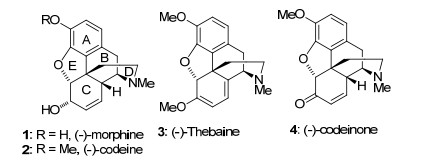
 图 1
天然的吗啡类生物碱
Figure 1.
Natural opium alkaloids
图 1
天然的吗啡类生物碱
Figure 1.
Natural opium alkaloids
引用本文:
李其林, 张洪彬. 吗啡类生物碱的合成研究进展[J]. 有机化学,
2017, 37(7): 1629-1652.
doi:
10.6023/cjoc201702048

Citation: Li Qilin, Zhang Hongbin. Research Progress on the Synthesis of Morphine Alkaloids[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1629-1652. doi: 10.6023/cjoc201702048
Citation: Li Qilin, Zhang Hongbin. Research Progress on the Synthesis of Morphine Alkaloids[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1629-1652. doi: 10.6023/cjoc201702048
吗啡类生物碱的合成研究进展
English
Research Progress on the Synthesis of Morphine Alkaloids
Abstract:
The morphine alkaloids constitute a class of structurally related natural products isolated from opium poppy, Papaver somniferum. The synthesis of morphine and its derivatives has attracted the attention of many generations of synthetic chemists due to their highly challenging molecular architecture and biological activities. Progresses toward the synthesis of the morphine alkaloids are reviewed in terms of chronological order.
-
Key words:
- morphine
- / morphine alkaloids
- / synthesis
-
吗啡1 (Morphine)是从鸦片中分离得到的生物碱, 其重量约占鸦片重量的14%~17%[1], 是人们熟知的一种麻醉剂及镇痛药物, 但具有成瘾性.其他天然的吗啡类生物碱还包括可待因2 (Codeine)、蒂巴因3 (Thebaine)和可待因酮4 (Codeinone)等(图 1).吗啡及其类似物独特的结构和生物活性引起了合成化学家高度的兴趣.天然的(-)-morphine是具有ABCDE五个环稠合而成的复杂立体结构分子, 其含有一个五元二氢呋喃环、一个含氮的桥环、五个连续的手性中心, 其中一个是苄位的季碳中心.吗啡被认为是合成化学家们心目中极具重要意义的合成目标.距盖茨第一次合成吗啡到今天的60多年里, 至少有30个研究组致力于吗啡及其类似物的全合成研究工作, 吗啡全合成的相关报道已有近50余篇, 关于吗啡合成的综述也有近8篇[2~9].本文按合成的时间顺序分类综述了吗啡类生物碱的全合成研究进展.
图 1
天然的吗啡类生物碱
Figure 1.
Natural opium alkaloids
1 吗啡类生物碱的合成
距离1806年吗啡首次从罂粟中成功分离出来到今天, 已经过去了两百多个年头.吗啡的首次全合成于1952年由Gates课题组完成, 在此之后, 每一个十年中均有关于吗啡的全合成文章发表.在众多吗啡的合成研究中, 涌现了相当大批的新的有效合成方法和新颖的合成策略.
1.1 Gates研究组的合成方法
1952年, 盖茨(Gates)对吗啡的[10, 11]首次合成(Scheme 1)被视为全合成领域中的经典之作.盖茨以化合物5作为起始原料经9步反应合成Diels-Alder反应的前体6, 化合物6与丁二烯在加热条件下发生分子间的Diels-Alder反应形成化合物7.再经铜-铬催化加氢反应获得关键中间体8, 但很遗憾C(14) 位上氢的相对立体构型与天然产物相反.关键中间体8经Wolf-Kishner-黄鸣龙还原, N上甲基化和LiAlH4还原酰胺得到化合物9.消旋的化合物9经酒石酸拆分得C(9) 和C(13) 立体构型与天然产物一致但C(14) 与天然产物相反的中间体.该中间体构型经天然Codeine的降解物得以证实, 这也进一步证实了Robinson对吗啡结构的推测[12].而后的合成步骤以天然Codeine的降解物为原料出发经区域选择性水合反应, 脱甲基反应和Oppennauer氧化反应得化合物10.之后经羰基α位溴代和苯环溴代, 再与2, 4-二硝基苯肼反应成腙后水解得到化合物11, 此时C(14) 的立体构型翻转为热力学稳定的正确天然产物构型.化合物11经双键氢化、溴代, 再与2, 4-二硝基苯肼反应构筑了二氢呋喃环, 水解腙得到α, β-不饱和酮化合物, LiAlH4还原羰基及苯基溴, 最终以31步0.06%的总收率第一次合成天然产物吗啡, 用全合成的方式证明了吗啡的结构.
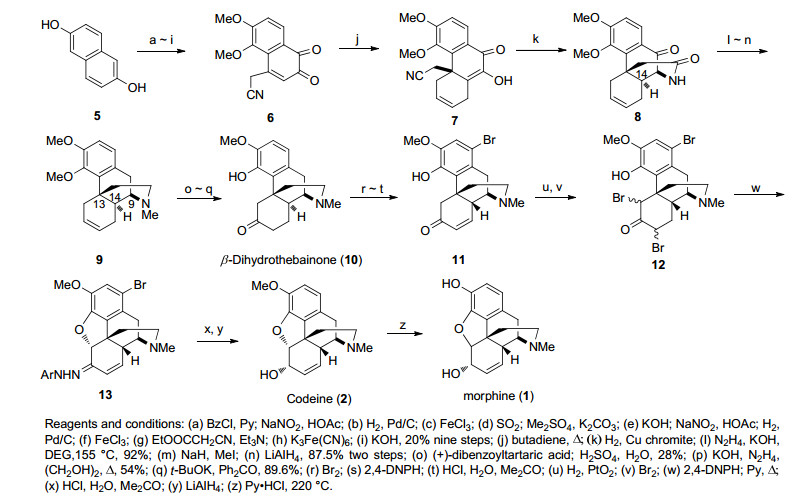 图式1
Gates研究组的合成方法
图式1.
Synthesis by Gates's group
图式1
Gates研究组的合成方法
图式1.
Synthesis by Gates's group
1.2 Ginsburg研究组的合成方法
继Gates之后, 1954年Ginsburg研究组[13]报道了合成吗啡的另一路线(Scheme 2), 尽管只完成了吗啡的形式合成, 但为合成吗啡及其类似物提供了新的策略.该路线在化合物16的基础上利用迈克尔加成反应及分子内的傅克酰基化反应为关键步骤合成化合物19.出其不意的是在将酮羰基选择性保护为缩酮后, 苯环上的甲氧基被切断, 直链上的乙酰基失去发生了亲核取代反应, 从而构建了含芳环季碳中心, 同时建立了吗啡的四环骨架结构得到化合物20.之后经保护、去保护和还原作用合成盖茨的中间体10 (dihydrothebainone).
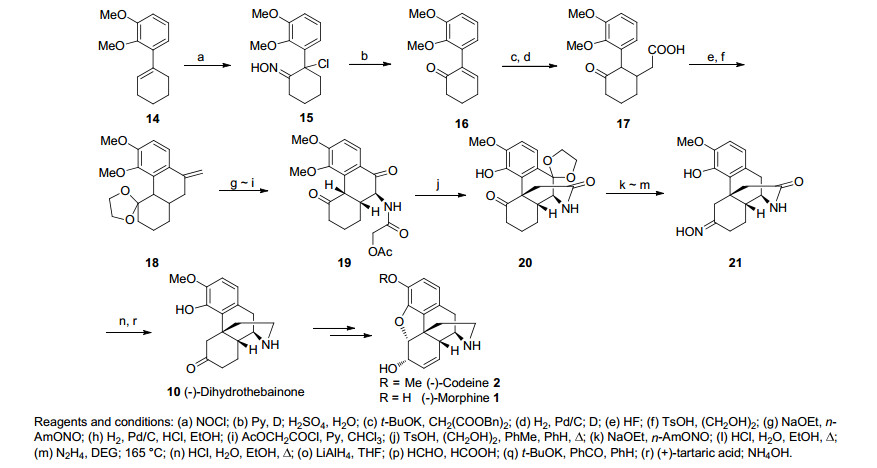 图式2
Ginsburg研究组的合成方法
图式2.
Synthesis by Ginsburg's group
图式2
Ginsburg研究组的合成方法
图式2.
Synthesis by Ginsburg's group
1.3 Barton、Szantay和White研究组的合成方法
1964年, Barton课题小组[14] (Scheme 3)从吗啡可能的生源途径[15](图 2)中(R)-网脉碱25经邻对位苯酚的氧化自由基偶联反应生成沙罗泰里啶26的过程得到启发, 第一次利用化学方法模拟了这一生源合成途径, 尽管这个反应只有0.02%的收率.之后Szantay和White等[16~18](Scheme 3)纷纷采用苯酚的氧化自由基偶联的策略对吗啡进行了合成.
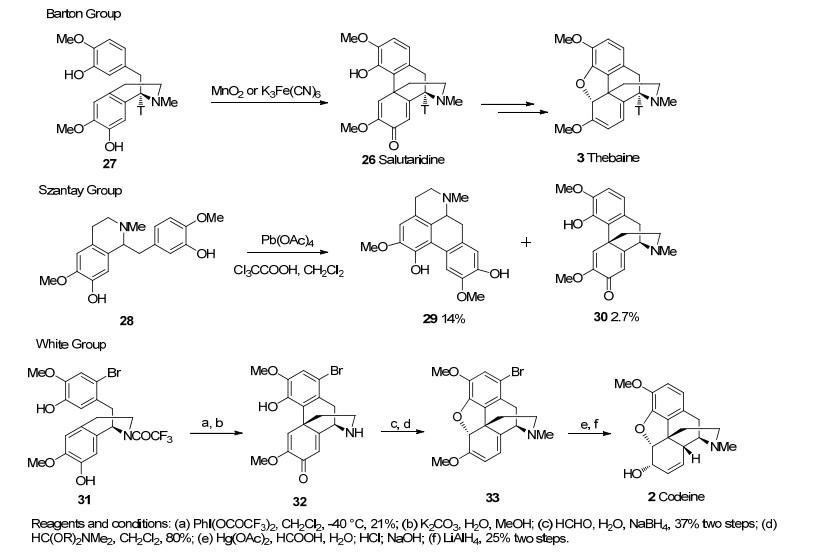 图式3
Barton、Szantay和White研究组的合成方法
图式3.
Synthesis by Barton, Szantay and White's group
图式3
Barton、Szantay和White研究组的合成方法
图式3.
Synthesis by Barton, Szantay and White's group
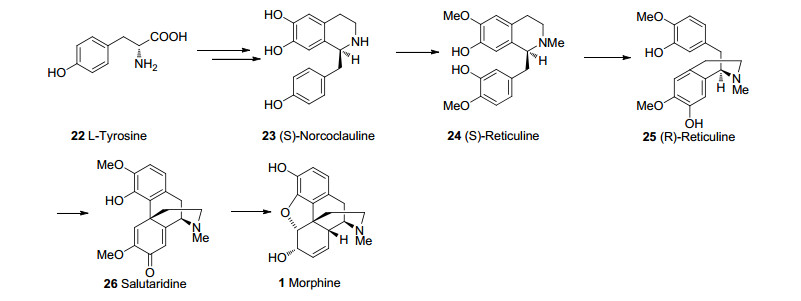 图 2
吗啡可能的生源途径
Figure 2.
Biosynthesis of morphine alkaloids
图 2
吗啡可能的生源途径
Figure 2.
Biosynthesis of morphine alkaloids
1.4 Rice研究组的合成方法
另外Grewe, Beyerman, Rice和Schafer[19~23]等课题小组也对吗啡的仿生合成产生了浓厚的兴趣, 以Grewe环化为关键步骤来合成吗啡类生物碱(Scheme 3).其中, Rice课题小组在1980年完成的合成路线最具代表性.从原料酸化合物34和胺化合物35出发经过一步缩合成酰胺, 之后发生一步Bischler-Napieralski反应和氰基硼氢化钠还原得到化合物36.伯奇Birch还原和N上甲酰化得到化合物37, 缩酮保护和芳香环溴代以占位的方式保护芳环对位不会受接下来的反应影响, 两步反应得到Grewe环化的前体38, 在甲酸的作用下形成β, γ-不饱和酮之后在氢氟酸/氟化铵的条件下得到正离子促进的关环产物, 后经去甲酰化和还原胺化得到化合物39, 随后经过一步羰基α位溴代和苯酚去质子化作用关环构筑二氢呋喃环, 芳环上的溴原子在甲醛存在下加氢移除后得到二氢可待因酮40 (dihydrocodeinone). Rice小组以29.7%的总收率共用14步最终完成了吗啡的形式全合成(Scheme 4).
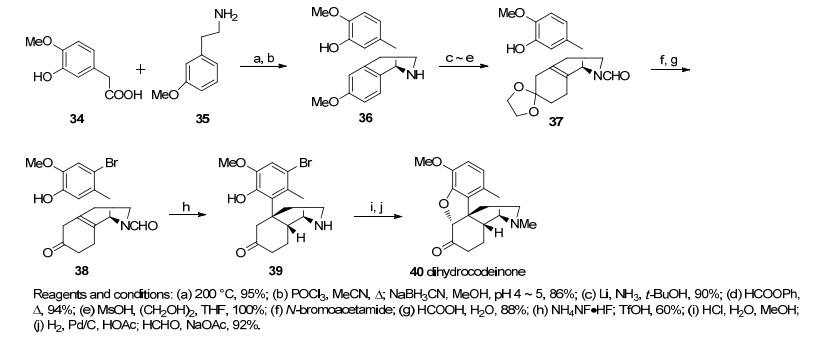 图式4
Rice研究组的合成方法
图式4.
Synthesis by Rice's group
图式4
Rice研究组的合成方法
图式4.
Synthesis by Rice's group
1.5 Evans研究组的合成方法
1982年, Evans小组[24]合成吗啡类生物碱的路线是先利用芳基锂试剂与羰基反应合成化合物43, 继而烯丙位上的氢去质子化后双键迁移与二溴代物发生亲核取代、烯胺烷基化得化合物45.烯胺质子化的亚胺在环丙烷重氮甲烷的作用下发生环丙烷化后经二甲亚砜氧化开环得关键中间体化合物48.在路易斯酸催化下分子内傅克反应构筑了吗啡骨架的B环, 羟基磺酰化被还原脱除, 端烯被氧化切断得到H构型与Gates的中间体相反的化合物50, 完成了吗啡的形式全合成(Scheme 5).
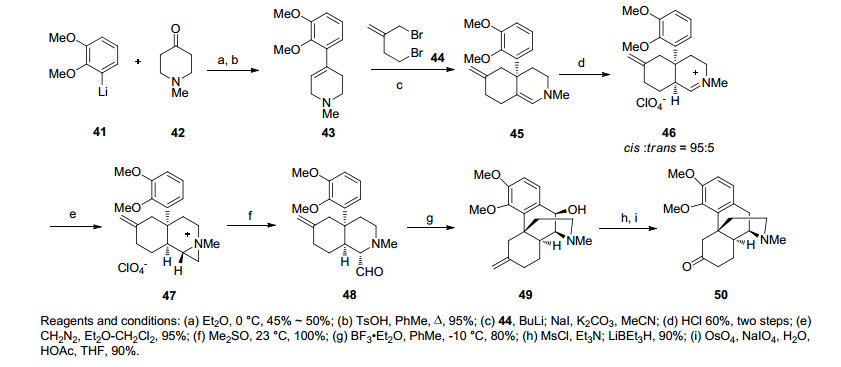 图式5
Evans研究组的合成方法
图式5.
Synthesis by Evans' group
图式5
Evans研究组的合成方法
图式5.
Synthesis by Evans' group
1.6 Rapport研究组的合成方法
Rapport研究组[25]于1983年试图合成吗啡, 但在合成过程中遇到了许多立体化学的问题, 导致最终合成只能止步于Evans的合成中间体.首先不饱和化合物51发生Michael加成反应和还原氰基酰胺化得到化合物52, 随后甲基化、选择性还原酰胺得到六氢吡啶-3-羧酸酯化合物53.六氢吡啶-3-羧酸酯发生Methylene-lactam重排、烯丙位氧化和酯交换后得Claisen重排前体55, 化合物55继而Claisen重排、酯同系化、Michael加成和脱羧后合成化合物57.之后Rapport曾尝试利用亲核反应关吗啡骨架B环, 但因立体化学始终不对的问题, 只能合成得到Evans的合成中间体(Scheme 6).
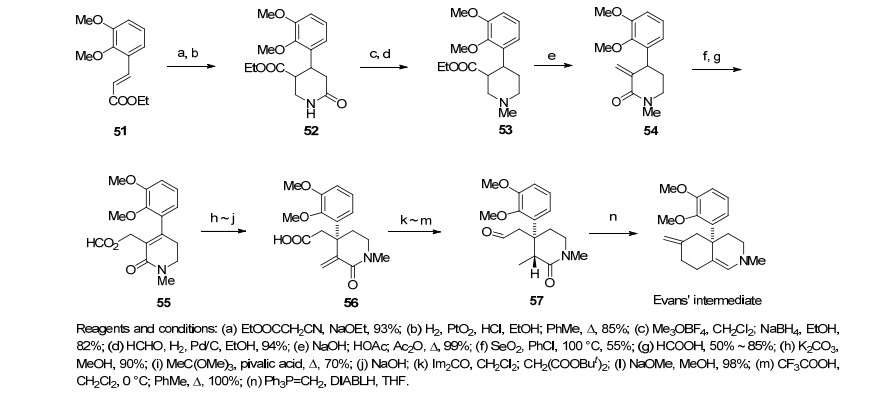 图式6
Rapport研究组的合成方法
图式6.
Synthesis by Rapport's group
图式6
Rapport研究组的合成方法
图式6.
Synthesis by Rapport's group
1.7 Schultz研究组的合成方法
1985年, Schultz等[26]合成吗啡的亮点在于利用光反应合成吗啡骨架二氢呋喃E环的同时建立了正确立体构型的含芳环季碳中心.该路线首先合成得到环氧化合物59, 在碱性条件下苯酚对环氧开环并脱除成双键得到醚类化合物60.之后在光反应条件下成功构筑了苯并呋喃环, 后将羰基保护、还原酰胺脱除成双键, 随后氰基被甲基锂亲核加成成甲基酮化合物65.化合物 65发生傅克反应关吗啡骨架B环得到化合物66, 随后脱苄基保护、上氰基和端烯被氧化切断合成化合物67, 再经一系列官能团转化即可合成吗啡(Scheme 7).
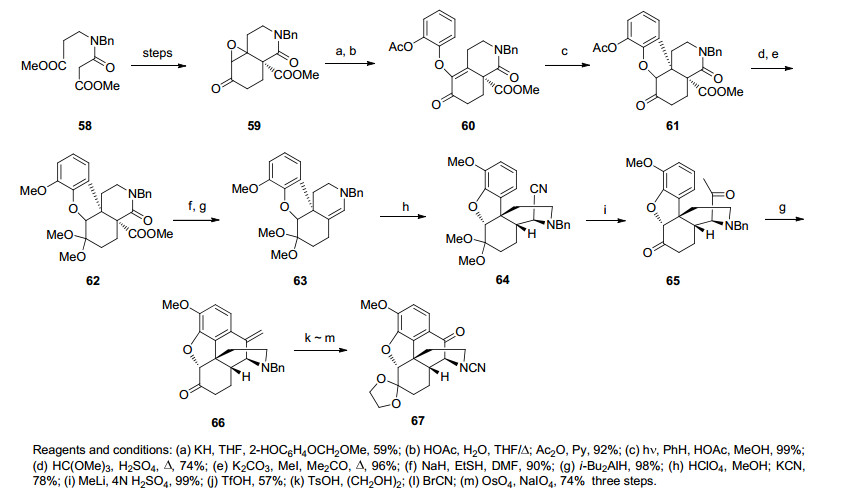 图式7
Schultz研究组的合成方法
图式7.
Synthesis by Schultz's group
图式7
Schultz研究组的合成方法
图式7.
Synthesis by Schultz's group
1.8 Fuchs研究组的合成方法
1987年, Fuchs研究组[27]利用串联的分子内加成-烷基化反应作为合成吗啡的关键步骤.该合成路线先用Mitsunobu反应连接两个片段68, 69得到醚化合物70, 脱除TBS保护基、随后将醇氧化成羰基、DIBAL-H还原羰基得到构型翻转的环化反应前体71.在丁基锂作用下发生锂交换随后发生分子内的加成-烷基化反应, 一步成功构建了吗啡的四环骨架和含芳环季碳中心.末端双键双羟化反应、氧化切断为醛基合成化合物73, 还原胺化和保护胺基得到74.经Swern氧化、烯醇甲醚化、再次保护由于上一步骤脱除保护基的胺基、消除成双键和DDQ氧化得到双稀产物77, 随后脱除氮上的保护基, 发生氮杂的1, 6-加成反应得到混合物78和79.化合物78可以经过双键异构化反应转化为79, 之后NaBH4还原得到可待因, 去甲基化后合成得到吗啡(Scheme 8).
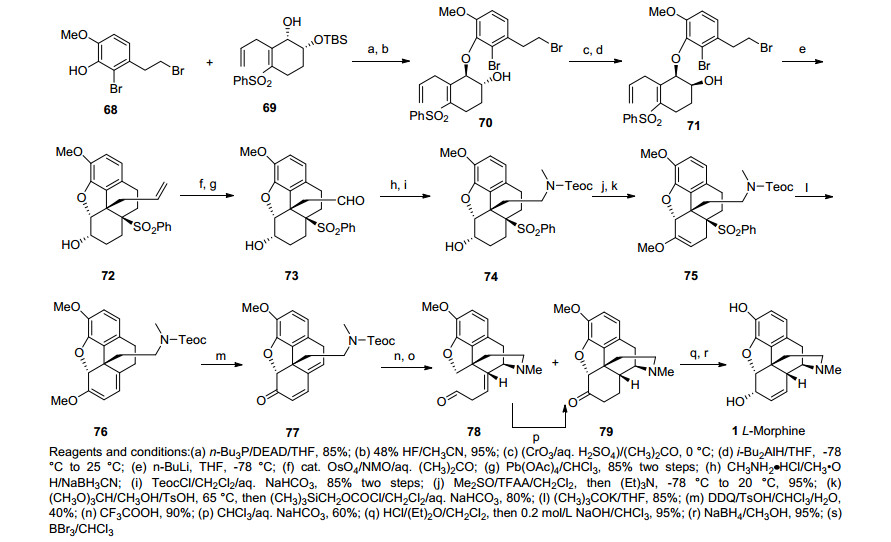 图式8
Fuchs研究组的合成方法
图式8.
Synthesis by Fuchs's group
图式8
Fuchs研究组的合成方法
图式8.
Synthesis by Fuchs's group
1.9 Tius研究组的合成方法
1992年, Tius研究组[28]首次以Diels-Alder反应作为关键步骤合成吗啡类生物碱.其将对苯二醌化合物80与81发生分子间的Diels-Alder反应得到化合物82.硒基环化氧化消除得到的氧负离子进攻亚胺离子、酸性条件下缩酮去保护、Davis试剂氧化羰基α位得羟基后还原消除成双键, Swern氧化羟基得到85.路易斯酸作用下芳环芳构化形成化合物86.硒基加成氧化消除、NaBH4立体选择性还原、甲基锂脱保护然后还原胺化、Dess-Martin氧化烯丙醇得到88, 锌粉还原断开胺缩酮并Michael加成环化、还原羰基并酸化得到化合物90, 其后的合成采用Gates的合成路线(Scheme 9).
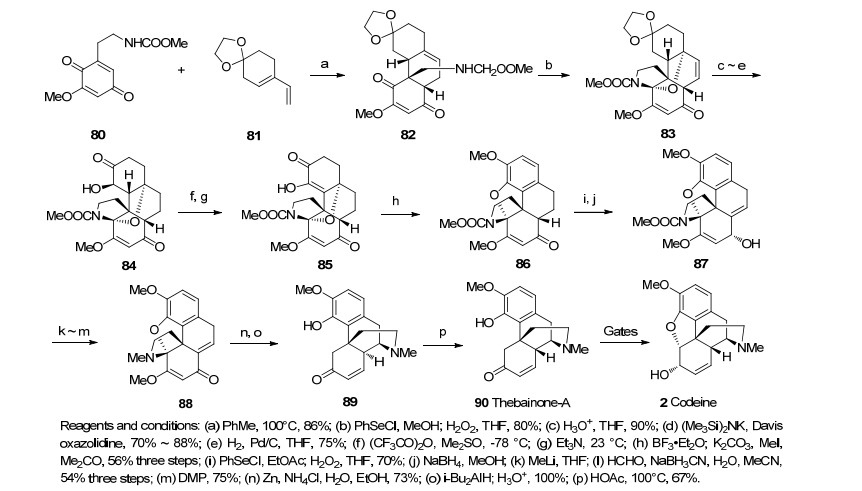 图式9
Tius研究组的合成方法
图式9.
Synthesis by Tius's group
图式9
Tius研究组的合成方法
图式9.
Synthesis by Tius's group
1.10 Parker研究组的合成方法
1992年, Parker课题小组[29, 30]以芳基溴的自由基环化反应作为关键步骤仅用11步就合成了消旋的二氢可待因, 完成了可待因和吗啡的形式全合成.在该合成路线中, 经Birch还原, 一系列官能团转化以及CBS催化的不对称还原后得到的化合物91经间氯过氧苯甲酸(m-CPBA)的氧化后得到环氧化合物92, 化合物92在路易斯酸的作用下环氧开环并用叔丁基二甲基硅烷基(TBDMS)保护基保护得到化合物93, 化合物93和化合物94经一步Mitsunobu反应随后脱去硅基保护得到进行关键步骤自由基环化反应的前体95.化合物95经自由基引发剂偶氮二异丁腈(AIBN)和Bu3SnH作用后经历一系列自由基环化/消除等过程一步构建了ABCE的吗啡环系统得到化合物96, 化合物96在Birch还原的条件下氢胺化构筑了D环, 最后经Swern氧化以75% ee值得到二氢可待因酮40 [(-)-dihydrocodeinone].该路线共用11步, 总收率为11.1% (Scheme 10).
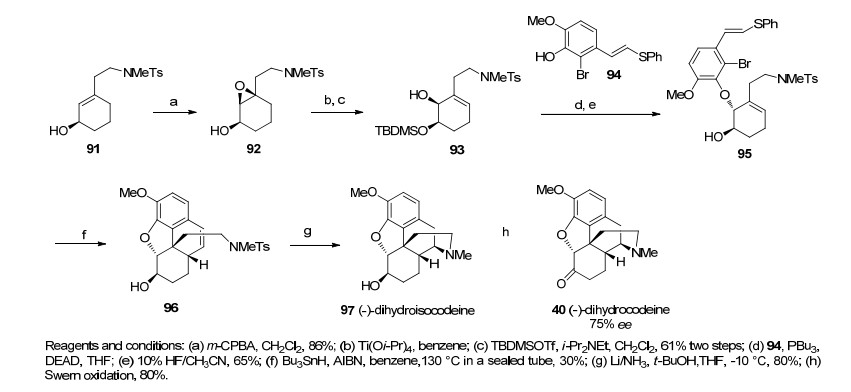 图式10
Parker研究组的合成方法
图式10.
Synthesis by Parker's group
图式10
Parker研究组的合成方法
图式10.
Synthesis by Parker's group
1.11 Overman研究组的合成方法
Heck反应至1972年被美国化学家理查德•赫克发现以来, 其无疑是受到最多合成化学家青睐的有效形成新C—C键的合成方法, 因此, Heck反应在天然产物吗啡的全合成中也得到了广泛的运用.在1993年, Overman课题小组[31]通过一步钯催化的Heck反应快速地构建了天然产物吗啡中含芳环的季碳手性中心, 之后再经一步环氧开环构筑二氢呋喃环, 最终以1.9%的收率, 共用14步合成二氢可待因酮(Dihydrocodeinone) (Scheme 11).
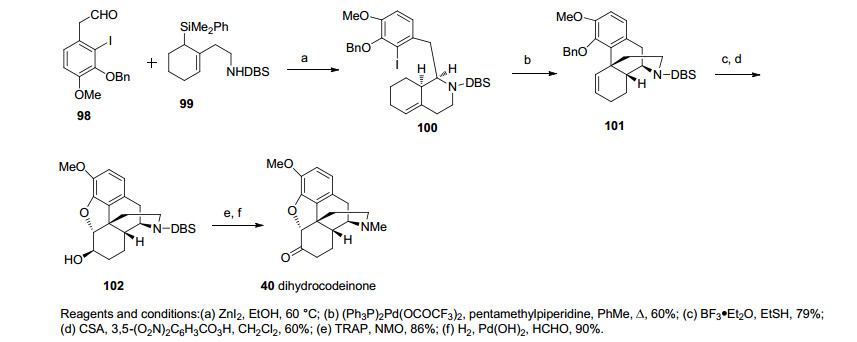 图式11
Overman研究组的合成方法
图式11.
Synthesis by Overman's group
图式11
Overman研究组的合成方法
图式11.
Synthesis by Overman's group
1.12 Mulzer研究组的合成方法
1998年, Mulzer等[32]利用Robinson环化合成吗啡骨架C环得到化合物104, 之后用格式试剂的Michael加成反应成功构筑含芳环的季碳手性中心, 再在羰基α位上溴合成化合物105.分子内的亲核取代反应合成吗啡骨架E环得到化合物106.随后发生脱卤反应, 端烯硼氢化氧化, 取代反应等一系列转化合成二氢可待因酮(Scheme 12).
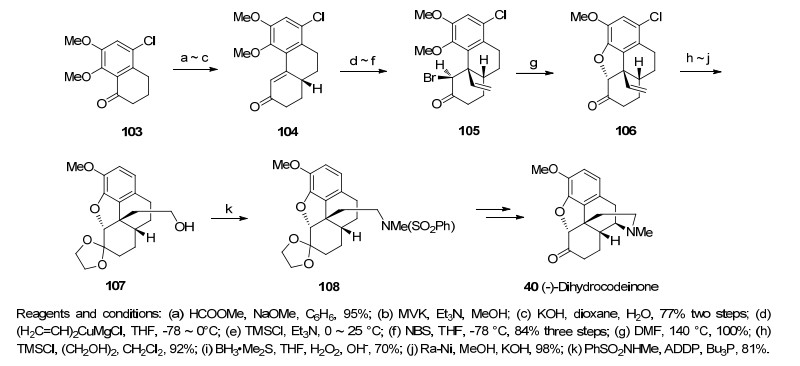 图式12
Mulzer研究组的合成方法
图式12.
Synthesis by Mulzer's group
图式12
Mulzer研究组的合成方法
图式12.
Synthesis by Mulzer's group
1.13 White研究组的合成方法
1999年, White小组[33]也设计了合成吗啡的路线, 虽然最终与天然产物构型相反, 但在该路线中充分看出其善用分子内反应合成天然产物的本领.从异香兰素出发, 依次经Stobbe缩合、分子内Friedel-Crafts反应、Robinson环化和分子内亲核取代反应构建了吗啡的ABCE四环骨架, 得到化合物110.保护羟基, 随后制成的酰氯和重氮甲烷反应得到卡宾前体111, 在二价铑的作用下构筑了含芳环的季碳手性中心.化合物113在发生贝克曼重排后合成了吗啡桥环D环, 再多次转化后得到了右旋的可待因, 因与天然产物构型恰好相反, 所以该合成是能算是一个形式合成(Scheme 13).
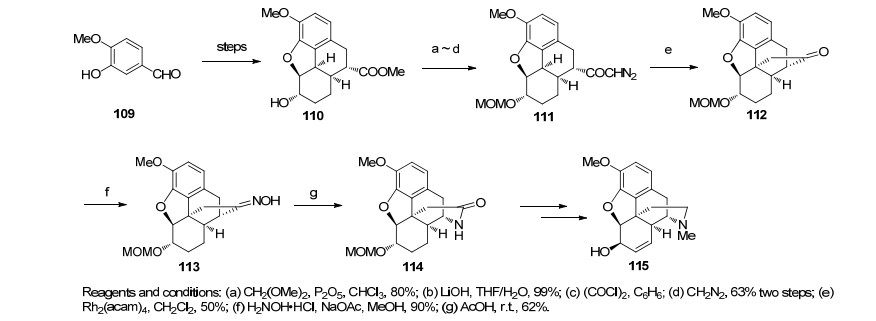 图式13
White研究组的合成方法
图式13.
Synthesis by White's group
图式13
White研究组的合成方法
图式13.
Synthesis by White's group
1.14 Cheng研究组的合成方法
2000年, Cheng课题组[34]采用钯催化环化和Stevens重排作为关键步骤合成吗啡.从四氧异喹啉衍生物116经甲基化、NaBH4还原、甲酸乙酷保护氮、二级醇去保护得到醇117, 随后经过Mitsunobu反应得到芳基酸118.还原苯甲醛并硅醚保护得到化合物119, 发生分子内Heck反应构建吗啡骨架中二氢呋喃环, 得到化合物120.化合物120脱除硅基保护基并氯代得到化合物121, 之后钯催化的环化反应得到7元环的产物122.苯基锂作用下的甲基季按盐产物发生Stevens重排得到最终产物123.该合成路线虽没最终合成吗啡, 但钯催化环化和Stevens重排合成吗啡骨架B环的过程确实十分新颖(Scheme 14).
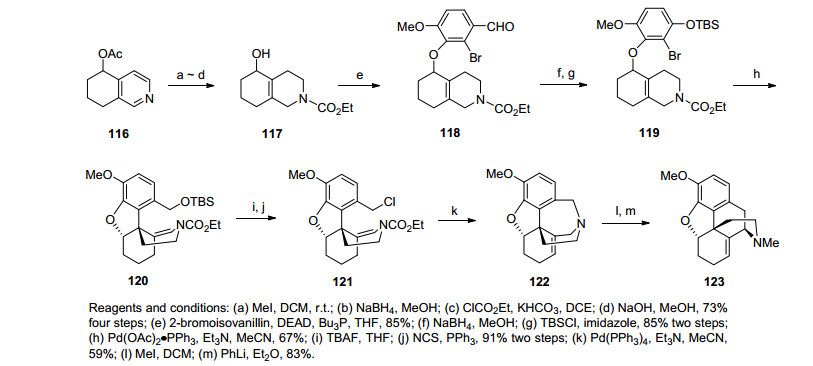 图式14
Cheng研究组的合成方法
图式14.
Synthesis by Cheng's group
图式14
Cheng研究组的合成方法
图式14.
Synthesis by Cheng's group
1.15 Ogasawara研究组的合成方法
2000年, Ogasawara研究组[35]从化合物124出发通过自由基环化反应构筑了含芳环季碳手性中心, 随后经还原切断内酯、选择性保护伯醇和仲醇消除成双键得到化合物125.化合物125在Sakurai条件下与烯丙基三甲基硅反应生成化合物126.羰基乙二醇保护和氧化切断端烯成醛, 之后发生傅克反应构建吗啡骨架B环, 得到化合物128.经还原去保护, Mitsunobu条件下伯醇转化为化合物129, 利用Parker路线中自由基环化合成吗啡桥环D环.整个路线从化合物124到Gates中间体共用23步, 总收率0.8% (Scheme 15).
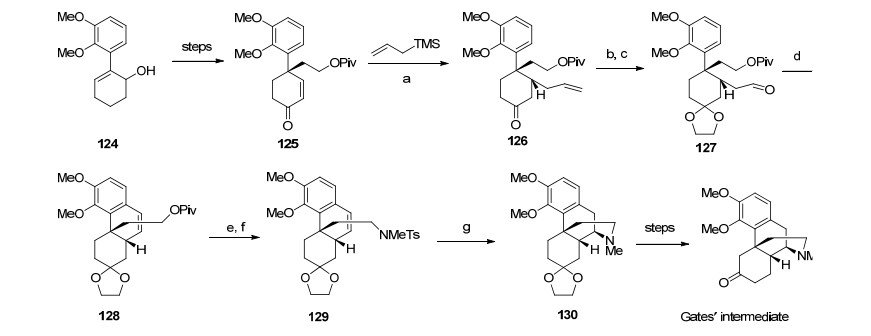 图式15
Ogasawara研究组的合成方法
图式15.
Synthesis by Ogasawara's group
图式15
Ogasawara研究组的合成方法
图式15.
Synthesis by Ogasawara's group
Ogasawara等[36]于2001年报道了其另一条合成吗啡的路线.该路线利用光学纯化合物131与醚锂试剂反应再经PCC氧化得到不饱和烯酮132, 随后与乙稀基格式试剂发生1, 4-加成、N-溴代琥珀酰亚胺(NBS)溴代、N, N-二甲基甲酰胺(DMF)中加热得到二氧苯并呋喃化合物, 用乙二醇保护羰基、硼氢化氧化、Piv保护得到串联环化反应前体136.化合物136在酸性条件下发生氧鎓离子促进的关键步骤逆Aldol反应构建吗啡环.还原除去保护基、Mitsunobu反应、锂氧条件下环化得到化合物139, 之后利用Mulzer的路线合成吗啡(Scheme 16).
 图式16
Ogasawara研究组的合成方法
图式16.
Synthesis by Ogasawara's group
图式16
Ogasawara研究组的合成方法
图式16.
Synthesis by Ogasawara's group
1.16 Taber研究组的合成方法
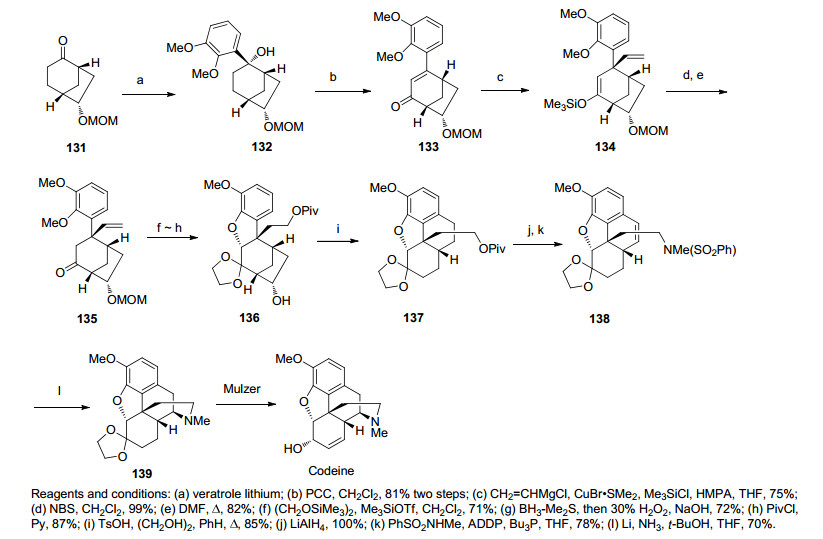
2002年, Taber等[37]发表了吗啡的合成路线, 该路线中的消旋化合物酮140与(S, S)-二苯基乙二醇反应得到非对映异构体142和143, 这两种化合物可通过柱色谱分离分开.化合物143通过苄位的亲核取代反应和缩酮水解得到化合物144.随后经立体选择性的还原羰基、Mitsunobu反应反转构型、还原叠氮化得到化合物146, 引入烷基化得到关键中间体147.经臭氧切断反应、碱性条件下发生分子内的烷基化和Robinson环化得到不饱和酮148, 一举构建了吗啡骨架的ABCD环.后经还原和BBr3脱除甲基生成二氢呋喃环产物149.继而通过一系列转换得到吗啡(Scheme 17).
 图式17
Taber研究组的合成方法
图式17.
Synthesis by Taber's group
图式17
Taber研究组的合成方法
图式17.
Synthesis by Taber's group
1.17 Trost研究组的合成方法
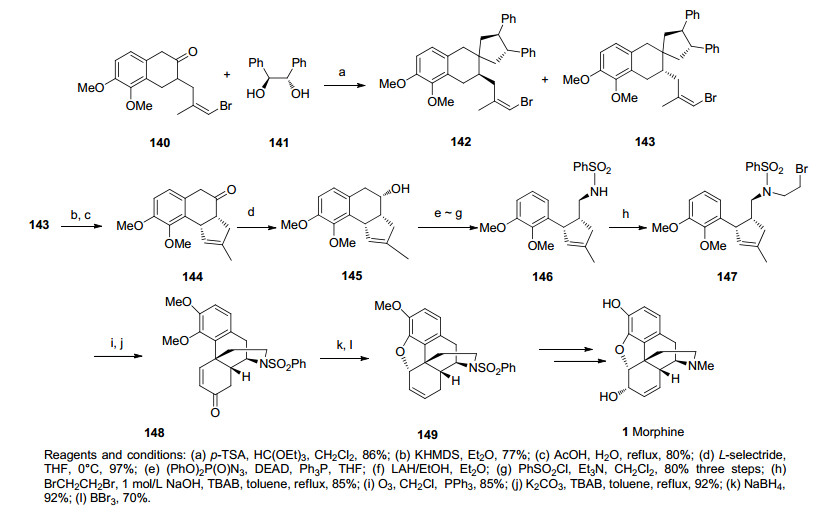
2002年, Trost研究小组[38, 39]报道了以钯催化的不对称烯丙基烷基化反应和Heck反应作为关键步骤不对称合成可待因[(-)-Codeine]的方法, 方法简单, 有效.合成路线中首先通过一步钯催化的不对称烯丙基烷基化反应高效立体专一性地合成了芳基醚化合物153, 化合物153在醛基被保护后, 酯基还原成伯醇, 经Mitsunobu反应生成氰基, 脱保护释放醛基成化合物154.化合物154在经第一次Heck反应后成功构建了ACE三环得到化合物155, 化合物155经Corey-Fucus烯化反应后生成烯基溴化合物156.随后化合物156通过第二次Heck反应构筑了B环得到苯并二氢呋喃的骨架157, 之后经一些简单氧化还原反应得到化合物160, 化合物160经一步氢胺化反应构筑D环得到目标分子可待因.该路线简洁高效仅用了15步, 总收率6.8% (Scheme 18).
 图式18
Trost研究组的合成方法
图式18.
Synthesis by Trost's group
图式18
Trost研究组的合成方法
图式18.
Synthesis by Trost's group
1.18 Fukuyama研究组的合成方法
2006年, Fukuyama课题小组[40]同样利用分子内的Heck反应作为合成可待因和吗啡的关键步骤.该路线先由碘代化合物162和环氧化合物163发生一步Tsuji-Trost反应形成醚键, 随后用Mitsunobu反应翻转仲醇的构型, 醇在与对硝基苯甲酸反应, 脱TBS保护, 伯醇转化为腈化合物167.化合物167还原脱去苄酯保护、TBS保护, 还原氰基再用氯甲酸甲酯保护氨基得关键步骤Heck反应的前体168.化合物168经Heck反应一步成功高效地构筑了二氢呋喃环和含芳环的季碳手性中心, TBS保护基脱除后得到单一构型的化合物169, 随后化合物169经过一步Mannich反应构筑B, C两环完成了吗啡所有环系的建立, 之后经几步官能团的转化和修饰最终共用25步, 6.7%的总收率完成了消旋吗啡(rac-Morphine)的全合成(Scheme 19).
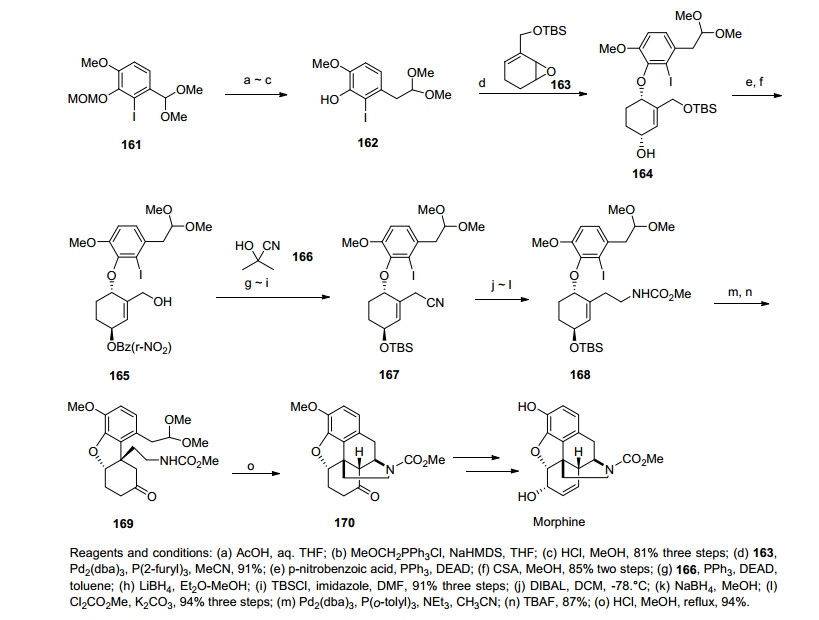 图式19
Fukuyama研究组的合成方法
图式19.
Synthesis by Fukuyama's group
图式19
Fukuyama研究组的合成方法
图式19.
Synthesis by Fukuyama's group
1.19 Hudlicky研究组的合成方法
2007年, Hudlicky研究小组[41]利用手性源化合物172作为反应原料, 通过Mitsunobu反应和Heck反应作为合成的关键步骤构建二氢呋喃以及含芳环季碳手性中心得到关键中间体178, 而后再经过第二次的Heck反应成功构建B环得到化合物180, 最后在醋酸汞的作用下关上D环.该路线共用15步, 0.23%的总收率完成了对映可待因(ent-Codeine)的全合成(Scheme 20).
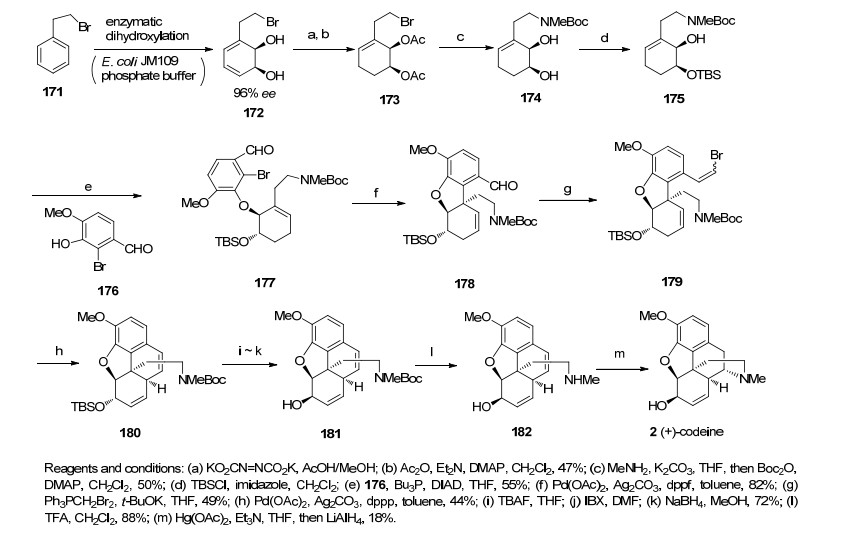 图式20
Hudlicky研究组的合成方法
图式20.
Synthesis by Hudlicky's group
图式20
Hudlicky研究组的合成方法
图式20.
Synthesis by Hudlicky's group
1.20 Chida研究组的合成方法
2008年, Chida课题小组[42]报道了以一系列Claisen重排反应作为关键步骤的合成二氢可待因(Dihydrocodeine)的方法.通过合成烯基三氟磺酸酯化合物189和硼酸化合物190发生Suzuki反应得到偶联产物, 并在二氯二氰基苯醌(DDQ)的作用下去除对甲氧基苄基(PMP)保护基得到化合物192, 接着脱叔丁基二甲基硅基(TBS)的化合物193发生连续两次Claisen重排反应构筑了含芳基季碳并得到化合物194, 化合物194经m-CPBA制备环氧后环氧开环构建二氢呋喃环, 而后还原酯基成醛发生傅克反应构筑B环, 再经Birch还原条件下构筑D环.合成路线共用24步, 3.8%的总收率合成了对映二氢化可待因(rac-Dihydroisocodeine) (Scheme 21).
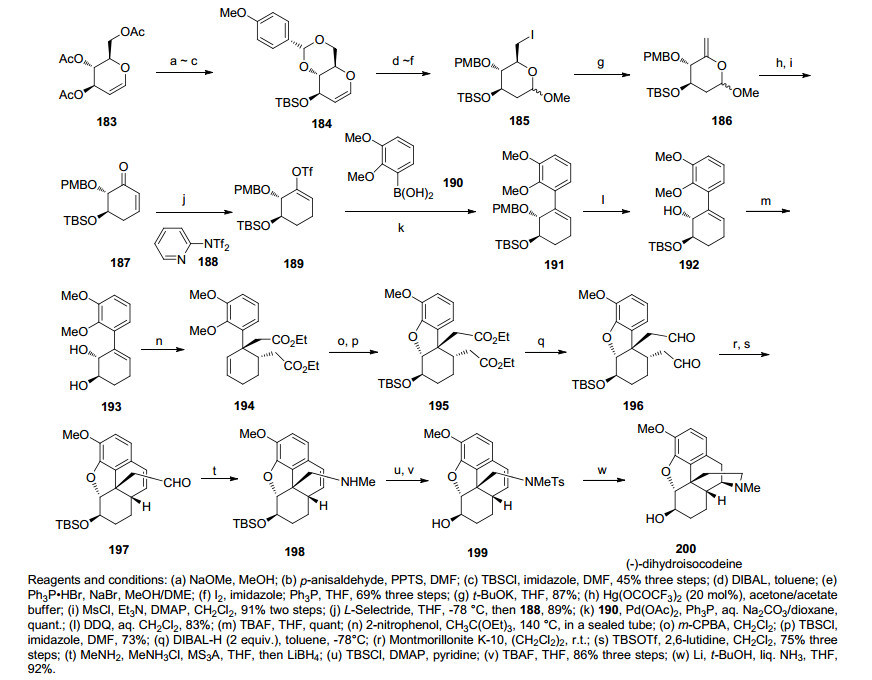 图式21
Chida研究组的合成方法
图式21.
Synthesis by Chida's group
图式21
Chida研究组的合成方法
图式21.
Synthesis by Chida's group
1.21 Guillou研究组的合成方法
同年, Guillou课题小组[43]利用内酯开环的Michael加成反应和Eschenmoser-Claisen重排反应作为关键步骤完成了可待因的全合成.该合成路线首先通过一步酯化得到化合物201, 化合物201经Heck反应形成内酯化合物202, 化合物202再经氧化脱除双键形成α, β不饱和酮化合物203, 随后经过胺解, 还原酰胺得到化合物205.化合物205经一步Eschenmoser-Claisen重排反应建立了正确立体构型的C(14).接下来还原酰胺, 傅克反应构建C环, 再通过几步官能团转化经Birch还原条件下构筑D环, 最后以17步, 0.64%的总收率完成了可待因的全合成(Scheme 22).
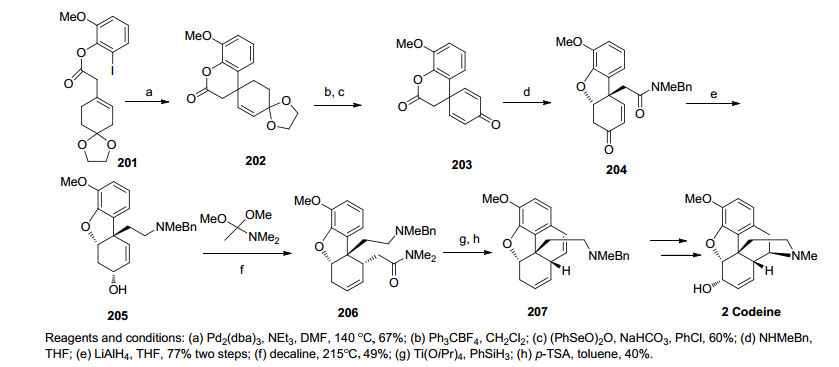 图式22
Guillou研究组的合成方法
图式22.
Synthesis by Guillou's group
图式22
Guillou研究组的合成方法
图式22.
Synthesis by Guillou's group
1.22 Stork研究组的合成方法
2009年Stork课题小组[44]以苯并呋喃的DA环化反应作为关键步骤一步构建了吗啡的B环和C环, 最终完成了吗啡的消旋合成.合成路线从化合物208经多步反应得到化合物209.化合物209和端醛化合物在Schwartz试剂的作用下反应得到仲醇化合物, 再经三乙基硅醚(TES)保护得到DA反应前体化合物211.化合物211经过分子内的DA反应得到环化产物212α和212β, 其中212α为主要产物.化合物212α和212β经超氢(三乙基硼氢化锂)还原, Dess-Martin (DMP)氧化成醛进行Witting反应得烯醇醚, 脱TES保护基, DMP氧化成酮化合物213, 化合物213被还原成醇化合物并和甲基磺酰氯反应生成甲基磺酸酯, 然后还原胺化成甲胺化合物215.最后D环的构筑由化合物215经过一步SN2的取代反应得到, 之后再经一系列官能团转化得到可待因.该路线历经22步, 以2.0%的总收率完成了消旋可待因(rac-Codeine)的全合成(Scheme 23).
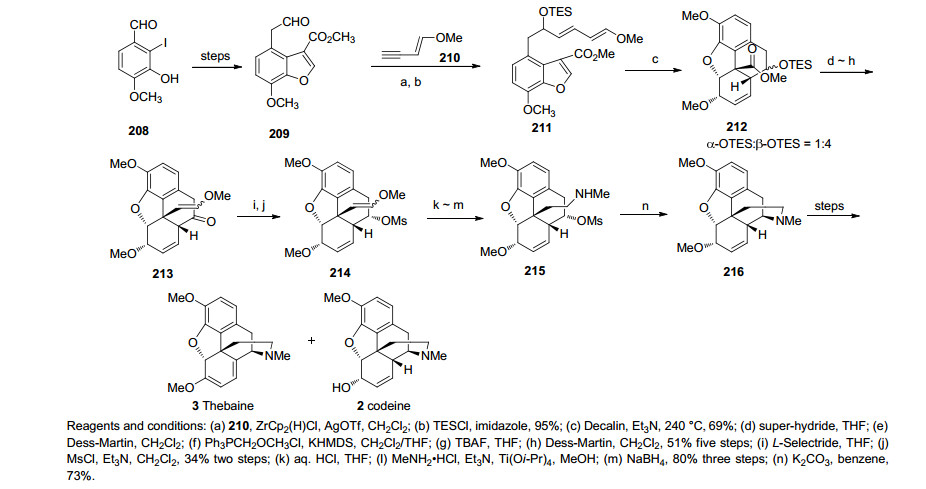 图式23
Stork研究组的合成方法
图式23.
Synthesis by Stork's group
图式23
Stork研究组的合成方法
图式23.
Synthesis by Stork's group
1.23 Magnus研究组的合成方法
2009年, Magnus课题小组[45]以苯酚烷基化反应为关键步骤完成了可待因的消旋全合成.该路线从芳基溴化合物217和硼酸化合物218出发经过一步Suzuki偶联反应得到化合物219, 化合物219转化为溴代乙缩醛化合物220.化合物220和CsF反应生成化合物221构筑了含芳环的手性季碳.化合物221经Henry反应脱水和随后的Michael加成反应一步构建了吗啡的ABC三环得到化合物222.化合物222经还原硝基烯得到化合物223.化合物223经LiAlH4还原成醇化合物224, 化合物224在酸性条件下还原胺化关上D环, 与此同时二氢呋喃环经酚羟基进攻烯丙基正离子关环得到, 之后脱水得到双键化合物225, 此时吗啡的五环体系已经全部建立.接下来氮上受酰基保护得化合物226到目标分子可待因的转化过程皆与Taber等[37]在2002年发表的吗啡全合成路线相同. Magnus课题小组的可待因消旋合成总收率为20.1%, 总共用13步(Scheme 24).
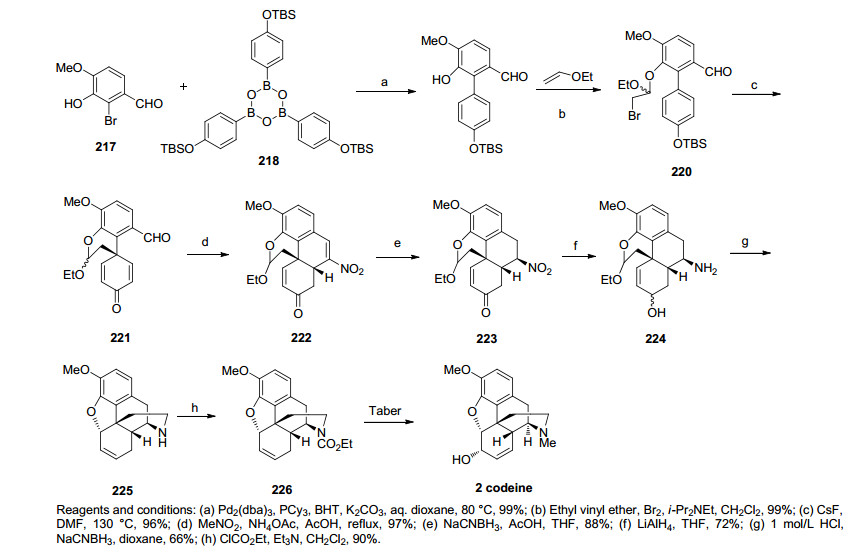 图式24
Magnus研究组的合成方法
图式24.
Synthesis by Magnus' s group
图式24
Magnus研究组的合成方法
图式24.
Synthesis by Magnus' s group
1.24 Metz研究组的合成方法
2011年, Metz课题组[46]利用1, 3-偶极环加成作为关键步骤完成了可待因的消旋全合成.该合成从商品化原料异香兰素出发, 通过酚羟基导向的苯环溴代, 甲基化作用和Wittig反应合成化合物228.化合物228经一步Suzuki偶联生成化合物230, 化合物230通过三氟醋酸碘苯氧化苯酚随后脱去缩醛保护得到化合物231, 化合物231和硝酮进行1, 3偶极环加成反应, 之后经L-selectride还原成羟基, 羟基由TBS保护得到化合物232, 化合物232经三氯化硼诱导的烯丙基重排, 随后溶剂二氯甲烷中的氯代反应和羟基取代反应得到化合物233, 化合物233通过Eschenmoser-Claisen重排反应构建了含芳环的手性季碳得到化合物234, 化合物234经还原成伯醇, 随后Ts保护成化合物235.化合物235在Raney镍的作用下自发地发生了分子内的烷基化反应得到桥环化合物236.化合物236在脱除TBS后, 三溴化硼的作用下醚键断裂, 新生的酚羟基进攻烯丙位构筑了二氢呋喃环, 随后再甲基化得到化合物237.化合物237最后经烯丙基迁移和DMP氧化得到可待因酮, 可待因酮通过硼氢化钠还原羰基得到目标分子可待因(Scheme 25).
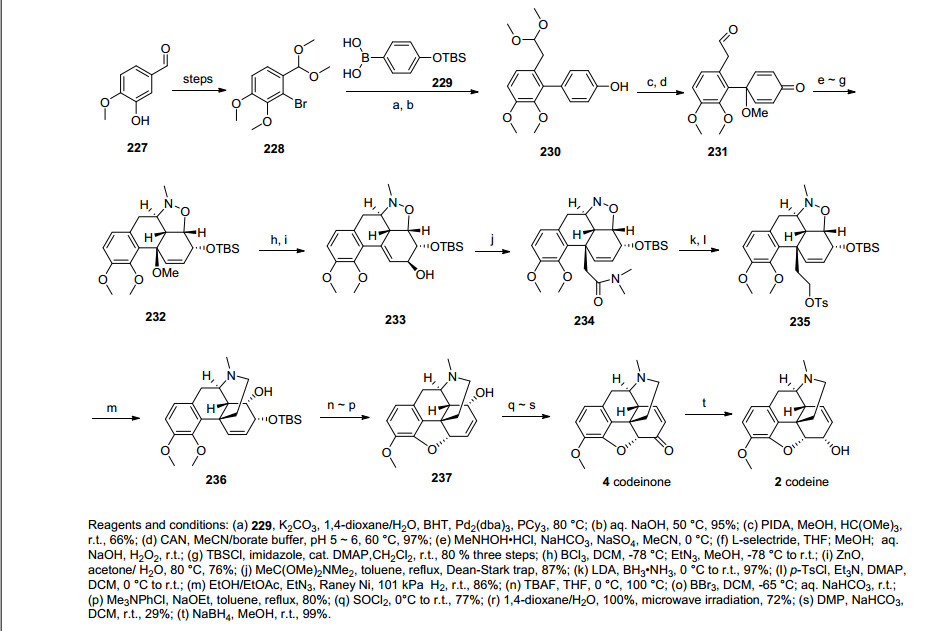 图式25
Metz研究组的合成方法
图式25.
Synthesis by Metz's group
图式25
Metz研究组的合成方法
图式25.
Synthesis by Metz's group
1.25 樊春安研究组的合成方法
2013年樊春安课题组[47]设计了串联醇解/氧杂Michael加成反应和SmI2促进的自由基还原偶联/去硫化反应为关键步骤的合成路线合成了Hudlicky路线的前体, 完成了吗啡的形式全合成.该路线合成化合物239后经醋酸碘苯的氧化偶联得到化合物240, 化合物240在氯铬酸吡啶(PCC)的氧化作用后得到内酯化合物241, 内酯化合物241经一步串联醇解/氧杂Michael加成反应成功构建了吗啡的三环骨架, 随后经一步非立体选择性的还原后得到化合物242, 化合物242经历苯硫酚的亲核取代反应后得到硫醚化合物243, 硫醚化合物243经酯基的还原氧化得到化合物244, 化合物244经还原胺化再上Boc保护基之后脱除TBS保护, DMP氧化成醛得到化合物245, 化合物245在经关键步骤SmI2促进的自由基还原偶联/去硫化反应后成功构建了吗啡的四环骨架246.化合物246在之后的官能团转化和修饰后得到Hudlicky路线的前体化合物, 完成了吗啡的形式全合成(Scheme 26).
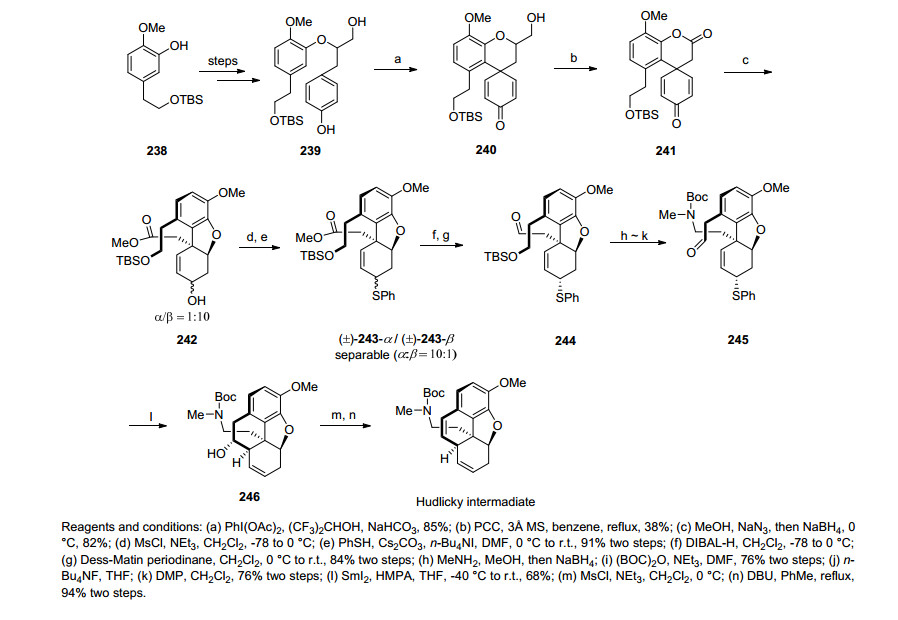 图式26
樊春安研究组的合成方法
图式26.
Synthesis by Fan's group
图式26
樊春安研究组的合成方法
图式26.
Synthesis by Fan's group
1.26 Fukuyama研究组的合成方法
2014年Fukuyama课题小组[48]以钯催化分子内溴苯的芳基偶联反应和分子内迈克尔加成反应作为关键步骤合成了一个半合成类鸦片镇痛剂(-)-Oxycodone.该合成路线利用化合物247和248在强碱的作用下制备化合物249.化合物249移去手性辅基后甲氧基化以高收率得到化合物250.化合物250经一步钯催化分子内溴苯的芳基偶联反应得到化合物251和少量二溴代的产物252.化合物251经还原、醋酸酯保护、脱出苄基得到化合物253.化合物253在醋酸碘苯氧化去芳化的作用下得到化合物254.化合物254经氢化反应和单酯单酰氯保护得到化合物256.化合物256在碳酸铯的作用下进行了分子内的迈克尔加成反应得到化合物257.化合物257脱酯和脱MOM保护基得到化合物258.化合物258在羰基α位溴代后在碱性条件下环合构筑吗啡的二氢呋喃环结构得到化合物259.化合物259经脱保护缩合过程得到酰胺化合物260.酰胺化合物260经水解过程自发生成内酰胺化合物261.内酰胺化合物261经还原氧化等过程最终获得半合成类鸦片镇痛剂(-)-Oxycodone (Scheme 27).
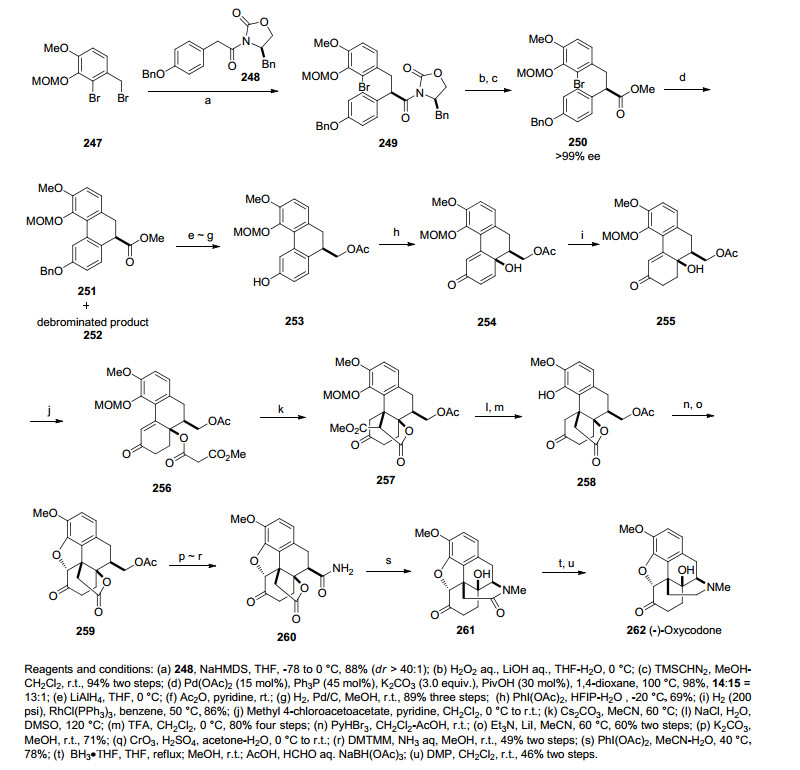 图式27
Fukuyama研究组的合成方法
图式27.
Synthesis by Fukuyama's group
图式27
Fukuyama研究组的合成方法
图式27.
Synthesis by Fukuyama's group
1.27 Gaunt研究组的合成方法
2014年, Gaunt研究小组[49]报道了以邻对位苯酚的氧化自由基偶联为策略立体专一性高效合成吗啡类生物碱衍生物的方法.该方法以化合物268作为进行邻对位苯酚的氧化自由基偶联反应的前体, 在醋酸碘苯的作用下将化合物268氧化偶联生成一个中间体, 这个中间体并未进行纯化, 在DBU的作用下, 实现了去对称化的区域选择性Michael加成反应, 以一锅法48%的收率得到化合物269.随后化合物269经Picnic氧化成酸, Curtius重排得到Boc-保护的胺化合物270.之后脱除硅保护基, DBU作用下甲磺酸酯脱双键得到烯醇醚化合物271.化合物271经Luche还原成醇, 在微波的酸性条件下烯醇醚键水解, 酚羟基进攻烯丙位的正离子构筑了二氢呋喃环, 与此同时Boc-脱除的胺与醛形成亚胺, 亚胺还原胺化再经保护得到吗啡类生物碱衍生物272.本路线立体选择性地以4.3%的收率历经18步得到吗啡类生物碱衍生物272完成了吗啡的形式全合成(Scheme 28).
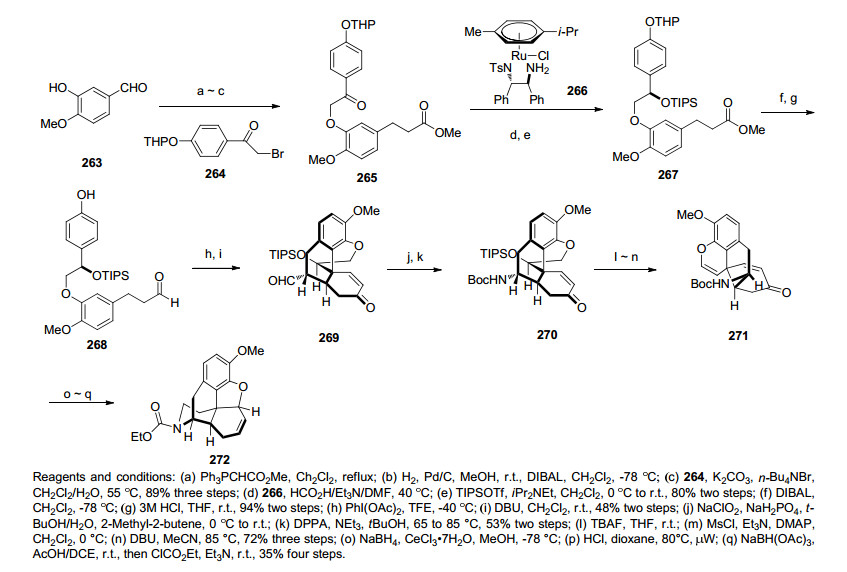 图式28
Gaunt研究组的合成方法
图式28.
Synthesis by Gaunt's group
图式28
Gaunt研究组的合成方法
图式28.
Synthesis by Gaunt's group
1.28 Hudlicky研究组的合成方法
2014年, Hudlicky课题小组[50]报道了以氧化去芳化和分子内DA反应为关键步骤合成对映二氢吗啡酮(ent-Hydromorphone)的简短合成路线.路线首先利用Mitsunobu反应将化合物273和化合物274偶联形成化合物275, 化合物275中的醛基进行一步Wittig反应转化为烯基官能团生成化合物276, 化合物276在温和的条件下移除MOM保护基得到化合物277, 化合物277通过关键步骤氧化去芳化和DA反应经去芳化的中间体278转化为化合物279, 这样就成功构建了吗啡骨架中的B环.化合物279在三氟乙酸的作用下再次芳构化得到中间体化合物280, 化合物280经Ts保护和TBS保护基脱出后得到胺化的前体化合物281, 化合物281经Birch还原条件下胺化构筑D环得到化合物282, 化合物282最后通过一步苯甲酮和叔丁醇钾的氧化得到对映二氢吗啡酮(ent-Hydromorphone).该合成路线用简短的12步就高效地合成了对映二氢吗啡酮(ent-Hydromor-phone) (Scheme 29).
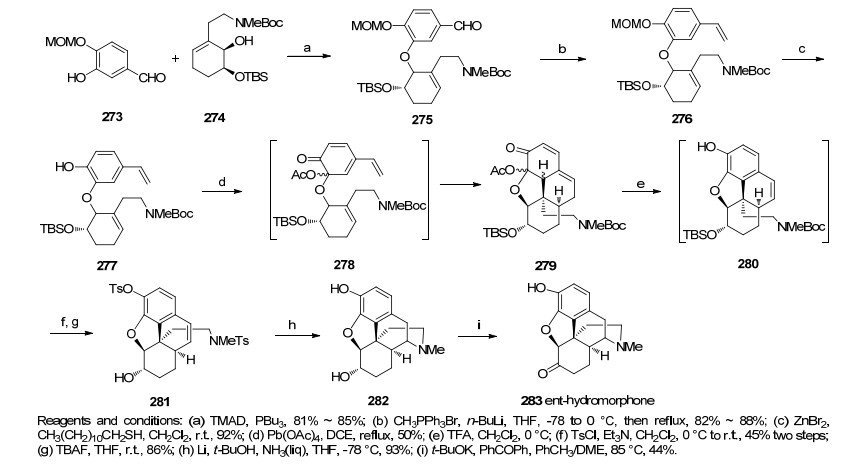 图式29
Hudlicky研究组的合成方法
图式29.
Synthesis by Hudlicky's group
图式29
Hudlicky研究组的合成方法
图式29.
Synthesis by Hudlicky's group
1.30 Opatz研究组的合成方法
2014年, Opatz课题组[51]是基于生源途径的启发来合成吗啡类生物碱的, 路线中利用Noyori不对称催化剂催化氢化反应实现了化合物286向化合物287的转化, 之后用Grewe环化反应作为合成的关键步骤从化合物290生成化合物291成功地构建了吗啡的ABCD四环骨架, 立体选择性地合成了二氢化可待因[(-)-dihy-drocodeine] (Scheme 30).
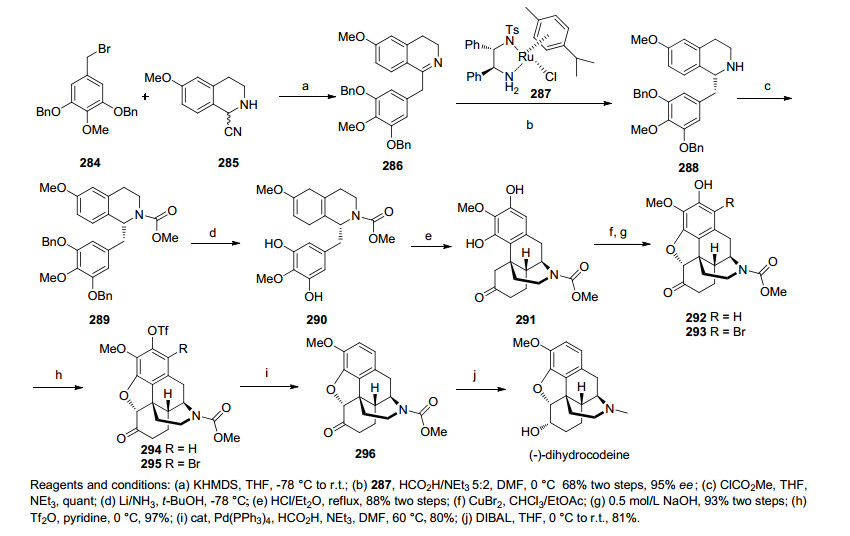 图式30
Opatz研究组的合成方法
图式30.
Synthesis by Opatz's group
图式30
Opatz研究组的合成方法
图式30.
Synthesis by Opatz's group
1.31 张洪彬研究组的合成方法
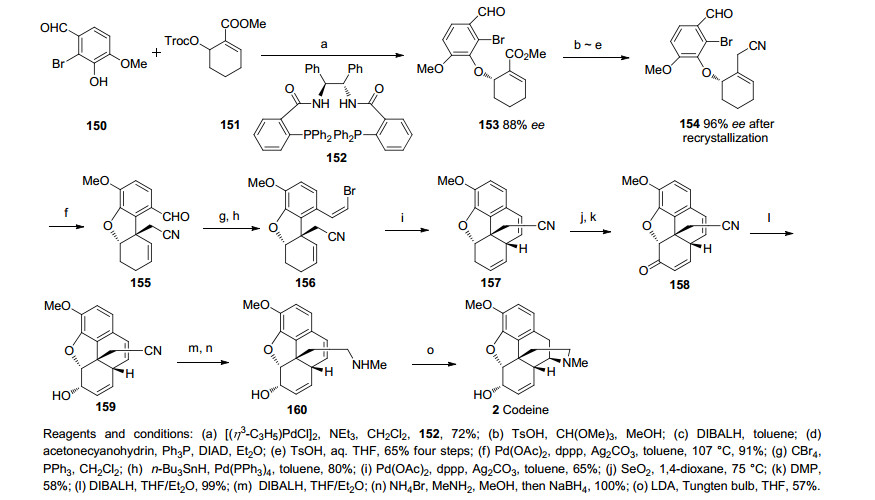
2015年, 本研究组[52]从商品化原料苯硼酸的衍生物出发, 首先一步Suzuki偶联反应合成化合物299, 经分子内烷基化构建含芳基季碳中心化合物302, 经选择性环氧化引入吗啡骨架C环官能团合成化合物305, 继而串联环合反应构筑苯并二氢呋喃环得化合物306, 利用Pd催化的分子内C-H烯基化反应构建吗啡B环, 确立了吗啡的基本骨架得化合物307, 之后经一系列官能团化合成311(Guillou中间体), 共14步完成了天然产物可待因(±)-Codeine的全合成(Scheme 31).
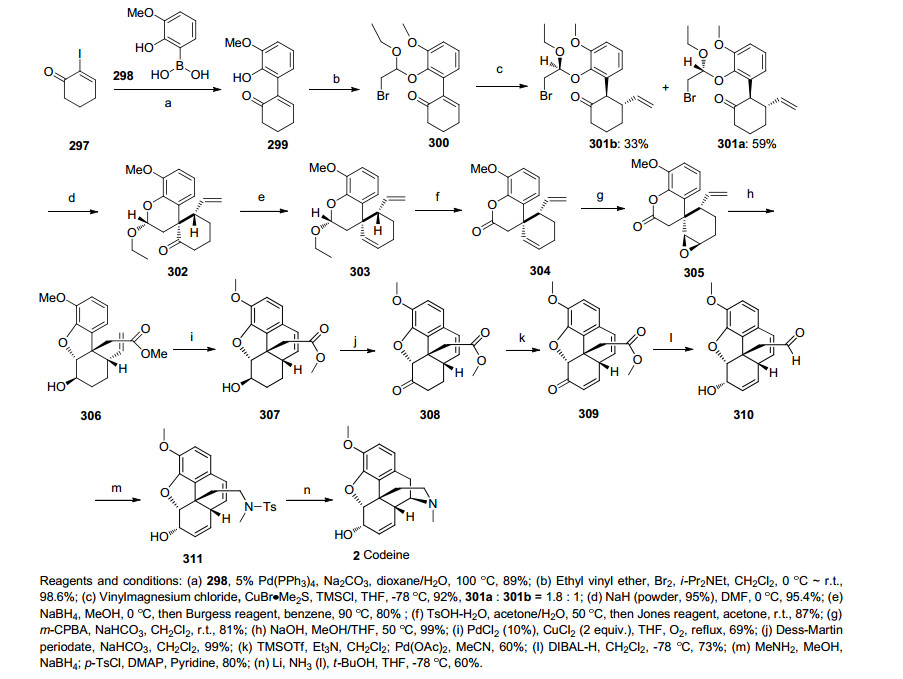 图式31
张洪彬研究组的合成方法
图式31.
Synthesis by Zhang's group
图式31
张洪彬研究组的合成方法
图式31.
Synthesis by Zhang's group
1.32 Smith研究组的合成方法
2016年, Smith研究组[53]报道了一条简短地、立体选择性地合成吗啡的路线.该路线从化合物312出发, 经苯酚烯基化和钯催化的sp3-sp2偶联反应得化合物316, 利用光环化反应立体选择性地构建了吗啡骨架中的苯并呋喃环化合物317, 继而通过一系列烯烃-炔烃-烯烃的环化反应作为关键步骤高效地合成吗啡的基本骨架的BC两环得到化合物320, 化合物320脱去Boc保护基经一步氮杂的1, 6加成反应构筑吗啡骨架D环, 合成天然产物可待因, 最后一步去甲基化完成吗啡的消旋合成, 总收率6.6% (Scheme 32).
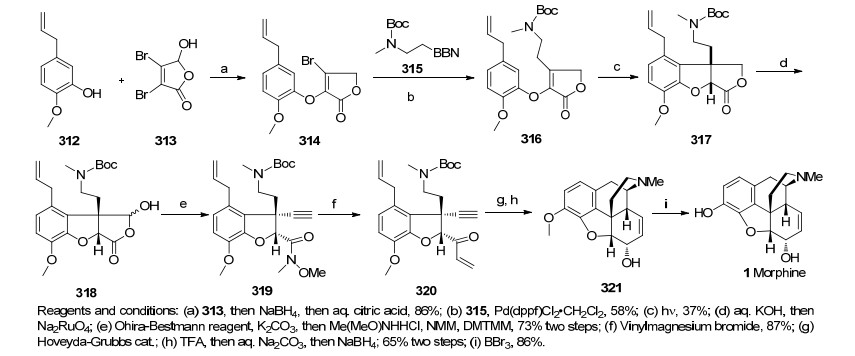 图式32
Smith研究组的合成方法
图式32.
Synthesis by Smith's group
图式32
Smith研究组的合成方法
图式32.
Synthesis by Smith's group
2 结语与展望
吗啡及其类生物碱的全合成, 长期以来受到了合成化学家们的关注和重视, 吗啡虽然不是一个庞大的目标分子, 但它拥有一个高度紧凑的五元环和五个连续的手性立体化学中心, 优秀的生物活性, 因而不论从合成的挑战性及药物化学的角度而言, 吗啡及其类似物都是非常好的合成目标.为了构建吗啡骨架, 不同的合成路线及在其合成中发展出的不同合成方法学被运用, 在此把至今合成吗啡类生物碱的有关信息列成表格(Table 1), 其中不乏优秀的代表之作:例如Fukuyama、Magnus、Hudlicky和Metz等的工作, 这些工作及其使用的合成方法和策略不管是对吗啡及其生物碱的全合成还是对整个有机合成化学的发展都起到了重要的意义.迄今为止, 吗啡的全合成路线还不具备实用意义和生产价值, 现今使用的吗啡仍需要从天然产物中获取, 因此, 现代有机合成家们还需要为寻找到一个更为简单, 新颖和快速合成吗啡的方法继续努力.
 表 1
吗啡类生物碱的合成总结
Table 1.
Summary of syntheses of morphine alkaloids
表 1
吗啡类生物碱的合成总结
Table 1.
Summary of syntheses of morphine alkaloids
Principle author Year Target Steps Overall yield/% Gates 1952 Morphine 31 0.06 Ginsberg 1954 rac-Dihydrothebainone 21 8.9 Grewe 1967 rac-Dihydrothebainone 9 0.81 Rice 1980 Dihydrocodeinone 14 29.7 Evans 1982 rac-O-Me-thebainone A 12 16.7 White 1983 Codeine 8 1.8 Rapoport 1983 rac-Codeine 26 1.2 Fuchs 1987 rac-Codeine 23 1.3 Tius 1992 rac-Thebainone-A 24 1.1 Parker 1992 rac-Dihydrocodeineone 11 11.1 Overman 1993 Dihydrocodeinone 14 1.9 Mulzer 1998 Dihydrocodeinone 15 9.1 White 1999 ent-Morphine 28 3.0 Cheng 2000 rac-Desoxycodeine-D 15 13.26 Ogasawara 2000 rac-3, 4-Dimethoxy-6-morphinanone 29 0.25 Ogasawara 2001 Dihydrocodeinone ethylene ketal 21 1.5 Taber 2002 Morphine 27 0.51 Trost 2002 Codeine 15 6.8 Fukuyama 2006 rac-Morphine 25 6.7 Hudlicky 2007 ent-Codeine 15 0.23 Iorga/Guillou 2008 rac-Codeine 17 0.64 Chida 2008 rac-Dihydroisocodeine 24 3.8 Hudlicky 2009 Codeine 18 0.19 Magnus 2009 rac-Codeine 13 20.1 Stork 2009 rac-Codeine 22 2.0 Fukuyama 2010 Morphine 18 4.8 Metz 2011 rac-Codeine 20 2.8 Fukuyama 2014 (-)-Oxycodone 21 2.4 Hudlicky 2014 ent-Hydromorphone 12 4.8 Opatz 2014 (-)-dihydrocodeine 10 35 Zhang 2015 rac-Codeine 14 3.6 Smith 2016 rac-Morphine 10 6.6 -
-
[1]
Herbert, R. B.; Venter, H.; Pos, S. Nat. Prod. Rep. 2000, 17, 317. doi: 10.1039/a809409h
-
[2]
Reed, J. W.; Hudlicky, T. Acc. Chem. Res. 2015, 48, 674. doi: 10.1021/ar500427k
-
[3]
Gum, A.; Stabile, M. In Studies in Natural Products Chemistry, Vol. 18, Elsevier, Amsterdam, 1996, pp. 43~154.
-
[4]
Novak, B. H.; Hudlicky, T.; Reed, J. W.; Mulzer, J.; Trauner, D. Curr. Org. Chem. 2000, 4, 343. doi: 10.2174/1385272003376292
-
[5]
Blakemore, P. R.; White, J. D. Chem. Commun. 2002, 1159. http://www.ncbi.nlm.nih.gov/pubmed/12109065
-
[6]
Zezula, J.; Hudlicky, T. Synlett 2005, 388. doi: 10.1002/chin.200524286/full
-
[7]
Taber, D. F.; Neubert, T. D.; Schlecht, M. F. In Strategies and Tactics in Organic Synthesis, Vol. 5, Ed.:Harmata, M., Elsevier, London, 2004, pp. 353~389.
-
[8]
Chida, N. Top. Curr. Chem. 2010, 299, 1. doi: 10.1007/978-3-642-18107-8
-
[9]
Rinner, U.; Hudlicky, T. Top. Curr. Chem. 2012, 309, 33. http://www.ncbi.nlm.nih.gov/pubmed/21547687
-
[10]
Gates, M.; Tschudi, G. J. Am. Chem. Soc. 1952, 74, 1109. http://www.researchgate.net/publication/231485484_THE_SYNTHESIS_OF_MORPHINE
-
[11]
Gates, M.; Tschudi, G. J. Am. Chem. Soc. 1956, 78, 1380. doi: 10.1021/ja01588a033
-
[12]
Gulland, J. M.; Robinson, R. Mem. Proc. Manchester Lit. Philos. Soc. 1925, 69, 79.
-
[13]
Elad, D.; Ginsburg, D. J. Chem. Soc. 1954, 3052.
-
[14]
Barton, D. H. R. Pure Appl. Chem. 1964, 9, 35. http://www.degruyter.com/view/j/pac.1964.9.issue-1/pac196409010035/pac196409010035.xml
-
[15]
Kutchan, T. M. Alkaloids Chem. Biol. 1998, 50, 257. doi: 10.1016/S1099-4831(08)60045-0
-
[16]
Szantay, C.; Barczai, B. M.; Pechy, P.; Blasko, G.; Dornyei, G. J. Org. Chem. 1982, 47, 594. doi: 10.1021/jo00342a051
-
[17]
White, J. D.; Caravatti, G.; Kline, T. B.; Edstrom, E.; Rice, K. C.; Brossi, A. Tetrahedron 1983, 39, 2393. doi: 10.1016/S0040-4020(01)91965-9
-
[18]
Schwartz, M. A. J. Org. Chem. 1988, 53, 2318. doi: 10.1021/jo00245a035
-
[19]
Grewe, R.; Friedriccsen, W. Chem. Ber. Recl. 1967, 100, 1550. doi: 10.1002/(ISSN)1099-0682
-
[20]
Grewe, R.; Fischer, H.; Friedric, W. Chem. Ber. Recl. 1967, 100, 1. doi: 10.1002/(ISSN)1099-0682
-
[21]
Rice, K. C. J. Org. Chem. 1980, 45, 3135. doi: 10.1021/jo01303a045
-
[22]
White, J. D.; Caravatti, G.; Kline, T. B.; Edstrom, E.; Rice, K. C.; Brossi, A. Tetrahedron 1983, 39, 2393. doi: 10.1016/S0040-4020(01)91965-9
-
[23]
Ludwig, W.; Schafer, H. J. Angew. Chem., Int. Ed. Engl. 1986, 25, 1025. doi: 10.1002/anie.198610251
-
[24]
Evans, D. A.; Mitch, C. H. Tetrahedron Lett. 1982, 23, 285. doi: 10.1016/S0040-4039(00)86810-0
-
[25]
Moos, W. H.; Gless, R. D.; Rapoport, H. J. Org. Chem. 1983, 48, 227. doi: 10.1021/jo00150a017
-
[26]
Schultz, A. D.; Lucci, R. D.; Napier, J. J.; Kinoshita, H.; Ravichan-dran, R.; Shannon, P.; Yee, Y. K. J. Org. Chem. 1985, 50, 217. doi: 10.1021/jo00202a014
-
[27]
Toth, J. E.; Fuchs, P. L. J. Org. Chem. 1987, 52, 473. doi: 10.1021/jo00379a038
-
[28]
Tius, M. A.; Kerr, M. A. J. Am. Chem. Soc. 1992, 114, 5959. doi: 10.1021/ja00041a008
-
[29]
Parker, K. A.; Fokas, D. J. Am. Chem. Soc. 1992, 114, 9688. doi: 10.1021/ja00050a075
-
[30]
Parker, K. A.; Fokas, D. J. Org. Chem. 2006, 71, 449. doi: 10.1021/jo0513008
-
[31]
Hong, C. Y.; Kado, N.; Overman, L. E. J. Am. Chem. Soc. 1993, 115, 11028. doi: 10.1021/ja00076a086
-
[32]
Trauner, D.; Bats, J. W.; Werner, A.; Mulzer, J. J. Org. Chem. 1998, 63, 5908. doi: 10.1021/jo9805394
-
[33]
White, J. D.; Hrnciar, P.; Stappenbeck, F. J. Org. Chem. 1999, 64, 7871. doi: 10.1021/jo990905z
-
[34]
Liou, J. P.; Cheng, C. Y. Tetrahedron Lett. 2000, 41, 915. doi: 10.1016/S0040-4039(99)02188-7
-
[35]
Yamada, O.; Ogasawara, K. Org. Lett. 2000, 2, 2785. doi: 10.1021/ol006172i
-
[36]
Nagata, H.; Miyazawa, N.; Ogasawara, K. Chem. Commun. 2001, 1094.
-
[37]
Taber, D. F.; Neubert, T. D.; Rheingold, A. L. J. Am. Chem. Soc. 2002, 124, 12416. doi: 10.1021/ja027882h
-
[38]
Trost, B. M.; Tang, W. P. J. Am. Chem. Soc. 2002, 124, 14542. doi: 10.1021/ja0283394
-
[39]
Trost, B. M.; Tang, W. P.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 14785. doi: 10.1021/ja054449+
-
[40]
Uchida, K.; Yokoshima, S.; Kan, T.; Fukuyama, T. Org. Lett. 2006, 8, 5311. doi: 10.1021/ol062112m
-
[41]
Omori, A. T.; Finn, K. J.; Leisch, H.; Carroll, R. J.; Hudlicky, T. Synlett 2007, 2859.
-
[42]
Tanimoto, H.; Saito, R.; Chida, N. Tetrahedron. Lett. 2008, 49, 358. doi: 10.1016/j.tetlet.2007.11.037
-
[43]
Varin, M.; Barre, E.; Iorga, B.; Guillou, C. Chem. Eur. J. 2008, 14, 6606. doi: 10.1002/chem.v14:22
-
[44]
Stork, G.; Yamashita, A.; Adams, J.; Schulte, G. R.; Chesworth, R.; Miyazaki, Y.; Farmer, J. J. J. Am. Chem. Soc. 2009, 131, 11402. doi: 10.1021/ja9038505
-
[45]
Magnus, P.; Sane, N.; Fauber, B. P.; Lynch, V. J. Am. Chem. Soc. 2009, 131, 16045. doi: 10.1021/ja9085534
-
[46]
Erhard, T.; Ehrlich, G.; Metz, P. Angew. Chem., Int. Ed. 2011, 50, 3892. doi: 10.1002/anie.201007448
-
[47]
Li, J.; Liu, G. L.; Zhao, X. H.; Du, J. Y.; Qu, H.; Chu, W. D.; Ding, M.; Jin, C. Y.; Wei, M. X.; Fan, C. A. Chem.-Asian J. 2013, 8, 1105. doi: 10.1002/asia.201300139
-
[48]
Kimishima, A.; Umihara, H.; Mizoguchi, A.; Yokoshima, S.; Fukuyama, T. Org. Lett. 2014, 16, 6244. doi: 10.1021/ol503175n
-
[49]
Tissot, M.; Phipps, R. J.; Lucas, C.; Leon, R. M.; Pace, R. D.; Ngouansavanh, T.; Gaunt, M. J. Angew. Chem., Int. Ed. 2014, 53, 13498. doi: 10.1002/anie.201408435
-
[50]
Arghese, V.; Hudlicky, T. Angew. Chem., Int. Ed. 2014, 53, 4355. doi: 10.1002/anie.201400286
-
[51]
Geffe, M.; Opatz, T. Org. Lett. 2014, 16, 5282. doi: 10.1021/ol5023849
-
[52]
Li, Q. L.; Zhang, H. B. Chem. Eur. J. 2015, 21, 16379. doi: 10.1002/chem.v21.46
-
[53]
Chu, S.; Mgnster, N.; Balan, T.; Smith, M. D. Angew. Chem., Int. Ed. 2016, 55, 14306. doi: 10.1002/anie.v55.46
-
[1]
-

图式3 Barton、Szantay和White研究组的合成方法
Scheme 3 Synthesis by Barton, Szantay and White's group
表 1 吗啡类生物碱的合成总结
Table 1. Summary of syntheses of morphine alkaloids
Principle author Year Target Steps Overall yield/% Gates 1952 Morphine 31 0.06 Ginsberg 1954 rac-Dihydrothebainone 21 8.9 Grewe 1967 rac-Dihydrothebainone 9 0.81 Rice 1980 Dihydrocodeinone 14 29.7 Evans 1982 rac-O-Me-thebainone A 12 16.7 White 1983 Codeine 8 1.8 Rapoport 1983 rac-Codeine 26 1.2 Fuchs 1987 rac-Codeine 23 1.3 Tius 1992 rac-Thebainone-A 24 1.1 Parker 1992 rac-Dihydrocodeineone 11 11.1 Overman 1993 Dihydrocodeinone 14 1.9 Mulzer 1998 Dihydrocodeinone 15 9.1 White 1999 ent-Morphine 28 3.0 Cheng 2000 rac-Desoxycodeine-D 15 13.26 Ogasawara 2000 rac-3, 4-Dimethoxy-6-morphinanone 29 0.25 Ogasawara 2001 Dihydrocodeinone ethylene ketal 21 1.5 Taber 2002 Morphine 27 0.51 Trost 2002 Codeine 15 6.8 Fukuyama 2006 rac-Morphine 25 6.7 Hudlicky 2007 ent-Codeine 15 0.23 Iorga/Guillou 2008 rac-Codeine 17 0.64 Chida 2008 rac-Dihydroisocodeine 24 3.8 Hudlicky 2009 Codeine 18 0.19 Magnus 2009 rac-Codeine 13 20.1 Stork 2009 rac-Codeine 22 2.0 Fukuyama 2010 Morphine 18 4.8 Metz 2011 rac-Codeine 20 2.8 Fukuyama 2014 (-)-Oxycodone 21 2.4 Hudlicky 2014 ent-Hydromorphone 12 4.8 Opatz 2014 (-)-dihydrocodeine 10 35 Zhang 2015 rac-Codeine 14 3.6 Smith 2016 rac-Morphine 10 6.6  下载: 导出CSV
下载: 导出CSV
-



































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 168
- 文章访问数: 10963
- HTML全文浏览量: 4890
 下载:
下载:
