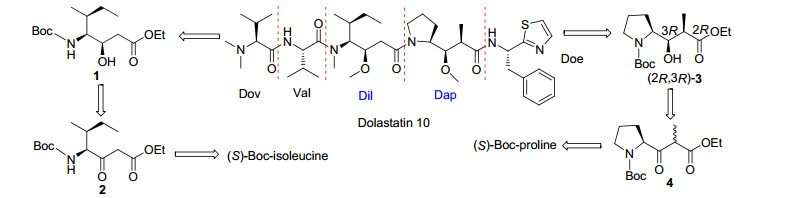
 图式1
Dolastatin 10的逆合成分析
图式1.
Retrosynthetic analysis of dolastatin 10
图式1
Dolastatin 10的逆合成分析
图式1.
Retrosynthetic analysis of dolastatin 10
引用本文:
鲁文玉, 于文静, 孙德群. 海洋生物活性肽在药物研发中的应用进展[J]. 有机化学,
2017, 37(7): 1681-1700.
doi:
10.6023/cjoc201612062

Citation: Lu Wenyu, Yu Wenjing, Sun Dequn. Advances in Application of Marine Bioactive Peptides in Drug Development[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1681-1700. doi: 10.6023/cjoc201612062
Citation: Lu Wenyu, Yu Wenjing, Sun Dequn. Advances in Application of Marine Bioactive Peptides in Drug Development[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1681-1700. doi: 10.6023/cjoc201612062
海洋生物活性肽在药物研发中的应用进展
摘要:
海洋是地球上资源最为丰富的领域.海洋生物生存环境复杂,使它们形成了具有特殊结构的活性物质;加之科技的进步,人们也日益关注如何从海洋生物中寻获新的活性物质,这使得海洋生物活性肽受到广泛关注.生物活性肽具有多种活性,经研究表明,该类物质具有抗氧化、抗高血压、抗病毒、抗肿瘤等活性;且具有低毒、高度靶向专一性和生物活性强等优点.目前,从海兔、芋螺、海鞘、海绵、海洋真菌、软体动物中提取出的生物活性肽或类似物,部分已上市或已进入临床试验阶段.针对这些海洋生物活性肽的研究现状,就其来源、合成方法、化学结构特点、活性、作用机制以及临床有效性及安全性等方面进行综述,并展望了该领域今后的发展方向.
English
Advances in Application of Marine Bioactive Peptides in Drug Development
Abstract:
The ocean is the most resource-rich area on earth. The living environment of marine life is complicated, so that they form a special structure of the active substances. Coupled with the progress of science and technology, people are increasingly concerned about how to find new active substances from marine life, which makes the marine bioactive peptide is widely concerned. Bioactive peptides have a variety of activities, the study shows that these substances have antioxidant, antihypertensive, anti-virus and anti-tumor activity. It has the advantages of low toxicity, high targeting specificity and strong biological activity. At present, some of bioactive peptides extracted from sea cucumbers, conus, ascidians, sponges, marine fungi, molluscs, or analogues derived from these compounds have entered the market or clinical stage. The current research status of marine bioactive peptides is reviewed including their source, synthetic method, chemical structure, activity, mechanism, clinical efficacy and safety. Research prospects in this field are discussed.
-
Key words:
- marine peptide
- / bioactivity
- / pharmaceutical
- / clinical trial
-
海洋面积约占全球总面积的70%, 占据90%的生物圈, 海洋中生物种类达到全球生物多样性的半数, 因此海洋是一个丰富的天然活性化合物库[1].海洋具有特殊的物理和化学条件[2], 如高盐、弱碱, 深海区甚至具有黑暗、寒冷、高压等复杂特点; 加之物种之间强烈的生存竞争, 使得海洋生物常采取化学手段进行自我保护[3]; 逐渐形成了不同于陆地生物的活性物质, 如活性肽、多元不饱和脂肪酸、固醇类、酶、多糖、抗氧化剂和色素等[4].随着培养技术、分子生物学技术的发展[5], 从海洋生物中获取新的活性化合物不断取得新进展, 尤其是内源性多肽活性作用的发现以及天然生物活性肽的靶向作用机制的研究, 使得肽类成为海洋活性物质的首要候选资源[6].
生物活性肽由3~20个氨基酸残基构成, 具有多种生物活性[7], 如抗氧化性、抗高血压、抗HIV病毒、抗增殖、抗凝血、钙离子螯合、抗肥胖、抗糖尿病等活性, 其活性由分子大小、氨基酸种类和次序决定[8].因其具有低毒、高效、高选择性等优点而被应用于食品与制药领域[9].目前, 国内外对海洋生物活性肽进行了大量研究.海洋生物活性肽来源主要有海绵、海鞘、海葵、芋螺、海藻、鱼类、软体动物、甲壳类动物, 海洋细菌和真菌等[6, 10].现在许多研究人员从海洋生物中提取了许多活性较高的天然活性肽, 如Lee等[11]从大马哈鱼中通过胰蛋白酶水解分离得到具有抑制血管紧张素I转化酶的活性肽, 其氨基酸序列为Gly-Leu-Pro-Leu-Asn-Leu-Pro, 后又以天然的活性肽为基础合成了活性更高的三肽Gly-Leu-Pro, IC50值为9.08 μmol•L-1; Zhan等[12]从海绵中提取出五种新型环形肽reniochalistatins A~E, 其中reniochalistatin E对RPMI-8226细胞和MGC-803细胞有细胞毒性作用, IC50值分别为4.9和9.7 μmol•L-1.由此可以看出, 一些海洋生物活性肽有一定的成药潜力.尤其是近年来, 海洋探测技术的发展, 使得海洋药物的开发得到了巨大进展.目前, 从海兔、芋螺、海鞘、海绵、海洋真菌、软体动物中提取的具有活性的天然肽, 或以天然肽为母体合成的衍生物已进入全球的药物市场及临床试验阶段[13].本文将综述已上市或处于临床试验阶段的海洋生物活性肽, 并按照活性化合物来源分类, 介绍活性化合物化学结构特点、合成、活性、作用机制以及耐受性和毒副作用等, 希望对从事此类研究的科研人员提供一定的参考依据.
1 海兔毒素
Pettit等[14]于1972年开始展开了对印度洋中海兔的提取物所含抗肿瘤活性成分的研究, 他们发现其提取物可以治疗小鼠P388淋巴细胞白血病, 延长小鼠寿命.随后他们又分离得到一系列具有抑制细胞增殖、抗肿瘤活性的肽类化合物, 命名为Dolastatin, 其中活性最强的是Dolastatin 10和Dolastatin 15.此后, 许多研究者对Dolastatin 10和Dolastatin 15展开了进一步研究, 并合成了一系列衍生物.其中, 以auristatin系列、Tasidotin和LU-103793活性最好, 于1995年进入临床试验.
1.1 Dolastatin 10
Dolastatin 10是一种线性五肽, 包含五个子单元, dolavaline (Dov), valine (Val), dolaisoleuine (Dil), dolaproine (Dap)和dolaphemine (Doe)[15]. Mordant等[16]通过逆合成分析发现Dap和Dil是合成的两个关键片段(Scheme 1), 因此他们先合成了N-Boc-Dil (9) (Scheme 2)和N-Boc-(2R, 3R)-Dap (11) (Scheme 3).再以9和11为原料合成Dolastatin 10 (Scheme 4), 在合成过程中焦炭酸二乙酯(DEPC)和三氟乙酸(TFC)主要用于偶联和脱保护. Dolastatin 10作用机制主要是通过与β微管蛋白结合, 使细胞分裂停滞, 最终导致细胞死亡.在某些临床前试验中, Dolastatin 10对一些实体瘤表现出了较好活性, 如对四种非小细胞肺癌(NCI-H69, NCI-H82, NCI-H446, NCI-H510) IC50值范围为0.032~0.184 nmol•L-1[17].
图式1
Dolastatin 10的逆合成分析
图式1.
Retrosynthetic analysis of dolastatin 10
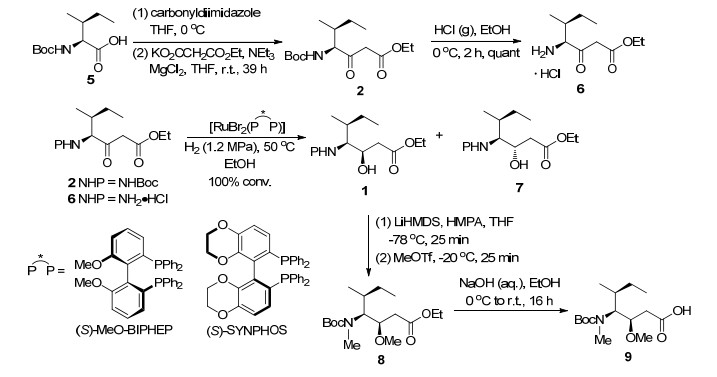 图式2
N-Boc-Dil的合成
图式2.
Synthesis of N-Boc-Dil
图式2
N-Boc-Dil的合成
图式2.
Synthesis of N-Boc-Dil
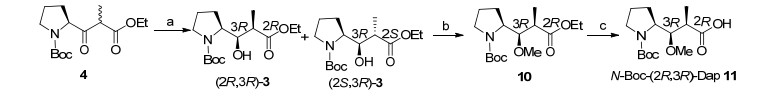 图式3
N-Boc-(2R, 3R)-Dap的合成
图式3.
Synthesis of N-Boc-(2R, 3R)-Dap
图式3
N-Boc-(2R, 3R)-Dap的合成
图式3.
Synthesis of N-Boc-(2R, 3R)-Dap
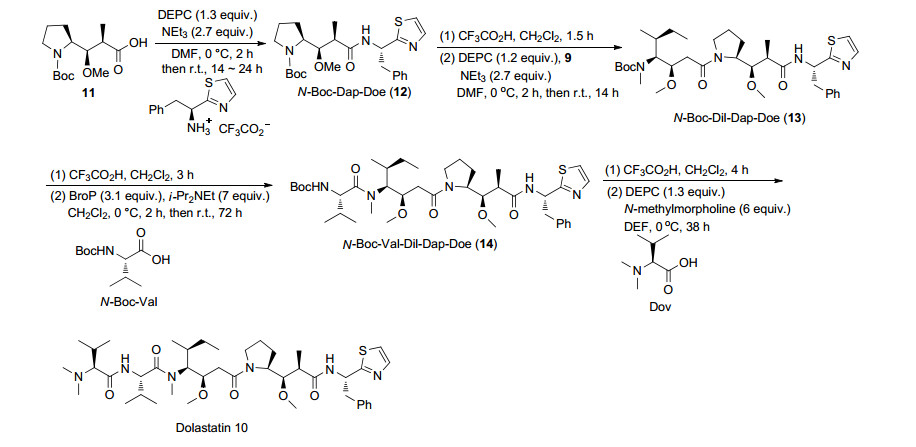 图式4
Dolastatin 10的合成
图式4.
Synthesis of dolastatin 10
图式4
Dolastatin 10的合成
图式4.
Synthesis of dolastatin 10
Perez等[18]和Kindler等[19]对Dolastatin 10治疗晚期乳腺癌和晚期胰胆癌开展了Ⅱ期临床试验.虽然, Dolastatin 10在体外试验效果较好, 但临床试验结果表明, 实验组患者的中位生存期较短, 治疗效果不佳.随后, Pettit和Miyazaki等为了检验结构改变对活性的影响, 对Dolastatin 10的C端或N端进行修饰, 合成了一系列Dolastatin 10的衍生物, 命名为auristatins系列, 如auristatin E, monomethyl auristatin D (MMAD)和monomethyl auristatin E等化合物(图 1), 但MMAD单药治疗的体内实验表明其抗肿瘤活性不佳.之后, Senter等发现Auristatins的N端仲胺可加载连接体以偶合单克隆抗体, 形成高效、高靶向性的抗体-药物偶合物[20].
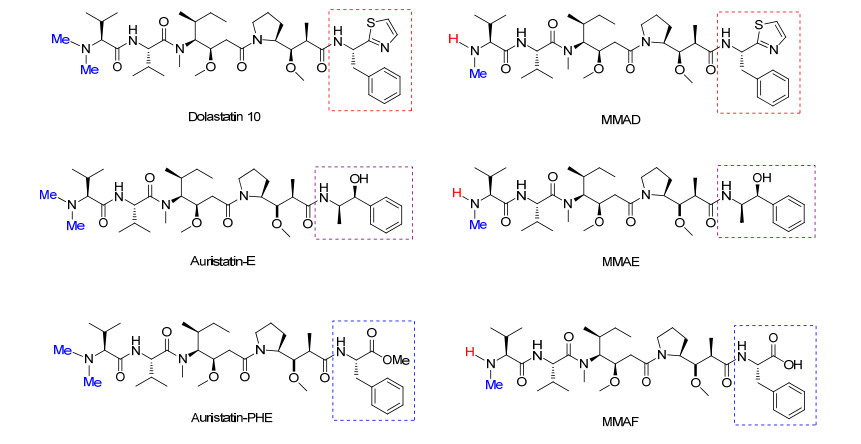 图 1
Dolastatin 10及衍生物的结构
Figure 1.
Structures of dolastatin 10 and derivatives
图 1
Dolastatin 10及衍生物的结构
Figure 1.
Structures of dolastatin 10 and derivatives
1.2 基于auristatins的抗体-药物偶合物
抗体-药物偶合物(ADC)是具有高活性、高靶向性的新兴抗癌治疗试剂.它由抗体、连接体、细胞毒性试剂三部分构成.细胞毒性试剂的效力、靶向的选择和连接体的稳定性等是影响新型ADC的因素.目前, 连接体技术的发展和新型高效的细胞毒性试剂的出现都极大地促进了ADC的发展[21].在海洋生物活性肽中, 以Dolastatin 10为母体化合物合成的auristatins系列被广泛应用于ADC[22].现约有45种Auristatins类的ADC处于临床研究阶段[23].下面将以泊仁妥西凡多汀, Depatuxizumab mafodotin, Glembatumumab Vedotin, Polatuzumabvedotin及AGS-67E为例介绍auristatins类ADC.
1.2.1 泊仁妥西凡多汀
伯仁妥西凡多汀(Brentuximab vedotin, SGN-35) 由日本武田(Takeda)制药公司及美国西雅图遗传学公司(Seattle Genetics)联合研发, 于2011年由美国食品药品管理局(FDA)批准上市, 用于治疗霍奇金淋巴瘤(Hodgkin’s lymphoma, HL)和系统性间变性大细胞淋巴瘤(Anaplastic large cell lymphoma, ALCL)[24].
伯仁妥西凡多汀可与表达CD30的肿瘤细胞高度特异性结合, 它由cAC10 (CD30抗体)、连接体和细胞毒试剂monomethyl auristatin E (MMAE)三部分组成.伯仁妥西凡多汀所使用的连接体为缬氨酸-瓜氨酸(vc), 为便于连接体(vc)和毒性试剂水解且使水解部位和细胞毒试剂的活性部位远离, 故用对氨基苄氧羰基(PABC)作为间隔体键合于MMAE与连接体之间, 形成vc-PABC-MMAE结构, 再将其通过马来酰亚胺己酰基(mc)与单克隆抗体中的半胱氨酸连接[25]. CD30是膜上的糖蛋白, 属于肿瘤坏死因子(TNF)受体家族成员, 它在HL, ALCL等多种淋巴瘤表面高度表达. cAC10本身具有对抗CD30的活性, 但在临床Ⅱ期时活性消失, 为增强其活性, 合成了伯仁妥西凡多汀.细胞毒试剂MMAE是从印度洋无壳软体动物截尾海兔Dolabella auricularia分离得到Dolastatin 10的合成衍生物.在生理条件下, MMAE具有高效、溶解度高、稳定性好等优点. MMAE的药理作用与Dolastatin 10相似, 通过抑制微管蛋白的聚合, 使表达CD30的淋巴瘤细胞在G2-M阶段增长停滞, 从而使细胞凋亡.泊仁妥西凡多汀作用机制是通过cAC10靶向识别肿瘤细胞上的CD30, 与CD30结合, 然后经细胞网格蛋白的调节, 泊仁妥西凡多汀被內吞进入细胞, 在溶酶体中被蛋白酶水解释放MMAE, MMAE在细胞内发挥作用, 杀死细胞[26]. Mc-vc-PBAC-MMAE的合成是通过用N-甲基缬氨酸类化合物15代替N, N-二甲基缬氨酸类化合物来保护auristatain E, 从而形成MMAE (18).用mc-vc-PBAC (19)进一步修饰化合物18得到mc-vc-PBAC-MMAE (20), 最后使还原的cAC10与化合物20结合即可得到伯仁妥西凡多汀(Scheme 5)[27].
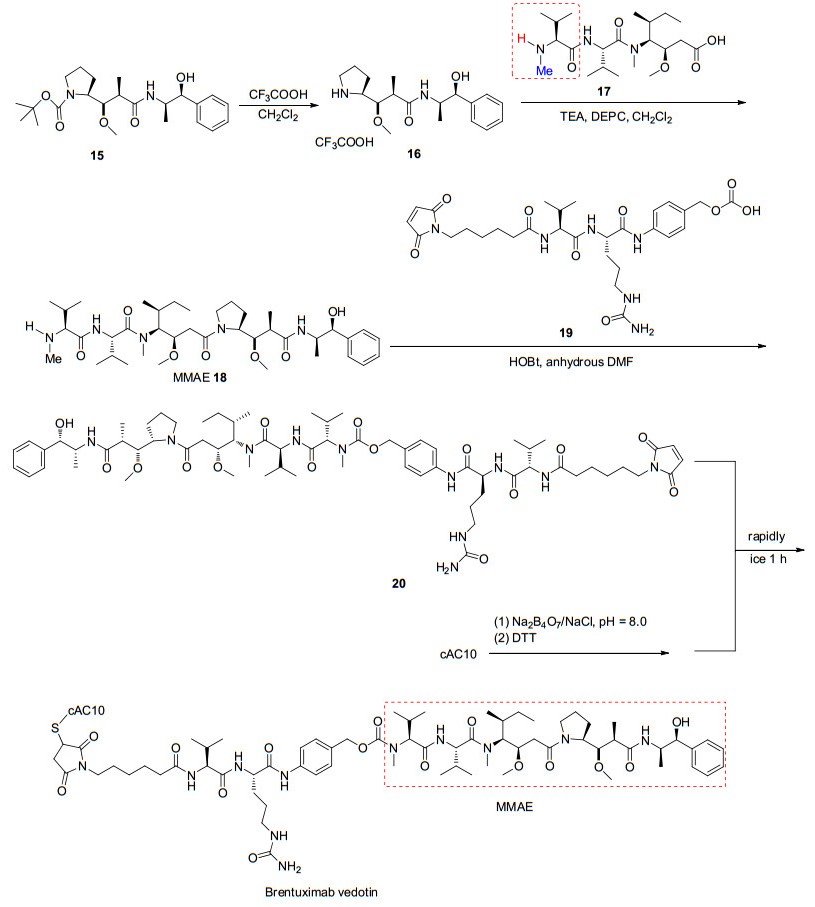 图式5
Brentuximab vedotin的合成
图式5.
Synthesis of brentuximab vedotin
图式5
Brentuximab vedotin的合成
图式5.
Synthesis of brentuximab vedotin
为评价Brentuxmab vedotin对自身干细胞移植后或两次既往化疗后疾病有进展不能接受移植的HL患者和1次既往化疗后疾病进展ALCL患者的治疗效果, 在北美和欧洲的78个地点进行了Ⅲ期临床试验.试验选择了329例符合要求的患者随机分配至Brentuxmab vedotin组(n=165) 和安慰剂组(n=164).结果显示, 与安慰剂组相比, Brentuxmab vedotin组的患者无进展生存期得到显著改善([HR] 0.57, 95% CI 0.40~0.81; p=0.0013), Brentuxmab vedotin组患者, 中位无进展生存期平均约为42.9个月(95% CI 30.4~42.9), 安慰剂组的患者, 中位无进展生存期平均约为24.1个月(11.5~not estimabl). Ⅲ期临床安全性试验统计167名Brentuximab vedotin组患者和160名安慰剂组患者的不良反应(Adversedrug reaction, ADR)发生情况, 两组ADR发生率分别为98%和89%, 严重不良反应率(≥Grade 3) 分别为56%和32%.在Brentuximab vedotin组中, 最常见的不良反应是外周感觉神经病变, 发生率为30.5%;因外周神经病变导致51人停止使用Brentuximab vedotin治疗.其它常见的ADR有中性粒细胞减少、上呼吸道感染、疲劳、外周运动神经病、恶心、咳嗽、腹泻、发热、呕吐等[28].
1.2.2 Depatuxizumab mafodotin (ABT-414)
Depatuxizumab mafodotin靶向作用于表皮因子受体(EGFR), 由抗体(ABT-806)、连接体和细胞毒试剂monomethyl auristatin F (MMAF)构成(图 2)[29].在Depatuxizumab mafodotin中, 使用的连接体是稳定性较高的马来酰亚胺己酰基(mc). FDA和欧洲药物管理局(EMA)于2014年批准Depatuxizumab mafodotin作为治疗胶质细胞瘤的孤儿药[30].
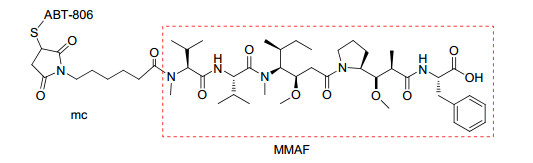 图 2
Depatuxizumab mafodotin的结构
Figure 2.
Structure of depatuxizumab mafodotin
图 2
Depatuxizumab mafodotin的结构
Figure 2.
Structure of depatuxizumab mafodotin
EGFR是表皮生长因子受体(HER)家族成员之一, 与细胞的生长、增殖和分化有关. EGFR在许多肿瘤细胞表面存在高表达或异常表达现象, 研究发现它与肿瘤细胞的增殖、血管生成、肿瘤侵袭、转移及细胞凋亡的抑制有关.小鼠抗体mAb 806是以EGFRvⅢ(缺少外显子2~7的配体结构域并保留组成型激酶活性的EGFR的最常见缺失突变体)或过表达的原生型EGFR为靶标, 为降低鼠源抗体可能引发的过敏反应并实现临床开发, 对mAb 806进行人源化得到重组IgG1/k mAb ABT-806. ABT-806对EGFRv Ⅲ或过表达的EGFR结合力更强, 特异性更高, 降低正常组织的ADR发生率[31]. MMAF同样是Dolastatin 10的合成衍生物, Doronina等[32]报道了MMAF的固相合成路线, 以Phe-2-氯三苯甲基树脂21为原料, 经过偶联脱保护得到MMAF. MeVal-Val-Dil-Dap-Phe-2-氯三苯甲基树脂若直接与mc偶联, 脱掉氯三苯甲基树脂则得到mcMMAF (Scheme 6).
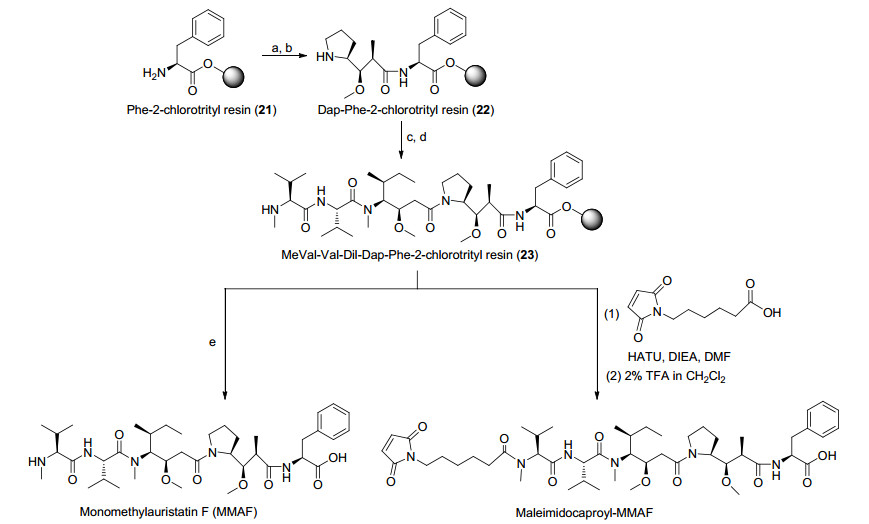 图式6
MMAF和mcMMAF的合成
图式6.
Synthesis of MMAF and mcMMAF
图式6
MMAF和mcMMAF的合成
图式6.
Synthesis of MMAF and mcMMAF
为评估ABT-414与替莫唑胺联合治疗复发性或难以切除恶性胶质瘤的安全性和有效性, 对12位受试者开展了Ⅰ期临床试验. 9位患者治疗效果较好, 其中1位完全缓解, 2位部分缓解.发现一般ADR (≥3名患者)包括视力模糊(n=5), 角膜沉积(n=4)、眼部异物感、恶心、发热和头痛(每项n=3).严重ADR包括淋巴球减少症、角膜沉积、皮肤感染和血液胆固醇升高(每项n=1).在一定剂量范围内(0.5~1.0 mg/kg)药代动力学(PK)参数与剂量成比例, 半衰期为7~8 d.其结果表明ABT-414对于治疗复发性或难以切除恶性胶质瘤值得进一步的研究[33].
1.2.3 Glembatumumab Vedotin (CDX-011)
Glembatumumab Vedotin是由CuraGen公司研发的ADC, 目前处于治疗转移性乳腺癌、黑色素瘤、骨肉瘤的临床Ⅱ期试验以及治疗鳞状细胞癌的临床Ⅰ/Ⅱ期试验阶段. 2010年FDA将Glembatumumab Vedotin加入快速审批名单, 以治疗晚期、顽固性、高表达非转移性黑色素瘤糖蛋白B (GPNMB)乳腺癌.
Glembatumumab Vedotin靶向作用于GPNMB, 由抗体CR011(以GPNMB为靶点的人源化IgG2单克隆抗体)、连接体和MMAE构成(图 3). GPNMB是一种跨膜糖蛋白, 在多种正常组织中均有表达, 如骨骼系统、造血系统、皮肤等, 与成骨形成和黑色素细胞生成有关; 但GPNMB高度表达于乳腺癌、黑色素瘤、骨肉瘤等多种恶性肿瘤中, 在癌症转移、肿瘤细胞侵袭和迁移中发挥重要作用. Glembatumumab Vedotin的作用机制与Brentuximab vedotin相似, 首先通过抗体CR011识别靶点, 使其与GPNMB形成复合物进入细胞, 再在溶酶体中被蛋白酶水解, 释放MMAE, MMAE通过抑制微管蛋白聚合, 使有丝分裂停滞, 导致细胞凋亡[34].
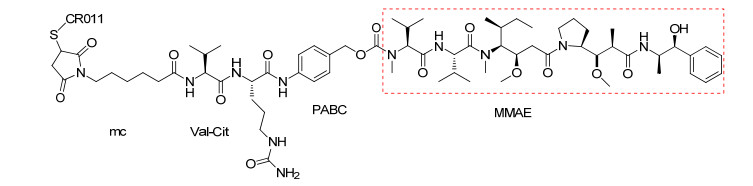 图 3
Glembatumumab Vedotin的结构
Figure 3.
Structure of glembatumumab vedotin
图 3
Glembatumumab Vedotin的结构
Figure 3.
Structure of glembatumumab vedotin
为评估Glembatumumab Vedotin治疗晚期、顽固性、高表达GPNMB乳腺癌的安全性和有效性, 对124名患者进行了Ⅱ期临床试验.将所有受试者以2:1的比例随机分成两组, 一组为Glembatumumab Vedotin组(n=83), 另一组由研究者选择(IC)药物(艾日布林、卡培他滨、长春瑞滨、吉西他滨、紫杉醇、紫衫特尔、盐酸多柔比星、多柔比星脂质体、紫杉醇与白蛋白结合型混悬液), 单药化疗(n=41).其结果表明, 与IC药物相比, Glembatumumab Vedotin表现出较好的耐受性, 较低的血液毒性; 出现的一般ADR有皮疹、瘙痒、神经病和脱发.在初级终点, Glembatumumab Vedotin组与IC化疗组相比, 客观缓解率没有显著性差异.但在次级终点, Glembatumumab Vedotin组中高表达GPNMB (≥25%的肿瘤细胞)患者的客观缓解率(ORR)为30%, 明显高于IC化疗组的9%.在此次试验中, 还测得了患有三阴性乳腺癌患者的ORR, Glembatumumab Vedotin组为18%, IC化疗组为0%.对于过度表达GPNMB的三阴性乳腺癌患者, Glembatumumab Vedotin组ORR为40%, IC化疗组为0%.此结果表明, Glembatumumab Vedotin对于治疗过度表达GPNMB的乳腺癌患者, 具有较好的疗效, 尤其对于三阴性乳腺癌展现出较高的活性[35].
1.2.4 Polatuzumab vedotin (RG-7596)
Polatuzumab vedotin是靶向作用于CD79b的ADC, 由人源化的抗体、连接体和MMAE构成.现在处于治疗弥漫大B细胞淋巴瘤和其他种类非霍奇金淋巴瘤的临床Ⅱ期研究、治疗滤泡性淋巴瘤的临床Ⅰ/Ⅱ期试验以及治疗慢性淋巴细胞白血病的临床Ⅰ期研究阶段. CD79b在B细胞、慢性淋巴细胞白血病(CLL)、B细胞非霍奇金淋巴瘤(B-NHLs)中表达, 是治疗非霍奇金氏淋巴瘤的良好靶标. Polatuzumab vedotin所使用的连接体、细胞毒试剂与Brentuximab vedotin相同, 为mc-vc-PABC-MMAE(图 4, 抗体为人源化抗体)[36].
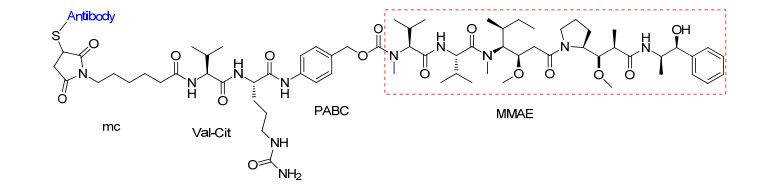 图 4
Polatuzumab vedotin和AGS-67E的结构
Figure 4.
Structure of polatuzumab vedotin and AGS-67E
图 4
Polatuzumab vedotin和AGS-67E的结构
Figure 4.
Structure of polatuzumab vedotin and AGS-67E
为进一步评估Polatuzumab vedotin治疗NHL和CLL的安全性及有效性, Palanca-Wessels及其同事[37]纳入95例患者进行Ⅱ期临床试验, 这些患者经过Ⅰ期、多中心、非盲研究, 表达CD79B, 难以治疗且治疗意义不大.试验组别设置为: NHL剂量递增组34名, CLL剂量递增组18名, 以确定Polatuzumab vedotin的最大耐受剂量; Ⅱ期推荐剂量NHL扩展组34名, NHL利妥昔单抗联合组9名(由于剂量递增组缺乏疗效未设置CLL扩展组). NHL患者最常见的ADR为中性粒细胞减少症, 贫血和周围神经病变.在42名可评估的NHL患者中, 23名客观缓解, 其中7名完全缓解、16名部分缓解. Polatuzumab vedotin和利妥昔单抗联合治疗的9名患者中7名患者客观缓解, 包括2名完全缓解和5名部分缓解.临床试验结果表明, Polatuzumab vedotin治疗NHL患者安全性和耐受性良好, 但对CLL患者则无效, 因此这一疗效应在NHL方面进行进一步评估.
1.2.5 AGS-67E
AGS-67E是由Agensys公司(安斯泰来的子公司)研发用于治疗急性髓性白血病(AML)、淋巴细胞白血病等血液恶性肿瘤的ADC.现处于临床Ⅰ期的研究阶段.
AGS-67E靶向于CD37, 由完全人源化IgG2型单克隆抗体、连接体和MMAE构成(图 4, 抗体为人源化IgG2抗体). CD37是四跨膜蛋白超家族(TM4SF)的成员, 具有四个潜在的跨膜区. CD37通过C和N末端结构域可以直接介导双信号转导. CD37在由原始B细胞到外周成熟B细胞的发育过程中表达量逐渐增加, 但在浆细胞、T细胞及单核细胞中不表达, 在自然杀伤(NK)细胞中表达水平非常低, 在血小板和红细胞上不存在.鉴于CD37选择性表达于B细胞, CD37成为治疗B细胞恶性肿瘤(CLL)的良好靶标.后来有人发现, CD37也可在T细胞淋巴瘤、AML和表达CD34+CD38-的AML干细胞中表达, 但在正常的表达的CD34+CD38-干细胞很少或没有表达. AGS-67E的作用机制与Brentuxmab Vedotin相似, 首先通过抗体识别靶点, 形成复合物进入细胞内, 再在溶酶体中被蛋白酶水解, 释放MMAE, 使MMAE在细胞内发挥毒性作用.体内外试验表明, AGS-67E可以抑制AML细胞系.以上结果均表明AGS-67E以CD37为靶标治疗AML、淋巴细胞白血病等血液恶性肿瘤前景光明[38].
为评价在有/无生长因子(GF)预防情况下, AGS-67E治疗复发性/难治性非霍奇金淋巴瘤患者的安全性、有效性及药代动力学(PK), 进行了剂量递增、多中心、Ⅰ期临床试验[39]. 13名患者分为7个剂量组(无GF: 0.05~1.2 mg/kg, 有GF: 1.2 mg/kg)进行单药治疗, 3周/次.没有GF预防患者的最大耐受剂量(MTD)超过1.2 mg/kg, 在第一次给药后的8~15 d观察到3名患者出现严重中性粒细胞减少.在给予1.2 mg/kg剂量组中一名患有弥漫性大B细胞淋巴瘤的患者完全缓解.在1.2 mg/kg时, AG S-67E和游离MMAE的半衰期分别为1.44~3.08和2.34~3.64 d.其结果表明3周/给药AGS-67E具有治疗淋巴瘤的活性且安全性良好.
1.3 Solidotin (TZT-1027)
Solidotin是Dolastatin 10的合成衍生物, 最初由Teikoku Hormone开发治疗晚期或转移的软组织肉瘤(STS)或非小细胞肺癌(NSCLC), 处于临床Ⅱ期研究阶段. Solidotin通过与微管蛋白结合, 抑制微管蛋白的聚合, 使细胞G2-M期分化停滞, 导致细胞凋亡[40].并且, Solidotin可以攻击晚期肿瘤良好的血管系统, 阻止血管生成, 杀死肿瘤细胞[41]. Watanabe等[42]发现Solidotin的抗肿瘤活性要优于长春新碱、5-氟尿嘧啶和顺氯氨铂.在结构上, Solidotin是用苯乙胺取代Dolastatin 10的Doe片段而得到的, 它的合成是以与Dil片段相关联的化合物(24)合成Dov-Val-Dil-Dap-OBzl (32), 三肽32进行脱保护再与苯乙胺(33)反应得到Solidotin (Scheme 7)[15].
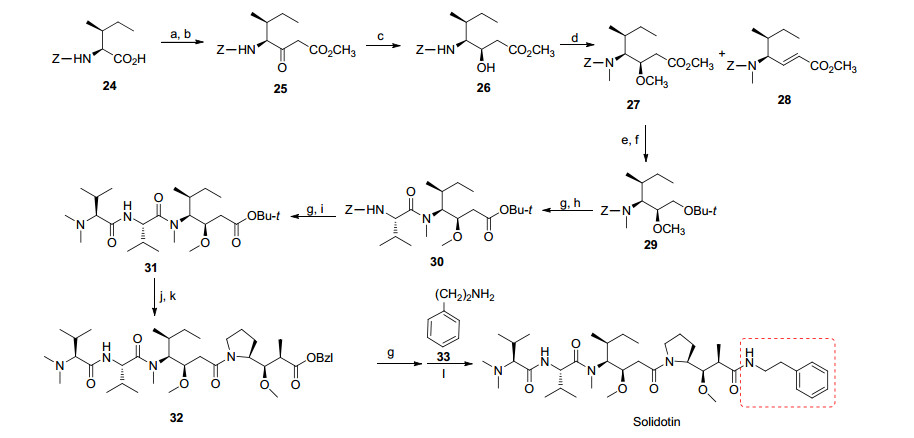 图式7
Solidotin的合成
图式7.
Synthesis of solidotin
图式7
Solidotin的合成
图式7.
Synthesis of solidotin
为评价Solidotin治疗晚期或转移软组织肉瘤的安全性及有效性, Patel等[43]纳入28名符合患者进行Ⅱ期临床试验. Solidotin组, 肿瘤进展的中值时间为44 d (95%置信区间[95% CI], 43.0~54.0), 中位生存期为178 d (95% CI, 134.0~317.0).常见的不良反应是中性粒细胞减少、疲劳和便秘.本试验结果表明, Solidotin安全性及耐受性良好, 但并不能有效缓解肿瘤进展.另一项由Riely等[44]组织的Ⅱ期临床试验选择了32名符合条件的患者.以28 d为一给药周期, 在第1 d和第8 d给予患者2.4 mg/m2 Solidotin.在试验中, 最常见的严重不良反应是白细胞减少和中性粒细胞减少. 4名患者在接受Solidotin的30 d内死亡, 3名患者病情进行, 1名患者出现肺炎和中性粒细胞减少.试验结果表明, 以28 d为一周期, 在第1 d和第8 d给予患者2.4 mg/m2 Solidotin, 患者病情未出现客观缓解, 未达到预期治疗效果.
1.4 Dolastatin 15衍生物
Dolastatin 15(图 5)可以有效抑制肿瘤生长, 但由于其结构复杂、化学合成产率低及水溶性差等原因限制了Dolastatin 15的客观临床评价.为了解决Dolastatin 15的这些弊端, 合成了Dolastatin 15的衍生物[45].其中, Tasidotin和Cemadotin已进入临床试验.
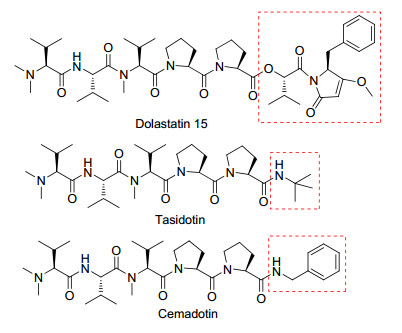 图 5
Dolatatin 15、Tasidotin和Cemadotin的结构
Figure 5.
Sructures of dolatatin 15, tasidotin and cemadotin
图 5
Dolatatin 15、Tasidotin和Cemadotin的结构
Figure 5.
Sructures of dolatatin 15, tasidotin and cemadotin
1.4.1 Tasidotin, Synthadotin (IXL-651)
Tasidotin是Dolastatin 15的第三代合成衍生物.在结构上, Tasidotin在羧基末端用叔丁基胺取代Dolastatin 15的酯基团(图 5). Tasidotin的主要作用机制是抑制纺锤体的形成. Tasidotin在体内的主要代谢产物是N, N-二甲基缬氨酸-缬氨酸-N-甲基缬氨酸-脯氨酰基-脯氨酸(P5), P5抑制微管蛋白聚合的活性要比Tasidotin高, 但作为细胞毒性剂的活性要比Tasidotin低[46].
为评价Tasidotin的安全性、耐受性和药代动力学, Mita等[47]纳入30名受试者进行Ⅰ期临床试验.在7.8至62.2 mg/m2的剂量范围设置6个剂量组, 1次/周给药, 持续82个疗程.在所有剂量水平上, 发生一般ADR有腹泻和呕吐, 非血液学毒性通常为轻至中度且易于控制. Tasidotin药代动力学是轻度非线性的, 而代谢动力学是线性的.非小细胞肺癌患者服药后病情轻微缓解, 肝细胞癌患者在11个月内病情稳定.
1.4.2 Cemadotin (LU-103793)
Cemadotin是Dolastatin 15的水溶性合成衍生物.由Abbott公司研发, 用于治疗癌症, 曾处于临床Ⅱ期, 现已被终止.在结构上, Cemadotin在C端用苯甲胺取代了Dolastatin 15的酯基团(图 5)[45]. Cemadotin的结合位点区别于长春碱, 其进入细胞后, 抑制微管蛋白的聚合, 使细胞周期停滞在G2-M阶段[48].
一项Ⅱ期临床试验评估了Cemadotin在转移性乳腺癌患者中的安全性和有效性.在可评价的23名患者中, 11位患者发生了严重中性粒细胞减少症, 其它一般ADR有乏力、口腔炎、肌痛及血清胆红素升高, 主要ADR为高血压.在试验期间, 患者病情无客观缓解, 故终止试验[49].
2 芋螺毒素
锥形蜗牛主要生活在热带海域, 它们通过形成毒液来保护自身及猎食.在毒液中包含致死的毒素——芋螺毒素.芋螺毒素是一种小分子, 结构稳定, 具有高度靶向特异性、富含有二硫键的活性多肽, 通常由12到41个氨基酸构成, 且易于合成[50].它主要靶向于膜蛋白, 特别是电压或配体门控离子通道, 转运蛋白或G蛋白偶联受体.它可以用于治疗神经性疼痛、癌症晚期的慢性疼痛以及其他疼痛.由于它具有较高的生物活性及靶向特异性且易于合成, 使得芋螺毒素有望成为先导化合物或药物[51]. 21世纪初期, 我国戚正武院士课题组开始了对各类芋螺毒素的研究, 进行翻译后修饰, 探测其功能, 为开发高效而特异的神经药物打下基础.目前, ω-芋螺毒素的合成形式——齐考诺肽(Ziconotide, Scheme 8)已上市.
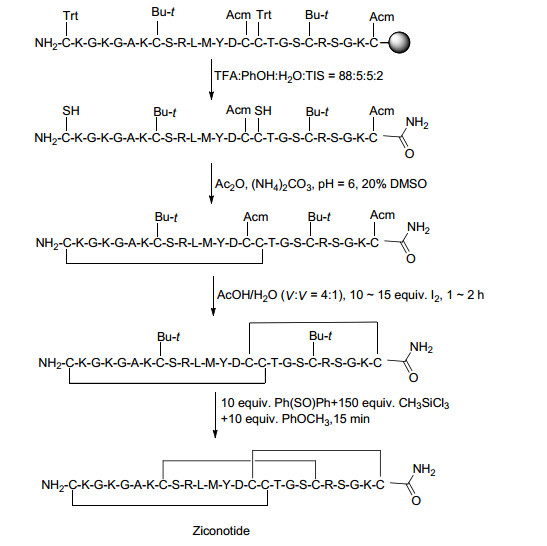 图式8
齐考若肽的合成
图式8.
Synthesis of ziconotide
图式8
齐考若肽的合成
图式8.
Synthesis of ziconotide
齐考诺肽(以前称SNX-111) 是来源于太平洋的幻芋螺毒液中具有亲水性ω-MVIIA芋螺毒素的合成形式.它于2004年12月经FDA批准上市, 用于常规治疗不耐受或难于治疗的慢性严重性连续鞘内病变患者的疼痛[52].一项专利[53]报道了其合成路线(Scheme 8), 以Fmoc-氨基树脂为固相载体, 从C端到N端依次用缩合反应连接25个侧链保护的氨基酸, 形成二硫键的三组Cys分别连接Trt, Acm和t-Bu保护基, 将树脂进行线性切割, 同时脱去除Cys (Acm)和Cys (t-Bu)外的所有氨基酸的侧链保护基, 氧化线性肽形成第一对二硫键, 得到单二硫环肽, 然后依次脱除Cys (Acm)和Cys (t-Bu)中的Acm和t-Bu, 在脱除时同时环化, 得到三二硫环肽, 最后经HPLC纯化, 冻干得到齐考诺肽.
齐考诺肽包含25个氨基酸, 富含有三个二硫键, 其药理活性依赖于这些完整的二硫键, 这些二硫键也是分子三维结构的决定因素. 3股β折叠可进一步稳定空间结构, 增强肽对酶的抗性, 也可能增强结合力和效力.蛋白表面与带电和极性氨基酸侧链连接, 使得齐考诺肽高度亲水.齐考诺肽的结构暗示其可以用于成药[54].齐考诺肽是一种效果非常好的非阿片类镇痛药, 它对N型电压敏感钙通道具有较高的亲和力, 可以有效地阻断钙离子的流通.因此, 它可以通过有效和选择性阻断N性钙离子通道, 从而控制多种突触释放的神经递质.它可能在脊髓水平上中断了疼痛信号的传递[55].
为评估鞘内齐考诺肽对常规治疗难治的疼痛患者的安全性和有效性进行了双盲、安慰剂随机对照试验[56], 试验选择111例癌症或AIDS患者, 以2:1的比例随机分配至齐考诺肽组和安慰剂组.在可评价的患者中, 齐考诺肽组中52.9%的患者疼痛缓解, 在安慰剂组中, 只有17.5% (P<0.001), 并且在接受齐考诺肽的患者中, 有5位患者达到了完全缓解.结果表明齐考若肽对于缓解难治型疼痛具有很好的疗效.
3 海鞘素
1981年Rinehart等从膜海鞘中提取出一系列环形缩肽类活性物质, 命名为Didemnins A, B, C, 它们具有较高的抗肿瘤活性.其中活性最高的是Didemnin B, 它成为美国第一个进入临床试验的源自海洋的抗肿瘤天然产物.第二代Didemnin——脱氢Didemnin B (Plitidepsin)也正在进行临床试验[57]. Plitidepsin与Didemnin B在分子结构上仅差两个氢(图 6).
 图 6
Didemnin B和Plitidepsin的结构
Figure 6.
Structure of didemnin B and plitidepsin
图 6
Didemnin B和Plitidepsin的结构
Figure 6.
Structure of didemnin B and plitidepsin
3.1 Didemnin B
研究人员对Didemnin B治疗前列腺癌、非小细胞肺癌、骨髓瘤和黑素瘤等多种肿瘤进行了Ⅰ期、Ⅱ期临床试验.其作用机制现在还未明确阐述, 但它似乎与抑制蛋白质合成有关, 在较小程度上可能与抑制DNA和RNA合成有关[58]. Didemnin B具有新型的环状结构, 包括一个抑胃酶氨酸, 增加了Didemnin B的稳定性[59]. Hamada等[60]报道其合成路线(Scheme 9), 通过(2RS, 4S)-Hip-(S)-Leu-(S)-Pro-OBzl (42)和Boc-(R)-MeLeu-(S)-Thr[Z-(S)-MeTyr(Me)]-(3S, 4R, 5S)-Ist(TBDMS)-OH (38)偶联并环化, 然后去保护, 得到Didemnin A, 然后将其转化为Didemnin B.
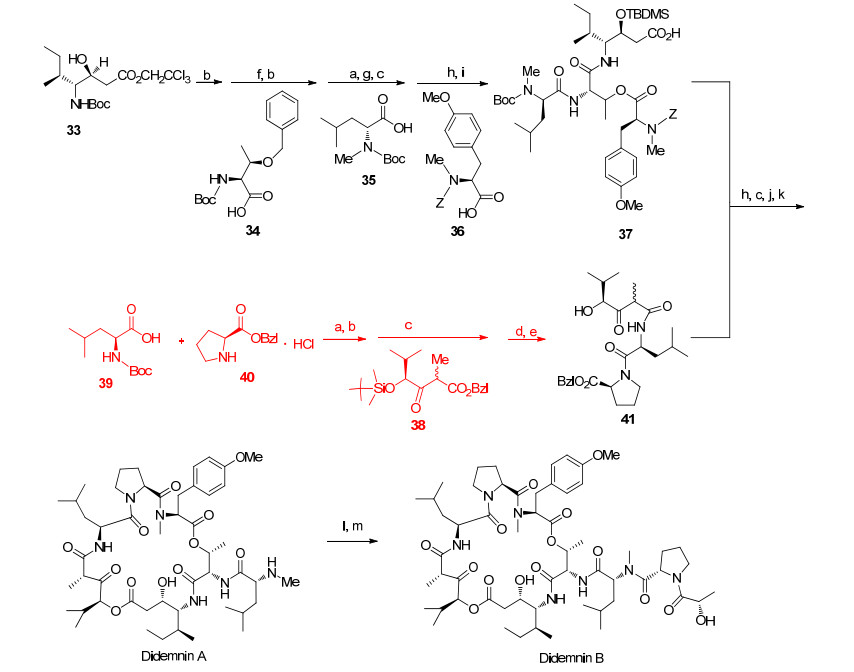 图式9
Didemnin B的合成
图式9.
Synthesis of didemnin B
图式9
Didemnin B的合成
图式9.
Synthesis of didemnin B
为评价Didemnin B治疗复发或发展的NHL患者的功效和毒性, 进行了临床Ⅱ期试验.试验选择了51名患者.在试验中, 患者都产生了3~4级的不良反应, 毒性较大, 因此停止了实验[61].
3.2 Plitidepsin (Aplidin)
Plitidepsin亦称Aplidin, 由PharmaMar研发.它是从地中海海鞘Aplidium albicans中分离得到的抗肿瘤活性天然产物的合成形式.它经FDA和欧洲委员会批准为治疗多发性骨髓瘤的孤儿药.在活性浓度时, Plitidepsin显示比Didemnin B更低的毒性[62]. Plitidepsin具有更高的治疗价值, 现处于治疗多发性骨髓瘤的临床Ⅲ期阶段.
Plitidepsin作用机制是通过与细胞膜表面高亲和力的位点结合后, 激活Rac1, P38/MAPK和JNK信号传导, Fas/CD95转移至脂筏处, 最终引起半胱氨酸蛋白酶的激活, 使细胞凋亡[63]. Plitidepsin通过(S)-Pro-Pyr (43)与Didemnin A中的(R)-N-甲基-Leu侧链中的氨基结合形成(Eq. 1)[64].
一项多中心、单组、开放且具有前瞻性的Ⅱ期临床试验[65]评估了plitidepsin治疗复发/难治性多发性骨髓瘤(MM)患者的安全性和有效性.在可评价的47名患者中, 单独使用plitidepsin时的总缓解率(OR)为13%, 与地塞米松联合使用时为22%.一般ADR有贫血(29%)、血小板减少症(18%)、疲劳(16%)、肌肉毒性(6%)和瞬时丙氨酸氨基转移酶/天冬氨酸氨基转移酶(27%)和肌酸磷酸激酶(23%)增加.这表明单用或与地塞米松联用plitidepsin治疗复发/难治性多发性骨髓瘤患者是安全、有效的.
4 海绵活性肽
Hemiasterlin(图 7)最初是从南非海绵Hemiasterella minor中提取出的三肽[66].其作用机制是通过与微管蛋白结合, 使微管解聚, 影响纺锤体的形成而产生细胞毒性.它在体外对小鼠白血病细胞P388 (ED50=4.57×10-5 μg/mL)、人乳腺癌细胞MCF-7 (ED50=0.089 μg/mL)、人成胶质瘤细胞/星形胶质瘤细胞U373 (ED50=0.012 μg/mL)、人卵巢癌细胞HEY(ED50=0.0014 μg/mL)和体内对小鼠的P388细胞具有活性[67].它良好的活性使得它有望成为先导化合物.人们为了探测它的结构与活性的关系, 合成了一系列衍生物, 其中HTI-286的活性比天然产物Hemiasterlin活性好, 现已经进入临床试验阶段[68].
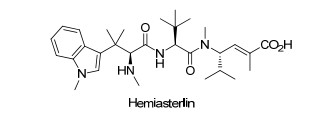 图 7
Hemiasterlin的结构
Figure 7.
Sructure of hemiasterlin
图 7
Hemiasterlin的结构
Figure 7.
Sructure of hemiasterlin
4.1 HTI-286
HTI-286由辉瑞公司研发, 用于治疗癌症. Niu等[69]报道了其合成路线(Scheme 10), 它可以甲基化的丙酮酸衍生物44作为原料, 在氢化钠存在下, 与碘甲烷反应得到偕二甲基化合物45, 45进行还原氨化得到片段46, 46与二肽47偶联得到非对应异构体48, 将其通过HPLC分离得到(S, S, S)-48, 在LiOH存在下, 将(S, S, S)-48进行水解即可得到HTI-286.在研究HTI-286结构与活性的关系时发现, HTI-286结构中最左侧氨基酸中的碱性基团是关键基团, 这一发现为新型衍生物的合成打下基础[70]. HTI-286通过抑制微管蛋白的聚合, 破坏细胞中的微管组织, 并诱导有丝分裂停滞, 使细胞凋亡, 并且它并不与多药耐药蛋白P-糖蛋白结合, 比紫杉醇、多西他赛、长春碱等抗肿瘤药物效果好[71].临床前研究表明, 在体外和体内HTI-286对于治疗肝癌、前列腺癌和膀胱癌是有效的[72].这也表明HTI-286具有作为治疗多种恶性肿瘤药物的潜力.
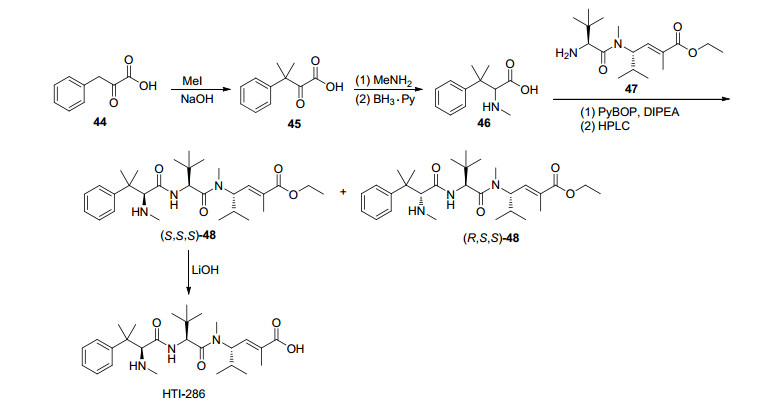 图式10
HTI-286的合成
图式10.
Synthesis of HTI-286
图式10
HTI-286的合成
图式10.
Synthesis of HTI-286
4.2 E7974
E7974同样是Hemiasterlin的衍生物, 由卫材公司研发, 用于治疗实体瘤.曾处于临床Ⅰ期, 现已终止试验.其作用机制是通过与α-微管蛋白结合, 抑制微管蛋白聚合.在体外细胞实验中, E7974可以诱导细胞停滞在G2-M期, 并靶向结合纺锤体, 抑制其形成[73].一项专利报道了其合成路线[74], 以N-Boc-N-Me-L-Val-OH (49)为起始原料, 经过反应合成化合物53, 54与55进行偶联, 脱保护盐酸化得到57. 57与N-异丙基哌可酸(58)经过亲核加成-取代反应, 水解即可得到目标产物(Scheme 11).
 图式11
E7974的合成
图式11.
Synthesis of E7974
图式11
E7974的合成
图式11.
Synthesis of E7974
为评价了E7974治疗晚期实体瘤的安全性和有效性, 进行了Ⅰ期临床试验.试验选择了28名患者, 分为5个剂量组(0.18, 0.27, 0.36, 0.45和0.56 mg/m2; MTD为0.45 mg/m2), 28天/次, 静脉注射给药(2至5 min).在17名患有难治性结肠癌患者中, 7名(41%)患者病情得以稳定.中位无进展生存期为1.2个月, 中位总生存期为6.7个月.出现血液ADR有嗜中性白血球减少症、贫血、白血球减少症; 一般ADR有疲劳、秃头症、恶心、呕吐、便秘[75].
5 海洋真菌活性肽
Kanoh等从曲霉属真菌的琼脂培养基中发现了一对对映异构体(phenylahistin), 之后Fenical等筛选分离出具有抑制细胞周期的活性化合物Halimide. Halimide可以作用于微管蛋白上的秋水仙素结合位点, 使微管解聚.体外试验表明, 该化合物可抑制肿瘤细胞生长, 活性较高.结构上, Halimide(图 8)是由L-苯丙氨酸和(Z)-异戊二烯化的脱氢组氨酸残基组成的环缩二氨酸(DKP)的衍生物.在对Halimide进行活性与结构关系分析时发现, DKP提供了用于诱导抗微管活性的新型杂环和相对亲水的模板, 并且L-苯丙氨酸、刚性和通过DKP与咪唑环之间的氢键形成的平面假三分结构以及咪唑环的5位上的偕二甲基结构对Halimide产生较高的细胞毒性具有重要作用, 同时为合成Halimide的衍生物提供了思路[76].在Halimide的衍生物中, plinabulin已进入临床阶段[77].
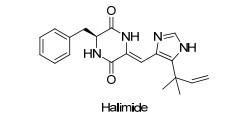 图 8
Halimide的结构
Figure 8.
Structure of halimide
图 8
Halimide的结构
Figure 8.
Structure of halimide
Plinabulin (NPI-2358) 由Beyondspring公司研发作为二线化疗药物结合多烯紫杉醇治疗晚期或转移性非小细胞肺癌患者.目前处于临床Ⅲ期试验阶段.它的作用机制与Halimide类似, 选择性作用于内皮微管蛋白秋水仙碱结合位点附近的α-和β-微管蛋白之间的边界区域结合[78].体外试验表明, Plinabulin对各种人类肿瘤细胞具有活性, 并且对各种多药耐药(MDR)谱的肿瘤细胞系也具有活性.在体内, Plinabulin可干扰血管内皮的正常功能, 使内皮细胞骨架紊乱, 抑制肿瘤细胞血液的流通[79]. Plinabulin的合成可以通过两种途径, 差异在于化合物62与5-叔丁基咪唑-4-甲醛(63)和苯甲醛(65)进行缩合反应的顺序(Scheme 12)[80].
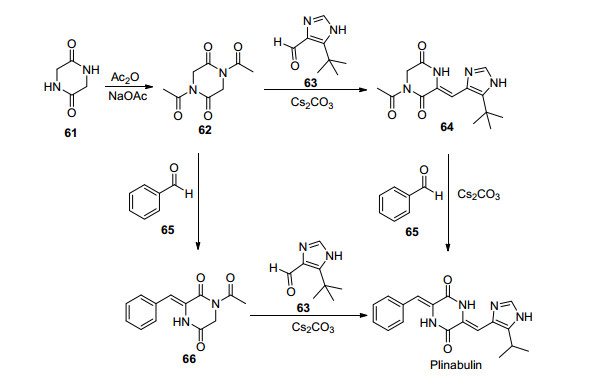 图式12
Plinabulin的合成
图式12.
Synthesis of plinabulin
图式12
Plinabulin的合成
图式12.
Synthesis of plinabulin
一项Ⅱ期临床试验, 对比了Plinabulin (N)联合多烯紫杉醇(D)治疗和单用多烯紫杉醇(D)治疗非小细胞肺癌NSCLC患者的效果.将172名患者随机分成两个不同剂量水平的大组, 163个治疗组, 第一大剂量组由30个小组构成, 给药剂量为50DN, 55D;第二大剂量组由20个小组构成, 给药剂量为40DN, 18D.在第一大组中, 使用DN联合治疗的总生存期为8.7个月, 单独使用D治疗是7.5个月; DN组的缓解率为14%, D组为14.5%; DN组的药效持续时间为12.7个月, D组为1.5个月; 第一大组治疗效果优于第二大组.此次试验中出现最常见的ADR为恶心、疲劳、腹泻、便秘和厌食.在两个给药大组中, DN组中性粒细胞减少发生率较低, 严重ADR发生率较低.此试验表明具有大的肺肿瘤(>3 cm)和之前经过标准化疗(ChRx)的患者在DN组存活几率比D组高[81].
6 软体动物活性肽
6.1 Kahalalide F
Kahalalide F(图 9)属于Kahalalide家族, 从食草的软体动物Elysia rufescens和绿藻Bryopsis sp中提取出来. Kahalalide F是分子量为1478的十三肽, 由大分子区域和线性区域组成, 在N-末端含有13个氨基酸和5-甲基己酸, 还包含Z-二脱氢氨基丁酸(Dhb)[82]. LópezMacià等[82b]报道了Kahalalide F的固相合成方法(Scheme 13), 其合成策略是使链在固相上延伸, 被保护的肽在树脂上裂解, 随后环化, 最终在溶液中去保护.在策略a中, 加上Fmoc-Val-OH并没有得到较好的产率, 推测是由于肽链的疏水性使其有利于链间聚集.因此, 结合保护的Val应当在前面的步骤(策略b和c)中.策略b和c都可以得到理想的产物, 但通过策略b所得到的粗产物的HPLC质量更优.
 图 9
Kahalalide F的结构
Figure 9.
Structure of kahalalide F
图 9
Kahalalide F的结构
Figure 9.
Structure of kahalalide F
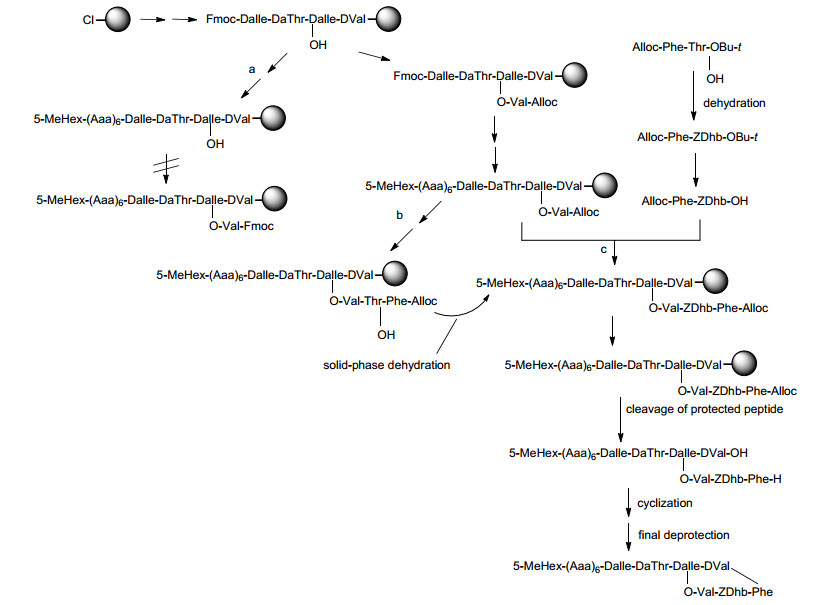 图式13
Kahalalide F的合成路线
图式13.
Synthesis route of kahalalide F
图式13
Kahalalide F的合成路线
图式13.
Synthesis route of kahalalide F
在体外, Kahalalide F作用的发挥与ErbB3和Akt信号有关[83].体外实验表明, Kahalalide F对前列腺癌和乳腺癌细胞显示出较强的细胞毒活性, IC50范围为0.07 μmol•L-1 (PC3)~0.28 μmol•L-1 (DU145, LNCaP, SKBR-3, BT474, MCF7).重要的是, 人类正常细胞(MCF10A, HUVEC, HMEC-1, IMR90) 对药物的敏感性低5~40倍(IC50=1.6~3.1 μmol•L-1). Kahalalide F在细胞内发挥作用时, 会发生一系列的变化:细胞质肿胀和空泡化, 内质网的扩张和囊泡形成, 线粒体损伤和质膜破裂.细胞核内不规则染色质聚集成小的块状, 而染色质从其他核域消失, 但核包膜被保留, 没有检测到DNA降解[84].
为评价Kahalalide F治疗晚期恶性黑素瘤患者的效果.试验选择了24例符合条件的患者, 1次/周注射给药. 14例患者有化疗或生物治疗史, 但实体瘤的疗效评价标准(RECIST)评价较差.在5名患有皮肤黑色素瘤化疗患者中, 疾病稳定为3个月, 中位无进展生存期为1.7个月(95% CI, 1.2~1.9个月), 中位总生存期为10.8个月(95% CI, 5.0-上限未达到).患者服药期间最常见的生化指标变化是转氨酶(ALT/AST)和γ-谷氨酰转移酶(GGT)的非累积增加.在研究期间患者未出现白细胞减少和血小板减少. Kahalalide F是一种安全性良好的化疗物质, 但由于恶性黑素瘤患者缺乏客观缓解, 该试验在第一阶段后停止[85].
6.2 Elisidepsin
Elisidepsin是Kahalalide F的合成衍生物, 由PharmaMar研发, 用于治疗实体瘤, 曾处于临床Ⅱ期, 现已被终止.合成过程为在PyAOP和DIEA存在下, 在DMF中线型多肽67与环状缩肽68的偶联产生侧链保护的赖氨酸蛋白酶69, 然后通过在CH2Cl2中的TFA脱保护得到Elisidepsin.或者, 通过在CH2Cl2中的DIC, HOBt和DIEA使缩肽70环化, 获得受保护的前体69, 然后进行脱保护即可得到Elisidepsin (Scheme 14)[86]. Elisidepsin的效力与HER3, ErbB3、糖基神经酰胺和羟基化脂肪酸(由FA2H产生)表达有关[87].
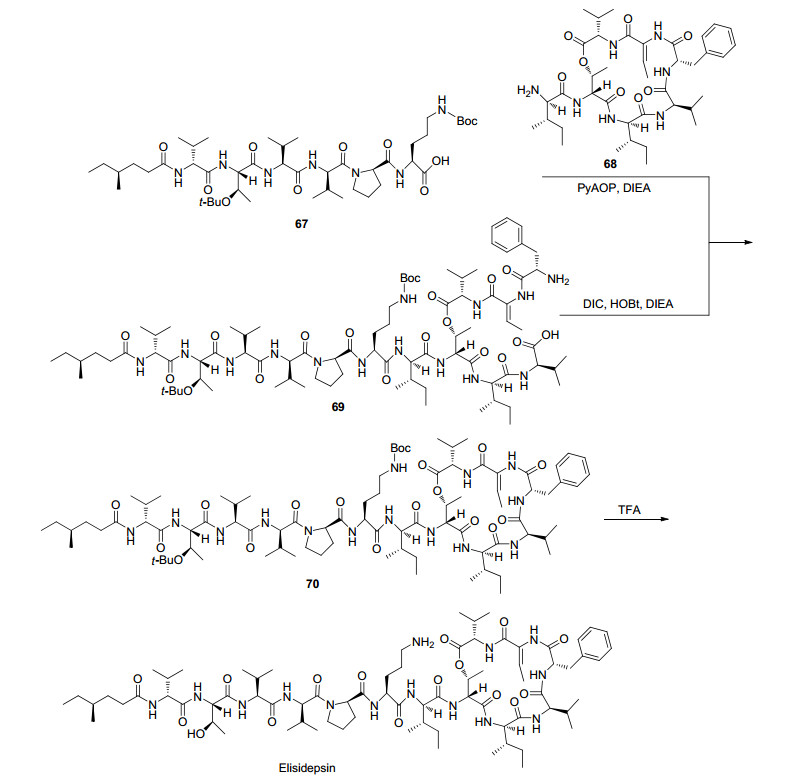 图式14
Elisidepsin的合成
图式14.
Synthesis route of elisidepsin
图式14
Elisidepsin的合成
图式14.
Synthesis route of elisidepsin
为评价Elisidepsin治疗晚期胃食管癌的有效性, 进行了Ib/Ⅱ期临床试验.将44名患者随机分配至两个剂量组.其中12名患者处于Ib期临床试验, 32名患者处于Ⅱ期临床试验.在试验期间, 较少发生ADR, 但是由于疗效不佳没有进行下一步试验[88].
7 展望
随着海洋开发的步伐加快及对生物活性肽认识的不断深入, 海洋生物活性肽是目前药物研发的一个重要方向.虽然对于来自于海洋生物的活性肽研究历程较短, 但由于其结构的特殊性吸引了众多的科研工作者, 开展了化学合成、构效关系、药理毒理、临床及相关的不良反应等方面的研究.但由于海洋生物活性肽结构复杂、活性成分含量低及封闭的N末端等特殊结构, 使得给开发研制增添了诸多困难.从目前上市药物和处于临床试验阶段的活性化合物来看, 对于海洋生物活性肽的开发, 主要在抗肿瘤药物研发领域, 化合物来源局限于海兔、芋螺、海绵、海鞘、软体动物等少数几种生物体中, 绝大多数海洋生物尚未得到开发.
为将更多的海洋生物活性肽类化合物开发为药物, 我们应从三个方向进行: (1) 以天然肽为先导化合物, 对其进行功能化修饰, 研究其构效关系, 以筛选有更好药效的活性肽类似物; (2) 海洋药物的研究需要探测、现代生物、分离纯化和产品制备等相关技术, 故需加大力度建设活性肽的分离鉴定需要的高效灵敏的技术平台. (3) 海洋是一个巨大的宝库, 还存在着许多未开发利用的生物活性肽类化合物, 应进行更多的广泛深入研究, 以寻找更多和更有价值的新型海洋药物.
-
-
[1]
Cheung, R. C.; Ng, T. B.; Wong, J. H. Mar. Drugs 2015, 13, 4006. doi: 10.3390/md13074006
-
[2]
Jin, L.; Quan, C.; Hou, X.; Fan, S. Mar. Drugs 2016, 14, 76. doi: 10.3390/md14040076
-
[3]
(a) Suarez-Jimenez, G. M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J. M. Mar. Drugs 2012, 10, 963.
(b) Ponnappan, N.; Budagavi, D. P.; Yadav, B. K.; Chugh, A. Probiotics Antimicro. Proteins 2015, 7, 75. -
[4]
(a) Lordan, S.; Ross, R. P.; Stanton, C. Mar. Drugs 2011, 9, 1056.
(b) Kim, S. K.; Wijesekara, I. J. Funct. Foods 2010, 2, 1. -
[5]
Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Mar. Environ. Res. 2017, 128, 58. doi: 10.1016/j.marenvres.2016.05.002
-
[6]
Aneiros, A.; Garateix, A. J. Chromatogr. B:Anal. Technol. Biomed. Life Sci. 2004, 803, 41. doi: 10.1016/j.jchromb.2003.11.005
-
[7]
Pan, X.; Zhao, Y. Q.; Hu, F. Y.; Wang, B. J. Funct. Foods 2016, 25, 220. doi: 10.1016/j.jff.2016.06.008
-
[8]
Ngo, D. H.; Vo, T. S.; Ngo, D. N.; Wijesekara, I.; Kim, S. K. Int. J. Biol. Macromol. 2012, 51, 378. doi: 10.1016/j.ijbiomac.2012.06.001
-
[9]
Castro, R. J. S. D.; Sato, H. H. Food Res. Int. 2015, 74, 185. doi: 10.1016/j.foodres.2015.05.013
-
[10]
Kang, H. K.; Seo, C. H.; Park, Y. Mar. Drugs 2015, 13, 618. doi: 10.3390/md13010618
-
[11]
Lee, J. K.; Jeon, J. K.; Byun, H. G. J. Funct. Foods 2014, 7, 381. doi: 10.1016/j.jff.2014.01.021
-
[12]
Zhan, K. X.; Jiao, W. H.; Yang, F.; Li, J.; Wang, S. P.; Li, Y. S.; Han, B. N.; Lin, H. W. J. Nat. Prod. 2014, 77, 2678. doi: 10.1021/np5006778
-
[13]
刘宸畅, 徐雪莲, 孙延龙, 张秀丽, 王聪, 庄波, 范迎方, 张广桃, 王成, 中国海洋药物, 2015, 34, 73.Liu, C.-C.; Xu, X.-L.; Kong, Y.-L.; Zhang, X.-L.; Wang, C.; Zhuang, B.; Fan, Y.-F.; Zhang, G.-T.; Wang, C. Chin. J. Mar. Drugs 2015, 34, 73.
-
[14]
(a) Pettit, G. R.; Kamano, Y.; Herald, C. L.; Tuinman, A. A.; Boettner, F. E.; Kizu, H.; Schmidt, J. M.; Baczynskyj, L.; Tomer, K. B.; Bontems, R. J. J. Am. Chem. Soc. 1987, 109, 6883.
(b) Pettit, G. R. Fortschritte der Chemie Organischer Naturstoffe: Progress in the Chemistry of Organic Natural Products, Vol. 70, Springer-Verlag, Wien, 1997, pp. 1~99.
(c) Pettit, G. R.; Kamano, Y.; Herald, C. L.; Fujii, Y.; Kizu, H.; Boyd, M. R.; Boettner, F. E.; Doubek, D. L.; Schmidt, J. M.; Chapuis, J. C. Tetrahedron 1993, 49, 9151. -
[15]
Miyazaki, K.; Kobayashi, M.; Natsume, T.; Gondo, M.; Mikami, T.; Sakakibara, K.; Tsukagoshi, S. Chem. Pharm. Bull. 1995, 43, 1706. doi: 10.1248/cpb.43.1706
-
[16]
Mordant, C.; Reymond, S.; Tone, H.; Lavergne, D.; Touati, R.; Hassine, B. B.; Ratovelomanana-Vidal, V.; Genet, J. P. Tetrahedron 2007, 63, 6115. doi: 10.1016/j.tet.2007.03.036
-
[17]
(a) Kalemkerian, G. P.; Ou, X.; Adil, M. R.; Rosati, R.; Khoulani, M. M.; Madan, S. K.; Pettit, G. R. Cancer Chemother. Pharmacol. 1999, 43, 507.
(b) Pettit, R. K.; Pettit, G. R.; Hazen, K. C. Antimicrob. Agents Chemother. 1998, 42, 2961.
(c) Poncet, J.; Busquet, M.; Roux, F.; Pierr, A., Atassi, G.; Jouin, P. J. Med. Chem. 1998, 41, 1524. -
[18]
Perez, E. A.; Hillman, D. W.; Fishkin, P. A.; Krook, J. E.; Tan, W. W.; Kuriakose, P. A.; Alberts, S. R.; Dakhil, S. R. Invest. New Drugs 2005, 23, 257. doi: 10.1007/s10637-005-6735-y
-
[19]
Kindler, H. L.; Tothy, P. K.; Wolff, R.; Mccormack, R. A.; Abbruzzese, J. L.; Mani, S.; Wade-Oliver, K. T.; Vokes, E. E. Invest. New Drugs 2005, 23, 489. doi: 10.1007/s10637-005-2909-x
-
[20]
Maderna, A.; Doroski, M.; Subramanyam, C.; Porte, A.; Leverett, C. A.; Vetelino, B. C.; Chen, Z.; Risley, H.; Parris, K.; Pandit, J. J. Med. Chem. 2014, 57, 10527. doi: 10.1021/jm501649k
-
[21]
(a) Diamantis, N.; Banerji, U. Br. J. Cancer 2016, 114, 362.
(b) Bakhtiar, R. Biotechnol. Lett. 2016, 38, 1.
(c) Gébleux, R.; Casi, G. Pharmacol. Ther. 2016, 167, 48 -
[22]
Thomas, A.; Teicher, B. A.; Hassan, R. Lancet Oncol. 2016, 17, e254. doi: 10.1016/S1470-2045(16)30030-4
-
[23]
王彦明, 郝伯钧, 钟武, 周辛波, 李松, 国际药学研究杂志, 2015, 42, 427.Wang, Y.-M.; Hao, B.-J.; Zhong, W.; Li, S. J. Int. Pharm. Res. 2015, 42, 427.
-
[24]
Newman, D. J.; Cragg, G. M. Mar. Drugs 2014, 12, 255. doi: 10.3390/md12010255
-
[25]
Senter, P. D.; Sievers, E. L. Nat. Biotechnol. 2012, 30, 631. doi: 10.1038/nbt.2289
-
[26]
(a) Ansell, S. M. Blood 2014, 124, 3197.
(b) Deng, C.; Pan, B.; O'Connor, O. A. Clin. Cancer Res. 2013, 19, 22.
(c) Okeley, N. M.; Miyamoto, J. B.; Zhang, X.; Sanderson, R. J.; Benjamin, D. R.; Sievers, E. L.; Senter, P. D.; Alley, S. C. Clin. Cancer Res. 2010, 16, 888. -
[27]
(a) Francisco, J. A.; Cerveny, C. G.; Meyer, D. L.; Mixan, B. J.; Klussman, K.; Chace, D. F.; Rejniak, S. X.; Gordon, K. A.; Deblanc, R.; Toki, B. E.; Law, C. L. Blood 2003, 102, 1458.
(b) Pettit, G. R.; Barkoczy, J. US 5635483, 1997 [Chem. Abstr. 1997, 122, 133848]. -
[28]
Moskowitz, C. H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M. H.; Chen, A. I.; Stiff, P.; Gianni, A. M.; Carella, A. Lancet 2015, 385, 1853. doi: 10.1016/S0140-6736(15)60165-9
-
[29]
Reilly, E. B.; Phillips, A. C.; Boghaert, E. R.; Vaidya, K. S.; Mitten, M. J.; Norvell, S.; Falls, H. D.; Devries, P. J.; Cheng, D.; Meulbroek, J. A. Mol. Cancer Ther. 2016, 15, 661. doi: 10.1158/1535-7163.MCT-15-0901
- [30]
-
[31]
Reilly, E. B.; Phillips, A. C.; Buchanan, F. G.; Kingsbury, G.; Zhang, Y.; Meulbroek, J. A.; Cole, T. B.; Devries, P. J.; Falls, H. D.; Beam, C. Mol. Cancer Ther. 2015, 14, 1141. doi: 10.1158/1535-7163.MCT-14-0820
-
[32]
Doronina, S. O.; Mendelsohn, B. A.; Bovee, T. D.; Cerveny, C. G.; Alley, S. C.; Meyer, D. L.; Oflazoglu, E.; Toki, B. E.; Sanderson, R. J.; Zabinski, R. F. Bioconjugate Chem. 2006, 17, 114. doi: 10.1021/bc0502917
-
[33]
Gan, H. K.; Fichtel, L.; Lassman, A. B.; Merrell, R.; Van, D. B., Martin J.; Kumthekar, P.; Scott, A. M.; Pedersen, M.; Gomez, E.; Fischer, J. D. J. Clinical Oncol. 2014, 32, 2021
-
[34]
(a) Vaklavas, C.; Forero, A. BioDrugs 2014, 28, 253.
(b) Roth, M.; Barris, D. M.; Piperdi, S.; Kuo, V.; Everts, S.; Geller, D.; Houghton, P.; Kolb, E. A.; Hawthorne, T.; Gill, J. Pediatr. Blood Cancer 2016, 63, 32.
(c) Keir, C. H.; Vahdat, L. T. Expert Opin. Biol. Ther. 2012, 12, 259. -
[35]
Yardley, D. A.; Weaver, R.; Melisko, M. E.; Saleh, M. N.; Arena, F. P.; Forero, A.; Cigler, T.; Stopeck, A.; Citrin, D.; Oliff, I. J. Clin. Oncol. 2015, 33, 1609. doi: 10.1200/JCO.2014.56.2959
-
[36]
Mehta, A.; Forero-Torres, A. Curr. Oncol. Rep. 2015, 17, 1. doi: 10.1007/s11912-014-0425-x
-
[37]
Palanca-Wessels, M. C. A.; Czuczman, M.; Salles, G.; Assouline, S.; Sehn, L. H.; Flinn, I.; Patel, M. R.; Sangha, R.; Hagenbeek, A.; Advani, R. Lancet Oncol. 2015, 16, 704. doi: 10.1016/S1470-2045(15)70128-2
-
[38]
(a) Pereira, D. S.; Guevara, C. I.; Jin, L.; Mbong, N.; Verlinsky, A.; Hsu, S. J.; Aviña, H.; Karki, S.; Abad, J. D.; Yang, P. Mol. Cancer Ther. 2015, 14, 1650.
(b) Lapalombella, R.; Yeh, Y. Y.; Wang, L.; Ramanunni, A.; Rafiq, S.; Jha, S.; Staubli, J.; Lucas, D. M.; Mani, R.; Herman, S. E. M. Cancer Cell 2012, 21, 694. -
[39]
Sawas, A.; Savage, K. J.; Perez, R.; Advani, R. H.; Butturini, A.; Lackey, J.; Trave, F.; Anand, B.; Huang, Y.; Reyno, L. Nature 2015, 128, 413.
-
[40]
Akashi, Y.; Okamoto, I.; Suzuki, M.; Tamura, K.; Iwasa, T.; Hisada, S.; Satoh, T.; Nakagawa, K.; Ono, K.; Fukuoka, M. Br. J. Cancer 2007, 96, 1532. doi: 10.1038/sj.bjc.6603769
-
[41]
Otani, M.; Natsume, T.; Watanabe, J. I.; Kobayashi, M.; Murakoshi, M.; Mikami, T.; Nakayama, T. Cancer Sci. 2000, 91, 837.
-
[42]
Watanabe, J.; Natsume, T.; Fujio, N.; Miyasaka, K.; Kobayashi, M. Apoptosis 2000, 5, 345. doi: 10.1023/A:1009687609330
-
[43]
Patel, S.; Keohan, M. L.; Saif, M. W.; Rushing, D.; Baez, L.; Feit, K.; Dejager, R.; Anderson, S. Cancer 2006, 107, 2881. doi: 10.1002/(ISSN)1097-0142
-
[44]
Riely, G. J.; Gadgeel, S.; Rothman, I.; Saidman, B.; Sabbath, K.; Feit, K.; Kris, M. G.; Rizvi, N. A. Lung Cancer 2007, 55, 181. doi: 10.1016/j.lungcan.2006.10.002
-
[45]
Jordan, M. A.; Walker, D.; Arruda, M. D.; Barlozzari, T.; Panda, D. Biochemistry 1999, 37, 17571.
-
[46]
(a) Ray, A.; Okouneva, T.; Manna, T.; Miller, H. P.; Schmid, S.; Arthaud, L.; Luduena, R.; Jordan, M. A.; Wilson, L. Cancer Res. 2007, 67, 3767.
(b) Bai, R.; Edler, M. C.; Bonate, P. L.; Copeland, T. D.; Pettit, G. R.; Ludueña, R. F.; Hamel, E. Mol. Pharmacol. 2009, 75, 218. -
[47]
Mita, A. C.; Hammond, L. A.; Bonate, P. L.; Weiss, G.; Mccreery, H.; Syed, S.; Garrison, M.; Chu, Q. S.; Debono, J. S.; Jones, C. B. Clin. Cancer Res. 2006, 12, 5207. doi: 10.1158/1078-0432.CCR-06-0179
-
[48]
De, A. M.; Cocchiaro, C. A.; Nelson, C. M.; Grinnell, C. M.; Janssen, B.; Haupt, A.; Barlozzari, T. Cancer Res. 1995, 55, 3085.
-
[49]
Kerbrat, P.; Dieras, V.; Pavlidis, N.; Ravaud, A.; Wanders, J.; Fumoleau, P. Eur. J. Cancer 2003, 39, 317. doi: 10.1016/S0959-8049(02)00531-2
-
[50]
(a) Schroeder, C. I.; Craik, D. J. Future Med. Chem. 2012, 4, 1243.
(b) Vetter, I.; Lewis, R. J. Curr. Top. Med. Chem. 2012, 12, 1546. -
[51]
(a) Durek, T.; Craik, D. J. Expert Opin. Ther. Pat. 2015, 25, 1.
(b) Livett, B. G.; Gayler, K. R.; Khalil, Z. Curr. Med. Chem. 2004, 11, 1715. -
[52]
Schmidtko, A.; Lajevardi, J.; Freynhagen, R.; Geisslinger, G. Lancet 2010, 375, 1569. doi: 10.1016/S0140-6736(10)60354-6
-
[53]
Dong, S.-L.; Cao, S.; Chen, Q.; Chang, M.; Peng, Y.-L. CN 103242441, 2013[Chem. Abstr. 2013, 159, 371454].
-
[54]
Miljanich, G. P. Curr. Med. Chem. 2004, 11, 3029. doi: 10.2174/0929867043363884
-
[55]
Williams, J. A.; Day, M.; Heavner, J. E. Expert Opin. Pharmacother. 2008, 9, 1575. doi: 10.1517/14656566.9.9.1575
-
[56]
Staats, P. S.; Yearwood, T.; Charapata, S. G.; Presley, R. W.; Wallace, M. S.; Byas-Smith, M.; Fisher, R.; Bryce, D. A.; Mangieri, E. A.; Luther, R. R. J. Am. Med. Assoc. 2004, 291, 63. doi: 10.1001/jama.291.1.63
-
[57]
Vera, M. D.; Joullié, M. M. Med. Res. Rev. 2002, 22, 102. doi: 10.1002/med.v22:2
-
[58]
Geldof, A. A.; Mastbergen, S. C.; Henrar, R. E.; Faircloth, G. T. Cancer Chemother. Pharmacol. 1999, 44, 312. doi: 10.1007/s002800050983
-
[59]
Stewart, J. A.; Low, J. B.; Roberts, J. D.; Blow, A. Cancer 1991, 68, 2550. doi: 10.1002/(ISSN)1097-0142
-
[60]
Hamada, Y.; Kondo, Y.; Shibata, M.; Shioiri, T. J. Am. Chem. Soc. 1989, 111, 669. doi: 10.1021/ja00184a041
-
[61]
Kucuk, O.; Young, M. L.; Habermann, T. M.; Wolf, B. C.; Jimeno, J.; Cassileth, P. A. Am. J. Clin. Oncol. 2000, 23, 273. doi: 10.1097/00000421-200006000-00013
-
[62]
Danu, A.; Willekens, C.; Ribrag, V. Expert Opin. Orphan Drugs 2013, 1, 569. doi: 10.1517/21678707.2013.808995
-
[63]
(a) Galmarini, C. M.; D'Incalci, M.; Allavena, P. Mar. Drugs 2014, 12, 719.
(b) Mitsiades, C. S.; Ocio, E. M.; Pandiella, A.; Maiso, P.; Gajate, C.; Garayoa, M.; Vilanova, D.; Montero, J. C.; Mitsiades, N.; Mcmullan, C. J. Cancer Res. 2008, 68, 5216. -
[64]
Jou, G.; Gonzalez, I.; Albericio, F.; Lloyd-Williams, P.; Giralt, E. J. Org. Chem. 1997, 62, 354. doi: 10.1021/jo961932h
-
[65]
Mateos, M. V.; Cibeira, M. T.; Richardson, P. G.; Prosper, F.; Oriol, A.; De, l. R. J.; Lahuerta, J. J.; García-Sanz, R.; Extremera, S.; Szyldergemajn, S. Clin. Cancer Res. 2010, 16, 3260. doi: 10.1158/1078-0432.CCR-10-0469
-
[66]
Rawat, D. S.; Joshi, M. C.; Joshi, P.; Atheaya, H. Anti-Cancer Agents Med. Chem. 2006, 6, 33. doi: 10.2174/187152006774755519
-
[67]
(a) Anderson, H. J.; Coleman, J. E.; Andersen, R. J.; Roberge, M. Cancer Chemother. Pharmacol. 1996, 39, 223.
(b) Coleman, J. E.; Silva, E. D. D.; Kong, F.; Andersen, R. J.; Allen, T. M. J. Nat. Prod. 1996, 27, 10653. -
[68]
Nieman, J. A.; Coleman, J. E.; Wallace, D. J.; Piers, E.; Lim, L. Y.; Roberge, M.; Andersen, R. J. J. Nat. Prod. 2003, 66, 183. doi: 10.1021/np020375t
-
[69]
Niu, C.; Ho, D. M.; Williamson, R. T.; Zask, A.; Ayral-Kaloustian, S. Bioorg. Med. Chem. Lett. 2010, 20, 1535. doi: 10.1016/j.bmcl.2010.01.047
-
[70]
Zask, A.; Birnberg, G.; Cheung, K.; Kaplan, J.; Niu, C.; Norton, E.; Suayan, R.; Yamashita, A.; Cole, D.; Tang, Z.; Krishnamurthy, G.; Williamson, R.; Khafizova, G.; Musto, S.; Hernandez, R.; Annable, T.; Yang, X.; Discafani, C.; Beyer, C.; Greenberger, L. M.; Loganzo, F.; Ayral-Kaloustian, S. J. Med. Chem. 2004, 47, 4774. doi: 10.1021/jm040056u
-
[71]
Loganzo, F.; Discafani, C. M.; Annable, T.; Beyer, C.; Musto, S.; Hari, M.; Tan, X.; Hardy, C.; Hernandez, R.; Baxter, M. Cancer Res. 2003, 63, 1838.
-
[72]
(a) Vashist, Y. K.; Tiffon, C.; Stoupis, C.; Redaelli, C. A. World J. Gastroenterol. 2006, 12, 6771.
(b) Hadaschik, B. A.; Ettinger, S.; Sowery, R. D.; Zoubeidi, A.; Andersen, R. J.; Roberge, M.; Gleave, M. E. Int. J. Cancer 2008, 122, 2368.
(c) Hadaschik, B. A.; Adomat, H.; Fazli, L.; Fradet, Y.; Andersen, R. J.; Gleave, M. E.; So, A. I. Clin. Cancer Res. 2008, 14, 1510. -
[73]
Kuznetsov, G.; Tendyke, K.; Towle, M. J.; Cheng, H.; Liu, J.; Marsh, J. P.; Schiller, S. E.; Spyvee, M. R.; Yang, H.; Seletsky, B. M. Mol. Cancer Ther. 2009, 8, 2852. doi: 10.1158/1535-7163.MCT-09-0301
-
[74]
Kowalczyk, J. J.; Kuznetsov, G.; Schiller, S.; Seletsky, B. M.; Spyvee, M.; Yang, H. WO 2003082268, 2003[Chem. Abstr. 2003, 139, 308008].
-
[75]
Rochalima, C. M.; Bayraktar, S.; Macintyre, J.; Raez, L.; Flores, A. M.; Ferrell, A.; Rubin, E. H.; Poplin, E. A.; Tan, A. R.; Lucarelli, A. Cancer 2012, 118, 4262. doi: 10.1002/cncr.27428
-
[76]
(a) Yamazaki, Y.; Tanaka, K.; Nicholson, B.; Deyanatyazdi, G.; Potts, B.; Yoshida, T.; Oda, A.; Kitagawa, T.; Orikasa, S.; Kiso, Y. J. Med. Chem. 2012, 55, 1056.
(b) Hayashi, Y.; Orikasa, S.; Tanaka, K.; Kanoh, K.; Kiso, Y. J. Org. Chem. 2000, 65, 8402. -
[77]
Kingston, D. G. I. J. Nat. Prod. 2009, 72, 507. doi: 10.1021/np800568j
-
[78]
Yamazaki, Y.; Sumikura, M.; Hidaka, K.; Yasui, H.; Kiso, Y.; Yakushiji, F.; Hayashi, Y. Bioorg. Med. Chem. 2010, 18, 3169. doi: 10.1016/j.bmc.2010.03.037
-
[79]
Nicholson, B.; Lloyd, G. K.; Miller, B. R.; Palladino, M. A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S. T. Anti-Cancer Drugs 2006, 17, 25. doi: 10.1097/01.cad.0000182745.01612.8a
-
[80]
Ferrer, E.; Bolos, J.; Castañer, R. Drugs Future 2010, 35, 11. doi: 10.1358/dof.2010.035.01.1439262
-
[81]
Heist, R. S.; Aren, O. R.; Mita, A. C.; Polikoff, J.; Bazhenova, L.; Lloyd, G. K.; Mikrut, W.; Reich, S. D.; Spear, M. A.; Huang, L. J. Clin. Oncol. 2014, 8054.
-
[82]
(a) Stokvis, E.; Rosing, H.; López-Lázaro, L.; Rodriguez, I.; Jimeno, J. M.; Supko, J. G.; Schellens, J. H.; Beijnen, J. H. J. Mass Spectrom. 2002, 37, 992.
(b) Lopezmacia, A.; Jimenez, J. C.; Royo, M.; Giralt, E.; Albericio, F. J. Am. Chem. Soc. 2001, 123, 11398. -
[83]
Janmaat, M. L.; Rodriguez, J. A.; Jimeno, J.; Kruyt, F. A.; Giaccone, G. Mol. Pharmacol. 2005, 68, 502.
-
[84]
Suarez, Y.; Gonzalez, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Munoz, A. Mol. Cancer Ther. 2003, 2, 863.
-
[85]
Martin-Algarra, S.; Espinosa, E.; Rubio, J.; Lopez Lopez, J. J.; Manzano, J. L.; Carrion, L. A.; Plazaola, A.; Tanovic, A.; Pazares, L. Eur. J. Cancer 2009, 45, 732. doi: 10.1016/j.ejca.2008.12.005
-
[86]
Coronado, C.; Galmarini, C. M.; Alfaro, V.; Yovine, A. Drugs Future 2010, 35, 287. doi: 10.1358/dof.2010.035.04.1497635
-
[87]
(a) Teixido, C.; Mares, R.; Aracil, M.; Ramon, Y. C. S.; Hernandezlosa, J. PloS One 2013, 8, e53645.
(b) Serova, M.; Gramont, A. D.; Bieche, I.; Riveiro, M. E.; Galmarini, C. M.; Aracil, M.; Jimeno, J.; Faivre, S.; Raymond, E. Mar. Drugs 2013, 11, 944.
(c) Molinaguijarro, J. M.; Garcia, C.; Macias, A.; Moreno, C.; Reyes, F.; Martínezleal, J. F.; Fernandez, R.; Martinez, V.; Valenzuela, C.; Lillo, M. P. PloS One 2015, 10, e0140782.
(d) Hajdu, T. Ageing Soc. 2013, 20, 633. -
[88]
Petty, R.; Anthoney, A.; Metges, J. P.; Alsina, M.; Gonçalves, A.; Brown, J.; Montagut, C.; Gunzer, K.; Laus, G.; Iglesias Dios, J. L. Cancer Chemother. Pharmacol. 2016, 77, 819. doi: 10.1007/s00280-016-2991-0
-
[1]
-

图式3 N-Boc-(2R, 3R)-Dap的合成
Scheme 3 Synthesis of N-Boc-(2R, 3R)-Dap
Reagents and conditions: (a) [Ru]/(P×P)=3 mol%, [RuCl2(pcymene)]2/(S)-SYNPHOS, H2, 13 MPa, 50 ℃, 117 h; (b) (1) LiHMDS, HMPA, THF, -78 ℃, 25 min; (2) MeOTf, -20 ℃, 15 min; (c) LiOH, EtOH/H2O, overnight.

图式6 MMAF和mcMMAF的合成
Scheme 6 Synthesis of MMAF and mcMMAF
Reagents and conditions: (a) Fmoc-Dap (2 equiv.), HATU (2 equiv.), DIEA (4 equiv.); (b) 20% piperidine in DMF; (c) Fmoc-MeVal-Val-Dil-OH (2 equiv.), HATU (2 equiv.), DIEA (4 equiv.); (d) 20% piperidine in DMF; (e) 2% TFA in CH2Cl2.

图 4 Polatuzumab vedotin和AGS-67E的结构
Figure 4 Structure of polatuzumab vedotin and AGS-67E

图式7 Solidotin的合成
Scheme 7 Synthesis of solidotin
Reagents and conditions: (a) CDI, THF; (b) CH2(CO2CH3)CO2K, MgCl2, THF; (c) NaBH4, MeOH; (d) CH3I, Ag2O, DMF; (e) aqueous NaOH, dioxane; (f) isobutene, H+; (g) H2, Pd-C, t-BuOH/H2O; (h) Z-Val-OH, DCC, CH2Cl2; (i) Dov, DEPC, Et3N, DMF; (j) TFA, CH2Cl2; (k) H-Dap-OBzl•HCl, DEPC, Et3N, DMF; (l) DEPC, Et3N, DMF.

图 5 Dolatatin 15、Tasidotin和Cemadotin的结构
Figure 5 Sructures of dolatatin 15, tasidotin and cemadotin

图式9 Didemnin B的合成
Scheme 9 Synthesis of didemnin B
Reagents and conditions: (a) (1) DEPC, Et3N, DMF, 0 ℃, 4 h; (2) r.t., 20 h; (b) 4 mol•L-1 HCl/dioxane, r.t., 1 h; (c) H2, 5% Pd-C, THF, r.t., 2~3 h; (d) (1) DCC, 1-hydroxybenzotriazole, N-methylmorpholine, THF/DMF (V:V=2:1), 0 ℃, 3 h; (2) r.t., 26 h; (e) Bu4N+F-, THF, r.t., 1 h; (f) (1) DEPC, i-Pr2EtN, DMF, 0 ℃, 4 h; (2) r.t., 1 h; (g) TBDMS-Cl, imidazole, DMF, r.t., 37 h; (h) (1) DCC, DMAP, CH2Cl2, 0 ℃, 3 h; (2) r.t., 16~22 h; (i) Zn, 1 mol•L-1 NH4OAc (aqueous), THF, r.t., 30 h; (j) Bop-Cl, Et3N, CH2Cl2, 2 ℃, 3 d; (k) TMSOTf, CH2Cl2, 0 ℃, 3 h; (m) Pd, HCO2H, MeOH.
-


 下载:
下载:






















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 114
- 文章访问数: 10866
- HTML全文浏览量: 3278
 下载:
下载:
