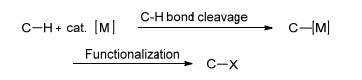
 图式1
内层机理反应途径
Scheme1.
Inner mechanism of the reaction pathway
图式1
内层机理反应途径
Scheme1.
Inner mechanism of the reaction pathway
引用本文:
任兰会, 高爽. C—H键氧化生成酮的研究进展[J]. 有机化学,
2017, 37(6): 1338-1351.
doi:
10.6023/cjoc201702022

Citation: Ren Lanhui, Gao Shuang. Recent Advances of the Oxidation of C—H Bonds to Ketones[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1338-1351. doi: 10.6023/cjoc201702022
Citation: Ren Lanhui, Gao Shuang. Recent Advances of the Oxidation of C—H Bonds to Ketones[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1338-1351. doi: 10.6023/cjoc201702022
C—H键氧化生成酮的研究进展
English
Recent Advances of the Oxidation of C—H Bonds to Ketones
Abstract:
The ketones are important intermediates for the synthesis of fine chemicals, such as pharmaceuticals, natural products, agricultural chemicals, dyes, etc. The oxidation of C—H bonds is one of the most direct and efficient synthetic methods for the preparation of ketones. In this review, the oxidation of C—H bonds to ketones is reviewed.
-
C—H键是有机化合物中最基本的化学键之一, 碳氢化合物来源广泛、种类繁多、价格便宜, 其官能团化是生产其它化学品最简洁、最高效的方法之一[1~11].通常所说的C—H键官能团化是指:在金属试剂或者金属催化剂的作用下, C—H键断裂, 生成碳金属键、自由基、卡宾等活性中间体, 这些活性中间体与其它的化学物质反应生成新的官能团.金属试剂或者金属催化剂不仅可以有效地降低反应所需的活化能, 还能提高活性中间体的选择性[12~14].
根据C—H键是否直接和金属中心作用, 可以将过渡金属催化剂参与的C—H键官能团化反应分为两种不同的反应机理:内层机理和外层机理.内层机理反应途径如Scheme 1所示, 首先在过渡金属作用下, C—H键断裂, 生成的金属烷基或芳基中间体与其它的反应物反应, 最终实现C—H键官能团化.这类反应的选择性取决于形成的金属烷基或芳基中间体的结构和性质, 位阻较小的C—H键往往具有较好的选择性.外层机理反应途径如Scheme 2所示, 科学家们通过对生物酶催化反应历程的研究提出了外层机理.首先氧化剂将过渡金属氧化成高氧化态的金属络合物(如金属酰氧、金属酰亚胺或金属卡宾等), 然后高活性、高氧化态的金属络合物与C—H键发生反应(直接插入或攫氢/自由基复合), 最终实现C—H键的官能团化.经历外层机理的C—H键官能团化反应, 选择性和键能的大小有关, 键能小, 选择性好, 这主要是因为键能小的碳氢化合物在官能团化反应中生成的碳自由基或离子等中间体比较稳定[15].
图式1
内层机理反应途径
Scheme1.
Inner mechanism of the reaction pathway
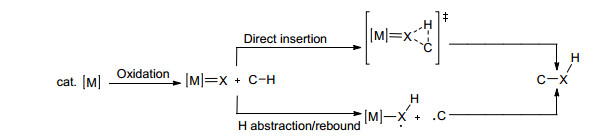 图式2
外层机理反应途径
Scheme2.
Outer mechanism of the reaction pathway
图式2
外层机理反应途径
Scheme2.
Outer mechanism of the reaction pathway
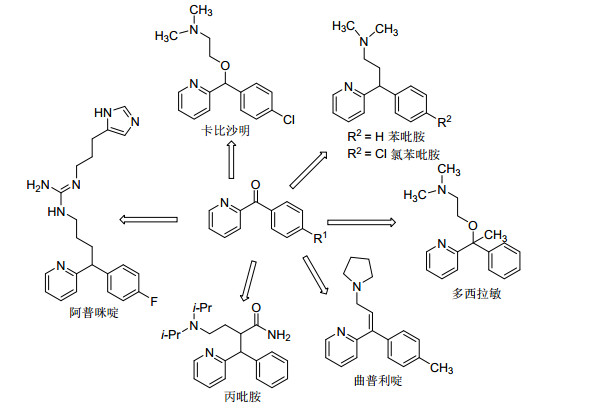 图式3
基于苯甲酰基吡啶类化合物开发的医药化合物
Scheme3.
Pharmaceutical compounds coming from benzoylpyridines
图式3
基于苯甲酰基吡啶类化合物开发的医药化合物
Scheme3.
Pharmaceutical compounds coming from benzoylpyridines
C—H键氧化生成酮是C—H键官能团化的重要研究内容之一.酮是合成医药、天然产物、农用化学品、染料等精细化学品的重要中间体, 例如苯甲酰基吡啶类化合物是合成众多医药产品的关键中间体(Scheme 1)[16~19].
在传统的方法中, 化学计量的强氧化剂被用于C—H键氧化生成酮的反应, 例如高锰酸钾、铬试剂等.使用这些氧化剂往往会产生大量副产品, 后处理麻烦, 对环境造成很大的污染.目前环境问题日益突出, 开发绿色、高效、高选择性的C—H键氧化生成酮的催化体系已成为人们研究的热点[20, 21].
本文主要综述比较了C—H键氧化生成酮的不同催化体系.
1 Fe催化体系
C—H键氧化是自然界中普遍存在的过程, 很多自然界中C—H键氧化反应都需要含有Fe的酶来催化. 20世纪80年代, 在自然界中氧化反应的启发下, 科学家Gifsur-Yvette提出了Gif化学体系[22]. Gif化学体系使用过渡金属(主要是铁)作为催化剂, 氧气作为氧化剂, 在吡啶中进行, 该体系必须加入还原剂. 20世纪80年代, Barton首次将Gif化学体系用于温和条件下饱和烃氧化生成酮的反应, 并且发现直链烷烃中不同C—H键发生氧化反应的选择性为3'>2'>1'[23~30].
2005年, 王彦广等[31]使用金属卟啉作为催化剂, 研究氯胺参与的四氢化萘的酰胺化反应时发现, 使用四苯基卟啉铁作为催化剂时, 四氢化萘被氧化生成四氢萘酮.在进行深入研究后, 王彦广等[31]报道了一种在氯胺的参与下, 以氧气作为氧化剂, Fe(TPP)Cl催化烷基苯、二苯基甲烷类化合物、1, 2, 3, 4-四氢化萘类化合物、2, 3-二氢-1H-茚、芴和环己烯氧化生成酮的方法, 收率为52%~87%, Fe(TPP)Cl还可以催化环己烷氧化生成环己酮, 收率为13% (Eq. 1).
2010年, White等[32]报道了一种Fe(S, S-PDP)催化剂, 这种简单的铁络合物催化剂表现出和酶催化剂相当的选择性, 可以催化天然产物和饱和烷烃中C—H键生成酮, 氧化位点在一定条件下可以预测(Scheme 4).
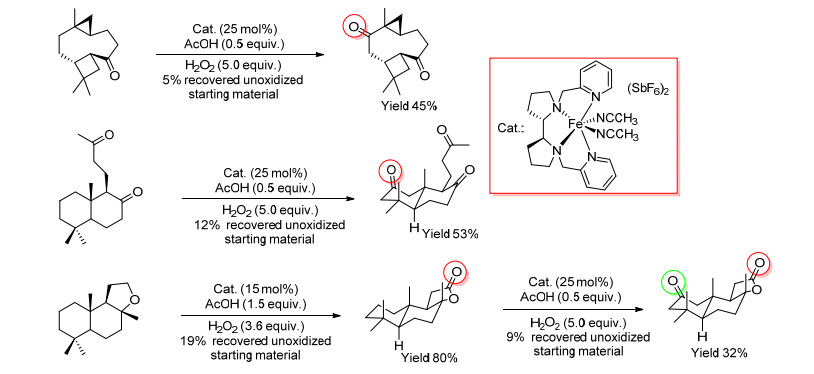 图式4
Fe(S, S-PDP)催化天然产物氧化生成酮
Scheme4.
Oxidation of natural product to ketone with Fe(S, S-PDP) as catalyst
图式4
Fe(S, S-PDP)催化天然产物氧化生成酮
Scheme4.
Oxidation of natural product to ketone with Fe(S, S-PDP) as catalyst
2013年, Kappe等[33]将微反应器应用于苄基吡啶类化合物氧化生成酮的反应.使用碳酸丙烯酯作为反应溶剂, 空气作为氧化剂, FeCl3作为催化剂, 在200 ℃下反应13 min, 可以将2-(4-甲基苄基)吡啶、2-(4-氯苄基)吡啶和2-(4-氟苄基)吡啶氧化生成酮, 收率分别是69%, 81%, 83% (Eq. 2).微反应器在很大程度上提高了反应速率和催化剂的活性.
2013年, Novák等[34]报道了一种FeCl3或Fe2(SO4)3催化芴类化合物、二苯基甲烷类化合物、烷基苯、2-乙基噻吩、1, 2, 3, 4-四氢化萘、氧杂蒽和9, 10-二氢蒽氧化生成酮的方法, 收率为21%~98%.作者发现表面活性剂可以显著地加快反应速率, 而且表面活性剂分子中亲油部分的碳链越长, 效果越明显.
2015年, Draye等[35]报道了一种Fe(TAML)Li催化剂(Eq. 3), 使用叔丁基过氧化氢作为氧化剂, 在-10 ℃时, 可以将烷基苯、二苯基甲烷、氧杂蒽、1, 2, 3, 4-四氢化萘、乙基吡啶类化合物和苄基吡啶类化合物氧化生成酮, 收率为8%~99%.但是, Fe(TAML)Li催化剂对反应液的pH值要求较高.
2015年, 王戈等[36]将FeCl3固定到聚(4-乙烯吡啶)上制备了一种Fe-P4VP催化剂, 使用叔丁基过氧化氢作为氧化剂, 在80 ℃下反应24 h, 可以将烷基苯、芴和双(4-氟苯基)甲烷氧化生成酮, 转化率为73%~99%, 选择性为90%~99%(Eq. 4). Fe-P4VP催化剂可以溶解在有机溶剂中, 二乙烯苯的含量影响反应体系中催化剂的活性.
2016年, Repo等[37]报道了一种Fe(Ⅲ)-THA(胸腺嘧啶-1-乙酸)催化剂, 使用过氧化氢作为氧化剂, 可以将饱和烷烃、2, 3-二氢-1H-茚、1, 2, 3, 4-四氢化萘、二苯基甲烷、乙苯、2-苄基吡啶、环己烯和环辛烯氧化生成酮, 收率为4%~95% (Eq. 5).
2016年, Wolf等[38]报道了一种核黄素四乙酸酯(RFT)和[Fe(TPA)-(MeCN)2](ClO4)2 [TPA:三(2-吡啶基甲基)胺]组成的催化体系, 可以催化乙苯类化合物、氧杂蒽、硫杂蒽、2, 3-二氢-1H-茚和1, 2, 3, 4-四氢化萘氧化生成酮, 收率为70%~99% (Eq. 6).核黄素四乙酸酯在440 nm蓝色光照射下会原位产生过氧化氢, 避免了直接使用容易爆炸的过氧化氢.过氧化氢在金属Fe的存在下会迅速分解, 所以使用过氧化氢作为氧化剂时, 一般会过量使用, 而且需要慢慢滴加.这种原位产生过氧化氢的方法可以彻底避免上述问题, 降低了使用成本.
2 Cu催化体系
铜作为一种廉价易得的金属, 在氧化反应中有广泛的用途, 很多在生物体中进行的氧化反应也是由含铜的酶催化的.
1996年, Murahashi等[39]报道了一种由CuCl2和18-冠-6组成的Cu-冠醚催化剂, 在乙醛存在下, 使用氧气作为氧化剂, 可以将正己烷、环己烷和乙苯氧化生成酮, 收率分别为44%、61%、13%.
2003年, Punniyamurthy等[40]报道了一种Salen-H4/Cu催化剂(Eq. 7), 使用30%过氧化氢水溶液作为氧化剂, 可以将乙苯、丁苯、苯乙酸乙酯、1, 2, 3, 4-四氢化萘和环己烷氧化生成酮, 收率分别为86%、88%、82%、89%、18%.
2009年, Gorden等[41]报道了一种Benzyl-salquCu催化剂(图 1), 使用3 equiv.叔丁基过氧化氢作为氧化剂, 以乙腈作为反应溶剂, 回流18 h, 可以氧化烷基苯、二苯基甲烷、1, 2, 3, 4-四氢化萘和2-乙基萘生成酮, 收率为66%~99%.当发生反应的亚甲基邻位为供电子取代基时, 反应结果较好; 为吸电子取代基时, 反应结果较差.
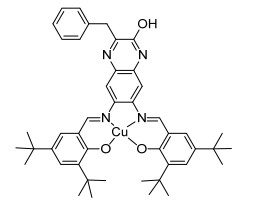 图 1
Benzyl-salquCu的结构
Figure Figure1.
Structure of benzyl-salquCu
图 1
Benzyl-salquCu的结构
Figure Figure1.
Structure of benzyl-salquCu
2012年, Gorden等[42]在之前工作的基础上, 报道了一种isopropyl-salquCu催化剂, 使用叔丁基过氧化氢作为氧化剂, 可以氧化环戊烯类化合物和环己烯类化合物生成酮, 收率为11%~99% (Eq. 8).
2012年, Maes等[43]使用CuI作为催化剂, 以氧气为氧化剂, 在酸性条件(AcOH)下氧化苄基吡啶类化合物和苄基嘧啶类化合物生成酮, 收率为61%~85% (Eq. 9).即使底物中含有容易被氧化的甲硫基时, CuI/AcOH/O2体系依然能高选择性地氧化相应的亚甲基.
2014年, 张致慧等[44]将原位产生的Cu-MOF(Cu-(NO3)2-BTC为前体, BTC: 1, 3, 5-苯三羧酸)负载到多孔二氧化硅材料的孔中, 制备了一种多相铜催化剂, 以叔丁基过氧化氢为氧化剂, 可以氧化乙苯、丙苯、二苯基甲烷、芴和1, 2, 3, 4-四氢化萘生成酮, 转化率分别为99%, 96%、29%、18%、95%, 选择性均大于99% (Eq. 10).作者用DMSO替换DMF/水/乙醇混合体系, 避免了Cu-MOF快速聚集, 同时也避免了Cu-MOF在二氧化硅材料的表面结晶.循环12次后, 催化活性没有降低.
2015年, Lam等[45]报道了一种含氟有机配体(Eq. 11), 使用该配体和CuI的络合物作为催化剂, 叔丁基过氧化氢作为氧化剂, 可以氧化环己烯类化合物、烷基苯、芴、二苯基甲烷、2, 3-二氢-1H-茚、9, 10-二氢蒽和1-对甲苯磺酰基-1, 2, 3, 4-四氢喹啉生成酮, 收率为51%~88%.这种含氟有机配体在经历了5次循环实验后, 活性只有少许下降.
2015年, 雷爱文等[46]首次将活化剂引入到C—H键氧化反应中.使用氯乙酸乙酯作为活化剂, 一水合氯化铜作为催化剂, 氧气作为氧化剂, 在DMF中, 130 ℃反应24 h, 可以氧化吡啶侧链C—H键、喹啉侧链C—H键、苯并咪唑侧链C—H键、苯并异噁唑侧链C—H键和喹喔啉侧链C—H键生成酮, 收率为30%~92% (Eq. 12).
3 Ru催化体系
1995年, Mak等[47]报道了一种钌氧化物, 在三氟乙酸、2, 2'-联吡啶和二氯甲烷的参与下, 这种钌氧化物可以氧化环己烷、正己烷、2-甲基丁烷和乙苯生成酮, 收率分别为64%、31%、11%、10%. 2000年, Mak等[48]优化了该氧化体系, 将2, 2'-联吡啶换成6, 6'-二氯-2, 2'-联吡啶, 改进后的氧化体系不仅可以氧化C—H键生成酮, 还可以将伯醇或仲醇氧化生成醛或酮.
2009年, Lee等[49]报道了一种阳离子络合物[(pymox-Me2)RuCl2]+BF4-催化剂(Scheme 5), 使用叔丁基过氧化氢作为氧化剂, 水作为溶剂, 在室温下可以氧化烷基苯、2-乙基萘、9, 10-二氢蒽、2, 3-二氢-1H-茚、氧杂蒽、芴、环己烷和环辛烷生成酮, 收率为41%~89%.
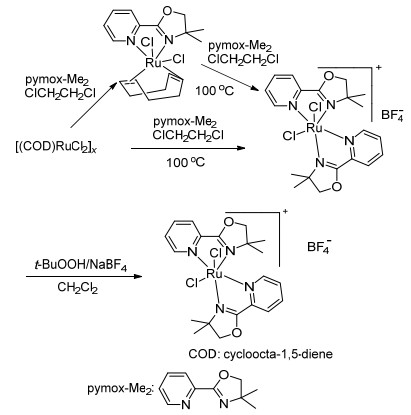 图式5
(pymox-Me2)RuCl2]+BF4-的合成路线
Scheme5.
The synthetic route of [(pymox-Me2)RuCl2]+BF4-
图式5
(pymox-Me2)RuCl2]+BF4-的合成路线
Scheme5.
The synthetic route of [(pymox-Me2)RuCl2]+BF4-
2009年, Yusubov等[50]报道了一种串联催化体系, 在钌催化剂的作用下, 过硫酸氢钾将碘苯氧化成亚碘酰苯, 然后亚碘酰苯氧化烷基苯、9, 10-二氢蒽、2, 3-二氢-1H-茚和1, 2, 3, 4-四氢化萘生成酮, 收率为69%~95% (Scheme 6).
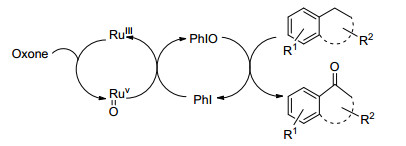 图式6
串联的碘苯和三氯化钌催化C—H键氧化生成酮的过程
Scheme6.
Process of the oxidation of C—H bonds to ketones with tandem iodobenzene and RuCl3 as catalyst
图式6
串联的碘苯和三氯化钌催化C—H键氧化生成酮的过程
Scheme6.
Process of the oxidation of C—H bonds to ketones with tandem iodobenzene and RuCl3 as catalyst
2014年, Oisaki等[51]报道了一种以Ru(bpy)32+-Cu(Ⅰ)作为光敏剂的光催化体系.使用氧气作为氧化剂, 在可见光的照射下, 可以氧化芴类化合物生成酮, 收率为41%~95% (Eq. 12).
4 Mn催化体系
含金属锰的催化剂被广泛地应用于氧化反应, 特别是烯烃环氧化反应[52~54]、醇氧化反应[55~57]和硫醚氧化反应[58].
1998年, Lee等[59]报道了一种Mn(Ⅲ) salen催化剂(图 2), 使用亚碘酰苯或次氯酸钠作为氧化剂, 可以氧化烷基苯、二苯基甲烷、氧杂蒽、2, 3-二氢-1H-茚类化合物、1, 2, 3, 4-四氢化萘和9, 10-二氢蒽生成酮, 收率为22%~92%.作者认为反应底物先被氧化生成仲醇, 仲醇进一步被氧化生成酮.
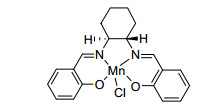 图 2
Mn(Ⅲ) salen的结构
Figure Figure2.
Structure of Mn(Ⅲ) salen
图 2
Mn(Ⅲ) salen的结构
Figure Figure2.
Structure of Mn(Ⅲ) salen
2001年, 陈焜铭等[60]报道了一种莰酮衍生物类配体(图 3), 使用该配体和四水合乙酸锰在氯仿溶液中回流12 h后, 减压蒸出溶剂, 固体直接用于氧化反应.使用2 equiv.叔丁基过氧化氢作为氧化剂, 0 ℃下, 可以氧化丙苯、1, 2, 3, 4-四氢化萘、芴和9, 10-二氢蒽生成酮, 收率分别为91%、88%、63%、52%.
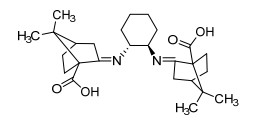 图 3
莰酮衍生物类配体的结构
Figure Figure3.
Structure of camphanone derivative ligand
图 3
莰酮衍生物类配体的结构
Figure Figure3.
Structure of camphanone derivative ligand
2006年, Shing等[61]使用Mn(OAc)3作为催化剂, 以叔丁基过氧化氢为氧化剂, 在氮气氛围中, 可以氧化△5-类固醇类化合物生成具有优异抗癌活性的△5-en-7-酮类化合物(Eq. 13).在氧气氛围中, 可以氧化环己烯类化合物生成酮(Eq. 14).由于三价锰离子在水溶液中容易发生歧化反应生成二价锰离子和四价锰离子, 作者加入3Å分子筛抑制三价锰离子的歧化反应.
2012年, Jayaram等[62]还原高锰酸钾制备了一种纳米MnO2催化剂, 使用3 equiv.叔丁基过氧化氢作为氧化剂, 在80 ℃下反应10 h, 可以氧化二苯基甲烷、1, 2, 3, 4-四氢化萘、2, 3-二氢-1H-茚、芴、1, 2-二氢苊、蒽-9(10H)-酮、异丁基苯和氧杂蒽生成酮, 转化率分别为92%、92%、89%、100%、88%、100%、85%、100%, 选择性分别为100%、80%、100%、100%、100%、100%、95%、100%.催化剂经历6次循环后, 活性没有降低.
2014年, 孙伟等[63]报道了一种仿生的S-PMB->Mn(OTf)2催化剂(图 4), 使用过氧化氢作为氧化剂, 在酸性条件(乙酸)下, 可以氧化烷基苯、二苯基甲烷、1, 2, 3, 4-四氢化萘、氧杂蒽、环己烷和环辛烷生成酮, 收率为15%~82%.
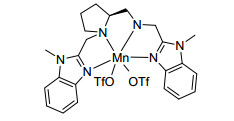 图 4
S-PMB-Mn(OTf)2的结构
Figure Figure4.
Structure of S-PMB-Mn(OTf)2
图 4
S-PMB-Mn(OTf)2的结构
Figure Figure4.
Structure of S-PMB-Mn(OTf)2
2015年, 吴传德等[64]使用四苯基氨基卟啉和三聚氰酰氯反应, 将产物和四水合氯化锰络合制备了一种多相锰卟啉催化剂, 使用叔丁基过氧化氢作为氧化剂, 可以氧化乙苯、丙苯、1, 2, 3, 4-四氢化萘、二苯基甲烷和4-乙基-1, 1'-联苯生成酮, 收率分别为99%、91%、73%、57%、76%.
2015年, Shaabani等[65]报道了一种天然白羊毛负载的MnO2催化剂.首先用氢氧化钠溶液洗涤羊毛, 然后分别用蒸馏水和甲醇洗涤, 干燥后, 将处理后的羊毛切割成微粒(20~500 μm).将羊毛微粒放入到高锰酸钾水溶液中搅拌12 h, 过滤, 干燥后得到天然白羊毛负载的MnO2催化剂.使用空气作为氧化剂, 天然白羊毛负载的MnO2可以催化烷基苯、二苯基甲烷、2, 3-二氢-1H-茚、1, 2, 3, 4-四氢化萘、9, 10-二氢蒽、芴和氧杂蒽氧化生成酮, 收率为86%~100%.
2015年, 高爽等[66]报道了一种双三氟甲磺酸锰催化吡啶侧链C—H键氧化生成酮的方法.作者选择苄基吡啶类化合物、6, 7-二氢-5H-环戊基[b]吡啶类化合物和2-乙基吡啶作为反应底物, 优化后的反应条件为: 0.5 mol% Mn(OTf)2, 5 equiv.叔丁基过氧化氢, 2.5 mL水, 25 ℃反应24 h.在优化的反应条件下, 多种6, 7-二氢-5H-环戊基[b]吡啶类化合物可以被氧化生成酮, 收率为73%~88% (Eq. 15).相比文献报道的方法, 作者使用了更加绿色环保的氧化剂(65%叔丁基过氧化氢水溶液)替代了CrO3.文献报道的方法使用的溶剂为H2SO4-HOAc, 不仅对环境污染大, 而且对设备腐蚀严重, 作者使用H2O为溶剂, 彻底解决了上述问题.使用叔丁醇作为反应溶剂, Mn(OTf)2还能催化苄基吡啶类化合物和2-乙基吡啶氧化生成酮, 收率为31%~87%.
2016年, 高爽等[67]将Mn(NO3)2和1, 10-菲啰啉的络合物在氮气氛围中煅烧制备了多种MnOx-N@C, 选择苄基吡啶类化合物和6, 7-二氢-5H-环戊基[b]吡啶类化合物作为反应底物, 研究了MnOx-N@C催化吡啶侧链C—H键氧化生成酮的方法(Scheme 7).优化后的反应条件为: 1 mg MnOx-N@C (600 ℃), 3 equiv.叔丁基过氧化氢, 无溶剂条件下, 60 ℃反应12 h.在优化的反应条件下, 多种苄基吡啶类化合物和6, 7-二氢-5H-环戊基[b]吡啶类化合物均能顺利地发生氧化反应, 生成相应的酮, 收率为38%~95% (Eq. 15).利用透射电子显微镜(TEM)和X射线光电子能谱分析(XPS)对MnOx-N@C (600 ℃)进行了初步表征. TEM谱图表明MnOx-N@C (600 ℃)中MnOx粒子的大小为1.7~6.6 nm. N1s的谱图表明MnOx-N@C (600 ℃)中的N主要以C—N、C=N和吡咯型N的形式存在. Mn2p3/2的谱图表明MnOx-N@C (600 ℃)中存在多种价态的Mn, 分别为Mn(Ⅱ)、Mn(Ⅲ)和Mn(Ⅳ).在连续循环6次后, MnOx-N@C (600 ℃)依然具有很高的催化活性, 这说明MnOx-N@C (600 ℃)的稳定性很好. MnOx-N@C (600 ℃)能催化克级规模的放大实验.
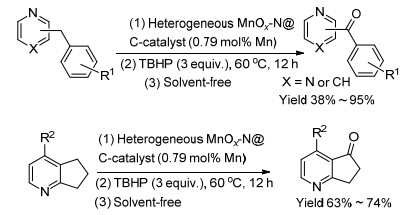 图式7
双三氟甲磺酸锰催化C—H键氧化生成酮的方法
Scheme7.
Oxidation of C—H bonds to ketones with Mn(OTf)2 as catalyst
图式7
双三氟甲磺酸锰催化C—H键氧化生成酮的方法
Scheme7.
Oxidation of C—H bonds to ketones with Mn(OTf)2 as catalyst
5 Co催化体系
1996年, Ishii等[68]使用N-羟基邻苯二甲酰亚胺(NHPI)作为主催化剂, 以Co(acac)n (n=2, 3) 作为助催化剂, 进行了环己烷和烷基苯氧化的研究.环己烷的转化率为43%, 环己酮的产率为31%, 反应中生成了己二酸.烷基苯的氧化产物主要是苯甲酸, 难以停留在酮产物阶段.
2002年, Minisci等[69]报道了一种NHPI-Co(Ⅱ)催化剂, 使用氧气作为氧化剂, 在极性溶剂中, 可以氧化N邻位C—H键生成羰基化合物(醛、酮、羧酸、酰胺), 产物的类型取决于底物的类型和反应条件.
2008年, Shaabani等[70]报道了一种酞菁Co(Ⅱ)催化剂, 以溴化1-丁基-3-甲基咪唑为溶剂, O2为氧化剂, 可以催化乙苯、丙苯、丁苯、二苯基甲烷、芴、2, 3-二氢-1H-茚、1, 2, 3, 4-四氢化萘和氧杂蒽氧化生成酮, 收率分别为77%、78%、81%、83%、91%、84%、89%、93%.
2009年, 何良年等[71]使用醋酸钴作为催化剂, 氧气作为氧化剂, 氧化芴、1, 2-二氢苊、1, 2, 3, 4-四氢化萘、乙苯、丁苯和环己烷生成酮, 收率分别为83%、58%、73%、20%、13%、4%.作者使用聚乙二醇氧化裂解产生自由基, 进而引发该氧化反应.作者还发现超临界CO2可以促进该氧化反应.
2013年, 李斌栋等[72]报道了一种二氧化硅负载的西弗碱钴, 在乙酸溶液中, 使用该席夫碱钴和NHPI作为催化剂, 以氧气为氧化剂, 可以氧化二苯基甲烷、2, 3-二氢-1H-茚、1, 2, 3, 4-四氢化萘、芴、1, 2-二氢苊、乙苯、丙苯和丁苯生成酮, 转化率分别为83%、70%、68%、70%、66%、98%、58%、52%, 选择性分别为95%、99%、94%、96%、99%、99%、90%、91%.在循环5次后, 催化剂的活性没有降低.
2014年, Islam等[73]报道了一种聚合物固定的多相钴催化剂.首先, 4-(2-吡啶偶氮)间苯二酚(PAR)和氯甲基聚苯乙烯反应得到固定的有机配体(PS-PAR), 然后固定的有机配体和乙酸钴络合得到多相的PS-PAR-Co (Scheme 8).同时使用过氧化氢和氧气作为氧化剂, 可以氧化烷基苯、二苯基甲烷、芴、1, 2-二氢苊、1, 2, 3, 4-四氢化萘、2, 3-二氢-1H-茚、蒽-9(10H)-酮和氧杂蒽生成酮, 转化率为80%~93%, 选择性为90%~100%.循环6次后, 催化剂的活性没有降低.此外, 该催化剂能用于烯烃环氧化反应.
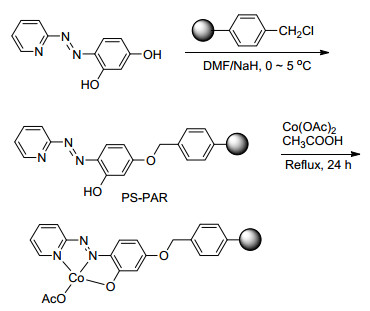 图式8
PS-PAR-Co催化剂的制备方法
Scheme8.
The preparation of PS-PAR-Co catalyst
图式8
PS-PAR-Co催化剂的制备方法
Scheme8.
The preparation of PS-PAR-Co catalyst
2014年, Shaabani等[74]通过一系列化学反应将4, 9, 16, 23-四氨基酞菁钴固定到纤维素上制备了一种多相钴催化剂.在碱性条件(KOH)下, 以NHPI和该多相钴作为催化剂, 以邻二甲苯为溶剂, 以氧气为氧化剂, 加热回流, 可以氧化乙苯、丁苯、庚苯、氧杂蒽、1, 2, 3, 4-四氢化萘、苯并二氢吡喃、2, 3-二氢-1H-茚、二苯基甲烷和芴生成酮, 收率分别为89%、85%、85%、91%、88%、91%、87%、87%、82%.循环6次后, 催化剂的活性没有降低.
2017年, Stahl等[75]将NHPI/Co(OAc)2/O2体系应用于苄基型C—H键氧化生成酮的反应.该体系可用于含有吡啶、苯并咪唑、噻吩等杂环的底物氧化生成酮的反应, 收率为50%~94%.
6 Zn催化体系
2012年, 吴晓峰[76]使用ZnBr2作为催化剂, 过氧化氢作为氧化剂, 在酸性环境(三氟乙酸)中, 氧化二苯基甲烷类化合物、烷基苯、9, 10-二氢蒽、氧杂蒽、4-苄基吡啶、2, 3-二氢-1H-茚和芴生成酮, 收率为18%~93% (Eq. 16).
7 Ce催化体系
铈作为重要的稀土金属, 被广泛地应用于氧化反应.二氧化铈可以通过Ce3+和Ce4+之间的氧化还原转变存储氧, 这种储氧能力和它的粒径大小、形状、形态以及表面积有关. 2012年, Akhlaghinia等[77]报道了一种纳米二氧化铈催化剂, 以溴酸钾作为氧化剂, 可以将乙苯、1-乙基-4-甲基苯、对溴乙苯、二苯基甲烷、9, 10-二氢蒽和2-苄基吡啶氧化生成酮, 收率分别为80%、85%、90%、95%、95%、95%.
通过在CeO2中掺杂低价态的金属可以产生氧负离子. 2015年, Chowdhury等[78]制备了一系列Sm掺杂的CeO2催化剂(Ce1-xSmxO2, x=0~0.1), 作者发现Sm的掺杂量为5%时, Ce0.95Sm0.05O2对乙苯氧化具有最高的催化活性.作者将Ce0.95Sm0.05O2应用于丁苯、二苯基甲烷、1, 2, 3, 4-四氢化萘、环己烯和环戊烯氧化生成酮的反应, 转化率分别为60%、71%、76%、98%、73%, 选择性分别为100%、84%、90%、65%、100%.
8 Rh催化体系
一般来讲, 可以发生自身单电子氧化还原过程的金属多被用来催化C—H键氧化反应, 比如Fe (Fe2+⇌Fe3+), Cu (Cu+⇌Cu2+), Co (Co2+⇌Co3+)[79~85]. 2004年, Doyle团队[86]基于Rh24+和Rh25+之间的氧化还原转换, 开发了一种Rh2(cap)4催化剂.使用叔丁基过氧化氢作为氧化剂, 以二氯甲烷作为溶剂, 在碱性条件下, Rh2(cap)4可以催化环己烯类化合物和环戊烯类化合物氧化生成酮, 收率为60%~94% (Scheme 9). 2005年, Doyle团队[87]在以往工作的基础上, 开发了Rh2(cap)4/ TBHP/KHCO3体系, 实现了乙苯类化合物、1, 2, 3, 4-四氢化萘类化合物、2, 3-二氢-1H-茚类化合物、1, 2, 3, 4-四氢喹啉类化合物、苯并二氢吡喃、二苯基甲烷、芴和氧杂蒽氧化成酮的反应, 收率为20%~99%.
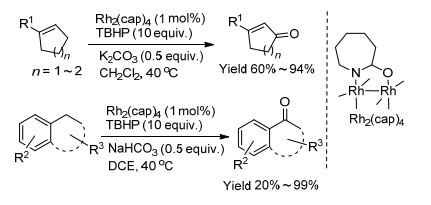 图式9
Rh2(cap)4催化C—H键氧化生成酮
Scheme9.
The oxidation of C—H bonds to ketones with Rh2(cap)4 as catalyst
图式9
Rh2(cap)4催化C—H键氧化生成酮
Scheme9.
The oxidation of C—H bonds to ketones with Rh2(cap)4 as catalyst
2012年, 卢崇道等[88]在Doyle团队的工作的启发下, 设计、合成了一系列新型铑催化剂Rh2(sip)4, 作者依据相关实验数据给出了Rh2(sip)4的立体结构(Eq. 17).Rh2(sip)4能催化烷基苯、1, 2, 3, 4-四氢化萘类化合物、二苯基甲烷、氧杂蒽和9, 10-二氢蒽氧化生成酮, 收率为41%~97%.
2015年, 王元桦等[89]报道了一种Rh2(esp)2催化剂.在Doyle团队报道的工作中, 催化剂Rh2(cap)4经历了Rh24+/Rh25+单电子氧化还原转变, 进而催化C—H键氧化生成酮. Rh2(esp)2并未被氧化成Rh2(esp)2+, Rh2(esp)2催化C—H键氧化生成酮, 并且Rh2(esp)2更稳定.作者分离回收的Rh2(esp)2依然表现出很高的催化活性.使用叔丁基过氧化氢(70%水溶液)作为氧化剂, Rh2(esp)2可以催化烯丙基型和苄基型C—H键氧化生成酮.
9 Bi催化体系
金属铋虽然是一种重金属, 但是铋具有很低的毒性[90~92]. 1998年, Banik等[93]将铋酸钠用于苄基型C—H键的氧化, 铋酸钠作为钠盐沉淀之一, 溶解性很差, 这导致其用量大, 反应时间长. 2005年, Barrent等[94]报道了一种Bi/吡啶-2-甲酸/叔丁基过氧化氢体系.以吡啶和乙酸混合液作为反应溶剂, 可以氧化烷基苯、烷基吡啶、2-乙基噻吩、1, 2, 3, 4-四氢化萘、二苯基甲烷、芴类化合物、氧杂蒽、10, 11-二氢-5H-二苯并[a, d][7]轮烯和2, 3-二氢-1H-茚生成酮, 收率为48%~99%.吡啶-2-甲酸和金属铋形成络合物, 促进了金属铋的溶解.作者认为该氧化过程是一个自由基过程, 不经历Bi(Ⅲ)-Bi(Ⅴ)的循环, 反应机理类似于Gif-GoAgg氧化反应.
10 Au催化体系
金虽然是一种稀有元素, 但是其储量比钯、铂、铑等金属的储量大, 价格也相对便宜.近年来, 金络合物被广泛地应用于C—C键、C—O键、C—N键、C—S键和C—F键等化学键的构筑[95~111]. 2009年, 李焕荣等[112]使用氯金酸钾作为催化剂, 以叔丁基过氧化氢为氧化剂, 氧化二苯基甲烷类化合物、芴、1, 2, 3, 4-四氢化萘和10, 11-二氢-5H-二苯并[a, d][7]轮烯生成酮, 收率为58%~99%.
11 Re催化体系
甲基三氧化铼作为高效的催化剂被用于许多氧化反应中, 但是甲基三氧化铼使用麻烦, 并且价格昂贵[113~116]. 2011年, 朱成建等[117]报道了一种ReOCl3(O-PPh3)(SMe2)催化剂, 使用叔丁基过氧化氢作为氧化剂, 该催化剂可以催化烷基苯、二苯基甲烷类化合物、芴、2, 3-二氢-1H-茚和1, 2, 3, 4-四氢化萘氧化生成酮, 收率为34%~97%.
12 Pd催化体系
2002年, Corey等[118]使用Pd(OAc)2作为催化剂, 以叔丁基过氧化氢作为氧化剂, 在碱性条件(K2CO3)下, 氧化环戊-2-烯-1-基苯、2, 3, 4, 5-四氢-1, 1'-联苯、1-(叔丁基)环己-1-烯、环戊烯和环己烯生成酮, 收率分别为71%、75%、85%、71%、79%.
2011年, SanMartin等[119]报道了两种钯络合物(图 5), 分别为NCN型(Cat. 1)和CNC型(Cat. 2).使用这两种钯络合物中的一种作为催化剂, 以氧气为氧化剂, 在聚乙二醇400中, 可以氧化乙苯、1, 2-二苯基乙烷、氧杂蒽、9, 10-二氢蒽、二苯基甲烷和芴生成酮, 使用NCN型钯络合物作为催化剂时, 收率分别为93%、92%、76%、90%、60%、98%, 使用CNC型钯络合物作为催化剂时, 收率分别为98%、89%、77%、95%、60%、94%.
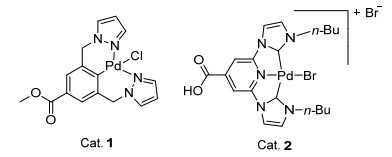 图 5
NCN型和CNC型钯络合物的结构
Figure Figure5.
Structures of NCN and CNC pincer palladium complexes
图 5
NCN型和CNC型钯络合物的结构
Figure Figure5.
Structures of NCN and CNC pincer palladium complexes
13 Ag催化体系
2012年, Burri等[120]报道了一种Ag/SBA-15催化剂, 使用叔丁基过氧化氢作为氧化剂, 可以氧化烷基苯、2-乙基萘、2-乙基蒽、二苯基甲烷、2, 3-二氢-1H-茚、芴、环己烷、环庚烷和环辛烷生成酮, 转化率为40%~99%, 选择性为69%~99%.
14 KBr催化体系
2008年, Kita等[121]报道了一种在KBr和蒙脱石-K-10的存在下, 亚碘酰苯氧化烷基苯、2, 3-二氢-1H-茚、1, 2, 3, 4-四氢化萘类化合物、二苯基甲烷、芴、氧杂蒽、蒽-9(10H)-酮、硫杂蒽和4-苄基吡啶生成酮的方法, 收率为43%~94%.作者认为亚碘酰苯和溴化钾反应生成活性物种1, 活性物种1分解产生溴自由基和自由基A, 在蒙脱石-K-10催化下, 自由基A将底物转变成碳自由基B, 碳自由基B和溴自由基反应生成苄溴中间体2, 苄溴中间体2水解产生仲醇, 仲醇被氧化生成酮(Scheme 10).
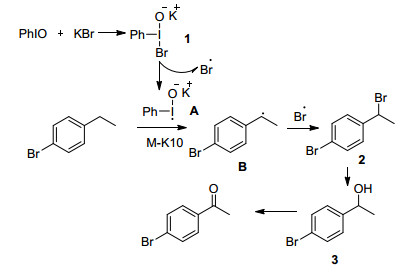 图式10
KBr和蒙脱石-K-10催化C—H键氧化反应的机理
Scheme10.
The reaction mechanism of the oxidation of C—H bonds with KBr and montmorillonite as catalyst
图式10
KBr和蒙脱石-K-10催化C—H键氧化反应的机理
Scheme10.
The reaction mechanism of the oxidation of C—H bonds with KBr and montmorillonite as catalyst
2012年, Togo等[122]报道了两种基于溴化钾和过硫酸氢钾的催化氧化体系(Scheme 11).两种方法的反应条件为: (1) 0.5 equiv.溴化钾, 3.0 equiv.过硫酸氢钾作为氧化剂, 硝基甲烷作为反应溶剂, 50 ℃反应24 h. (2) 1.2 equiv.溴化钾, 1.2 equiv.过硫酸氢钾作为氧化剂, 二氯甲烷和水的混合溶液作为反应溶剂(二氯甲烷:水=9:1), 光照, 室温反应24 h.方法(2) 对于含有吸电子基的乙苯类化合物的氧化效果好于方法(1), 但是方法(2) 对于甲苯类化合物氧化的选择性不好, 不能将氧化产物停留在醛, 而是氧化生成酸.
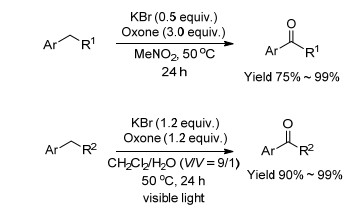 图式11
基于溴化钾和过硫酸氢钾的C—H键的氧化
Scheme11.
The oxidation of C—H bonds based on KBr and oxone
图式11
基于溴化钾和过硫酸氢钾的C—H键的氧化
Scheme11.
The oxidation of C—H bonds based on KBr and oxone
15 非金属催化体系
2004年, Hayashi等[123]使用活性炭作为催化剂, 用氧气作为氧化剂, 氧化9H-芴-2-甲醛、2-硝基-9H-芴、N-(9H-芴基-2-基)乙酰胺、蒽-9(10H)-酮、9H-氧杂蒽、9H-硫杂蒽、二苯基甲烷生成酮, 收率分别为77%、77%、71%、84%、94%、88%、26%.
2009年, 王志勇等[124]开发一种碘-吡啶催化体系, 使用叔丁基过氧化氢作为氧化剂, 在碱性环境(碳酸钾)中, 可以氧化二苯基甲烷类化合物、芴、2-苄基吡啶、烷基苯类化合物氧化生成酮, 收率为42%~99%.
2013年, 马丁等[125]报道了一种氮掺杂的碳材料, 使用叔丁基过氧化氢作为氧化剂, 可以氧化烷基苯类化合物、二苯基甲烷、芴、正己烷、环己烷生成酮, 收率为7%~99%.作者发现这种碳材料中氮含量对催化活性影响很大.
2015年, 李浩等[126]报道了一种含有磺酸钠基团的TEMPO催化剂, 使用NaNO2和HCl作为共催化剂, 以氧气作为氧化剂, 乙腈作为反应溶剂, 可以氧化氧杂蒽类化合物、吖啶类化合物、硫杂蒽和二苯基甲烷类化合物生成酮, 收率为52%~92% (Eq. 18).这种TEMPO催化剂能通过萃取回收, 作者对回收的TEMPO催化剂进行了6次循环试验, 催化活性没有降低.
2015年, 沈振陆等[127]报道了一种DDQ/TBN/O2 (TBN:亚硝酸叔丁酯)催化氧化体系. DDQ为催化剂, TBN为NO来源, 活化分子氧.该体系可以催化氧化二苯基甲烷类化合物生成酮, 收率为65%~99% (Eq. 19).
2015年, 高爽课等[128]选择苄基吡啶类化合物作为反应底物, 以氧气作为氧化剂, 研究了H4NI-AcOH协同催化吡啶侧链C—H键氧化生成酮的方法.优化后的反应条件为: 5 mol% H4NI, 10 mol% AcOH, 0.1 MPa氧气, 无溶剂条件下, 100 ℃反应24 h.在优化的反应条件下, 多种2-苄基吡啶类化合物和4-苄基吡啶类化合物均能顺利地发生氧化反应, 生成相应的酮, 收率为61%~97% (Scheme 12).该方法具有优异的化学选择性, 不氧化底物中的苄位甲基、苄位乙基和甲硫基.通过对反应机理的研究, 发现AcOH和苄基吡啶类化合物反应生成的盐促进了该氧化反应.对照实验证明了H4NI和AcOH协同催化的机理:在AcOH的参与下, H4NI被氧气氧化成I2, I2催化苄基吡啶类化合物氧化生成酮, AcOH不仅参与了H4NI被氧化成I2的过程, 还提高了该氧化反应的选择性.氧化剂是氧气, 而且不需要溶剂, 所以该氧化反应是非常绿色环保的. H4NI-AcOH可以催化克级规模的放大实验, 具有非常好的实用性.
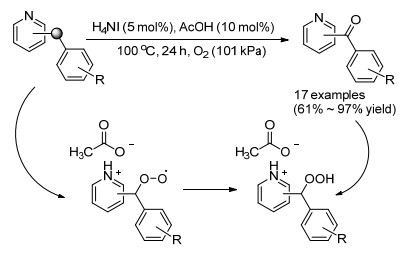 图式12
H4NI-AcOH协同催化C—H键氧化生成酮
Scheme12.
Oxidation of C—H bonds to ketones with H4NI-AcOH as synergistic catalyst
图式12
H4NI-AcOH协同催化C—H键氧化生成酮
Scheme12.
Oxidation of C—H bonds to ketones with H4NI-AcOH as synergistic catalyst
2015年, 雷爱文等[129]使用9-苯基-10-甲基吡啶盐作为光催化剂, 氧气作为氧化剂, 在可见光的照射下, 氧化二苯基甲烷类化合物生成酮, 收率为28%~77% (Eq. 20).作者通过同位素标记实验证明了产物酮中的O来自于氧气.
2016年, Shaabani等[130]使用氯磺酸和四甲基胍反应制备了一种TMG-SO3H催化剂, 使用溴酸钠作为氧化剂, TMG-SO3H可以催化2, 3-二氢-1H-茚、乙苯、氧杂蒽、1, 2, 3, 4-四氢化萘、二苯基甲烷和芴氧化生成酮, 收率分别为96%、90%、80%、88%、99%、90%.
16 结论和展望
酮是一类非常重要的化工中间体, 无论是在实验室基础研究中, 还是工业化生产中都占有非常重要的地位.虽然许多有机反应可以生成酮类化合物, 比如醇氧化反应、烯烃或炔烃氧化反应以及傅克反应等.相比上述反应, C—H键氧化生成酮具有反应路线短、原料价格低等优点.以往文献报道的C—H键氧化生成酮的研究主要是烷基苯、二苯基甲烷类化合物、芴类化合物、氧杂蒽类化合物、环己烯类化合物和饱和烷烃的氧化.迄今为止, 很少有文献报道杂环侧链C—H键氧化生成酮的方法, 含有杂环结构的酮又是十分重要的有机合成中间体, 因此杂环侧链C—H键氧化生成酮的研究具有重要的理论意义和应用背景.
-
-
[1]
Simandil, T. Catalytic Activation of Dioxygen by Metal Complexes, Kluwer Aeademie Publishers, Dordrecht, 1992, 1.
-
[2]
Suresh, A. K.; Sharmat, M. M.; Sridhar, T. Ind. Eng. Chem. Res. 2000, 39, 3958. doi: 10.1021/ie0002733
-
[3]
Ertl, G.; Knozinger, H.; Weitkam, P. J. Handbook of Heterogeneous Catalysis, Wiley & Sons-VCH Publishers, Weinheim, 1997.
-
[4]
王临红, 李红旗, 柯明, 赵锁奇, 范志明, 天然气化工, 2001, 16, 53. doi: 10.3969/j.issn.1007-3426.2001.02.001Wang, L. H.; Li, H. Q.; Ke, M.; Zhao, S. Q.; Fan, Z. M. Nat. Gas Chem. Ind. 2001, 16, 53. doi: 10.3969/j.issn.1007-3426.2001.02.001
-
[5]
Bernaard, M. Chem. Rev. 1992, 92, 1411. doi: 10.1021/cr00014a008
-
[6]
Dolphin, D.; Traylor, T. G.; Xie, L. Y. Acc. Chem. Res. 1997, 30, 251. doi: 10.1021/ar960126u
-
[7]
鲁平, 冯超, 罗德平, 化学学报, 2015, 73, 1315. doi: 10.3866/PKU.WHXB201504222Lu, P.; Feng, C.; Luo, D. P. Acta Chim. Sinica 2015, 73, 1315. doi: 10.3866/PKU.WHXB201504222
-
[8]
陈翠, 邱会华, 有机化学, 2016, 36, 826. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345348.shtmlChen, C.; Qiu, H. H. Chin. J. Org. Chem. 2016, 36, 826. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345348.shtml
-
[9]
徐星, 关耀辉, 徐东成, 李新生, 有机化学, 2016, 36, 850. http://sioc-journal.cn/Jwk_yjhx/CN/Y2016/V36/I4/850Xu, X.; Guan, Y. H.; Xu, D. C.; Li, X. S. Chin. J. Org. Chem. 2016, 36, 850. http://sioc-journal.cn/Jwk_yjhx/CN/Y2016/V36/I4/850
-
[10]
胡志勇, 童晓峰, 刘桂霞, 有机化学, 2015, 35, 539. http://sioc-journal.cn/Jwk_yjhx/CN/Y2015/V35/I3/539Hu, Z. Y.; Tong, X. F.; Liu, G. X. Chin. J. Org. Chem. 2015, 35, 539. http://sioc-journal.cn/Jwk_yjhx/CN/Y2015/V35/I3/539
-
[11]
陆庆全, 易红, 雷爱文, 化学学报, 2015, 73, 1245. doi: 10.3969/j.issn.0253-2409.2015.10.013Lu, Q. Q.; Yi, H.; Lei, A. W. Acta Chim. Sinica 2015, 73, 1245. doi: 10.3969/j.issn.0253-2409.2015.10.013
-
[12]
Shilov, A. E.; Shulpin, G. B. Chem. Rev. 1997, 97, 2879. doi: 10.1021/cr9411886
-
[13]
Crabtree, R. H. J. Organomet. Chem. 2004, 689, 4083. doi: 10.1016/j.jorganchem.2004.07.034
-
[14]
Giri, R.; Shi, B. R.; Engle, K. M.; Maugel, N.; Yu, J. Q. Chem. Soc. Rev. 2009, 38, 3242. doi: 10.1039/b816707a
-
[15]
Dick, A. R.; Sanford, M. S. Tetrahedron 2006, 62, 2439. doi: 10.1016/j.tet.2005.11.027
-
[16]
Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Syntheses, Patents, Applications, 4th ed., Georg Thieme, Stuttgart, 2001.
-
[17]
Reis, D. C.; Pinto, M. C. X.; Souza-Fagundes, E. M.; Wardell, S. M. S. V.; Wardell, J. L.; Beraldo, H. Eur. J. Med. Chem. 2010, 45, 3904. doi: 10.1016/j.ejmech.2010.05.044
-
[18]
Guyton, A. C.; Hall, J. E. Textbook of Medical Physiology, 11th ed., Philadelphia, PA, Elsevier Saunders, 2005.
-
[19]
Easmon, J.; Heinisch, G.; Pürstinger, G.; Langer, T.; Österreicher, J. K. J. Med. Chem. 1997, 40, 4420. doi: 10.1021/jm970255w
-
[20]
Crook, K. E.; Mcelval, S. M.; Mceivain, S. M. J. Am. Chem. Soc. 1930, 52, 4006. doi: 10.1021/ja01373a035
-
[21]
Huntress, E. H.; Walter, H. C. J. Am. Chem. Soc. 1948, 70, 3702. doi: 10.1021/ja01191a046
-
[22]
Watson, P. L. J. Am. Chem. Soc. 1983, 21, 6491.
-
[23]
Barton, D. H. R.; Taylor, D. K.; Hu, B. Tretrahetron Lett. 1996, 8, 1133.
-
[24]
Fridovich, I. Science 1978, 201, 875. doi: 10.1126/science.210504
-
[25]
Han, S.; Eltis, L. D.; Timmis, K. N.; Muchmore, S. W.; Bolin, J. T. Science 1995, 270, 976. doi: 10.1126/science.270.5238.976
-
[26]
Ford, P. C.; Fernandez, B. O.; Lim, M. D. Chem. Rev. 2005, 105, 2439. doi: 10.1021/cr0307289
-
[27]
Barton, D. H. R.; Doller, D. Acc. Chem. Res. 1992, 25, 504. doi: 10.1021/ar00023a004
-
[28]
Barton, D. H. R. Chem. Soc. Rev. 1996, 25, 237. doi: 10.1039/cs9962500237
-
[29]
Stavropoulos, P.; C-Elenligil-C-Etin, R.; Tapper, A. E. Acc. Chem. Res. 2001, 34, 745. doi: 10.1021/ar000100+
-
[30]
Perkins, M. J. Chem. Soc. Rev. 1996, 25, 229. doi: 10.1039/cs9962500229
-
[31]
Li, S. J.; Wang, Y. G. A. Tetrahedron Lett. 2005, 46, 8013. doi: 10.1016/j.tetlet.2005.09.055
-
[32]
Chen, M. S.; White, M. C. Science 2010, 327, 566. doi: 10.1126/science.1183602
-
[33]
Pieber, B.; Kappe, C. O. Green Chem. 2013, 15, 320. doi: 10.1039/c2gc36896j
-
[34]
Szabó, F.; Pethó, B.; Gonda, Z.; Novák, Z. RSC Adv. 2013, 3, 4903. doi: 10.1039/c3ra22856h
-
[35]
Napoly, F.; Kieffer, R.; Gérard, L. J.; Henry, C. G.; Draye, M.; Andrioletti, B. Tetrahedron Lett. 2015, 56, 2517. doi: 10.1016/j.tetlet.2015.03.115
-
[36]
Fan, S.; Luan, Y. Wang, J. J.; Gao, H. Y.; Zhang, X. W.; Wang, G. J. Mol. Catal. A: Chem. 2015, 404~405, 186.
-
[37]
Al-Hunaiti, A.; Raisanen, M.; Repo, T. Chem. Commun. 2016, 52, 2043. doi: 10.1039/C5CC07597A
-
[38]
Muhldorf, B.; Wolf, R. Angew. Chem., Int. Ed. 2016, 55, 427. doi: 10.1002/anie.201507170
-
[39]
Komiya, N.; Naota, T.; Murahashi, S. I. Tetrahedron Lett. 1996, 37, 1633. doi: 10.1016/0040-4039(96)00074-3
-
[40]
Velusamy, S.; Punniyamurthy, T. Tetrahedron Lett. 2003, 44, 8955. doi: 10.1016/j.tetlet.2003.10.016
-
[41]
Wu, X. H.; Gorden, A. E. V. Eur. J. Org. Chem. 2009, 503.
-
[42]
Li, Y. C.; Lee, T. B.; Wang, T. Y.; Gamble, A. V. Gorden, A. E. V. J. Org. Chem. 2012, 77, 4628. doi: 10.1021/jo300372q
-
[43]
Houwer, J. D.; Tehrani, K. A.; Maes, B. U. W. Angew. Chem., Int. Ed. 2012, 51, 2745. doi: 10.1002/anie.201108540
-
[44]
Song, G. Q.; Lu, Y. X.; Zhang, Q.; Wang, F.; Ma, X. K.; Huang, X. F.; Zhang, Z. H. RSC Adv. 2014, 4, 30221. doi: 10.1039/C4RA04076G
-
[45]
Ang, W. J.; Lam, Y. L. Org. Biomol. Chem. 2015, 13, 1048. doi: 10.1039/C4OB02017K
-
[46]
Liu, J. M.; Zhang, X.; Yi H.; Liu, C.; Liu, R.; Zhang, H.; Zhuo, K. L.; Lei, A. W. C. Angew. Chem., Int. Ed. 2015, 54, 1261. doi: 10.1002/anie.201409580
-
[47]
Lau, T. C.; Mak, C. K. Chem. Commun. 1995, 943.
-
[48]
Che, C. M.; Cheng, K. W.; Chan, M. C. W.; Lau, T. C.; Mak, C. K. J. Org. Chem. 2000, 65, 7996. doi: 10.1021/jo0010126
-
[49]
Yi, C. S.; Kwon, K. H.; Lee, D. W. Org. Lett. 2009, 11, 1567. doi: 10.1021/ol900097y
-
[50]
Yusubov, M. S.; Zagulyaeva, A. A.; Zhdankin, V. V. Chem. Eur. J. 2009, 15, 11091. doi: 10.1002/(ISSN)1521-3765
-
[51]
Kojima, M.; Oisaki, K.; Kanai, M. Tetrahedron Lett. 2014, 55, 4736. doi: 10.1016/j.tetlet.2014.06.038
-
[52]
Dai, W. Li, J.; Li, G. S.; Yang, H.; Wang, L. Y.; Gao, S. Org. Lett. 2013, 15, 4138. doi: 10.1021/ol401812h
-
[53]
Dai, W.; Shang, S. S.; Chen, B.; Li, s.; Wang, L. Y.; Ren, L. H.; Gao, S. J. Org. Chem. 2014, 79, 6688. doi: 10.1021/jo501178k
-
[54]
Dai, W.; Li, G. S.; Chen, B.; Wang, L. Y.; Gao, S. Org. Lett. 2015, 17, 904. doi: 10.1021/acs.orglett.5b00018
-
[55]
Dai, W.; Li, J.; Chen, B.; Li, G. S.; Lv, Y.; Wang, L. Y.; Gao, S. Org. Lett. 2013, 15, 5658. doi: 10.1021/ol402612x
-
[56]
Dai, W.; Li, G. S.; Wang, L. Y.; Chen, B.; Shang, S. S.; Lv, Y.; Gao, S. RSC Adv. 2014, 4, 46545. doi: 10.1039/C4RA09832C
-
[57]
Dai, W.; Mi, Y.; Lv, Y.; Chen, B.; Li, G. S.; Chen, G. G.; Gao, S. Adv. Synth. Catal. 2016, 4, 667.
-
[58]
Dai, W.; Lv, Y.; Wang, L. Y.; Shang, S. S.; Chen, B.; Li, G. S.; Gao, S. Chem. Commun. 2015, 51, 11268. doi: 10.1039/C5CC03657G
-
[59]
Lee, N. H.; Lee, C. S.; Jung, D. S. Tetrahedron Lett. 1998, 39, 1385. doi: 10.1016/S0040-4039(98)00030-6
-
[60]
Pan, J. F.; Chen, K. J. Mol. Catal. A: Chem. 2001, 176, 19. doi: 10.1016/S1381-1169(01)00238-2
-
[61]
Shing, T. K. M.; Yeung, Y. Y.; Su, P. L. Org. Lett. 2006, 8, 3149. doi: 10.1021/ol0612298
-
[62]
Burange, A. S.; Kale, S. R.; Jayaram, R. V. Tetrahedron Lett. 2012, 53, 2989. doi: 10.1016/j.tetlet.2012.03.091
-
[63]
Shen, D. Y.; Miao, C. X.; Wang, S. F. Xia, C. G.; Sun, W. Org. Lett. 2014, 16, 1108. doi: 10.1021/ol4037083
-
[64]
Zou, C.; Zhao, M.; Wu, C. D. Catal. Commun. 2015, 66, 116. doi: 10.1016/j.catcom.2015.03.031
-
[65]
Shaabani, A.; Hezarkhani, Z.; Badali, E. RSC Adv. 2015, 5, 61759. doi: 10.1039/C5RA10522F
-
[66]
Ren, L. H.; Wang, L. Y.; Lv, Y.; Shang, S. S.; Bo Chen, B.; Gao, S. Green Chem. 2015, 17, 2369. doi: 10.1039/C4GC02471K
-
[67]
任兰会, 王连月, 吕迎, 李国松, 高爽, 催化学报, 2016, 37, 1216.Ren, L. H.; Wang, L. Y.; Lü, Y.; Li, G. S.; Gao, S. Chin. J. Catal. 2016, 37, 1216.
-
[68]
Ishii, Y.; Iwahama, T.; Sakaguchi, S.; Nakayama, K.; Nishiyama, Y. J. Org. Chem. 1996, 61, 4520. doi: 10.1021/jo951970l
-
[69]
Minisci, F.; Punta, C.; Recupero, F.; Pedulli, F. F.; Franco, G. J. Org. Chem. 2002, 67, 2671. doi: 10.1021/jo016398e
-
[70]
Shaabani, A.; Farhangi, E.; Rahmati, A. Appl. Catal. A-Gen. 2008, 338, 14. doi: 10.1016/j.apcata.2007.12.014
-
[71]
Wang, J. Q.; He, L. N. New J. Chem. 2009, 33, 1637. doi: 10.1039/b908993d
-
[72]
陈磊, 李斌栋, 徐秀霞, 刘大斌, 中国化学快报, 2013, 24, 849.Chen, L.; Li, B. D.; Xu, Q. X.; Liu, D. B. Chin. Chem. Lett. 2013, 24, 849.
-
[73]
Islam, S. M.; Ghosh, K.; Molla, R. A.; Roy, S. A.; Salam, N.; Iqubal, M. A. J. Organomet. Chem. 2014, 774, 61. doi: 10.1016/j.jorganchem.2014.10.010
-
[74]
Shaabani, A.; Keshipour, S.; Hamidzad, M.; Shaabani, S. J. Mol. Catal. A: Chem. 2014, 395, 494. doi: 10.1016/j.molcata.2014.09.003
-
[75]
Hruszkewycz, D. P.; Miles, K. C.; Thielb, O. R.; Stahl, S. S. Chem. Sci. 2017, 8, 1282. doi: 10.1039/C6SC03831J
-
[76]
Wu, X. F. Tetrahedron Lett. 2012, 53, 6123. doi: 10.1016/j.tetlet.2012.08.149
-
[77]
Akhlaghinia, B.; Ebrahimabadi, H.; Goharshadi, E. K.; Samiee, S. Rezazadeh, S. J. Mol. Catal. A: Chem. 2012, 357, 67. doi: 10.1016/j.molcata.2012.01.020
-
[78]
Pahari, S. K.; Pal, P.; Sinhamahapatra, A.; Saha, A.; Santra, C.; Ghosh, S. C.; Chowdhury, B.; Panda, A. B. RSC Adv. 2015, 5, 45144. doi: 10.1039/C5RA05441A
-
[79]
Bien, S.; Segal, Y. J. Org. Chem. 1977, 42, 1685. doi: 10.1021/jo00430a003
-
[80]
Noels, A. F.; Hubert, A. J.; Teyssie, P. J. Organomet. Chem. 1979, 166, 79. doi: 10.1016/S0022-328X(00)91422-0
-
[81]
Moody, C. J.; Palmer, F. N. Tetrahedron Lett. 2001, 43, 139.
-
[82]
Doyle, M. P.; Westrum, L. J.; Wolthuis, W. N. E.; See, M. M.; Boone, W. P.; Bagheri, V.; Pearson, M. M. J. Am. Chem. Soc. 1993, 115, 958. doi: 10.1021/ja00056a021
-
[83]
Doyle, M. P.; Ren, T. Chiral Dirhodium(Ⅱ) Catalysts and Their Applications, Wiley, New York, 2001, 49, 113.
-
[84]
Das, K.; Kadish, K. M.; Bear, J. L. Inorg. Chem. 1978, 17, 930. doi: 10.1021/ic50182a027
-
[85]
Zhu, T. P.; Ahsan, M. Q.; Malinski, T.; Kadish, K. M.; Bear, J. L. Inorg. Chem. 1984, 23, 2. doi: 10.1021/ic00169a002
-
[86]
Catino, A. J.; Forslund, R. E.; Doyle, M. P. J. Am. Chem. Soc. 2004, 126, 13622. doi: 10.1021/ja045330o
-
[87]
Catino, A. J.; Nichols, J. M.; Choi, H.; Gottipamula, S.; Doyle, M. P. Org. Lett. 2005, 7, 5167. doi: 10.1021/ol0520020
-
[88]
Abudureheman, W.; Xiarepati, T.; Lu, C. D. Eur. J. Org. Chem. 2012, 3088.
-
[89]
Wang, Y.; Kuang, Y.; Wang, Y. H. Chem. Commun. 2015, 51, 5852. doi: 10.1039/C4CC10336J
-
[90]
Matano, Y.; Nomura, H. J. Am. Chem. Soc. 2001, 123, 6443. doi: 10.1021/ja010584k
-
[91]
Firouzabadi, H.; Iranpoor, N.; Amani, K. Synth. Commun. 2004, 34, 3587. doi: 10.1081/SCC-200031036
-
[92]
Salvador, J. A. R.; Silvestre, S. M. Tetrahedron Lett. 2005, 46, 2581. doi: 10.1016/j.tetlet.2005.02.080
-
[93]
Banik, B. K.; Venkatraman, M. S.; Mukhopadhyay, C.; Becker, F. F. Tetrahedron Lett. 1998, 39, 7247. doi: 10.1016/S0040-4039(98)01556-1
-
[94]
Bonvin, Y.; Callens, E.; Larrosa, I.; Henderson, D. A.; Oldham, J.; Burton, A. J.; Barrett, A. G. M. Org. Lett. 2005, 7, 4549. doi: 10.1021/ol051765k
-
[95]
Hoffmann-Roder, A.; Krause, N. Org. Biomol. Chem. 2005, 3, 387. doi: 10.1039/B416516K
-
[96]
Cinellu, M. A.; Minghetti, G.; Cocco, F.; Stoccoro, S.; Zucca, A.; Manassero, M. Angew. Chem., Int. Ed. 2005, 44, 6892. doi: 10.1002/(ISSN)1521-3773
-
[97]
Hashmia, S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896. doi: 10.1002/(ISSN)1521-3773
-
[98]
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239. doi: 10.1021/cr068434l
-
[99]
Yao, T.; Zhang, X.; Larock, R. C. J. Am. Chem. Soc. 2004, 126, 11164. doi: 10.1021/ja0466964
-
[100]
Sherry, B. D.; Toste, F. D. J. Am. Chem. Soc. 2004, 126, 15978. doi: 10.1021/ja044602k
-
[101]
Wei, C.; Li, C. J. J. Am. Chem. Soc. 2003, 125, 9584. doi: 10.1021/ja0359299
-
[102]
Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2004, 126, 11806. doi: 10.1021/ja046112y
-
[103]
Yao, X.; Li, C. J. J. Am. Chem. Soc. 2004, 126, 6884. doi: 10.1021/ja0482637
-
[104]
Teles, J. H.; Brode, S. Chabanas, M. Angew. Chem., Int. Ed. 1998, 37, 1415. doi: 10.1002/(ISSN)1521-3773
-
[105]
Arcadi, A.; Giuseppe, S. D.; Marinelli, F.; Rossi E. Adv. Synth. Catal. 2001, 343, 443. doi: 10.1002/(ISSN)1615-4169
-
[106]
Shi, X.; Gorin, D. J.; Tosite, F. D. J. Am. Chem. Soc. 2005, 127, 5802. doi: 10.1021/ja051689g
-
[107]
Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2005, 127, 6962. doi: 10.1021/ja051110e
-
[108]
Mamane, V.; Gress, T.; Krause, H.; Furstner, A. J. Am. Chem. Soc. 2004, 126, 8654. doi: 10.1021/ja048094q
-
[109]
Luzung, M. R.; Markham, J. P.; Toste, F. D. J. Am. Chem. Soc. 2004, 126, 10858. doi: 10.1021/ja046248w
-
[110]
Yang, C. G.; He, C. J. Am. Chem. Soc. 2005, 127, 6966. doi: 10.1021/ja050392f
-
[111]
Shapiro, N. D.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 4160. doi: 10.1021/ja070789e
-
[112]
Li, H. R.; Li, Z. P.; Shi, Z. J. Tetrahedron 2009, 65, 1856. doi: 10.1016/j.tet.2008.12.055
-
[113]
Kühn, F. E.; Santos, A. M.; Gonçalves, I. S.; Romão, C. C.; Lopes, A. D. Appl. Organomet. Chem. 2001, 15, 43. doi: 10.1002/(ISSN)1099-0739
-
[114]
Ghorai, P.; Dussault, P. H. Org. Lett. 2009, 11, 213. doi: 10.1021/ol8023874
-
[115]
Ghorai, P.; Dussault, P. H. Org. Lett. 2008, 10, 4577. doi: 10.1021/ol801859c
-
[116]
Bernini, R.; Mincione, E.; Barontini, M.; Fabrizi, G.; Pasqualetti, M.; Tempesta, S. Tetrahedron 2006, 62, 7733. doi: 10.1016/j.tet.2006.05.069
-
[117]
Peng, H.; Lin, A.; Zhang, Y.; Jiang, H. L.; Zhou, J. C.; Cheng, Y. X.; Zhu, C. J.; Hu, H. W. ACS Catal. 2012, 2, 163. doi: 10.1021/cs2003577
-
[118]
Yu, J. Q.; Corey, E. J. Org. Lett. 2002, 4, 2727. doi: 10.1021/ol0262340
-
[119]
Urgoitia, G.; SanMartin, R.; Herrero, M. T.; Domínguez, E. Green Chem. 2011, 13, 2161. doi: 10.1039/c1gc15390k
-
[120]
Anand, N.; Reddy, K. H. P.; Prasad, G. V. S.; Rao, K. S. R.; Burri, D. R. Catal. Commun. 2012, 23, 5. doi: 10.1016/j.catcom.2012.02.023
-
[121]
Dohi, T.; Takenaga, N.; Takenaga, G. A.; Fujioka, H.; Kita, Y. J. Org. Chem. 2008, 73, 7365. doi: 10.1021/jo8012435
-
[122]
Moriyama, K.; Takemura, M.; Togo, H. Org. Lett. 2012, 14, 2414. doi: 10.1021/ol300853z
-
[123]
Kawabata, H.; Hayashi, M. Tetrahedron Lett. 2004, 45, 5457. doi: 10.1016/j.tetlet.2004.05.030
-
[124]
Zhang, J. T.; Wang, Z. T.; Wang, Y.; Wan, C. F.; Zheng, X. Q.; Wang, Z. Y. Green Chem. 2009, 11, 1973. doi: 10.1039/b919346b
-
[125]
Gao, Y. J.; Hu, G.; Zhong, J.; Shi, Z. J.; Zhu, Y. S.; Su, D. S.; Wang, J. G.; Bao, X. H.; Ma, D. Angew. Chem., Int. Ed. 2013, 52, 2109. doi: 10.1002/anie.v52.7
-
[126]
Zhang, Z. G.; Gao, Y.; Liu, Y.; Li, J. J.; Xie, H. X.; Li, H.; Wang, W. Org. Lett. 2015, 17, 5492. doi: 10.1021/acs.orglett.5b02877
-
[127]
Ma, J. Q.; Hu, Z. M.; Li, M. C.; Zhao, W. J.; Hua, X. Q.; Mo, W. M.; Hua, B. X.; Sun, N.; Shen, Z. L. Tetrahedron 2015, 71, 6733. doi: 10.1016/j.tet.2015.07.042
-
[128]
Ren, L. H.; Wang, L. Y.; Lü, Y.; Li, G. S.; Gao, S. Org. Lett. 2015, 17, 2078. doi: 10.1021/acs.orglett.5b00602
-
[129]
Yi, H.; Bian, C. L.; Hu, X.; Niua, L. B.; Lei, A. W. Chem. Commun. 2015, 51, 14046. doi: 10.1039/C5CC06015J
-
[130]
Shaabani, A.; Laeini, M. S.; Shaabani, S.; Seyyedhamzeh, M. New J. Chem. 2016, 40, 2079. doi: 10.1039/C5NJ02215K
-
[1]
-

图式3 基于苯甲酰基吡啶类化合物开发的医药化合物
Scheme 3 Pharmaceutical compounds coming from benzoylpyridines

图式4 Fe(S, S-PDP)催化天然产物氧化生成酮
Scheme 4 Oxidation of natural product to ketone with Fe(S, S-PDP) as catalyst

图式5 (pymox-Me2)RuCl2]+BF4-的合成路线
Scheme 5 The synthetic route of [(pymox-Me2)RuCl2]+BF4-

图式6 串联的碘苯和三氯化钌催化C—H键氧化生成酮的过程
Scheme 6 Process of the oxidation of C—H bonds to ketones with tandem iodobenzene and RuCl3 as catalyst

图式7 双三氟甲磺酸锰催化C—H键氧化生成酮的方法
Scheme 7 Oxidation of C—H bonds to ketones with Mn(OTf)2 as catalyst

图式9 Rh2(cap)4催化C—H键氧化生成酮
Scheme 9 The oxidation of C—H bonds to ketones with Rh2(cap)4 as catalyst

图式10 KBr和蒙脱石-K-10催化C—H键氧化反应的机理
Scheme 10 The reaction mechanism of the oxidation of C—H bonds with KBr and montmorillonite as catalyst
-


 下载:
下载:
















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 88
- 文章访问数: 5717
- HTML全文浏览量: 1033
 下载:
下载:
