氢化合物可能在1O2作用下分别产生硫自由基F和磷自由基G.接着, 作者提出可能通过以下三条路径生成目标偶联产物: (1) 产生的硫自由基F和磷自由基G直接发生反应得到硫代磷酸酯20; (2) 产生的硫自由基F自身偶联得到二硫醚23后再与磷自由基G进行反应得到相应的硫代磷酸酯20; (3) 中间体二硫醚直接与磷-氢化合物的互变异构体2'反应得到相应的产物.
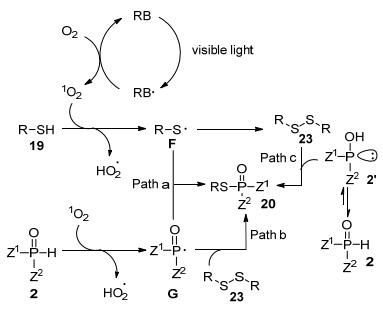 图 图式8
可见光催化S—H/P—H交叉偶联反应的可能机理
Figure 图式8.
Proposed mechanism of the visible light-catalyzed S—H/P—H cross coupling
图 图式8
可见光催化S—H/P—H交叉偶联反应的可能机理
Figure 图式8.
Proposed mechanism of the visible light-catalyzed S—H/P—H cross coupling
2017年, 张波和李萍课题组[34]又利用氧化性较弱的DMSO作为氧化剂实现了硫代磷酸酯的合成.该反应非常绿色、高效, 反应条件简单、温和, 同样也无需过渡金属催化剂和添加剂, 多种官能团取代的硫酚或硫醇类化合物与二苯基膦氧化物在DMSO中室温下反应12 h即可得到相应的硫代磷酸酯类化合物(Eq. 27).作者通过对照实验确认该反应可能是经过非自由基历程, 反应中硫酚先在DMSO的作用下生成二硫醚化合物, 然后再与二级膦氧类化合物反应得到偶联产物硫代磷酸酯.另外, 在该体系中, 直接利用二硫醚作为反应底物时也可得到目标产物.
同年, 焦宁和宋颂课题组[35]报道了一例简单、高效且实用性非常高的合成硫代磷酸酯的新方法, 利用Cs2CO3催化硫醇或硫酚与磷-氢化合物在氧气气氛下实现氧化偶联(Eq. 28).该体系官能团容忍性高, 酰胺基、氨基以及酯基等官能团取代的脂肪族硫醇作为底物时, 可得到良好收率的偶联产物, 强拉电子的三氟甲基取代的苯硫酚也可在该体系中顺利反应, 氨基和羟基这类活泼基团也能在反应中很好地耐受.另外, 该反应体系可放大至10 mmol, 且可应用于合成多种生物活性分子.
结合对照实验, 作者提出了可能的反应机理.首先, 亚磷酸二乙酯与Cs2CO3反应得到中间体(EtO)2P(O)Cs H.同时, 在Cs2CO3和氧气作用下, 硫醇或硫酚形成二硫醚23.接着, 在无需碳酸铯和氧气促进的情况下, 23再与中间体H直接反应得到硫代磷酸酯20并释放出C(RS—Cs+).最后, 氧气再氧化成二硫醚23.
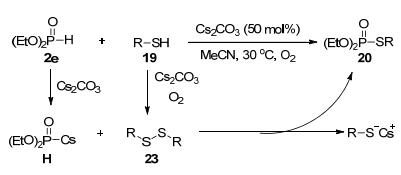 图 图式9
Cs2CO3催化S—H/P—H交叉偶联反应的可能机理
Figure 图式9.
Proposed mechanism of the Cs2CO3-catalyzed S—H/P—H cross coupling
图 图式9
Cs2CO3催化S—H/P—H交叉偶联反应的可能机理
Figure 图式9.
Proposed mechanism of the Cs2CO3-catalyzed S—H/P—H cross coupling
硫酚或硫醇类化合物是构建P—S键的常用试剂, 亚磺酸及其盐类化合物也可用于硫代磷酸酯的合成.Hong课题组[36]实现了在PPh3作用下苯基亚磺酸与磷-氢化合物的交叉偶联反应.该反应无需加入金属和氧化剂, 且底物适用范围非常广泛.在1 equiv. PPh3作用下, 亚磺酸类化合物可与磷-氢化合物在氮气气氛下室温中反应得到较高产率的硫代磷酸酯类化合物(Eq. 29).芳香族和脂肪族的亚磺酸类化合物均可在该体系中兼容.卤原子取代的芳基亚磺酸以及杂环取代的亚磺酸在该体系中均可取得非常好的反应效果.不同种类的磷-氢化合物, 如不同种类的二级膦氧化物、亚膦酸酯以及多种亚磷酸酯都可在该体系中耐受.特别是, 手性磷-氢化合物也可与亚磺酸反应得到手性保持的硫代磷酸酯类化合物.通过对照实验, 作者推测PPh3在该体系中作为还原剂.
易文斌和陆国平课题组[37]报道了通过简单的芳基亚磺酸钠与磷-氢化合物反应来构建硫代磷酸酯的新方法.在0.5 equiv. H2SO4作用下, 无臭的芳基亚磺酸钠与2 equiv.二芳基膦氧化物可在室温下, 以水为溶剂进行交叉偶联(Eq. 30).该反应无需过渡金属催化剂、添加剂和有机溶剂, 更为简单绿色.不同种类的二芳基膦氧化物均能在该体系中耐受.遗憾的是, 亚磷酸酯类化合物在该体系中活性较低.
此外, 许新华课题组[38]和陈建课题组[39]等还发展了利用简单易得的二硫醚化合物与磷-氢化合物偶联来构建P—S键. 2013年, 许新华课题组[38]发展了一例CsOH催化二硫醚与亚磷酸酯类化合物交叉偶联构建硫代磷酸酯的方法(Eq. 31).在10 mol% CsOH催化下, 亚磷酸酯与芳基二硫醚在室温下DMSO中反应得到硫代磷酸酯类化合物, 为硫代磷酸酯构建提供了一种简单便利途径. 2016年, 陈建课题组[39]又在不加任何催化剂和氧化剂的条件下, 实现了二硫醚与二级膦氧化物的偶联反应.二级膦氧化物与二硫醚在氮气气氛下THF中80 ℃反应4 h即可得到相应的硫代磷酸酯(Eq. 32), 非常简单高效, 且底物适用范围较广.卤原子、酯基以及活泼羟基取代的苯基二硫醚均能在该体系中兼容.除了芳基二硫醚外, 脂肪族的二硫醚也能与二苯基膦氧化物进行反应.
4 含氮有机化合物与磷-氢化合物交叉偶联构建sp2/sp3-C—P键或P—Z键
4.1 含氮有机化合物与磷-氢化合物交叉偶联构建sp2-C—P键
2014年, 肖君华课题组[40]实现了钯/银双金属催化邻硝基苯甲酸类化合物与亚磷酸酯脱羧偶联构建sp2-C—P键的方法, 为芳基膦酸酯类化合物提供了一种新的合成方法.在催化量Pd(OAc)2和2 equiv. Ag2CO3以及1 equiv. LiNO3作用下, 邻硝基苯甲酸与亚磷酸酯在微波辅助下120 ℃加热10 min, 即可得到中等到良好收率的芳基膦酸酯化合物(Eq. 33).该反应对许多常见官能团, 如甲氧基、硝基和卤素原子等, 都有着非常好的相容性.遗憾的是, 其他类型的羧酸类化合物在该体系中并不发生反应.
同年, 赵玉芬课题组[41]以简单的芳基肼类化合物为底物, 发展了钯催化氧气氧化C—N/P—H偶联构建芳基膦酸酯的新方法.在10 mol% Pd(OAc)2、30 mol% dppp以及1.5 equiv.三氟乙酸(TFA)作用下, 芳基肼类化合物在氧气气氛下DMF中90 ℃反应24 h可发生C —N键断裂与亚磷酸酯交叉偶联制备相应的芳基膦酸酯类化合物(Eq. 34), 反应简单, 无卤参与, 使用氧气为氧化剂, 主要副产物为氮气和水, 较为绿色环保.此外, 该体系的官能团容忍性较高, 氟、氯、羧基、磺酰胺基、氰基、三氟甲基等官能团都可在该体系中兼容.多种二烷基亚磷酸酯均可顺利发生交叉偶联反应.然而, 二苯基膦氧化物在该体系中会被氧化成膦酸而不能得到目标产物.
2015年, 我们课题组[42]报道了镍催化磷-氢化合物与简单的芳基腈类化合物P—H/C—CN交叉偶联构建芳基磷化合物的方法(Eq. 35).该反应可在Ni(cod)2/8-羟基喹啉的催化下高效进行, 避免了贵金属催化剂和膦配体的使用.反应条件温和, 底物范围较广, 不仅可以通过五价磷-氢化合物与芳基腈构建芳基膦氧类化合物, 还可利用三价的二苯基膦与芳基腈反应直接得到三价的有机膦配体, 为构建sp2-C—P键提供了一种有力的手段.遗憾的是, 亚磷酸酯不能在该体系中兼容.
氨基酸类化合物也可用于P—C键构建. 2011年, 王锐课题组[43]发展了一例CuI/DIPEA催化, 醛引发的α-氨基酸与磷-氢化合物串联脱羧偶联制备α-氨基膦化合物的方法(Eq. 36).该方法利用天然氨基酸为起始原料, 廉价铜为催化剂, 对特定位点进行官能团化, 官能团容忍性较好, 为合成一系列膦配体以及具有生物活性的非天然氨基酸衍生物提供了便利.在30 mol% CuI/DIPEA催化下, α-氨基酸类化合物在芳基醛的引发下可与多种类型的磷-氢化合物进行脱羧偶联, 其中包含多种亚磷酸酯与二级膦氧化物以及特殊的二烯丙基膦氧化物.此外, 不同官能团取代的芳基醛类化合物如对硝基苯甲醛可在该体系中取得较好的反应效果.
2015年, 我们课题组[44]实现了在无任何催化剂、添加剂和氧化剂的存在下, 通过胺类化合物选择性C—N键断裂与磷-氢化合物、CH2Cl2三组分偶联构建α-氨基膦化合物新方法.简单的胺类化合物与磷-氢化合物、CH2Cl2只需在DMF中加热过夜即可得到高产率的α-氨基膦氧类化合物(Eq. 37), 反应更为简单、绿色、高效, 且底物适用范围广.一级胺、二级胺和三级胺均能在该体系中兼容.不同种类的磷-氢化合物, 如二级膦氧化物、亚膦酸酯以及亚磷酸酯都可在该体系中耐受得到相应的α-氨基膦氧类化合物, 具有特殊结构的五元环状亚磷酸酯也可顺利进行反应.该反应体系还可用于高立体选择性地构建光学纯的磷手性氨基酸类似物, 反应过程中磷原子的绝对构型是保持的, 这也是首例三组分立体专一性偶联反应.
结合参考文献和对照实验, 我们还提出了可能的反应机理(Scheme 10).由胺类化合物与CH2Cl2形成的铵盐J分解产生亚胺盐K; 中间体K再对磷-氢化合物的互变异构体2'进行亲电进攻得到磷原子绝对构型保持的产物.
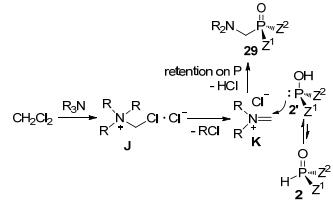 图 图式10
三组分偶联可能的反应机理
Figure 图式10.
Proposed mechanism of the three component coupling
图 图式10
三组分偶联可能的反应机理
Figure 图式10.
Proposed mechanism of the three component coupling
4.2 胺类化合物N—H/P—H交叉偶联构建P—N键
含有P(O)—N键的有机化合物在有机合成、生物和材料中有着广泛的应用[45].传统的构建该类化合物的方法[20b, 20c, 46]是通过磷卤试剂与胺反应或者是Todd反应:在四氯化碳的作用下磷-氢化合物与胺反应.
2013年, Prabhu课题组[21]发展了一例无金属催化的胺类化合物与亚磷酸二乙酯交叉偶联构建P—N键的新方法.在10 mol%碘单质和1 equiv. H2O2的作用下, 定量的胺可与亚磷酸二乙酯在CH2Cl2中室温下得到良好到高产率的脱氢偶联产物(Eq. 38).该反应无需金属催化剂、过量底物和外加碱, 反应简单、温和、高效、绿色, 一系列一级或二级胺都能在该体系中取得较好的反应效果.
同年, Hayes课题组[47]报道了铜催化胺类化合物与亚磷酸酯在空气中氧化脱氢偶联构建磷酰胺类化合物的新方法.在20 mol% CuI催化下, 空气中的氧气作为氧化剂, 2 equiv.胺类化合物与亚磷酸酯在乙腈中55 ℃反应可得到相应的磷酰胺化合物(Eq. 39).在该反应体系中, 甲氧基、酯基、苄基和烯丙基取代的初级胺可与亚磷酸酯反应得到中等到良好收率的偶联产物.位阻较大的叔丁胺和二级胺(吗啉和哌啶)反应效果较差.苯胺的反应效果比简单的环己胺要好.氨基酸衍生物如甘氨酸酯和苯丙氨酸酯类化合物在该体系中可与亚磷酸酯偶联得到高产率的目标产物.此外, 该方法还可用于合成磷酰胺类前药分子.
同时, 于晓琪和陈善勇课题组[48]也报道了一例铜催化空气氧化芳基胺类化合物与亚磷酸酯构建P—N键的方法.以5 mol% CuBr作为催化剂, 以乙酸乙酯为溶剂, 1.5 equiv.芳基胺类化合物与二烷基亚磷酸酯在空气中反应合成酰胺磷酸酯(Eq. 40).在该体系中, 甲基取代的苯胺卤原子、甲氧基等其它基团取代的苯胺可更高效地与亚磷酸酯反应, 而苄胺和二级苯胺活性较低.
2014年, 我们课题组[49]也发展了一例铜催化亚磷酸酯与胺类化合物氧化脱氢偶联构建磷酰胺类化合物的方法(Eq. 41), 并对该反应的立体化学进行了探索.在3 mol%的铜盐作用下, 胺类化合物与磷-氢化合物在空气中室温下反应3 h即可得到高产率的酰胺磷(膦)酸酯类化合物.不同种类的一级胺, 小位阻、大位阻及长直链的胺都表现出非常高的反应活性, 二级胺则在该体系中活性较低.不同种类的磷-氢化合物, 如亚磷酸酯、亚膦酸酯均能在该体系中兼容.核苷类似物也能在该体系中耐受得到高官能团化的有机磷化合物.立体化学研究表明, 该反应具有非常高的立体选择性, 磷原子的绝对构型在反应过程中发生翻转得到光学纯的手性磷化合物(Eq. 42).这是当时首例过渡金属催化立体专一性的脱氢偶联反应构建光学纯的磷酰胺类化合物的方法.该方法为合成酰胺磷酸酯类化合物提供了更为高效、直接的选择.
3 结论与展望
总结了一系列简单易得试剂, 如末端炔烃、含氧有机化合物、含硫有机化合物以及含氮有机化合物, 和磷-氢化合物交叉偶联反应高效地构建P—C键及P—Z键的方法及相关反应机理.反应类型涉及贵金属催化(如钯等)、廉价金属(如铜、铁等)催化、无金属催化、新型光催化以及无需任何催化剂和添加剂的反应.尽管有机磷化合物的绿色、高效合成研究取得一定进展, 但仍然存在一些问题和挑战:通过对简单底物的sp3-C—H键活化与磷-氢化合物交叉偶联来构建含sp3-C—P键的有机磷化合物的研究仍然存在一定困难; 在构建磷手性有机化合物方面的研究仍然较少, 发展简单、高效、立体专一性的构建手性有机磷化合物的反应可为复杂膦配体及生物活性有机磷化合物的合成提供新途径; 由于含磷化合物结构的多样性和特殊性, 大部分该类反应的机理还有待进一步的深入研究、探索.
-
-
[1]
(a) Quin, L. D. A Guide to Organophosphorus Chemistry, Wiley Interscience. New York, 2000.
(b) Majoral, J. -P. New Aspects in Phosphorus Chemistry, Springer, Berlin, Germany, Vol. 1~5, 2002~2005.
(c) Murphy, P. J. Organophosphorus Reagents, Oxford University Press, Oxford, U. K. , 2004.
(d) Tang, W. ; Zhang, X. Chem. Rev. 2003, 103, 3029.
(e) Baumgartner, T. ; Réau, R. Chem. Rev. 2006, 106, 4681.
(f) Queffélec, C. ; Petit, M. ; Janvier, P. ; Knight, D. A. ; Bujoli, B. Chem. Rev. 2012, 112, 3777.
(g) Yang, L. ; Ma J. Acta Chim. Sinica 2016, 74, 130 (in Chinese).
(杨丽军, 马军安, 化学学报, 2016, 74, 130. )
(h) Shi, B. ; Fang, Y. ; Zhang, L. ; Jin, X. ; Wu, Y. ; Fang, M. ; Yang, Y. ; Chen, C. Chin. J. Org. Chem. 2016, 36, 673 (in Chinese).
(施波超, 方烨汶, 张莉, 金小平, 武永辉, 方媚, 杨宇飞, 陈冲, 有机化学, 2016, 36, 673. )
(i) Jin, C. ; He, J. ; Wang, C. ; He, H. Chin. J. Org. Chem. 2010, 30, 1 (in Chinese).
(金传飞, 贺军波, 王储备, 贺红武, 有机化学, 2010, 30, 1. )
(j) Zhao, Y. ; Xiao, Q. ; Ju, Y. ; Li, Y. Chin. J. Org. Chem. 2001, 21, 869 (in Chinese).
(赵玉芬, 肖强, 巨勇, 李艳梅, 有机化学, 2001, 21, 869. ) -
[2]
(a) Engel, R. Handbook of Organophosphorus Chemistry, Marcel Dekker, New York, 1998.
(b) Jiménez, M. V.; Pérez-Torrente, J. J.; Bartolomé, M. I.; Oro, L. A. Synthesis 2009, 1916.
(c) Casey, C. P.; Paulsen, E. L.; Beuttenmueller, E. W.; Proft, B. R.; Petrovich, L. M.; Matter, B. A.; Powell, D. R. J. Am. Chem. Soc. 1997, 119, 11817.
(d) Kinas, R. W.; Okruszek, A.; Stec, W. J. Tetrahedron Lett.2002, 43, 7875. -
[3]
(a) Tappe, F. M. J. ; Trepohl, V. T. ; Oestreich, M. Synthesis 2010, 3037.
(b) Chen, T. ; Zhang, J. -S. ; Han, L. -B. Dalton Trans. 2016, 45, 1843.
(c) Jablonkai, E. ; Keglevich, G. Curr. Org. Synth. 2014, 11, 429.
(d) Shao, C. ; Xu, W. ; Li, L. ; Zhang, X. . Chin. J. Org. Chem. 2017, 37, 335 (in Chinese).
(邵长伟, 徐炜刚, 李亮, 张兴华, 有机化学, 2017, 37, 335. )
(e) Zhang, L. ; Zhang, P. ; Xue, J. ; Sun, W. ; Zou, J. Acta Chim. Sinica 2016, 74, 811 (in Chinese).
(张令, 张沛之, 薛剑飞, 孙网彬, 邹建平, 化学学报, 2016, 74, 811. )
(f) Xu, Q. ; Jia, X. ; Li, X. ; Sun, Q. ; Zhou, Y. -B. ; Yin, S. -F. ; Han, L. -B. Chin. J. Org. Chem. 2014, 34, 1340 (in Chinese).
(徐清, 贾小娟, 李晓慧, 孙清, 周永波, 尹双凤, 韩立彪, 有机化学, 2014, 34, 1340. ) -
[4]
(a) Mo, J.; Kang, D.; Eom, D.; Kim, S. H.; Lee, P. H. Org. Lett. 2013, 15, 26.
(b) Li, X.; Hu, G.; Luo, P.; Tang, G.; Gao, Y.; Xu, P.; Zhao, Y. Adv. Synth. Catal. 2012, 354, 2427.
(c) Ekkert, O.; Kehr, G.; Fröhlich, R.; Erker, G. J. Am. Chem. Soc. 2011, 133, 4610.
(d) Van derpoorten, K.; Migaud, M. E. Org. Lett. 2004, 6, 3461. -
[5]
Iorga, B.; Eymery, F.; Carmichael, D.; Savignac, P. Eur. J. Org. Chem. 2000, 65, 3103 and references cited therein.
-
[6]
(a) Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74.
(b) Waterman, R. Chem. Soc. Rev. 2013, 42, 5629.
(c) Liu, C.; Zhang, H.; Shi, W.; Lei, A. Chem. Rev. 2011, 111, 1780.
(d) Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215.
(e) Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Chem. Rev. 2011, 111, 1293. -
[7]
Gao, Y.; Wang, G.; Chen, L.; Xu, P.; Zhao, Y.; Zhou, Y.; Han, L.-B. J. Am. Chem. Soc. 2009, 131, 7956. doi: 10.1021/ja9023397
-
[8]
(a) Yang, J.; Chen, T.; Zhou, Y.; Yin, S.; Han, L.-B. Chem. Commun. 2015, 51, 3549.
(b) Yang, J.; Chen, T.; Zhou, Y.; Yin, S.-F.; Han, L.-B. Organometallics 2015, 34, 5095. -
[9]
Wang, T.; Chen, S.; Shao, A.; Gao, M.; Huang, Y.; Lei, A. Org. Lett. 2015, 17, 118. doi: 10.1021/ol503341t
-
[10]
(a) Bhattachary, A. K.; Thyarajan, G. Chem. Rev. 1981, 81, 415.
(b) Yang, G.; Shen, C.; Zhang, L.; Zhang, W. Tetrahedron Lett.
2011, 52, 5032.
(c) Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Tetrahedron Lett. 1980, 21, 3595.
(d) Berger, O.; Petit, C.; Deal, E. L.; Montchamp, J.-L. Adv. Synth. Catal. 2013, 355, 1361. -
[11]
Reviews for C—O bond activation, see:
(a) Tobisu, M.; Chatani, N. Acc. Chem. Res. 2015, 48, 1717.
(b) Chen, T.; Han, L.-B. Angew. Chem., Int. Ed. 2015, 54, 8600.
(c) Tasker, S. Z.; Standley, E. A.; Jamison, T. F. Nature 2014, 509. 299.
(d) Cornella, J.; Zarate, C.; Martin, R. Chem. Soc. Rev. 2014, 43, 8081. -
[12]
Zhao, Y.-L.; Wu, G.-J.; Han, F.-S. Chem. Commun. 2012, 48, 5868. doi: 10.1039/c2cc31718d
-
[13]
Shen, C.; Yang, G.; Zhang, W. Org. Biomol. Chem. 2012, 10, 3500. doi: 10.1039/c2ob25225b
-
[14]
Fu, W. C.; So, C. M.; Kwong, F. Y. Org. Lett. 2015, 17, 5906. doi: 10.1021/acs.orglett.5b03104
-
[15]
Yang, J.; Chen, T.; Han, L.-B. J. Am. Chem. Soc. 2015, 137, 1782. doi: 10.1021/ja512498u
-
[16]
Yang, J.; Xiao, J.; Chen, T.; Han, L.-B. J. Org. Chem. 2016, 81, 3911. doi: 10.1021/acs.joc.6b00289
-
[17]
Yang, J.; Xiao, J.; Chen, T.; Han, L.-B. J. Organomet. Chem. 2016, 820, 120. doi: 10.1016/j.jorganchem.2016.07.026
-
[18]
Li, X.; Chen, T.; Saga, Y.; Han, L.-B. Dalton Trans. 2016, 45, 1877. doi: 10.1039/C5DT02454D
-
[19]
(a) Steinbach, T.; Wurm, F. R. Angew. Chem., Int. Ed. 2015, 54, 6098.
(b) Bogomilova, A.; Hohn, M.; Gunther, M.; Troev, K.; Wagner, E.;
Schreiner, L. Eur. J. Pharm. Sci. 2013, 50, 410.
(c) Rectenwald, M. F.; Gaffen, J. R.; Rheingold, A. L.; Morgan A. B.; Protasiewicz, J. D. Angew. Chem., Int. Ed. 2014, 53, 4173. -
[20]
(a) Oliana, M.; King, F.; Horton, P. N.; Hursthouse, M. B.; Hii, K. K. J. Org. Chem. 2006, 71, 2472.
(b) Zhou, Y.; Wang, G.; Saga, Y.; Shen, R.; Goto, M.; Zhao, Y.; Han,
L.-B. J.Org. Chem. 2010, 75, 7924.
(c) Wang, G.; Shen, R.; Xu, Q.; Goto, M.; Zhao, Y.; Han, L.-B. J. Org. Chem. 2010, 75, 3890. -
[21]
Dhineshkumar, J.; Prabhu, K. R. Org. Lett. 2013, 15, 6062. doi: 10.1021/ol402956b
-
[22]
Li, S.; Chen, T.; Saga, Y.; Han, L.-B. RSC Adv. 2015, 5, 71544. doi: 10.1039/C5RA16015D
-
[23]
Li, C.; Chen, T.; Han, L.-B. Dalton Trans. 2016, 45, 14893. doi: 10.1039/C6DT02236G
-
[24]
Recent reviews on metal-catalyzed C—S transformation:
(a) Pan, F.; Shi, Z.-J. ACS Catal. 2014, 4, 280.
(b) Wang, L.; He, W.; Yu, Z. Chem. Soc. Rev. 2013, 42, 599.
(c) Modha, S. G.; Mehta, V. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2013, 42, 5042. -
[25]
Miao, T.; Wang, L. Adv. Synth. Catal. 2014, 356, 967. doi: 10.1002/adsc.v356.5
-
[26]
Li, J.; Bi, X.; Wang, H.; Xiao, J. RSC Adv. 2014, 4, 19214. doi: 10.1039/c4ra01270d
-
[27]
Yang, J.; Xiao, J.; Chen, T.; Yin, S.-F.; Han, L.-B. Chem. Commun. 2016, 52, 12233. doi: 10.1039/C6CC06048J
-
[28]
(a) Kumar, T. S.; Yang, T.; Mishra, S.; Cronin, C.; Chakraborty, S.;
Shen, J.-B.; Liang, B. T.; Jacobson, K. A. J. Med. Chem. 2013, 56, 902.
(b) Lauer, A. M.; Mahmud, F.; Wu, J. J. Am. Chem. Soc. 2011, 133, 9119.
(c) Lauer, A. M.; Wu, J. Org. Lett. 2012, 14, 5138. -
[29]
(a) Atherton, F. R.; Todd, A. R. J. Chem. Soc. 1947, 674.
(b) Xiong, B.; Zhou, Y.; Zhao, C.; Goto, M.; Yin, S.-F.; Han, L.-B.Tetrahedron 2013, 69, 9373. -
[30]
Wang, J.; Huang, X.; Ni, Z.; Wang, S.; Wu, J.; Pan, Y. Green Chem. 2015, 17, 314. doi: 10.1039/C4GC00944D
-
[31]
Bi, X.; Li, J.; Meng, F.; Wang, H.; Xiao, J. Tetrahedron 2016, 72, 706. doi: 10.1016/j.tet.2015.12.020
-
[32]
Zhu, Y.; Chen, T.; Li, S.; Shimada, S.; Han, L.-B. J. Am. Chem. Soc. 2016, 138, 5825. doi: 10.1021/jacs.6b03112
-
[33]
Sun, J.-G.; Yang, H.; Li, P.; Zhang, B. Org. Lett. 2016, 18, 5114. doi: 10.1021/acs.orglett.6b02563
-
[34]
Sun, J.-G.; Weng, W.-Z.; Li, P.; Zhang, B. Green Chem. 2017, 19, 1128. doi: 10.1039/C6GC03115C
-
[35]
Song, S.; Zhang, Y.; Yeerlan, A.; Zhu, B.; Liu, J.; Jiao, N. Angew. Chem., Int. Ed. 2017, 56, 2487. doi: 10.1002/anie.201612190
-
[36]
Moon, Y.; Moon, Y.; Choi, H.; Hong, S. Green Chem. 2017, 19, 1005. doi: 10.1039/C6GC03285K
-
[37]
Lin, Y.-M.; Lu, G.-P.; Wang, G.-X.; Yi, W.-B. J. Org. Chem. 2017, 82, 382. doi: 10.1021/acs.joc.6b02459
-
[38]
Ouyang, Y.-J.; Li, Y.-Y.; Li, N.-B.; Xu, X.-H. Chin. Chem. Lett. 2013, 24, 1103. doi: 10.1016/j.cclet.2013.06.020
-
[39]
Xia, M.; Chen, J. Tetrahedron Lett. 2016, 57, 4702. doi: 10.1016/j.tetlet.2016.09.021
-
[40]
Li, J.; Bi, X.; Wang, H.; Xiao, J. Asian J. Org. Chem. 2014, 3, 1113. doi: 10.1002/ajoc.v3.10
-
[41]
Xu, W.; Hu, G.; Xu, P.; Gao, Y.; Yin, Y.; Zhao, Y. Adv. Synth. Catal. 2014, 356, 2948. doi: 10.1002/adsc.201400155
-
[42]
Zhang, J.-S.; Chen, T.; Yang, J.; Han, L.-B. Chem. Commun. 2015, 51, 7540. doi: 10.1039/C5CC01182E
-
[43]
Yang, D.; Zhao, D.; Mao, L.; Wang, L.; Wang, R. J. Org. Chem. 2011, 76, 6426. doi: 10.1021/jo200981h
-
[44]
Zhao, Y.; Chen, X.; Chen, T.; Zhou, Y.; Yin, S.-F.; Han, L.-B. J. Org. Chem. 2015, 80, 62. doi: 10.1021/jo501961h
-
[45]
(a) Darcel, C.; Uziel, J.; Jugé, S. In Phosphorous Ligands in Asym-metric Catalysis, Ed.: Bö rner, A., Wiley-VCH, Weinheim, 2008, Vol. 3, p. 1211.
(b) Sawa, M.; Tsukamoto, T.; Kiyoi, T.; Kurokawa, K.; Nakajima,
F.; Nakada, Y.; Yokota, K.; Inoue, Y.; Kondo, H.; Yoshino, K. J. Med. Chem. 2002, 45, 930.
(c) Moonen, K.; Laureyn, I.; Stevens, C. V. Chem. Rev. 2004, 104, 6177. -
[46]
Atherton, F.; Openshaw, A.; Todd, A. J. Chem. Soc. 1945, 660.
-
[47]
Wang, G.; Yu, Q.-Y.; Chen, S.-Y.; Yu, X.-Q. Tetrahedron Lett. 2013, 54, 6230. doi: 10.1016/j.tetlet.2013.09.006
-
[48]
Fraser, J.; Wilson, L. J.; Blundell, R. K.; Hayes, C. J. Chem. Commun. 2013, 49, 8919. doi: 10.1039/c3cc45680c
-
[49]
Zhou, Y.; Yang, J.; Chen, T.; Yin, S.-F.; Han, L.-B. Bull. Chem. Soc. Jpn. 2014, 87, 400. doi: 10.1246/bcsj.20130310
-
[1]
-

图式1 钯催化C—H/P—H交叉偶联可能的反应机理
Scheme 1 roposed mechanism of the palladium-catalyzed C— H/P—H cross coupling

图式2 银媒介C—H/P—H交叉偶联可能的反应机理
Scheme 2 Proposed mechanism of the silver-mediated C—H/P—H cross coupling

图式3 传统的构建sp2/sp3-C—P键的方法
Scheme 3 Traditional method for the construction of sp2/sp3-C—P bond

图式4 镍催化C—O/P—H交叉偶联可能的反应机理
Scheme 4 Proposed mechanism of the nickel-catalyzed C—O/P—H cross coupling

图式5 镍催化C—S/P—H交叉偶联可能的反应机理
Scheme 5 Proposed mechanism of the nickel-catalyzed C—S/ P—H cross coupling

图式6 钯催化脱氢偶联的化学计量反应
Scheme 6 Stoichiometric reactions of the dehydrogenative coupling catalyzed by Pd

图式7 钯催化S—H/P—H交叉偶联反应的可能机理
Scheme 7 Proposed mechanism of the palladium-catalyzed S—H/P—H cross coupling

图式8 可见光催化S—H/P—H交叉偶联反应的可能机理
Scheme 8 Proposed mechanism of the visible light-catalyzed S—H/P—H cross coupling

图式9 Cs2CO3催化S—H/P—H交叉偶联反应的可能机理
Scheme 9 Proposed mechanism of the Cs2CO3-catalyzed S—H/P—H cross coupling
-


 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 45
- 文章访问数: 5503
- HTML全文浏览量: 1117
 下载:
下载:
