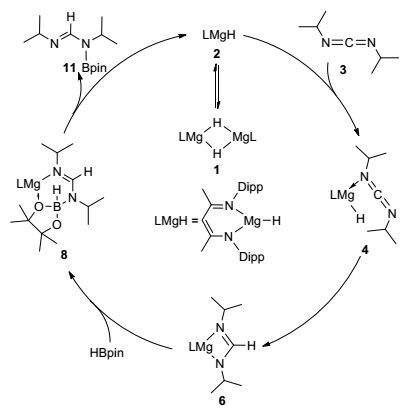
 图 2
镁催化碳二亚胺的硼氢化反应路径
Figure 2.
Proposed mechanism for magnesium catalysed hydroboration of carbodiimides
图 2
镁催化碳二亚胺的硼氢化反应路径
Figure 2.
Proposed mechanism for magnesium catalysed hydroboration of carbodiimides
引用本文:
徐冬冬, 单春晖, 白若鹏, 蓝宇. 碱土金属催化碳二亚胺硼氢化反应机理的理论研究[J]. 有机化学,
2017, 37(5): 1231-1236.
doi:
10.6023/cjoc201701033

Citation: Xu Dongdong, Shan Chunhui, Bai Ruopeng, Lan Yu. Mechanism of Alkaline Earth Metal Catalyzed Hydroboration of Carbodiimides: A Theoretical Study[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1231-1236. doi: 10.6023/cjoc201701033
Citation: Xu Dongdong, Shan Chunhui, Bai Ruopeng, Lan Yu. Mechanism of Alkaline Earth Metal Catalyzed Hydroboration of Carbodiimides: A Theoretical Study[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1231-1236. doi: 10.6023/cjoc201701033
碱土金属催化碳二亚胺硼氢化反应机理的理论研究
摘要:
本文运用密度泛函理论(Density functional theory,DFT)算法,研究了β-二亚胺氢化碱土金属复合物催化碳二亚胺硼氢化反应的机理.计算结果表明,当使用镁复合物作为催化剂时,该反应催化循环中的活性催化物种为镁氢复合物.从镁氢复合物的生成开始,该反应经过碳氮双键插入镁-氢共价键,硼氮偶联成键,以及从硼到镁的氢负离子转移等步骤,从而再生镁氢复合物并释放产物.该反应的决速步为负氢原子从硼到镁的转移过程.此外,通过理论计算拓展预测了钙、锶等其他碱土金属复合物催化剂的反应机理.研究结果表明,和镁催化氢化反应不同,钙、锶参与反应的活性催化物种是碳二亚胺插入相应氢化金属复合物后所生成的对应的氨基金属络合物.以此化合物作为催化循环的开始,反应经历硼氮偶联成键后,可直接与碳二亚胺发生硼碳之间的氢转移,得到产物并再生活性催化物种.钙、锶复合物作为催化剂时经历不同反应机理的原因是,它们的原子半径远远大于镁,因此可以与额外的碳二亚胺结合,并发生协同氢转移反应.理论计算表明,当使用钙、锶复合物作为催化剂时,反应决速步活化能低于镁催化的反应途径.因此,如果使用钙、锶复合物作为催化剂将会获得更温和的反应条件.
English
Mechanism of Alkaline Earth Metal Catalyzed Hydroboration of Carbodiimides: A Theoretical Study
Abstract:
Density functional theory (DFT) calculations are employed to study the mechanism of alkaline earth metal catalyzed hydroboration of carbodiimides. Our theoretical study revealed that the active catalytic species is a hydridemagnesium complex when magnesium is used as catalyst. The catalytic cycle starts with a C=N bond insertion into Mg-H bond followed by a B-N bond formation. A hydride transfer from boron to magnesium regenerates the active catalytic species and yields the hydroboration product. This process is considered to be the rate-determining step. Moreover, the mechanism of calcium or strontium catalyzed corresponding reactions was also studied theoretically. Alternatively, DFT calculations showed that the active catalytic species is amide-metal complex, which could be generated by the carbodiimide insertion into metal-hydride bond. In this catalytic cycle, amide-metal complex reacts with borane to form a B-N bond. After the coordination of another molecular carbodiimide, a concerted hydride transfer takes place from boron to carbon, which yields the final product and regenerates the active species amide-metal. The different reaction pathway with calcium or strontium catalyzed corresponding reactions could be attributed to that the radius of calcium or strontium is larger than that of magnesium significantly. Thus, those two metals would be coordinated with an extra carbodiimide molecule, which is the precursor for the concerted hydride transfer. The DFT calculations showed that the activation free energy for the rate-determining step with calcium or strontium catalyst is much lower than that with magnesium catalyst. Therefore, a mild reaction condition might be found with calcium or strontium as catalyst for corresponding reactions.
-
催化化学是世界化学工业的中心之一.与当前化学合成工业中的催化过程一样, 实验室中大多数的化学合成反应都需要如钌、铑、钯、金、银、铂等第五、六周期过渡金属作为催化剂[1].由于这些元素含量在自然界中比较稀少且开采难度大[2], 因此含有上述元素的催化剂往往价格相对昂贵, 这无疑限制了过渡金属催化反应在合成化学中的应用.为了解决这个问题, 进一步拓展金属催化反应的应用范围, 越来越多的化学家开始将目光转向元素周期表中的s区、p区元素.其中, 镁、钙等碱土金属元素由于在自然界中大量存在[2], 廉价易得, 并且其单质性质不像碱金属那样活泼难以利用, 近期引起了化学工作者的关注.
在过去几十年, 随着对碱土金属元素的结构和化学性质越发深入的认识, 科学家合成了一系列+2氧化态的有机碱土金属化合物[3].这些碱土金属化合物由于具有更低的价格和更小的毒性, 因此有着广泛的应用前景.在所有碱土金属中, 镁是最早、最广泛被应用于有机合成中的.自从格氏试剂被合成以来, 科学家们发展了大量的镁试剂, 并将其应用于各种有机合成反应中[4].例如, 有机镁试剂用于催化含硼化合物(HBpin、9-BBN等)与酯类、酮类、胺类、吡啶类、亚胺类、腈类、酰胺类的硼氢化加成反应[5].鉴于有机镁试剂催化范围之广, 并且其催化体系日渐复杂化, 对其反应机理的研究显得愈发重要.
2016年, Hill等[6]报道了镁催化碳二亚胺硼氢化反应, 其中使用了β-二亚胺烷基镁复合物作为催化剂.作者认为该催化剂首先与频哪醇硼烷反应生成活性催化物种β-二亚胺氢化镁.在催化循环中, β-二亚胺氢化镁真正催化碳二亚胺的硼氢化.他们尝试使用氢化镁物种探究了反应的机理.实验表明该物种同样能够与碳二亚胺进行反应并生成同样目标产物. β-二亚胺氢化镁表现出的特殊催化性质引起了我们的关注, 但该类反应机理仍未明确.在此, 我们使用密度泛函理论(DFT)方法对其反应机理进行理论计算研究.我们选取Hill课题组报道的镁催化碳二亚胺硼氢化反应作为机理研究的模板(Eq. 1).在此反应中, 二异丙基碳二亚胺(3)与频哪醇硼烷HBpin反应生成硼基甲脒11, 选用的有机镁催化剂为β-二亚胺氢化镁复合物1.
1 计算方法
本文中的计算均采用GAUSSIAN 09量子化学计算程序包完成[7].密度泛函方法B3-LYP[8]和基组6-31G(d)[9](对金属锶采用基组SDD[10])用来对反应势能曲线上的中间体和过渡态进行结构优化.简谐频率振动计算用来确认每一个驻点是稳定的中间体或者过渡态, 同时也用来得到每一个驻点焓和Gibbs自由能的校正值.为了得到更加准确的能量数值, 在气相驻点结构的基础上, 使用Truhlar课题组开发的M06-L[11]方法和6-311+G(d, p)[12]基组(对金属锶采用基组SDD)来计算各驻点的溶剂化单点能.溶剂化计算时采用连续可极化溶剂化模型SMD[13], 以苯作为溶剂.文中讨论的能量值是M06-L计算得到的溶剂中Gibbs自由能.关键反应中间体和过渡态的几何构型由CYLview[14]软件绘制.
2 结果与讨论
如图 2所示, 结合实验条件以及催化剂β-二亚胺氢化镁复合物的性质和通过实验推测的机理, 我们提出了一条镁催化碳二亚胺的硼氢化反应可能的反应路径.首先, 二聚的氢化镁复合物1解离为两个单体氢化镁2并进入催化循环.在催化循环中, 碳二亚胺3与单体氢化镁中的金属配位形成中间体4.接下来, 经过由镁到碳的氢转移后形成氨基镁复合物中间体6.随后中间体6与频哪醇硼烷HBpin反应得到含有氮—硼成键的中间体8, 最后, 经过一个从硼到镁的负氢转移生成目标产物硼基甲脒11, 而此处产生的β-二亚胺氢化镁2则作为活性催化物种进入下一个催化循环.
图 2
镁催化碳二亚胺的硼氢化反应路径
Figure 2.
Proposed mechanism for magnesium catalysed hydroboration of carbodiimides
我们通过理论计算模拟了我们推测反应机理中势能的变化趋势.如图 3所示, 我们选取二聚β-二亚胺氢化镁复合物1作为势能面的相对Gibbs自由能零点.二聚体1解离为单体2, 然后碳二亚胺3与2配位形成中间体4, 接着经过硼氢转移的过渡态5-ts(其相对Gibbs自由能为13.1 kcal/mol), 生成中间体6, 其几何结构如图 4所示.然后6与频哪醇硼烷反应, 经过活化能为16.6 kcal/mol硼氮偶联的过渡态7-ts, 生成的比较稳定硼化甲脒镁复合物中间体8(其相对Gibbs自由能为-36.7 kcal/mol).在最后的产物生成阶段, 我们发现硼氢转移过渡态9-ts的活化能高达26.4 kcal/mol, 随后生成的配合物10解离, 生成目标产物11, 而生成的2则作为活性催化物种进入下一个催化循环.虽然从理论计算的角度判断该反应是可能发生的, 但是我们仍然希望能找到其它活化能更低的反应路径.因此我们还考虑了另外三种硼氢转移的反应路径.
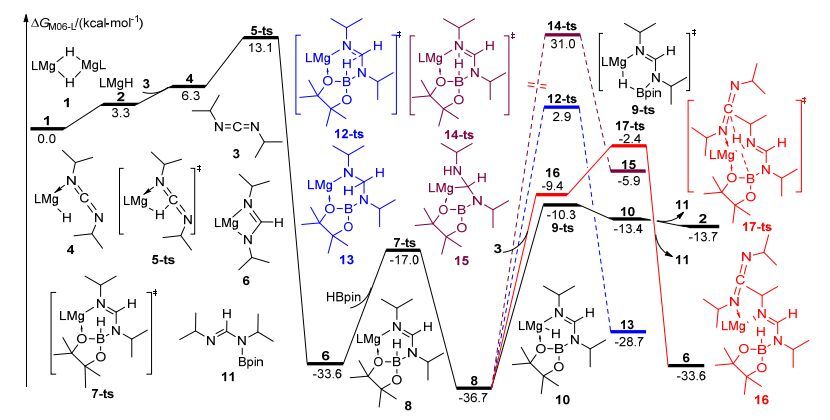 图 3
镁催化碳二亚胺的硼氢化反应势能面
Figure 3.
Potential energy surface for magnesium catalyzed hydroboration of carbodiimides
图 3
镁催化碳二亚胺的硼氢化反应势能面
Figure 3.
Potential energy surface for magnesium catalyzed hydroboration of carbodiimides
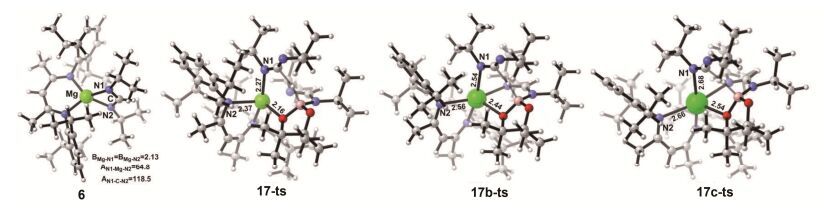 图 4
6、17-ts、17b-ts和17c-ts的几何结构
Figure 4.
Optimized structures of 6, 17-ts, 17b-ts and 17c-ts Bond lengths (Å) and bond angles (°)
图 4
6、17-ts、17b-ts和17c-ts的几何结构
Figure 4.
Optimized structures of 6, 17-ts, 17b-ts and 17c-ts Bond lengths (Å) and bond angles (°)
首先, 我们尝试计算了硼氢转移到不饱和碳上这条路径, 即中间体8经过B—H键断裂C—H键形成的过渡态12-ts生成中间体13的过程.结果表明, 生成中间体13需要的活化能为39.6 kcal/mol, 说明该反应难以完成.此外, 我们还研究了另外一条直接协同硼氢转移的反应路径, 即中间体8经过一个B—H键断裂N—H键形成的过渡态14-ts生成中间体15.我们发现其活化能高达67.7 kcal/mol, 证明该反应在动力学上是不可行的.此外, 我们还计算了中间体8中与硼相连的氢原子直接转移到另一分子的碳二亚胺的不饱和碳上这一可能性.即首先可逆生成碳二亚胺与8配位的中间体16, 然后经过一个B—H键断裂C—H键形成的氢转移过渡态17-ts直接得到中间体6和产物11.计算表明, 这一步的活化能高达34.3 kcal/mol.通过观察该过渡态结构(图 4), 我们认为活化能较高的原因是镁的原子半径较小并且催化剂周围的空间位阻较大.而导致空间位阻较大的主要基团是β-二亚胺镁复合物上的二异丙基苯基和碳二亚胺上的异丙基.综合以上对硼氢转移的不同反应路径的研究, 我们认为中间体8是经过分子内氢转移从而生成产物11, 并且分子内氢转移是整个反应的决速步.
根据元素周期律基本原理, 同一主族元素往往具有相似的化学性质.基于这一规律, 在确定了镁催化碳二亚胺的硼氢化反应机理之后, 尝试研究同族其他碱土金属钙和锶在类似情况下的催化性能.按照之前提出类似的反应路径, 我们计算了钙、锶催化碳二亚胺硼氢化反应的势能面(图 5).如图 5a所示, 当使用钙络合物1b作为催化剂时, 在类似的反应路径中, 经过四元环氢转移过渡态5b-ts生成中间体6b的活化能为15.3 kcal/mol, 比相应镁催化氢转移的活化能高了2.2 kcal/mol.中间体6b与频哪醇硼烷HBpin反应, 经过硼氮偶联过渡态7b-ts, 生成的比较稳定硼化甲脒镁复合物中间体8b, 其活化能为10.1 kcal/mol, 比镁催化的活化能低了6.5 kcal/mol.但是在产物生成阶段, 中间体8b中, 氢原子由硼转移到钙经过的过渡态9b-ts活化能为29.8 kcal/mol, 相比镁催化的过程更难发生.我们认为, 由于钙的金属性比镁略强且半径较大, 因此与负氢的结合相对较弱, 导致此过程活化能升高.考虑到钙的原子半径比镁大, 周围可以容纳更多配位原子, 因此我们研究了中间体8b和碳二亚胺直接发生氢转移的反应路径.中间体8b和碳二亚胺配位生成中间体16b.计算表明, 8b与碳二亚胺的结合焓为-13.3 kcal/mol, 说明金属钙与碳二亚胺有较强的相互作用.之前我们也曾尝试找过镁催化过程中对应的中间体, 但由于位阻原因并未优化得到相应结构.当中间体16b生成后, 与硼相连的氢可以经过一个六元环过渡态17b-ts转移至碳二亚胺中间不饱和碳原子上.之后释放产物11, 得到活性催化物种6b.这一过程活化能仅为21.9 kcal/mol, 比经过过渡态9b-ts发生分子内直接硼氢转移释放产物生成活性催化剂2b的活化能低了7.9 kcal/mol.因此我们认为在钙催化碳二亚胺的硼氢化反应过程中, 6b为实际的活性催化物种.当反应通过氢转移得到6b并启动后, 首先发生硼氮偶联, 得到中间体8b.接下来中间体8b与碳二亚胺直接发生氢转移, 释放产物11, 并再生活性催化物种6b, 完成一个催化循环.此过程中, 表观活化自由能仅为21.9 kcal/mol.
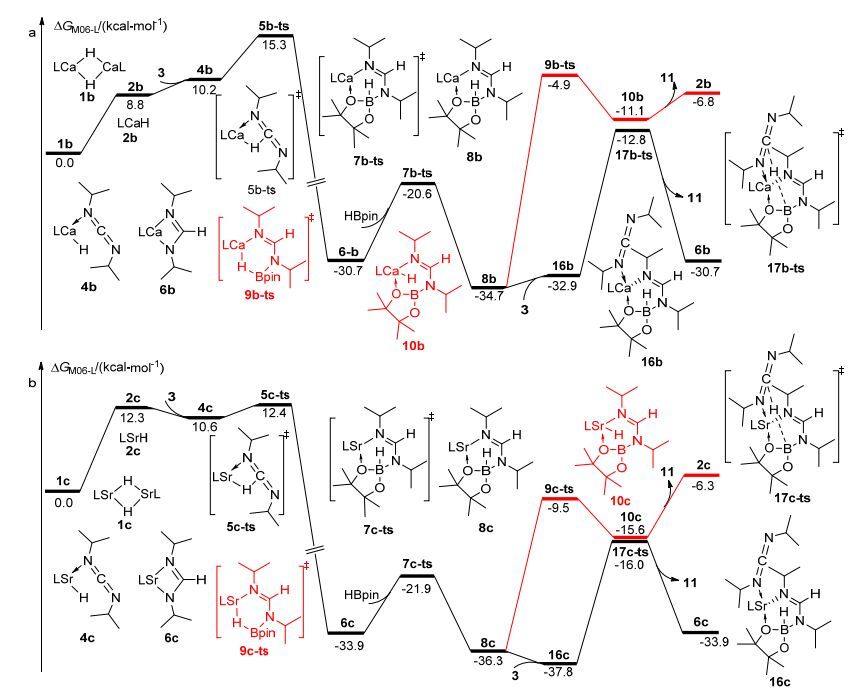 图 5
钙(a)或锶(b)催化碳二亚胺的硼氢化反应势能面
Figure 5.
Potential energy surface for calcium (a) or strontium (b) catalysed hydroboration of carbodiimides
图 5
钙(a)或锶(b)催化碳二亚胺的硼氢化反应势能面
Figure 5.
Potential energy surface for calcium (a) or strontium (b) catalysed hydroboration of carbodiimides
如图 5b所示, 我们也考虑了锶催化剂对于这类反应的催化作用.同样考虑了与钙、镁催化类似的两个途径.计算表明, 中间体6c的生成同样是一个不可逆的放热过程.在中间体6c与频哪醇硼烷HBpin反应生成硼化甲脒镁复合物中间体8c的过程中, 反应活化能为12.0 kcal/mol, 低于镁催化该过程的活化能.与钙催化反应机理类似, 锶活性催化物种8c同样更倾向于与碳二亚胺结合得到中间体16c, 并经历过渡态17c-ts发生类似的直接氢转移反应, 得到活性催化物种6c, 并释放出产物11.该反应决速步同样是从16c到6c的氢转移过程, 表观活化能为21.8 kcal/mol, 与钙催化过程类似.计算结果表明, 发生从硼到碳的直接氢转移时, 对于镁催化体系来说, 活化能高达34.3 kcal/mol; 而对于钙、锶催化体系, 相应步骤的活化能则分别为21.9和21.8 kcal/mol.该能量的差别体现在过渡态17-ts、17b-ts、17c-ts的几何结构差别上.如图 4所示, 在过渡态17-ts中, 由于镁原子半径较小, Mg—N(1)、Mg—N(2)、Mg—O的键长分别仅为2.27、2.37、2.16 Å.由于多个原子过于靠近金属中心, 这使得体系空间位阻很大, 因此对应很高的活化能.钙原子的半径比镁大, 在过渡态17b-ts中, Ca—N(1)、Ca—N(2)、Ca—O的键长分别为2.54、2.56、2.44 Å.因此, 过渡态17b-ts对应的能量较低.随着原子半径的增大, 在过渡态17c-ts中, 相应的Sr—N(1)、Sr—N(2)、Sr—O的键长分别为2.68、2.66、2.54 Å, 与钙催化体系类似.因此, 对应的活化能也与之接近.
3 结论
本文运用密度泛函理论计算, 系统研究了碱土金属催化碳二亚胺硼氢化反应的机理.计算结果表明, 当使用镁作为催化剂时, 活性催化物种为氢化镁中间体.催化循环中, 首先碳二亚胺插入镁-氢键, 然后发生硼氮偶联得到六元环中间体, 接下来的氢转移重新得到氢化镁中间体并释放产物.此外, 本文从元素周期律出发, 推测并利用密度泛函验证了碱土金属和锶催化二亚胺硼氢化的过程.研究表明, 对于钙和锶催化的过程, 活性催化物种是对应的氨基钙或氨基锶络合物.与镁催化体系不同, 由于钙和锶的半径更大, 导致此类化合物在形成六元环中间体后, 可以与配位的第二分子碳二亚胺发生氢转移反应, 并得到氢化产物.理论计算表明, 当使用钙、锶复合物作为催化剂时, 反应决速步活化能低于镁催化的反应途径.因此, 我们认为如果使用钙、锶复合物作为催化剂, 将会获得更温和的反应条件.我们希望这一结论能为后续碱土金属催化的不饱和键氢化反应的设计和应用提供指导.
辅助材料(Supporting Information) 所有结构的能量值和原子坐标.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
(a) Diez-Gonzalez, S. ; Marion, N. ; Nolan, S. P. Chem. Rev. 2009, 109, 3612. (b) Dobereiner, G. E. ; Crabtree, R. H. Chem. Rev. 2010, 110, 681. (c) Werkmeister, S. ; Junge, K. ; Beller, M. Org. Process Res. Dev. 2014, 18, 289. (d) Shao, Z. ; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745. (e) D'Souza, D. M. ; Muller, T. J. Chem. Soc. Rev. 2007, 36, 1095. (f) Zhang, H. ; Sun, H. ; Li, X. Chin. J. Org. Chem. 2016, 36, 2843 (in Chinese). (仉花, 孙宏建, 李晓燕, 有机化学, 2016, 36, 2843. ) (g) Dou, R. ; Tan, X. ; Fan, Y. ; Pei, Y. ; Qiao, M. ; Fan, K. ; Sun, B. ; Zong, B. Acta Chim. Sinica 2016, 74, 503 (in Chinese). (窦镕飞, 谭晓荷, 范义秋, 裴燕, 乔明华, 范康年, 孙斌, 宗保宁, 化学学报, 2016, 74, 503. ) (h) Gao, A. ; Ye, Q. ; Yu, J. ; Liu, W. Chin. J. Org. Chem. 2017, 37, 47 (in Chinese). (高安丽, 叶青松, 余娟, 刘伟平, 有机化学, 2017, 37, 47. ) (i) Wu, R. ; Yang, W. ; Cheng, G. ; Li, Y. ; Yang, D. Chin. J. Org. Chem. 2016, 36, 2368 (in Chinese). (仵瑞华, 杨文, 程果, 李玥, 杨定乔, 有机化学, 2016, 36, 2368. )
-
[2]
Yaroshevsky, A. A. Geochem. Int. 2006, 44, 48. doi: 10.1134/S001670290601006X
-
[3]
(a) Bonyhady, S. J.; Green, S. P.; Jones, C.; Nembenna, S.; Stasch, A. Angew. Chem. Int. Ed. 2009, 48, 2973. (b) Green, S. P.; Jones, C.; Stasch, A. Angew. Chem., Int. Ed. 2008, 47, 9079. (c) Arrowsmith, M.; Shepherd, W. M.; Hill, M. S.; Kociok-Kohn, G. Chem. Commun. 2014, 50, 12676. (d) Arrowsmith, M.; Hill, M. S.; Kociok-Kohn, G. Chem. Eur. J. 2015, 21, 10548. (e) Rochat, R.; Lopez, M. J.; Tsurugi, H.; Mashima, K. ChemCatChem 2016, 8, 10. (f) Arrowsmith, M.; Crimmin, M. R.; Hill, M. S.; Lomas, S. L.; Heng, M. S.; Hitchcock, P. B.; Kociok-Kohn, G. Dalton Trans. 2014, 43, 14249. (g) Barrett, A. G. M.; Crimmin, M. R.; Hill, M. S.; Hitchcock, P. B.; Procopiou, P. A. Organometallics 2007, 26, 4076.
-
[4]
(a) Liu, B. ; Roisnel, T. ; Carpentier, J. F. ; Sarazin, Y. Angew. Chem. , Int. Ed. 2012, 51, 4943. (b) Rossin, A. ; Peruzzini, M. Chem. Rev. 2016, 116, 8848. (c) Hill, M. S. ; Liptrot, D. J. ; Weetman, C. Chem. , Soc. Rev. 2016, 45, 972. (d) Schwamm, R. J. ; Day, B. M. ; Mansfield, N. E. ; Knowelden, W. ; Hitchcock, P. B. ; Coles, M. P. Dalton Trans. 2014, 43, 14302. (e) Intemann, J. ; Lutz, M. ; Harder, S. Organometallics 2014, 33, 5722. (f) Wang, J. ; Cui, D. Chin. J. Org. Chem. 2016, 36, 1163 (in Chinese). (王剑, 崔冬梅, 有机化学, 2016, 36, 1163. ) (g) Ma, M. ; Wang, W. ; Yao, W. Chin. J. Org. Chem. 2016, 36, 72 (in Chinese). (马猛涛, 王未凡, 姚薇薇, 有机化学, 2016, 36, 72. )
-
[5]
(a) Lampland, N. L.; Hovey, M.; Mukherjee, D.; Sadow, A. D. ACS Catal. 2015, 5, 4219. (b) Liptrot, D. J.; Hill, M. S.; Mahon, M. F.; Wilson, A. S. Angew. Chem. Int. Ed. 2015, 54, 13362. (c) Arrowsmith, M.; Hadlington, T. J.; Hill, M. S.; Kociok-Kohn, G. Chem., Commun. 2012, 48, 4567. (d) Arrowsmith, M.; Hill, M. S.; Kociok-Kohn, G. Chem. Eur. J. 2013, 19, 2776. (e) Mukherjee, D.; Ellern, A.; Sadow, A. D. Chem. Sci. 2014, 5, 959. (f) Weetman, C.; Anker, M. D.; Arrowsmith, M.; Hill, M. S.; Kociok-Köhn, G.; Liptrot, D. J.; Mahon, M. F. Chem. Sci. 2016, 7, 628. (g) Arrowsmith, M.; Hill, M. S.; Hadlington, T.; Kociok-Köhn, G.; Weetman, C. Organometallics 2011, 30, 5556.
-
[6]
Weetman, C.; Hill, M. S.; Mahon, M. F. Chem. Eur. J. 2016, 22, 7158. doi: 10.1002/chem.201600681
-
[7]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013.
-
[8]
(a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (b) Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623. (c) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785.
-
[9]
(a) Francl, M. M. J. Chem. Phys. 1982, 77, 3654. (b) Hehre, W. J. J. Chem. Phys. 1972, 56, 2257. (c) Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213.
-
[10]
(a) Dolg, M.; Stoll, H.; Preuss, H. J. Chem. Phys. 1989, 90, 1730. (b) Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. J. Chem. Phys. 1987, 86, 866.
-
[11]
(a) Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41, 157. (b) Qi, X.; Zhang, H.; Shao, A.; Zhu, L.; Xu, T.; Gao, M.; Liu, C.; Lan, Y. ACS Catal. 2015, 5, 6640. (c) Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101. (d) McDonald, T. M.; Mason, J. A.; Kong, X.; Bloch, E. D.; Gygi, D.; Dani, A.; Crocella, V.; Giordanino, F.; Odoh, S. O.; Drisdell, W. S.; Vlaisavljevich, B.; Dzubak, A. L.; Poloni, R.; Schnell, S. K.; Planas, N.; Lee, K.; Pascal, T.; Wan, L. F.; Prendergast, D.; Neaton, J. B.; Smit, B.; Kortright, J. B.; Gagliardi, L.; Bordiga, S.; Reimer, J. A.; Long, J. R. Nature 2015, 519, 303. (e) Jentzsch, A. V.; Emery, D.; Mareda, J.; Nayak, S. K.; Metrangolo, P.; Resnati, G.; Sakai, N.; Matile, S. Nat. Commun. 2012, 3, 905. (f) Suess, A. M.; Ertem, M. Z.; Cramer, C. J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 9797. (g) Li, Y. F.; Liu, Z. P.; Liu, L.; Gao, W. J. Am. Chem. Soc. 2010, 132, 13008. (h) Ferrighi, L.; Hammer, B.; Madsen, G. K. J. Am. Chem. Soc. 2009, 131, 10605.
-
[12]
(a) Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650. (b) McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639.
-
[13]
(a) Li, Y.; Liu, S.; Qi, Z.; Qi, X.; Li, X.; Lan, Y. Chem. Eur. J. 2015, 21, 10131. (b) Qi, X.; Li, Y.; Zhang, G.; Li, Y.; Lei, A.; Liu, C.; Lan, Y. Dalton Trans. 2015, 44, 11165. (c) Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378. (d) Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 4538. (e) Yu, Z.; Qi, X.; Li, Y.; Liu, S.; Lan, Y. Org. Chem. Front. 2016, 3, 209.
-
[14]
Legault, C. Y. CYLview 1.0b, Université de Sherbrooke, Canada, 2009; http://www.cylview.org.
-
[1]
-

图 2 镁催化碳二亚胺的硼氢化反应路径
Figure 2 Proposed mechanism for magnesium catalysed hydroboration of carbodiimides

图 3 镁催化碳二亚胺的硼氢化反应势能面
Figure 3 Potential energy surface for magnesium catalyzed hydroboration of carbodiimides

图 4 6、17-ts、17b-ts和17c-ts的几何结构
Figure 4 Optimized structures of 6, 17-ts, 17b-ts and 17c-ts Bond lengths (Å) and bond angles (°)
-


 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 18
- 文章访问数: 1565
- HTML全文浏览量: 304
 下载:
下载:
