 图式 1
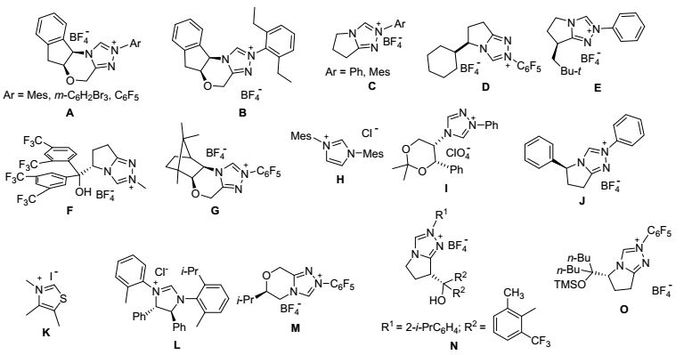
常见的NHC催化剂
Scheme1.
General N-hetercyclic carbenes
图式 1
常见的NHC催化剂
Scheme1.
General N-hetercyclic carbenes
引用本文:
王翱, 肖永龙, 周宇, 徐进宜, 柳红. 氮杂环卡宾催化有机反应的研究进展[J]. 有机化学,
2017, 37(10): 2590-2608.
doi:
10.6023/cjoc201702041

Citation: Wang Ao, Xiao Yonglong, Zhou Yu, Xu Jinyi, Liu Hong. Progress of Organic Reactions Catalyzed by N-Heterocyclic Carbenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2590-2608. doi: 10.6023/cjoc201702041
Citation: Wang Ao, Xiao Yonglong, Zhou Yu, Xu Jinyi, Liu Hong. Progress of Organic Reactions Catalyzed by N-Heterocyclic Carbenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2590-2608. doi: 10.6023/cjoc201702041
氮杂环卡宾催化有机反应的研究进展
English
Progress of Organic Reactions Catalyzed by N-Heterocyclic Carbenes
Abstract:
Since the first stable N-heterocyclic carbene was isolated by Arduengo in 1991, N-heterocyclic carbine has developed rapidly as a kind of efficient organic catalyst. It plays important role in building complex molecules in organic synthesis for the characteristics of umpolung. Some special Lewis bases and oxidants can induce carbine reaction with carbonyl to form Breslow intermediates, enol and homoenolate, which expand the reaction greatly. In this paper, the recent progresses in organic catalytic reactions including Stetter reaction, a3-d3 umpolung catalyzed by carbines are reviewed.
-
Key words:
- N-heterocyclic carbene
- / organic catalytic
- / umpolung
-
氮杂环卡宾(N-heterocyclic carbene, NHC)的研究历史最早要追溯到1832年, Wohler和Liebig[1]发现氰根负离子(CN-)可以催化苯甲醛的安息香缩合反应.随后Lapworth[2]对其机理进行研究后发现一分子的苯甲醛在氰根负离子的催化下, 羰基碳原子由传统的电正性转变为电负性(即醛基的反应极性发生了翻转), 进而对另一分子的苯甲醛进行亲核加成, 最终得到安息香产物.尽管如此, 直到20世纪后期, Bertrand和Arduengo等[3]报道了稳定的NHC之后, NHC才引起了众多化学家的兴趣, 如金属有机化学家们用NHC作为配体合成了多种金属催化剂, 催化了各式各样化学反应[4].近年来, 直接使用NHC作为催化剂也得到了很大的发展, NHC已经成为有机化学的研究热点之一, 尤其是其催化的反应往往具有不需要金属参与、价格相对低廉、反应体系环境友好等诸多特点, 在有机催化领域受到广泛的关注.根据催化底物类型的不同, 可将其氮杂卡宾催化的反应分为:羰基碳原子、羰基α碳原子、羰基β碳原子、羰基γ碳原子以及羰基δ碳原子活化参与的各种亲电和亲核反应.这些反应为有机合成提供了简单高效的C—C、C—N和C—O键的构建方法.因此, 在本文中我们基于NHC (Scheme 1)对活化羰基化合物碳原子位置的不同, 对近五年来NHC催化的有机化学反应进行简要综述.
图式 1
常见的NHC催化剂
Scheme1.
General N-hetercyclic carbenes
1 羰基不同位点与氮杂卡宾结合
1.1 羰基碳参与的亲核反应
1.1.1 羰基碳参与安息香反应
1832年, Wohler和Liebig[1]发现氰根负离子(CN-)可以催化苯甲醛的安息香缩合反应. 1943年, Ukai等[5]发现生物体内的维生素B1可以以辅酶形式催化多种重要生物化学反应, 其结构与氰根负离子相似, 也可以催化苯甲醛发生安息香缩合反应.因其操作简单污染小且对人体无毒害, 故成为了氰化物的最佳替代试剂.之后, Breslow等[6]对维生素B1催化的安息香缩合的机理做了详细的研究, 如Scheme 2所示.维生素B1 (2)中噻唑环1上碳原子的氢在相邻氮原子和硫原子的影响下, 具有一定的酸性; 在碱的作用下去质子化, 形成活化的氮杂卡宾Ⅰ, 其具有较强的亲核性, 进而对苯甲醛Ⅱ进行亲核加成, 而生成中间体Ⅲ; 后者经过质子迁移生成电荷更稳定的噻唑-烯胺中间体Ⅳ, 其也被称为Breslow中间体, 使得苯甲醛苄位的碳具有很强的亲核性, 可亲核进攻另外一分子的醛, 最终得到安息香产物Ⅵ.
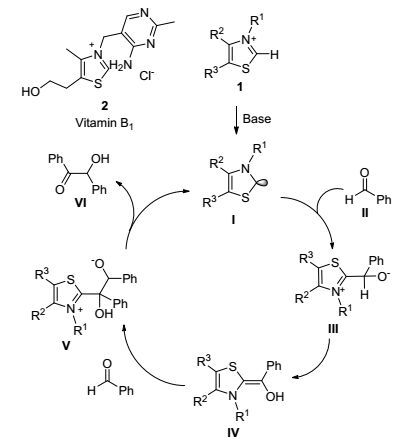 图式 2
维生素B1催化的安息香缩合反应机理
Scheme2.
Reaction mechanism of benzoin condensation catalyzed by vitamin B1
图式 2
维生素B1催化的安息香缩合反应机理
Scheme2.
Reaction mechanism of benzoin condensation catalyzed by vitamin B1
近年来, 利用交叉安息香反应构建α-羟基酮的方法也得到很大改进.早期该结构骨架的构建主要是通过对羰基的α位进行羟基化实现, 然而该方法产率低且手性选择性差.随后Enders等[7]利用卡宾与芳香醛形成的Breslow中间体与三氟甲基酮类衍生物结合来构建α-羟基酮, 成功解决了立体选择性问题. Gravel等在交叉安息香反应构建手型分子方面也有过杰出的工作.在2013年Gravel等[8]实现了脂肪醛3与二酮酯4之间的安息香反应并取得较好的产率及ee值(Eq. 1).相比于脂肪醛结构, 芳香环的结构活性明显降低, 这可能与芳香环形成Breslow中间体过程难于脂肪醛有关.而在2014年, Gravel等[9]成功地找到一类合适的稠环三氮唑卡宾催化剂, 其在脂肪醛5和芳香醛6之间具有很好的选择性(Eq. 2), 得到具有较好产率的α-羟基酮产物.脂肪醛之间的交叉安息香反应一直是该类偶联反应的难点, 尤其在底物选择性及手性控制方面难度较大.而在2016年Gravel等[10]通过在反应位点附近引入大体积吸电子基团很好地解决这类问题, N-Boc保护的α-氨基醛7由于具有较好的空间选择及电子配位能力成为了合适的底物(Eq. 3), 实现了脂肪醛之间的安息香反应并取得良好的产率及非对映选择性.
Murry等[11]报道了第一例卡宾催化羰基-亚胺的氮杂交叉安息香反应, 取得了较好的产率, 突破了传统羰基与羰基之间安息香反应. 2012年, Rovis等[12]在前人的研究基础上利用三氮唑催化剂成功实现了脂肪醛8与N-Boc亚胺9的偶联, 解决了氮杂交叉安息香反应中的立体选择性问题(Eq. 4).相比于饱和醛结构, α, β-不饱和醛10在卡宾催化中具有多个反应位点, 其中β位容易通过高烯醇中间体发生C(3) 加成, 而α位容易通过烯醇中间体发生C(2) 加成. Ye等[13]与Chi等[14]分别于2013年和2014年找到合适的手性卡宾催化体系、底物及反应条件, 使得反应优先进行C(1) 位的加成, 生成C—N键安息香产物, 如Eqs. 5, 6所示.
1.1.2 羰基碳参与stetter反应
20世纪70年代初, Stetter[15]报道了一例醛在噻唑盐的催化作用下对活化的α, β-不饱和化合物进行类Michael加成反应得到产物13的反应.极性翻转的醛作为亲核试剂参与了1, 4加成反应, 该反应随后也被命名为Stetter反应(Eq. 7).在Stetter反应被发现的20年后, Ciganek[16]于1995年报道了第一例分子内非手性的Stetter反应, 随后Enders等[17]完成第一例分子内具有顺反选择性的Stetter反应.在接下来一段时间, 分子内的Stetter反应得到迅速发展. 2008年, Rovis等[18]通过分子内的Stetter反应成功将杂原子引入苯并二氢吡喃酮15结构中(Eq. 8), 同时迈克尔受体种类也得到很大的丰富, 包括烯烃磷酸酯[19]受体(Eq. 9) 和炔基磷酸酯[20]受体等.且Law和McErlean等[21]研究发现可通过插烯Stetter反应将底物延展至1, 6受体. 2012年, Xu课题组[22]报道了一例在氮杂环卡宾催化下, 不饱和醛18的δ位碳原子参与的分子内Stetter环合反应制备环戊烯酮骨架19的反应, 获得较高的收率和较好的立体选择性.且当底物中的Ar基团为酯基20替代时, 反应会生成六元环结构的产物21 (Eqs. 10, 11).
分子间Stetter反应可用于合成多种分子的前体结构.从20世纪70年代起, Stetter着手研究手性催化剂对该反应的影响开始, 一系列新的偶联配体陆续被报道. 2009年, You课题组[23]发现醛类结构在卡宾催化剂作用下可以与芳基磺酰吲哚发生偶联.该反应的关键在于对甲苯磺酰基在碱的作用下容易离去, 产生相应的α, β-不饱和亚胺离子作为迈克尔受体参与Stetter反应, 这类型反应适用于大多数芳香醛, 而脂肪醛类的产率较低(Eq. 12).然而他们发现这些产率较低的脂肪醛的产物24, 都具有较好的立体选择性(90%~97% ee). Liu课题组[24]在2012年首次将Stetter反应运用在糖基化过程中(Eq. 13), 当卡宾与醛结合后可增强反应中心电性, 容易与硝基取代的糖结构发生迈克尔加成, 这为糖基化的发展提供了新思路.在不断探索中, 新的羰基供体也不断被发现. Massi等[25]发现二酮类结构可作为羰基供体与查尔酮发生Stetter反应. Chi等[26]发现碳水化合物28通过糖结构的羟醛缩合互变特点, 在三氮唑卡宾催化下可以与查尔酮29发生Stetter反应(Scheme 3), 他将传统意义上的羰基扩展到碳水化合物中的羟醛缩合, 丰富了卡宾的催化范围.
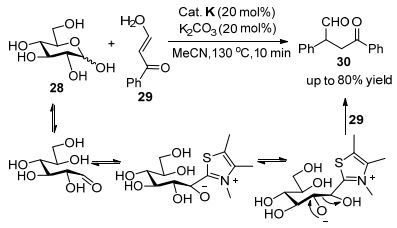 图式 3
卡宾催化下的分子间Stetter反应
Scheme3.
Intermolecular Stetter reaction catalyzed by N-hetercyclic carbenes
图式 3
卡宾催化下的分子间Stetter反应
Scheme3.
Intermolecular Stetter reaction catalyzed by N-hetercyclic carbenes
1.2 羰基化合物α碳参与的反应
羰基化合物α位碳原子是离羰基碳最近的碳原子, 较其它位置(β, γ, δ)而言, 该位置受羰基电子云影响最大, 形成烯醇化中间体的能力最强.因此, 通过控制外界条件来实现该位置的活化相对容易.早期关于卡宾催化羰基化合物反应的报道, 也主要集中于该位置碳原子的活化.实现α位碳原子活化的关键在于形成了酰基烯醇式中间体, 这也是区别其他反应位点活化的原因.
2011年, Chi课题组[27]报道了一种由α, β-不饱和醛18与α位具有吸电子基团取代的α, β不饱和酮32生成δ内酯33的方法.该方法不仅以较好的收率和对映选择性得到相应的内酯衍生物, 其最大的特点在于通过在α, β-不饱和酮的α引入合适的吸电子基团高效地避免了不饱和醛发生传统的高烯醇反应(β活化) (Eq. 14).取代咪唑类作为一种优势骨架经常出现于具有抗人类免疫缺陷病毒(HIV)、抗抑郁和抗病毒的生物活性分子结构中. 2013年, Scheidt课题组[28]通过在咪唑骨架34上引入适当的基团, 实现了与α, β-不饱和醛18中α-碳原子的偶联, 形成多元杂环产物35.该反应不仅产率高、对映选择性好, 且生成的目标产物可以通过开环反应得到具有生物活性的化合物结构(Eq. 15). 2015年, Chi课题组[29]首次报道了一例在氮杂环卡宾与布朗斯特酸共催化下, α, β-不饱和醛18的α-胺烷基化反应.该反应不仅具有较好的收率和对映选择性, 且反应生成的β-氨基酯37通过简单的转化可进一步生成具有重要功能的β-氨基酸(Eq. 16).
相比于不饱和醛, 饱和醛的α碳原子活化难度更大. 2012年, Rovis课题组[30]通过在反应体系中加入适当的氧化剂, 实现了在氮杂卡宾催化下, 饱和脂肪醛38与α, β-不饱和亚胺39的[4+2]环加成反应.该反应以高收率和高对映选择性得到了具有广泛生物学功能的δ-内酰胺骨架40 (Scheme 4).其机理大致为, 在氧化剂的作用下, 卡宾催化剂与饱和醛形成的中间体发生去质子化, 在碱的作用下形成关键烯醇中间体Ⅰ.基于类似的反应机理, Chi课题组[31]于2013年也报道了一例由饱和脂肪醛41与α, β-不饱和酮42合成δ内酯43的方法(Eq. 17).同样, 该反应也通过加入适当的氧化剂来生成反应必须的烯醇中间体.最近, NHC催化的氟化学也引起了化学家们的广泛兴趣.如Wang课题组[32]在2015年以N-氟代双苯磺酰胺(NFSI)为氟源, 成功实现了不饱和醛44的羰基α位碳原子的氟化(Eq. 18).在该反应中, NFSI不仅作为氟化试剂, 同时还作为氧化剂参与羰基α位碳原子的活化.从2012年Rovis第一次通过三氮唑实现饱和醛α碳的活化以来, 该类型的成环反应大多限定于[3+2]、[4+2]的反应形式, 分别用于构建五元环和六元环.通过[2+2]环加成构建四元环的难度要远大与构建六元环或五元环结构.首先, 相比于五/六元环, 四元环的表面张力较大, 稳定性较小; 其次, 在脂肪醛与α, β-不饱和羰基环化过程中, 四元环与六(五)元环的生成具有一定竞争性.而在2017年Xu等[33]成功实现了[2+2]构建四元环反应, 如Scheme 5所示.首先卡宾与脂肪醛在路易斯碱作用下形成Breslow中间体Ⅰ, 随后在氧化剂作用下去质子化形成中间体Ⅱ, 与酮亚胺结构发生曼尼希反应得到中间体Ⅲ, 最后经过分子内反应得到β-螺环吲哚产物. Ye等[34]在2014年也通过卡宾催化烯酮结构实现[2+2]环化反应.
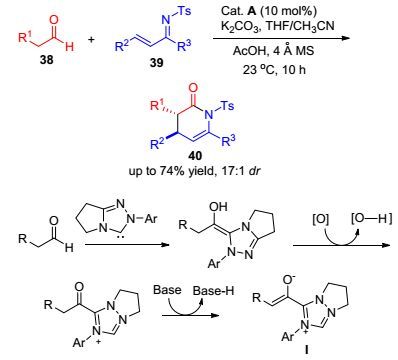 图式 4
饱和醛的α活化
Scheme4.
α-Activation of saturated aldehydes
图式 4
饱和醛的α活化
Scheme4.
α-Activation of saturated aldehydes
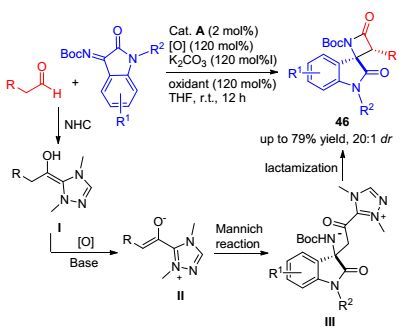 图式 5
NHC催化饱和醛的α活化
Scheme5.
α-Activation of saturated aldehydes catalyzed by NHC
图式 5
NHC催化饱和醛的α活化
Scheme5.
α-Activation of saturated aldehydes catalyzed by NHC
通过在分子中引入适当的离去基团, 在卡宾催化剂的作用下, 同样可以生成反应过程中所必需的烯醇中间体. 2012年, Sun课题组[35]通过在α, β-不饱和醛47的γ位引入适当的离去基团(MeO2CO), 成功实现了该底物α位的氟化.该反应的产率较好, 立体选择性较高.并且经过条件筛选, 以醋酸钠为碱, 三氯甲烷为溶剂, 成功阻断了二氟取代及无氟取代副产物的生成(Eq. 19). 2013年, Enders课题组[36]以α位引入氯原子的饱和醛49为底物, 实现了与吲哚取代的硝基烯50的反应(Eq. 20).基于相同的底物, Sun课题组[37]在随后又实现了α-氯代饱和醛52的α位氟取代反应(Eq. 21).
1.3 羰基化合物β-碳参与的反应
羰基化合物β位碳原子的活化主要以α, β-不饱和羰基化合物为底物.由于β位碳原子离羰基的距离较远, 受到羰基的影响相对较弱, 因此β位碳原子的活化较α位碳原子困难; 同时反应过程中还会受到来自α活化中间体的竞争.近年来, 有机化学家们通过选择适当的卡宾催化系统, 巧妙设计反应底物, 实现了羰基β位碳原子的活化.而α, β-不饱和酰基唑中间体的形成是实现β位碳原子活化的关键.
2011年, You课题组[38]首先报道了一例氮杂环卡宾催化α, β-不饱和醛53与1, 3-二羰基酮54之间的Michael加成反应.该反应条件温和, 以较高的收率和立体选择性得到1, 3-二氢吡喃酮类衍生物(Eq. 22).此后, Yu课题组[39]通过在α, β-不饱和醛的α位引入易离去的溴原子55, 也成功实现了与1, 3-二羰基酮之间的Michael加成反应, 制备了一系列1, 3-二氢吡喃酮衍生物56(Eq. 23).不同的是, Yu等采用了α位溴代的α, β不饱和醛为起始原料, 反应过程无需添加额外的氧化剂.
水相反应是绿色化学家追求的目标, 但是如何解决溶解性和反应性等问题是其最大的难点. 2013年, Chi课题组[40]首次实现了NHC在水中高效催化取代环戊烯59的合成.该反应不仅高效、绿色环保, 而且提高了卡宾催化剂的适用性(Scheme 6).随后他们通过使用氘代水为溶剂, 分析了各个反应中间体的氘代情况, 提出了如Scheme 6所示反应机理:首先, NHC与不饱和醛反应生成中间体Ⅰ, 该中间体与另一分子不饱和酮作用生成中间体Ⅱ, 中间体Ⅱ中与烯键相连的氢原子与氘代水分子中的氘原子互换形成中间体Ⅲ, 后者另一烯键上的氢原子再次与氘代水中的氘原子发生互换形成中间体Ⅳ, 该中间体随后发生分子内取代、脱CO2等反应生成终产物.在反应过程中, 双键的质子化是该反应的关键, 是推进后续反应的基础.
 图式 6
以水为溶剂的NHC催化不饱和醛与酮的反应
Scheme6.
Reaction of unsaturated aldehydes with ketones catalyzed by NHC in the solution of water
图式 6
以水为溶剂的NHC催化不饱和醛与酮的反应
Scheme6.
Reaction of unsaturated aldehydes with ketones catalyzed by NHC in the solution of water
通常情况下, 氮杂环卡宾作为一种路易斯碱, 与酸性试剂无法共同催化同一化学反应.但是, Scheidt课题组[41]首次实现了氮杂环卡宾与路易斯酸共同催化不饱和醛18与亚胺衍生物60之间的环加成反应.该反应不仅打破了传统意义上酸碱不共存的界限, 其最大意义在于他们提出了通过卡宾催化剂提升不饱和醛最高已占轨道(HOMO)的能级, 通过路易斯酸降低亚胺衍生物最低未占轨道(LUMO)能级提升反应动力的新反应理念(Eq. 24).随后, Rovis课题组[42]于2011年实现了氮杂环卡宾与布朗斯特酸共同催化α, β-不饱和醛62与不饱和亚胺63之间的环合反应, 得到γ-内酰胺骨架64.该反应获得了较好的产率和非对映选择性(Eq. 25).基于上述酸碱共催化反应机理, D'hooghe课题组[43]在NHC催化作用下, 以2-氯苯甲酸作为辅助催化剂, 高效地合成了一类γ-内酰胺骨架66 (Eq. 26).从反应热力学角度来看, 反式烯烃比顺式烯烃能量更低, 所得的产物相对更稳定, 因此该类反应更实用, 更易得到反式构型产物或者反式烯烃中间体.在顺反异构体中, 顺式结构势能高于反式, 使得其在底物的合成中具有一定难度; 此外顺式结构在反应过程中易转化为更稳定的反式构型, 因此合成具有手性值的顺式构型产物一直是有机合成构型的难点. 2013年, Chi课题组[44]通过合适的NHC催化剂体系, 实现了卡宾催化剂与顺式α, β不饱和醛67结合, 得到顺式高烯醇中间体, 克服了传统意义上不能实现的转化(Scheme 7).
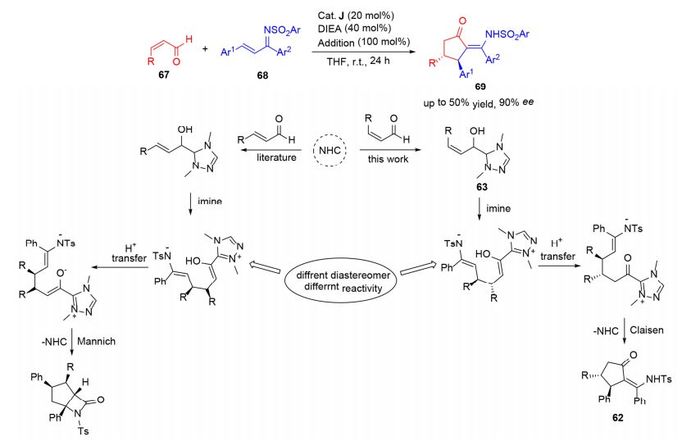 图7
NHC催化顺式构型的反应
Scheme7.
cis-Enals in N-heterocyclic carbene-catalyzed reactions
图7
NHC催化顺式构型的反应
Scheme7.
cis-Enals in N-heterocyclic carbene-catalyzed reactions
2009年, Nair等[45]首次报道了二烯醛与硝基烯在非手性咪唑鎓盐催化下通过高烯醇中间体生成目标产物, 并取得较好的产率.在此基础上, Liu等[46]首次提出利用手性三唑鎓盐催化二烯醛与硝基烯的不对称合成, 实现了芳香二烯醛及脂肪二烯醇69与芳香硝基烯70合成产物71 [Scheme 8(b)], 很好地解决了分子手性选择问题. 2013年Rovis课题组[47]报道了氮杂环卡宾催化α, β-不饱和醛72与硝基烯73的分子间加成反应, 得到高收率、高选择性的顺式产物.该方法的重要意义在于通过使用适当的催化剂, 可以选择性地使反应只沿着高烯醇途径发生, 避开了反应过程中更容易进行的Stetter反应途径[Scheme 8(a)].
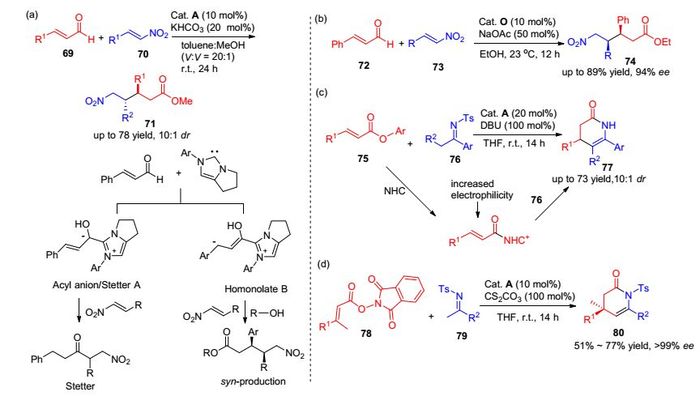 图式 8
NHC催化不饱和羰基化合物的极端反应
Scheme8.
Extreme reaction of unsaturated carbonyls catalyzed by NHC
图式 8
NHC催化不饱和羰基化合物的极端反应
Scheme8.
Extreme reaction of unsaturated carbonyls catalyzed by NHC
由于不饱和脂的羰基碳与氧原子发生共轭, 降低了其电正性, 因此相比于不饱和醛, 不饱和脂的活化难度更大. 2013年, Chi课题组[48]通过选择适当的手性氮杂环卡宾催化剂, 实现了α, β-不饱和酯75的活化.该方法最大的特点在于4-硝基苯酚作为离去基团, 具有较好的离去能力.另外, 通过加入卡宾催化剂可以增强不饱和酯β位碳原子的电正性, 活化后的中间体可以进一步与亚胺底物76发生环化反应, 高收率、高化学选择性地生成具有生物学功能的δ-内酰胺分子77 [Scheme 8(c)].随后在2015年, Zhong等[49]发现羟基邻苯二甲酰亚胺(NHPI) 78作为离去酯也具有较好的反应活性, 与N-Ts酮亚胺在三氮唑的催化下通过[3+3]环合生成具有较高手性及产率的环状内酯化合物80 [Scheme 8(d)].
2016年, 我们[50]采用1-羟基苯并三氮唑(HOBt)作为离去基团, 在氮杂卡宾的催化下, 成功实现了α, β-不饱和酯81 β碳的活化, 进一步通过与亚胺反应, 高效地制备了δ-内酰胺类化合物83 (Scheme 9). HOBt是常用的构建酰胺/酯的缩合剂, 具有易结合易离去特点. HOBt构建的酯与卡宾结合, 容易自身离去, 形成β位带有正电性电性酰基唑中间体, 后者易发生1, 2加成及1, 4加成, 这对构建环状酰胺起到关键作用.其机理推测如Scheme 9所示: NHC催化α, β-不饱和酯形成中间体Ⅱ, 经过1, 2加成和重排反应得到中间体Ⅲ, 再经过环内亲核加成离去NHC得到目标产物.
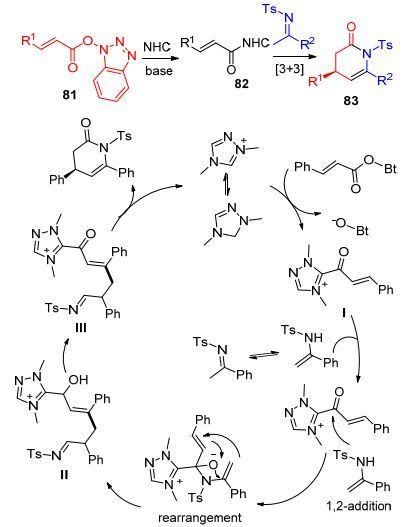 图式 9
NHC催化不饱和醛与亚胺的反应及机理
Scheme9.
Reaction and mechanism of unsaturated aldehydes with imine catalyzed by NHC
图式 9
NHC催化不饱和醛与亚胺的反应及机理
Scheme9.
Reaction and mechanism of unsaturated aldehydes with imine catalyzed by NHC
随着对卡宾催化剂催化反应特点不断地研究, 化学家发现氮杂环卡宾催化剂在构建复杂的并环、螺环骨架方面具有简单、高效及手性易控制等优势. 2012年, Chi课题组[51]采用NHC催化剂, 以靛红衍生物酮亚胺84和α, β-不饱和醛10为起始原料, 高效地实现了[3+2]环加成反应, 制备了一类含多氮的氧化吲哚螺环类化合物85 (Eq. 27).同年, 他们[52]又实现了通过α, β-不饱和醛10与甲基氧化吲哚衍生物86反应, 高效地合成吲哚螺环戊烯骨架87, 该骨架已经被证明具有重要生物学功能(Eq. 28). 2013年, Ye课题组[53]报道了一个氮杂环卡宾催化合成苯并环庚酯89的方法.该方法具有高产率和良好的立体选择性, 且生成的产物骨架广泛存在于活性天然产物分子结构中(Eq. 29).
通常情况下, 通过氮杂环卡宾催化所实现的羰基β位碳原子的活化仅适用于α, β-不饱和羰基化合物.但随着有机化学的不断发展, 科研人员对氮杂环卡宾的催化机理有了更深的认识. 2013年, Chi课题组[54]报道了氮杂环卡宾介导的饱和醛8与1, 3-二羰基化合物90的环合反应, 该反应通过引入氧化剂促进饱和醛8 β碳原子去质子化, 经[3+3]环合生成六元环, 这也是首次实现饱和醛β位碳原子的活化(Eq. 30).由于饱和脂的羰基碳受氧原子共轭的影响, 电性降低, 活化更困难.而在2013年, Chi课题组[55]首次报道了一种无需氧化剂参与, 实现饱和酯92 β位碳原子活化的方法.该方法不仅具有高收率、高立体选择性的特点, 而且底物的适用范围非常广泛.该反应成功的关键在于4-硝基苯酚羧酸酯为活化底物(Eq. 31), 其易离去特点省去了氧化剂的使用. 2015年, Yao课题组[56]首次实现了以饱和羧酸95为活化底物, 与靛红衍生物96发生[3+2]环化反应, 该反应突破了传统意义上的羰基作为底物的局限, 利用羧酸作为反应底物, 与加入的PCR试剂在反应过程中优先形成羧酸酯, 随后在卡宾催化作用下发生活化反应(Eq. 32).
1.4 羰基化合物γ-碳原子参与的反应
相对于α和β位碳原子的活化, 羰基γ位碳原子的活化无论是在活化程度还是手性控制上, 难度都远大于前者, 因此羰基γ位碳原子的活化是目前卡宾催化羰基化合物的反应中较难的一类反应.活化难度较大的原因一方面由于γ位碳原子离羰基较远, 受到羰基电子云的影响较低; 另一方面在于不对称合成过程中, γ位碳原子受到来自手性卡宾催化剂的手性信息较弱, 加大了手性合成的难度; 另外, 来自α和β位的竞争反应往往会阻断γ碳原子的活化.因此, γ位碳原子的活化相对于α和β位碳原子更具有的挑战性. γ位有烷基取代的α, β-不饱和酰基唑式中间体是活化该反应位点的关键.
2011年, Ye等[57]以α, β-不饱和酰氯98为起始原料, 在NHC催化剂的作用下, 将反应位点引入γ位, 与三氟苯乙酮99衍生物进行亲核加成, 以高收率和高立体选择性制备了取代δ代内酯100.该反应的意义在于他们克服了α和β位碳原子的竞争性反应, 首次实现了羰基化合物γ位碳原子的活化, 完成了NHC催化剂在反应位点上的突破(Eq. 33). 2015年, Yao等[58]在α, β-不饱和羧酸上引入了苯并三氮唑酯101, 以氧化吲哚102为Michael受体, 也实现了α, β-不饱和酯γ位碳原子的活化, 成功构建了具有广泛生物活性的氧化吲哚螺环类化合物103 (Eq. 34).
2016年, 我们课题组[59]通过以α, β-不饱和醛105为底物, 在NHC、碳酸铯和氧化剂催化体系中实现了γ位的sp3碳活化, 进一步与靛红104反应构建出一系列螺内酯类化合物106 (Scheme 10).与Ye等[57]利用酰氯实现γ碳活化策略不同, 我们采用卡宾和醌类氧化剂这一催化体系, 实现了α, β-不饱和醛的γ位sp3碳的活化, 丰富了底物的适应性.其机理如Scheme 10所示:卡宾与α, β-不饱和醛结合生成Breslow中间体Ⅰ, 在氧化剂的作用下形成中间体Ⅱ, 再在碳酸铯作用下离去γ位的质子形成中间体Ⅲ, 最后与靛红衍生物反应得到目标产物106.
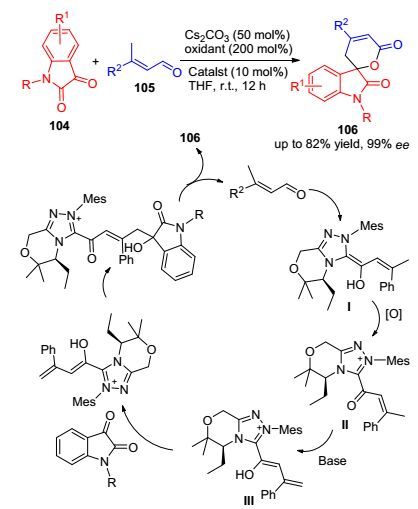 图式 10
NHC介导的α, β不饱和羰基化合物的γ位碳原子活化及机理
Scheme10.
Reaction and mechanism of NHC mediated γ-activation of α, β-unsaturated carbonyls
图式 10
NHC介导的α, β不饱和羰基化合物的γ位碳原子活化及机理
Scheme10.
Reaction and mechanism of NHC mediated γ-activation of α, β-unsaturated carbonyls
在之前工作的基础上, Chi课题组[60]于2012年实现了α, β-不饱和醛106末端γ位碳原子的活化.较以往工作不同的是, 该方法需要加入当量的醌类氧化剂和Sc类盐来促使γ位碳原子的去质子化.在该反应中, 他们发现采用Sc盐类路易斯酸可以提高反应的立体选择性(29%到94%).进一步的研究发现, Sc原子的空轨道与两个底物分子形成配位键, 拉近了两个反应分子的距离, 使得γ位碳原子得到更多卡宾催化剂的不对称信息, 提高了立体选择性(Eq. 35).基于类似机理, 他们[61]又于2014年报道了一个α, β-不饱和醛106与偶氮甲碱类亚胺108之间的[3+4]环合反应, 以高收率和高立体选择性构建了七元内酰胺骨架109, 该骨架可以通过简单的转化生成具有光学以及生物学活性的分子(Eq. 36).
此外, Ye等[53, 62]在α, β-不饱和醛的γ位碳原子引入适当的离去基团(OCO2Me)110, 通过该离去基团的离去而产生活化的烯醇中间体, 进而与另一分子偶氮底物111发生[4+2]环合反应, 该反应具有较高的收率和良好的立体选择性[Scheme 11, (a)].
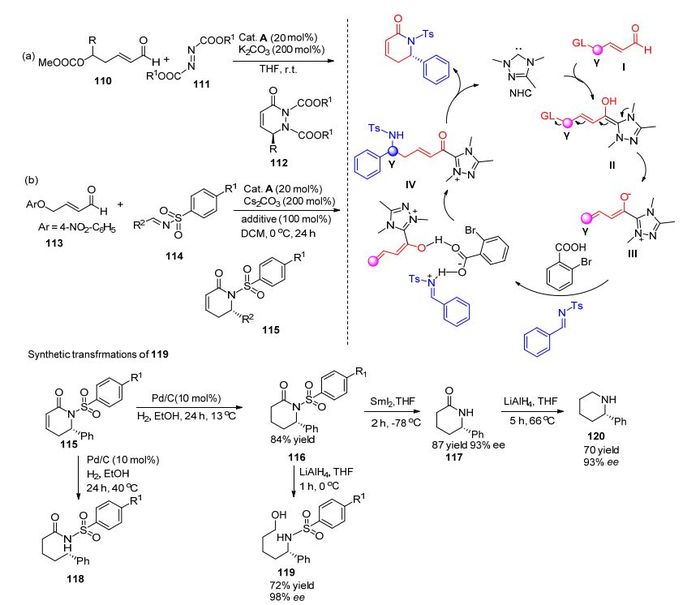 图式 11
离去基团介导的γ位碳原子的活化及机理
Scheme11.
Reaction and mechanism of leaving group mediated of γ-activation
图式 11
离去基团介导的γ位碳原子的活化及机理
Scheme11.
Reaction and mechanism of leaving group mediated of γ-activation
在此基础上, 我们课题组[63]在α, β-不饱和醛γ位碳原子引入新型的离去基团(4-NO2-C6H4) 113, 高效率、高选择性地实现了γ位碳原子的活化.与以前工作不同, 我们采用了离去能力较强的4-硝基苯酚作为离去基团; 另外, 在Ye等报道的反应中, γ位需要其他基团来稳定卡宾催化的活性中间体, 而我们实现了γ位无额外取代的不饱和醛的活化, 成功构建了一类内酰胺分子骨架115 [Scheme 11, (b)], 并可以通过一系列反应转化为多样化的药物合成中间体, 如吡啶120、氨基醇119等.该反应可能的机理如Scheme 11所示:首先, 一分子不饱和醛与一分子卡宾催化剂反应生成中间体Ⅱ, 该中间体通过离去基团的离去发生分子内消除反应生成中间体Ⅲ, 中间体Ⅲ在4-溴苯甲酸的作用下与另一底物亚胺作用生成中间体Ⅳ, 中间体Ⅳ发生分子内亲核取代反应, 释放产物分子以及卡宾催化剂参与下一个循环反应.
芳香并脂肪环的骨架广泛存在于生物、天然产物和聚合物材料中, 而活化芳基sp2碳转移到其支链的sp3碳变得尤为重要.该类结构的传统合成方式主要是通过过渡金属催化C—H活化实现的, 而Chi等[64]在2013年通过卡宾体系催化芳香醛实现了芳香支链sp3碳的活化, 其机理如Scheme 12所示.芳香醛121与卡宾形成中间体Ⅰ, 活化端基碳, 增强其酸性, 随后经过质子转移得到中间体Ⅱ, 后者与三氟甲基酮122发生1, 2加成取得了高产率和良好立体选择性的产物多元环分子123.随后在2015年, Xu等[62]将此反应拓展到将芳香酯与靛红酮亚胺衍生物结合, 构建复杂的多元多环结构化合物.
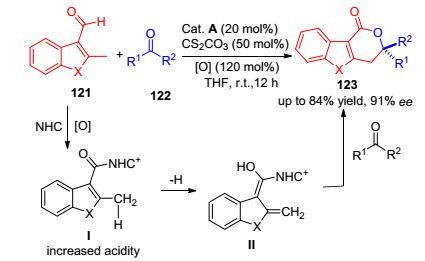 图式 12
芳基侧链sp3碳活化
Scheme12.
sp3-Carbon activation of aromatic chain
图式 12
芳基侧链sp3碳活化
Scheme12.
sp3-Carbon activation of aromatic chain
1.5 羰基化合物δ-碳原子参与的反应
羰基的δ位碳原子的活化是目前卡宾活化羰基化合物反应体系中最难的活化位点之一, 主要原因在于δ位碳原子与羰基碳原子的距离最远, 受到其电子云的影响最弱, 同时由于距离约束, 其手性也受到很大限制, 目前极少有文献报道.但是, 有机化学家通过巧妙地设计底物, 选择适当的反应条件, 也成功实现了部分羰基的δ位碳原子的活化.就仅有的实例来看, 形成α, β-γ, δ不饱和羰基唑中间体是该位点的反应关键.
2015年, Chi课题组[65]首次报道了通过氮杂环卡宾活化α, β-γ, δ双不饱和醛124的δ位碳原子, 与1, 3二羰基化合物125发生分子间的[4+2]环合反应.其推测机理为, 在氧化剂与三氮唑催化下, α, β-γ, δ不饱醛底物124生成128中间体, 后者与底物125经过1, 6加成形成双酮化合物129, 随后根据底物不同, 当取代基为非芳基酯基团时其经过羟醛反应和分子内β内酯引导生成分子131, 再经过去四元环化得到终产物126; 另一方面当取代基为芳基酯基团时, 经过分子内酯交换反应生成五元内酯134, 后者经过羟醛缩合反应得到目标产物127.该方法不仅高效地实现了不饱和醛δ位碳原子, 且他们通过对底物进行适当的改变, 可以使反应的路线发生改变(Scheme 13).
 图式 13
氮杂环卡宾介导的α, β, γ, δ-双不饱和醛的δ活化
Scheme13.
δ-Activation of α, β, γ, δ-unsaturated aldehydes catalyzed by NHC
图式 13
氮杂环卡宾介导的α, β, γ, δ-双不饱和醛的δ活化
Scheme13.
δ-Activation of α, β, γ, δ-unsaturated aldehydes catalyzed by NHC
1.6 NHC参与的动力学拆分及动态动力学拆分
随着化学家和药物学家对手性药物的逐渐重视, 拆分手段也得到广泛的探索. NHC参与的动力学拆分和动态动力学拆分[66]在近些年得到很大突破.
为实现手性3-羟基-3取代氧化吲哚衍生物的合成, Zhao等[67]利用手性卡宾催化α, β-不饱和醛138, 与羟基化合物137形成不对称酯139, 分离出另一种手性羟基化合物140.该反应的意义在于借用卡宾的手性及易于形成手性化合物的特点, 实现了三级醇的动力学拆分, 且拆分效率很高(ee值达到90%), 为动力学拆分提供了一种简单而高效的方法(Eq. 37).联苯在天然产物、药物以及材料应用方面起到了重要的作用, 且被广泛用于手性化合物催化合成[68], 而2014年Zhao课题组[69]再次利用NHC实现了联苯化合物141的拆分(Eq. 38).
动力学拆分自身存在不可跨越的缺陷, 即拆分光学对映体的理论产率只有50%.近几年来发展起来的动态动力学拆分(DKR)可以克服以上缺点, 使底物全部转化为单一光学对映体, 理论产率为100%.动力学与动态动力学的差别在于是否发生了外消旋化过程.动态动力学可通过外消旋化过程, 使两个对映体之间可以相互转化.
常见的动态动力学拆分方法包括酶催化、手性金属催化及过渡金属与酶配合催化等, 但这些方法价格相对昂贵[70].近些年发现的利用NHC实现动态动力学拆分很好地解决了这类问题. 2012年, Scheidt课题组[71]首先实现利用NHC完成动态动力学拆分, 构建出手性β内酯衍生物(Scheme 14).化合物144在卡宾诱导作用下, 形成Breslow中间体Ⅰ和Ⅱ, 通过β质子化发生外消旋化作用, 在温和条件下, 这二者可快速相互转化, NHC-烯醇结构与羰基结构易形成分子内氢键, 构建成六元环, 氢键构建成的六元环可强迫分子发生构型转变, 通过分子内相互作用形成化合物146~149, 由于形成146的速率k1远大于k2~k4, 所以其为优势产物.
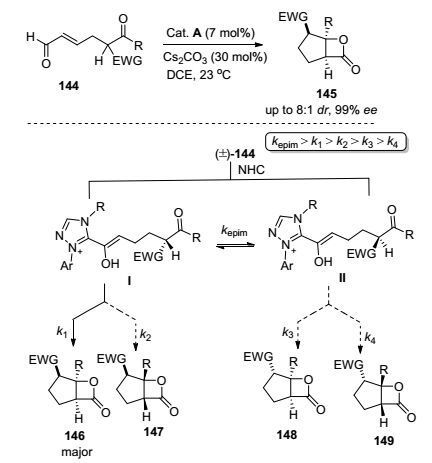 图式 14
氮杂环卡宾介导的β-内酯衍生物拆分
Scheme14.
Resolution of β-lactone derivative
图式 14
氮杂环卡宾介导的β-内酯衍生物拆分
Scheme14.
Resolution of β-lactone derivative
交叉偶联安息香反应产生的α-羟基酮类结构可被广泛地运用于生物、医药等领域, 同时该类反应的原子效率可达到100%.但最大缺陷在于在手性催化剂下只能产生一个手性中心, 多手性中心的构建是该类反应的难点. 2014年Johnson等[72]成功的实现了交叉安息香反应与动态动力学的结合, 通过三氮唑催化剂实现了羰基α位原子的动力学拆分, 实现了具有较好产率和对映选择性的双手性分子151构建(Eq. 39).在此工作基础上, Johnson等[73]发现用肉桂醛及衍生物与α-取代酮酯实现了三手性中心结构的构建, 取得良好的立体选择性(Eq. 40).
在2015年, Wang课题组[74]实现了双分子间的动态动力学拆分, 并推测反应机理如Scheme 15.其机理如图所示, 卡宾与羰基反应生成Breslow中间体Ⅰ在氧化剂及碱的作用下得到高烯醇化合物Ⅱ, 而路易斯酸可以调节高烯醇与羰基酮的协同作用, 如Ⅲ所示, 这种协同作用可能降低LUMO轨道能, 容易发生外消旋化, 利用卡宾空间位阻形成优势产物156.这是卡宾首次完成分子间的动态动力学拆分, 合成出δ-内酯衍生物.在此基础上, Wang课题组[75]于2016年又完成了对吡喃酮157动态动力学拆分(Scheme 16). 6-羟基-3吡喃酮作为碳水化合物合成的原料在药物化学中起到关键作用, 如O’Doherty课题组通过钯催化合成具有立体构型的糖苷化结构, 以及Sugawara课题组通过脂肪酶催化合成(R)-3-TBS, 但是大多数该类催化反应的反应步骤多(4 step), 且产率低(小于19%). Wang课题组通过卡宾和吡喃酮自身的烯醇互变特性, 克服了产率及效率的弊端, 获得了较好的产率和对应异构体.
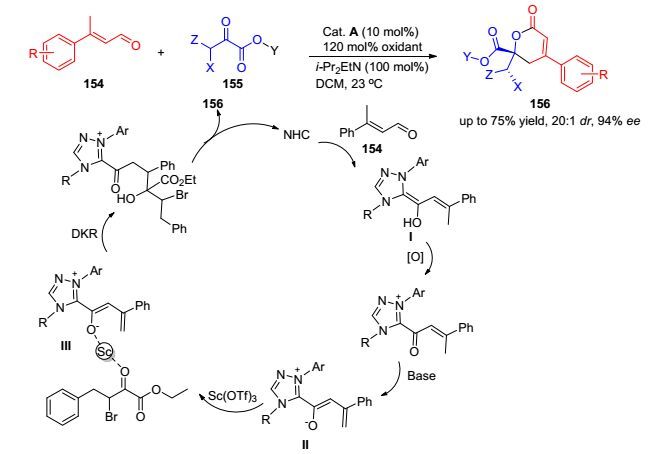 图式 15
氮杂环卡宾介导的分子间动态动力学拆分及机理
Scheme15.
Dynamic kinetic resolution and mechanism of intermolecular catalyzed by NHC
图式 15
氮杂环卡宾介导的分子间动态动力学拆分及机理
Scheme15.
Dynamic kinetic resolution and mechanism of intermolecular catalyzed by NHC
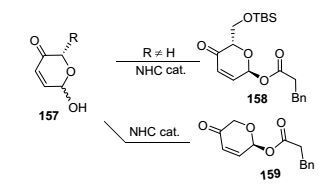 图式 16
氮杂环卡宾介导的吡喃酮动态动力学拆分
Scheme16.
Dynamic kinetic resolution of pyrone
图式 16
氮杂环卡宾介导的吡喃酮动态动力学拆分
Scheme16.
Dynamic kinetic resolution of pyrone
当醛或二烯醇的α位被烷基或者烷基取代物占据, 该结构的活性就会显著降低, 另外相对于醛来说, 酯的活性明显低于前者.这两点限制了NHC催化该类结构的反应, 而Chi课题组[78]在2016年通过筛选得到合适的底物, 成功解决这类问题(Eq. 41).底物160的酯基部分为4-硝基苯酚, 为活性较强的离去基团, 而亲核试剂为二苯基甲醇161, 是一种亲核性较强的试剂, 二者反应活性可调节到最优.首先卡宾与161底物反应, 生成Breslow中间体, 在碱的作用下, 该结构可发生外消旋化, 得到动力学产物162.
1.7 NHC参与的单电子转移途径
传统的NHC催化途径包括不对称stetter反应、不对称安息香反应、迈克尔加成反应等都是卡宾的孤对电子在反应中间体之间转移.而在2014年, Rovis等[79]发现二烯醇类结构通过电子富集的硝基化合物发生氧原子转移, 生成β-羟基衍生物的过程是经过单电子转移途径实现的[Scheme 17, (a)].这一方面拓展了NHC卡宾催化有机反应的范围, 同时也为卡宾催化的机理研究提供了新的见解.其机理如Scheme 17所示, α, β-不饱和醛结构与卡宾生成Breslow中间体Ⅱ, 随后该中间体通过硝基苯的单电子氧化形成Breslow自由基中间体Ⅲa和硝基苯自由基Ⅲb, 这些自由基相互结合形成中间体Ⅳ, 该中间体可以通过自身电子转移离去亚硝基苯Ⅴ, 随后甲醇通过亲核作用置换出卡宾催化剂生成目标产物Ⅵ.中间体Ⅲ的去质子化及单电子的离去形成的中间体Ⅶ, 在甲醇的作用下形成α, β-不饱和甲脂Ⅷ副产物.几乎同时Chi课题组[80]也发现了类似的氧化剂可引发单电子途径[Scheme 17, (b)].
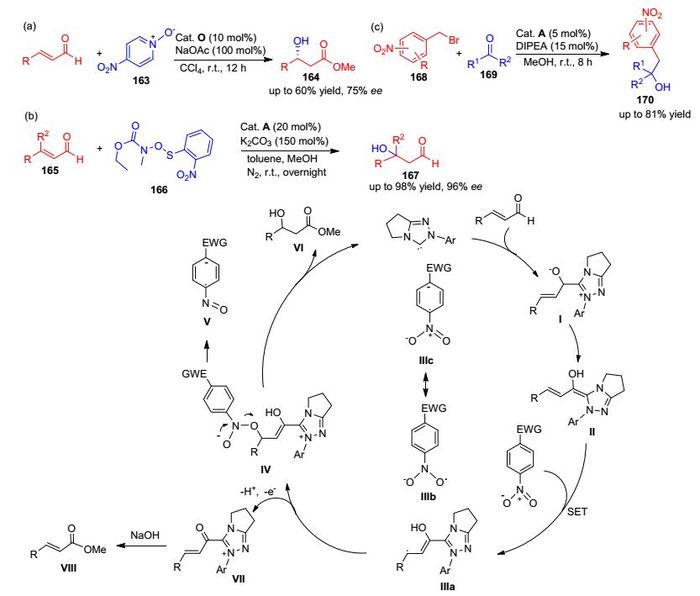 图式 17
NHC参与的单电子自由基反应
Scheme17.
Single-electron-transfer (SET) process for radical reactions mediated by NHC
图式 17
NHC参与的单电子自由基反应
Scheme17.
Single-electron-transfer (SET) process for radical reactions mediated by NHC
由于卤素的电负性, 溴苄等卤化物通常作为亲电试剂参与亲核取代反应.这类物质在光照条件下可通过自由基途径参与反应, 而在2016年, Chi等[81]通过NHC同样实现了溴苄自由基中间体.如Scheme 17 (c)所示, 苯环上硝基取代的溴苄168, 在醛作为还原剂条件下, 可以产生溴苄自由基中间体, 随后与酮类衍生物进行1, 2加成, 可得到三级醇产物170.
1.8 NHC参与的其他反应
随着研究的深入, 一些新的氮杂卡宾结合位点也逐渐被人们发现.例如2015年Sudalai课题组[82]报道了NHC偶联芳香烯172与醛类171化合物, 构建α, β-环氧酮173的方法(Eq. 42), 这是首次NHC催化α-溴酮与醛的达森反应.其巧妙地运用了N-溴代琥珀酰亚胺(NBS)/1, 8-二氮杂二环十一碳-7-烯(DBU)/二甲亚砜(DMSO)作为氧化系统, 与芳香烯构建溴鎓离子.更有意思的是, 在这里NHC优先与烯烃/溴酮反应, 而不是醛, 这为卡宾的应用拓展了方向.
2016年, Lupton等[83]发现α, β-不饱和酮175的端基烯键可以和氮杂卡宾结合(Eq. 43).它突破传统意义上氮杂卡宾与羰基结合方式.主要是卡宾与端基烯结合, 通过电子转移, 构建出β-阴离子结构, 发生1, 4加成成环.
除了端基烯结构外, Biju等[84]发现卡宾催化剂也可以作用于亚胺结构, 如Eq. 44所示.游离的卡宾亲核进攻分子176的亚胺部分, 产生的四面体中间体后经质子转移形成Breslow中间体, 随后进行1, 4加成和质子迁移过程得到目标化合物177.
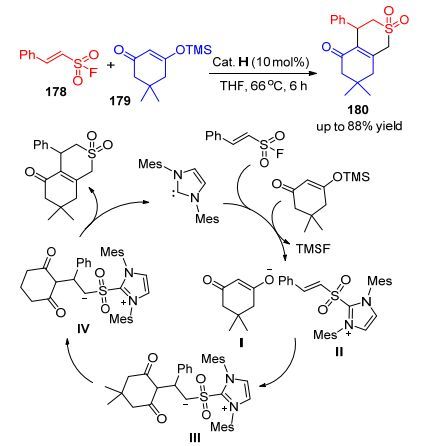
此外, Lupton等[85]也实现了以α, β-不饱和磺酰基178作为双电子亲体, 构建出一系列不饱和δ-磺酸内酯结构180 (Scheme 18).这也提示了α, β-不饱和“非碳”类似物即α, β-不饱和磺酰基也可逐渐被开发研究.最早研究磺酰基氟要追述到1954年, Truce和Hoerger报道的利用磺酰基氟实现D-A反应.其机理如Scheme 18所示:卡宾与不饱和磺酸基氟化物178结合, 后者脱去一个氟, 再与脱去TMS的TMS烯醇醚结合生成化合物Ⅰ和磺酰基唑Ⅱ.中间体Ⅲ与双甲酮阴离子结合得到磺酰基唑烯醇化物Ⅳ, 最后经过质子化重拍和移去卡宾可得到δ-磺内酯类化合物179.
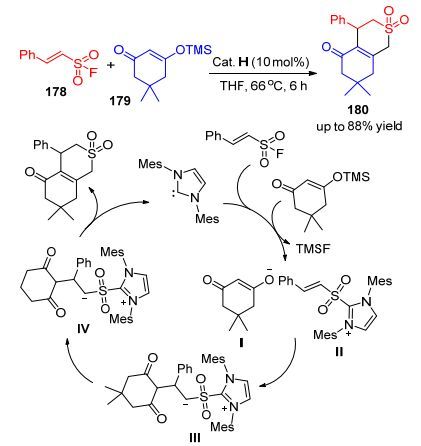 图式 18
NHC介导的δ-磺内酯衍生物活化及机理
Scheme18.
Reaction and mechanism of NHC mediated δ-sulfonated lactones
图式 18
NHC介导的δ-磺内酯衍生物活化及机理
Scheme18.
Reaction and mechanism of NHC mediated δ-sulfonated lactones
2 总结与讨论
从不同的反应位点总结了近年来氮杂环卡宾催化的研究进展(Scheme 19).羰基碳通过与氮杂环卡宾结合, 形成极性反转的碳原子, 其电子云密度对与之邻近的碳原子有着不同的影响, 通过适当的调节, 可以控制不同位点碳原子的活化能力.通常活化能力为α位碳原子>β位碳原子>γ位碳原子>δ位碳原子. α位碳原子由于与羰基碳距离最近, 受电子云影响最大, 反应活性最优, 成为了科学家们研究最多的位点.但是随着研究的深入, 羰基碳的β位和γ位碳原子的活化也逐渐被报道.由于这二者距离羰基碳中心有一定距离, 其活性有所下降, 同时来自α碳活性的竞争, 使得反应性也受到了较大的限制, 尤其是γ位碳活性更弱, 但是科学家们通过构建具有适当离去能力基团的底物或者运用合适的催化体系, 成功将反应位点引入到β/γ位, 实现了该类反应位点的活化.羰基碳δ位碳由于距离羰基最远, 且受空间位阻影响最大, 反应活性最弱, 但是近年来通过构建特殊的底物, 也成功地实现了该位点的活化.随着化学家们对氮杂环卡宾催化机理认识的不断加深, 氮杂环卡宾催化剂的适用范围也不断扩大, 反应类型不再局限于早期的安息香缩合和Stetter反应, 科学家们可以通过卡宾构建螺环、多环等复杂结构和实现(动态)动力学拆分; 除了催化活性较高的醛, 部分活性较弱的酯类、羧酸或亚胺结构已经逐渐成为卡宾催化的底物.随着对氮杂环卡宾催化有机反应机理的逐步阐明, 我们相信氮杂环卡宾作为优秀的催化剂必将发挥更大的作用, 可以被应用到更多的有机反应中.
 图图 9
羰基不同位点反应机理总览
Scheme19.
Overview of carbonyls activation
图图 9
羰基不同位点反应机理总览
Scheme19.
Overview of carbonyls activation
-
-
[1]
Nef, J. U. Justus Liebigs Ann. Chem. 1895, 287, 265. doi: 10.1002/(ISSN)1099-0690
-
[2]
Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B. Chem. Rev. 2009, 109, 3561. doi: 10.1021/cr8005153
-
[3]
Garrison, J. C.; Youngs, W. J. Chem. Rev. 2005, 105, 3978. doi: 10.1021/cr050004s
-
[4]
Peris, E.; Crabtree, R. H. Coord. Chem. Rev. 2004, 248, 2239. doi: 10.1016/j.ccr.2004.04.014
-
[5]
Ukai, T.; Tanaka, R.; Dokawa, T. J. Pharm. Soc. Jpn. 1943, 63, 296. doi: 10.1248/yakushi1881.63.6_296
-
[6]
Shapiro, L. R.; Samuels, S.; Breslow, L.; Camacho, T. Am. J. Public Health 1983, 73, 773. doi: 10.2105/AJPH.73.7.773
-
[7]
Enders, D.; Grossmann, A.; Fronert, J.; Raabe, G. Chem. Commun. 2010, 46, 6282. doi: 10.1039/c0cc02013c
-
[8]
Thai, K.; Langdon, S. M.; Bilodeau, F.; Gravel, M. Org. Lett. 2013, 15, 2214. doi: 10.1021/ol400769t
-
[9]
Langdon, S. M.; Wilde, M. M.; Thai, K.; Gravel, M. J. Am. Chem. Soc. 2014, 136, 7539. doi: 10.1021/ja501772m
-
[10]
Haghshenas, P.; Gravel, M. Org. Lett. 2016, 18, 4518. doi: 10.1021/acs.orglett.6b02123
-
[11]
Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowski, E. J. J.; Reider, P. J. J. Am. Chem. Soc. 2001, 123, 9696. doi: 10.1021/ja0165943
-
[12]
DiRocco, D. A.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 5904. doi: 10.1002/anie.201202442
-
[13]
Sun, L. H.; Liang, Z. Q.; Jia, W. Q.; Ye, S. Angew. Chem., Int. Ed. 2013, 52, 5803. doi: 10.1002/anie.201301304
-
[14]
Xu, J.; Mou, C.; Zhu, T.; Song, B. A.; Chi, Y. R. Org. Lett. 2014, 16, 3272. doi: 10.1021/ol501286e
-
[15]
Stetter, H. Angew. Chem., Int. Ed. Engl. 1976, 15.
-
[16]
Ciganek, E. Synthesis-Stuttgart 1995, 1311.
-
[17]
Enders, D.; Breuer, K.; Raabe, G.; Runsink, J.; Teles, J. H.; Melder, J. P.; Ebel, K.; Brode, S. Angew. Chem., Int. Ed. Engl. 1995, 34, 1021. doi: 10.1002/(ISSN)1521-3773
-
[18]
de Alaniz, J. R.; Kerr, M. S.; Moore, J. L.; Rovis, T. J. Org. Chem. 2008, 73, 2033. doi: 10.1021/jo702313f
-
[19]
Cullen, S. C.; Rovis, T. Org. Lett. 2008, 10, 3141. doi: 10.1021/ol801047k
-
[20]
Wang, Z.; Yu, Z.; Wang, Y.; Shi, D. Synthesis 2012, 44, 1559. doi: 10.1055/s-0031-1290976
-
[21]
Law, K. R.; McErlean, C. S. P. Chem.-Eur. J. 2013, 19, 15852. doi: 10.1002/chem.201303435
-
[22]
Liu, G.; Wilkerson, P. D.; Toth, C. A.; Xu, H. Org. Lett. 2012, 14, 858. doi: 10.1021/ol203375y
-
[23]
Li, Y.; Shi, F.-Q.; He, Q.-L.; You, S.-L. Org. Lett. 2009, 11, 3182. doi: 10.1021/ol9013238
-
[24]
Vedachalam, S.; Tan, S. M.; Teo, H. P.; Cai, S.; Liu, X.-W. Org. Lett. 2012, 14, 174. doi: 10.1021/ol202959y
-
[25]
Bortolini, O.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A.; Pacifico, S. Org. Biomol. Chem. 2011, 9, 8437. doi: 10.1039/c1ob06480k
-
[26]
Zhang, J.; Xing, C.; Tiwari, B.; Chi, Y. R. J. Am. Chem. Soc. 2013, 135, 8113. doi: 10.1021/ja401511r
-
[27]
Fang, X.; Chen, X.; Chi, Y. R. Org. Lett. 2011, 13, 4708. doi: 10.1021/ol201917u
-
[28]
McCusker, E. O.; Scheidt, K. A. Angew. Chem., Int. Ed. 2013, 52, 13616. doi: 10.1002/anie.201307292
-
[29]
Xu, J.; Chen, X.; Wang, M.; Zheng, P.; Song, B. A.; Chi, Y. R. Angew. Chem., Int. Ed. 2015, 54, 5161. doi: 10.1002/anie.v54.17
-
[30]
Zhao, X.; Ruhl, K. E.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 12330. doi: 10.1002/anie.v51.49
-
[31]
Mo, J.; Yang, R.; Chen, X.; Tiwari, B.; Chi, Y. R. Org. Lett. 2013, 15, 50. doi: 10.1021/ol303035r
-
[32]
Li, F.; Wu, Z.; Wang, J. Angew. Chem., Int. Ed. 2015, 54, 656.
-
[33]
Xu, J.; Yuan, S.; Peng, J.; Miao, M.; Chen, Z.; Ren, H. Chem. Commun. 2017, 53, 3430. doi: 10.1039/C7CC01232B
-
[34]
Zhang, H.-M.; Gao, Z.-H.; Ye, S. Org. Lett. 2014, 16, 3079. doi: 10.1021/ol501205v
-
[35]
Zhao, Y. M.; Cheung, M. S.; Lin, Z.; Sun, J. Angew. Chem., Int. Ed. 2012, 51, 10359. doi: 10.1002/anie.201204521
-
[36]
Ni, Q.; Zhang, H.; Grossmann, A.; Loh, C. C.; Merkens, C.; Enders, D. Angew. Chem., Int. Ed. 2013, 52, 13562. doi: 10.1002/anie.201305957
-
[37]
Dong, X.; Yang, W.; Hu, W.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 660.
-
[38]
Rong, Z. Q.; Jia, M. Q.; You, S. L. Org. Lett. 2011, 13, 4080. doi: 10.1021/ol201595f
-
[39]
Yao, C.; Wang, D.; Lu, J.; Li, T.; Jiao, W.; Yu, C. Chemistry 2012, 18, 1914. doi: 10.1002/chem.201103358
-
[40]
Leong, W. W. Y.; Chen, X.; Chi, Y. R. Green Chem. 2013, 15, 1505. doi: 10.1039/c3gc40397a
-
[41]
Raup, D. E.; Cardinal-David, B.; Holte, D.; Scheidt, K. A. Nat. Chem. 2010, 2, 766. doi: 10.1038/nchem.727
-
[42]
Zhao, X.; DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2011, 133, 12466. doi: 10.1021/ja205714g
-
[43]
De Vreese, R.; D'Hooghe, M. Beilstein J. Org. Chem. 2012, 8, 398. doi: 10.3762/bjoc.8.43
-
[44]
Chen, X.; Fang, X.; Chi, Y. R. Chem. Sci. 2013, 4, 2613. doi: 10.1039/c3sc50666e
-
[45]
Nair, V.; Sinu, C. R.; Babu, B. P.; Varghese, V.; Jose, A.; Suresh, E. Org. Lett. 2009, 11, 5570. doi: 10.1021/ol901918x
-
[46]
Maji, B.; Ji, L.; Wang, S.; Vedachalam, S.; Ganguly, R.; Liu, X. W. Angew. Chem., Int. Ed. 2012, 51, 8276. doi: 10.1002/anie.v51.33
-
[47]
White, N. A.; DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2013, 135, 8504. doi: 10.1021/ja403847e
-
[48]
Cheng, J.; Huang, Z.; Chi, Y. R. Angew. Chem., Int. Ed. 2013, 52, 8592. doi: 10.1002/anie.201303247
-
[49]
Zhang, Z.; Zeng, X.; Xie, D.; Chen, D.; Ding, L.; Wang, A.; Yang, L.; Zhong, G. Org. Lett. 2015, 17, 5052. doi: 10.1021/acs.orglett.5b02527
-
[50]
Xia, W.; Yao, H.; Liu, D.; Zhao, L.; Zhou, Y.; Liu, H. Adv. Synth. Catal. 2016, 358, 1864. doi: 10.1002/adsc.201600136
-
[51]
Lv, H.; Tiwari, B.; Mo, J.; Xing, C.; Chi, Y. R. Org. Lett. 2012, 14, 5412. doi: 10.1021/ol302475g
-
[52]
Jiang, K.; Tiwari, B.; Chi, Y. R. Org. Lett. 2012, 14, 2382. doi: 10.1021/ol3008028
-
[53]
Chen, X. Y.; Xia, F.; Cheng, J. T.; Ye, S. Angew. Chem., Int. Ed. 2013, 52, 10644. doi: 10.1002/anie.201305571
-
[54]
Mo, J.; Shen, L.; Chi, Y. R. Angew. Chem., Int. Ed. 2013, 52, 8588. doi: 10.1002/anie.201302152
-
[55]
Fu, Z.; Xu, J.; Zhu, T.; Leong, W. W.; Chi, Y. R. Nat. Chem. 2013, 5, 835. doi: 10.1038/nchem.1710
-
[56]
Xie, Y.; Yu, C.; Li, T.; Tu, S.; Yao, C. Chemistry 2015, 21, 5355. doi: 10.1002/chem.201500345
-
[57]
Shen, L.-T.; Shao, P.-L.; Ye, S. Adv. Synth. Catal. 2011, 353, 1943. doi: 10.1002/adsc.v353.11/12
-
[58]
Que, Y.; Li, T.; Yu, C.; Wang, X. S.; Yao, C. J. Org. Chem. 2015, 80, 3289. doi: 10.1021/jo502920w
-
[59]
Rong, X.; Yao, H.; Xia, W.; Du, Y.; Zhou, Y.; Liu, H. ACS Comb. Sci. 2016, 18, 220. doi: 10.1021/acscombsci.5b00197
-
[60]
Mo, J.; Chen, X.; Chi, Y. R. J. Am. Chem. Soc. 2012, 134, 8810. doi: 10.1021/ja303618z
-
[61]
Wang, M.; Huang, Z.; Xu, J.; Chi, Y. R. J. Am. Chem. Soc. 2014, 136, 1214. doi: 10.1021/ja411110f
-
[62]
Liang, Z.-Q.; Wang, D.-L.; Zhang, H.-M.; Ye, S. Org. Lett. 2015, 17, 5140. doi: 10.1021/acs.orglett.5b02695
-
[63]
Xu, J.; Yuan, S.; Miao, M. Org. Lett. 2016, 18, 3822. doi: 10.1021/acs.orglett.6b01831
-
[64]
Xiao, Y.; Wang, J.; Xia, W.; Shu, S.; Jiao, S.; Zhou, Y.; Liu, H. Org. Lett. 2015, 17, 3850. doi: 10.1021/acs.orglett.5b01827
-
[65]
Chen, X.; Yang, S.; Song, B. A.; Chi, Y. R. Angew. Chem., Int. Ed. 2013, 52, 11134. doi: 10.1002/anie.201305861
-
[66]
Zhu, T.; Mou, C.; Li, B.; Smetankova, M.; Song, B. A.; Chi, Y. R. J. Am. Chem. Soc. 2015, 137, 5658. doi: 10.1021/jacs.5b02219
-
[67]
Pellissier, H. Tetrahedron 2008, 64, 1563. doi: 10.1016/j.tet.2007.10.080
-
[68]
Lu, S.; Poh, S. B.; Siau, W. Y.; Zhao, Y. Angew. Chem., Int. Ed. 2013, 52, 1731. doi: 10.1002/anie.v52.6
-
[69]
Telfer, S. Coord. Chem. Rev. 2003, 242, 33. doi: 10.1016/S0010-8545(03)00026-2
-
[70]
Lu, S.; Poh, S. B.; Zhao, Y. Angew. Chem., Int. Ed. 2014, 53, 11041. doi: 10.1002/anie.201406192
-
[71]
Steinreiber, J.; Faber, K.; Griengl, H. Chemistry 2008, 14, 8060. doi: 10.1002/chem.v14:27
-
[72]
Cohen, D. T.; Eichman, C. C.; Phillips, E. M.; Zarefsky, E. R.; Scheidt, K. A. Angew. Chem., Int. Ed. 2012, 51, 7309. doi: 10.1002/anie.201203382
-
[73]
Goodman, C. G.; Johnson, J. S. J. Am. Chem. Soc. 2014, 136, 14698. doi: 10.1021/ja508521a
-
[74]
Goodman, C. G.; Walker, M. M.; Johnson, J. S. J. Am. Chem. Soc. 2015, 137, 122. doi: 10.1021/ja511701j
-
[75]
Wu, Z.; Li, F.; Wang, J. Angew. Chem., Int. Ed. 2015, 54, 1629. doi: 10.1002/anie.201410030
-
[76]
Babu, R. S.; O'Doherty, G. A. J. Am. Chem. Soc. 2003, 125, 12406. doi: 10.1021/ja037097k
-
[77]
Sugawara, K.; Imanishi, Y.; Hashiyama, T. Tetrahe-dron-Asymmetry 2000, 11, 4529. doi: 10.1016/S0957-4166(00)00424-9
-
[78]
Chen, X.; Fong, J. Z.; Xu, J.; Mou, C.; Lu, Y.; Yang, S.; Song, B. A.; Chi, Y. R. J. Am. Chem. Soc. 2016, 138, 7212. doi: 10.1021/jacs.6b00406
-
[79]
White, N. A.; Rovis, T. J. Am. Chem. Soc. 2014, 136, 14674. doi: 10.1021/ja5080739
-
[80]
Zhang, Y.; Du, Y.; Huang, Z.; Xu, J.; Wu, X.; Wang, Y.; Wang, M.; Yang, S.; Webster, R. D.; Chi, Y. R. J. Am. Chem. Soc. 2015, 137, 2416. doi: 10.1021/ja511371a
-
[81]
Li, B.-S.; Wang, Y.; Proctor, R. S. J.; Zhang, Y.; Webster, R. D.; Yang, S.; Song, B.; Chi, Y. R. Nat. Commun. 2016, 7.
-
[82]
Reddi, R. N.; Prasad, P. K.; Sudalai, A. Angew. Chem., Int. Ed. 2015, 54, 14150. doi: 10.1002/anie.201507363
-
[83]
Nakano, Y.; Lupton, D. W. Angew. Chem., Int. Ed. 2016, 55, 3135. doi: 10.1002/anie.201510106
-
[84]
Patra, A.; Mukherjee, S.; Das, T. K.; Jain, S.; Gonnade, R. G.; Biju, A. T. Angew. Chem., Int. Ed. 2017, 56, 2730. doi: 10.1002/anie.v56.10
-
[85]
Ungureanu, A.; Levens, A.; Candish, L.; Lupton, D. W. Angew. Chem., Int. Ed. 2015, 54, 11780. doi: 10.1002/anie.201504633
-
[1]
-

图式 2 维生素B1催化的安息香缩合反应机理
Scheme 2 Reaction mechanism of benzoin condensation catalyzed by vitamin B1

图式 3 卡宾催化下的分子间Stetter反应
Scheme 3 Intermolecular Stetter reaction catalyzed by N-hetercyclic carbenes

图式 6 以水为溶剂的NHC催化不饱和醛与酮的反应
Scheme 6 Reaction of unsaturated aldehydes with ketones catalyzed by NHC in the solution of water

图式 8 NHC催化不饱和羰基化合物的极端反应
Scheme 8 Extreme reaction of unsaturated carbonyls catalyzed by NHC

图式 9 NHC催化不饱和醛与亚胺的反应及机理
Scheme 9 Reaction and mechanism of unsaturated aldehydes with imine catalyzed by NHC

图式 10 NHC介导的α, β不饱和羰基化合物的γ位碳原子活化及机理
Scheme 10 Reaction and mechanism of NHC mediated γ-activation of α, β-unsaturated carbonyls

图式 11 离去基团介导的γ位碳原子的活化及机理
Scheme 11 Reaction and mechanism of leaving group mediated of γ-activation

图式 13 氮杂环卡宾介导的α, β, γ, δ-双不饱和醛的δ活化
Scheme 13 δ-Activation of α, β, γ, δ-unsaturated aldehydes catalyzed by NHC

图式 15 氮杂环卡宾介导的分子间动态动力学拆分及机理
Scheme 15 Dynamic kinetic resolution and mechanism of intermolecular catalyzed by NHC

图式 17 NHC参与的单电子自由基反应
Scheme 17 Single-electron-transfer (SET) process for radical reactions mediated by NHC

图式 18 NHC介导的δ-磺内酯衍生物活化及机理
Scheme 18 Reaction and mechanism of NHC mediated δ-sulfonated lactones
-


 下载:
下载:


















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 650
- 文章访问数: 19601
- HTML全文浏览量: 7378
 下载:
下载:
