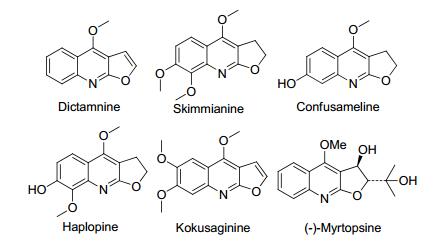
 图 1
几种代表性的含有呋喃[2, 3-b]喹啉母核结构的天然产物
Figure 1.
Several representative furo[2, 3-b]quinolines-con-taining natural products
图 1
几种代表性的含有呋喃[2, 3-b]喹啉母核结构的天然产物
Figure 1.
Several representative furo[2, 3-b]quinolines-con-taining natural products
引用本文:
张志国, 郑丹, 麻娜娜, 毕晶晶. 布朗斯特酸促进的呋喃[2, 3-b]喹啉类并环化合物的合成[J]. 有机化学,
2017, 37(7): 1824-1829.
doi:
10.6023/cjoc201612032

Citation: Zhang Zhiguo, Zheng Dan, Ma Na'na, Bi Jingjing. Brønsted Acid-Promoted the Synthesis of Furo[2, 3-b]quinolines[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1824-1829. doi: 10.6023/cjoc201612032
Citation: Zhang Zhiguo, Zheng Dan, Ma Na'na, Bi Jingjing. Brønsted Acid-Promoted the Synthesis of Furo[2, 3-b]quinolines[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1824-1829. doi: 10.6023/cjoc201612032
布朗斯特酸促进的呋喃[2, 3-b]喹啉类并环化合物的合成
摘要:
呋喃并喹啉结构单元广泛存在于很多天然产物中.介绍一种在加热的条件下,三氟甲磺酸促进的,通过多取代呋喃衍生物分子内环合反应制备呋喃[2,3-b]喹啉类化合物的方法.该方法具有操作简单、官能团兼容性好、区域选择性好和产品产率较高等优点.
-
关键词:
- 布朗斯特酸
- / 呋喃[2, 3-b]喹啉
- / 三氟甲磺酸
- / 傅-克反应
English
Brønsted Acid-Promoted the Synthesis of Furo[2, 3-b]quinolines
Abstract:
The furoquinoline unit is present in many natural products. Here, an approach is presented for the preparing of furo[2, 3-b]quinolines from readily available multi-substituted furans in the presence of Brønsted acid via an intramolecular cyclization under the heating conditions. Simple operation, good compatibility, high regioselectivity and morderate yields are the advantages of the method.
-
呋喃[2, 3-b]喹啉类化合物是一种重要的含氮和氧杂原子的三并杂环类化合物[1].含有这类母核结构的生物碱, 如白鲜碱、崖椒碱和吴萸春碱等, 广泛存在于芸香科植物中[2].它们拥有多种生物活性, 比如抗菌、抗病毒、抗人类免疫缺陷病毒、抗炎、抗肿瘤和抗细胞毒素等(图 1)[3].白鲜碱是含有呋喃[2, 3-b]喹啉结构单元的结构最简单的呋喃并喹啉类天然产物. 1956年, 呋喃并喹啉类化合物的合成方法首次在德国《应用化学》杂志上发表[4], 从此, 这类化合物的合成方法一直是有机合成化学的热点研究领域[5].目前, 文献报道的呋喃[2, 3-b]喹啉类化合物的合成方法大致可以分为两种合成策略: (a)通过3位官能化喹啉衍生物环合反应合成呋喃[2, 3-b]喹啉类化合物.这类反应一般是先在喹啉的3位构建一个端位带有炔基、羟基, 烯基或者卤素等官能团的烷基链, 然后再通过分子内环合反应得到呋喃[2, 3-b]喹啉类化合物的呋喃部分(Scheme 1, a)[6]. (b)通过官能化的呋喃衍生物环合反应合成呋喃[2, 3-b]喹啉类化合物.这类反应一般是以亚胺和富电子的呋喃为原料, 通过杂Diels-Alder反应先构建一个角型的呋喃并喹啉类前体化合物, 然后再经过呋喃部分的开环再环合反应得到呋喃[2, 3-b]喹啉类化合物(Scheme 1, b)[7].虽然通过上述两种策略都可以获得呋喃[2, 3-b]喹啉类化合物, 但是, 一些方法存在普适性差、区域选择性差(自由基环合反应)、原料合成局限性大、反应条件苛刻以及所用试剂昂贵等问题, 影响了这些反应的实用价值.因此, 发展新的呋喃并喹啉化合物的合成方法仍然具有重要的现实意义和理论研究价值.
图 1
几种代表性的含有呋喃[2, 3-b]喹啉母核结构的天然产物
Figure 1.
Several representative furo[2, 3-b]quinolines-con-taining natural products
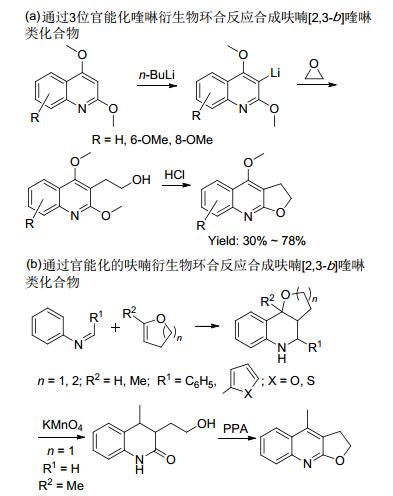 图式1
常见的呋喃[2, 3-b]喹啉类化合物的合成策略
图式1.
Strategies for the synthesis of furan[2, 3-b]quinolines
图式1
常见的呋喃[2, 3-b]喹啉类化合物的合成策略
图式1.
Strategies for the synthesis of furan[2, 3-b]quinolines
串联反应因具有高效和实用的特点被广泛应用于有机合成化学[8].据我们所知, 通过同时构建呋喃环和喹啉环的串联反应策略合成呋喃[2, 3-b]喹啉类化合物的文献报道不多[5a, 9]. 2016年, Shi等[5a]以邻氨基芳基亚烷基环丙烷衍生物为原料, 在三光气(BTC)和三乙胺的联合催化下, 通过形式上的分子内[3+2]环加成串联反应合成了呋喃[2, 3-b]喹啉类化合物(Eq. 1). 2013年, Ru等[9b]发现在卤化铜的作用下, 1-酰基-1-芳酰胺基环丙烷类化合物可以发生高选择性的开环加成反应, 生成一种双卤代的β-羰基酰胺类化合物, 这种酰胺衍生物可以在硫酸的促进下进一步得到呋喃[2, 3-b]喹啉类化合物(Scheme 2). 2007年, 我们课题组[9a]利用高效的串联反应, 通过先构建呋喃再构建喹啉环的新串联反应策略, 从1-酰基-1-芳酰胺基环丙烷类化合物出发, 在五水四氯化锡的催化下, 经过分子内开环/再环合反应, 合成了一系列呋喃[2, 3-b]喹啉类衍生物(Eq. 2).同年, 利用便宜易得的双活化的1-酰基-1-芳酰胺基环戊烷衍生物, 在硫酸的促进下, 合成了吡喃[2, 3-b]喹啉类生物碱[10].该反应具有原料易得、产物产率高、反应普适性好和易于操作等优点(Eq. 3).在上述研究工作的基础上[11], 我们希望能够发展更多的合成呋喃[2, 3-b]喹啉类化合物的新方法, 以满足这类化合物取代基多样性需要.本文介绍一种由简单易得的多取代呋喃为原料, 在三氟甲磺酸(CF3SO3H)的促进下, 经过分子内亲电环合反应一步生成呋喃[2, 3-b]喹啉类化合物的方法(Eq. 4).
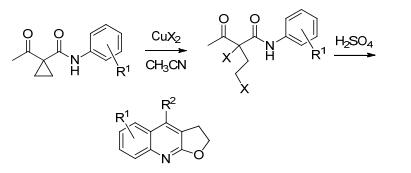 图式2
Ru等合成呋喃[2, 3-b]喹啉类化合物的工作
图式2.
Ru et al. developed the synthesis of furo[2, 3-b]-quinolines
图式2
Ru等合成呋喃[2, 3-b]喹啉类化合物的工作
图式2.
Ru et al. developed the synthesis of furo[2, 3-b]-quinolines
1 结果与讨论
首先, 以N, N-二甲基甲酰胺(DMF)为溶剂, 在无水K2CO3的作用下, 以乙酰乙酰苯胺和1, 2, 3-三溴丙烷为原料, 通过分子间C, O-双烷基化反应制得了多取代的呋喃衍生物1a[9a].在得到了化合物1a以后, 我们开始从催化剂的种类、用量、反应温度及溶剂种类等几个方面对该反应的条件进行了优化, 尝试合成目标化合物2a.主要的实验结果总结在表 1中. 2007年, Klumpp课题组[12]通过利用理论计算和核磁共振波谱等手段证明三氟甲磺酸是促进酰基和芳烃发生傅-克反应最有效的质子酸, 因此, 我们首先选择了三氟甲磺酸尝试促进该环合反应.经过多次尝试我们发现, 在80 ℃时, 在8倍量的三氟甲磺酸中, 10 h以后反应混合物经过中和, 萃取, 柱层析分离提纯后, 环合产物2a的产率可以达到76% (表 1, Entry 2).如果降低或者升高反应的温度, 产物2a的产率都会降低, 只是在越高的温度下反应的速度越快(表 1, Entries 1, 3).增加三氟甲磺酸的用量到10倍量也不能提高产物2a的产率(表 1, Entry 4).另外, 我们也尝试了使用低于8倍量的三氟甲磺酸促进这个反应, 但是由于三氟甲磺酸的用量太少, 底物1a不能很好地溶解.我们还尝试了其它的液体的布朗斯特酸, 比如:质子酸多聚磷酸(PPA)、浓硫酸(H2SO4)和路易斯酸三氟甲磺酸三甲基硅酯(TMSOTf).实验结果表明, 虽然PPA可以促进2a的收率达到71%, 但是反应时间需要24 h (表 1, Entry 5).浓硫酸也需要14 h才能得到68%的2a (表 1, Entry 6).虽然TMSOTf参与的反应速率较快, 但是产物产率只能达到57% (表 1, Entry 7).此外, 我们还尝试了对甲苯磺酸(p-TsOH)这种固体酸对该环合反应的影响.为了使固体的原料1a和对甲苯磺酸溶解形成均一的溶液, 我们使用了1 mL的1, 4-二氧六环作溶剂, 结果我们观察到反应产生了非常复杂的混合物(表 1, Entry 8).其它一些常见的质子酸, 如:醋酸(AcOH)、特戊酸(PivOH)、三氟乙酸(TFA)和三氯乙酸(TCA)在10 h内不能得到出目标化合物(表 1, Entries 9~12).
 表 1
优化条件表a
Table 1.
Survey of the reaction conditions
表 1
优化条件表a
Table 1.
Survey of the reaction conditions

Entry Acid (equiv.) T/℃ Time/h Yield of 2a/% 1 CF3SO3H (8) 60 14 59b 2 CF3SO3H (8) 80 10 76 3 CF3SO3H (8) 100 8 70 4 CF3SO3H (10) 80 10 67 5 PPA (8) 80 24 71 6 Con. H2SO4 (8) 80 14 68c 7 TMSOTf (8) 80 6 57d 8 p-TsOH (8) 80 10 0e 9 AcOH (8) 80 10 0f 10 PivOH (8) 80 10 0g 11 TFA (8) 80 10 0h 12 TCA (8) 80 10 0i a除非另外说明, 所有反应都是在空气氛围中进行, 反应物1a的用量为0.2 mmol, 收率为分离收率; b18%的1a被回收. c5%的1a被回收. dTMSOTf的沸点为77 ℃, 因此反应在密封管中进行. e20%的1a被回收. f16%的1a被回收. g30%的1a被回收. h5%的1a被回收. i12%的1a被回收. 在确定了最佳的反应条件以后(表 1, Entry 2), 我们对反应的适用范围进行了研究, 主要结果总结归纳在表 2中.首先, 我们研究了各种R2取代基对反应的影响.实验表明, 不但苯基取代的原料1a可以顺利地得到目标化合物2a(产率76%), 而且, 当R2取代基为甲氧基或者甲基时, 原料也可以顺利地生成相应的目标产物2.邻甲氧基取代的原料1b可以以75%的收率给出目标产物2b.邻位和对位甲基取代的底物1c和1e也可以分别生成目标化合物2c和2e (产率: 72%和77%).对于弱吸电子的取代基R2, 如Cl和Br等卤代基团, 反应1f~1i也可以顺利的进行, 邻、间和对位卤代的产物2f~2i的收率可以达到73%~79%.但是, 苯基对位带有酯基这种强吸电子基的原料1j不能得到环合产物2j, 78%的原料1j被回收.这一结果符合傅-克反应适用范围所描述的当芳基带有强吸电子基时傅-克反应不能发生这一结论, 因此硝基苯可以作为这类反应的溶剂[13].而化合物1f~1i能够发生该亲电环合反应的原因是氨基是带有供电子性质的基团, 它对傅-克反应的影响大于像卤素这样的弱吸电子基.还值得一提的是产物2i, 溴取代基能够被反应很好地兼容, 这有利于这类化合物进一步发生Heck反应等衍生化反应, 从而得到更加有用的分子.另外, 由于取代基R1的局限性, 我们只尝试了当R1为苯基取代基的原料是否能够发生这类反应.实验结果表明, 化合物1k可以顺利的发生亲电环合反应, 得到91%的目标产物2k.
表 2
底物扩展a
Table 2.
Extension of the reaction scope

2 反应机理的研究
根据近些年的文献报道[9a, 10, 14]和Combes喹啉类化合物合成原理[11], 我们认为这个反应经历了分子内芳基亲电环合芳构化的反应机理.首先, 在加热和布朗斯特酸的共同作用下, 多取代呋喃类化合物1的羰基上的氧原子被质子化, 形成氧鎓离子中间体A[12], 然后中间体A发生分子内亲电环合反应生成中间体B, 最后, 再经过质子转移和脱水反应得到呋喃[2, 3-b]喹啉类化合物2 (Scheme 3).
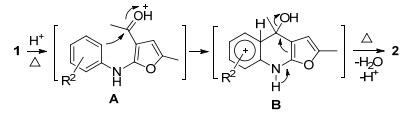 图式3
反应可能的机理
图式3.
Proposed mechanism
图式3
反应可能的机理
图式3.
Proposed mechanism
3 结论
总之, 发展了一种布朗斯特酸酸促进的以多取代呋喃为原料一步合成具有重要意义的呋喃[2, 3-b]喹啉类化合物的新方法.该方法的原料便宜易得, 条件温和, 反应易操作, 化学选择性和区域选择性好, 产物产率较高.该方法进一步丰富了先构建杂环, 再形成喹啉环的合成杂环并喹啉类生物碱的合成策略, 可以为设计与合成其它的杂环并喹啉类化合物提供实验支持和理论指导.
4 实验部分
4.1 仪器与试剂
13C NMR和1H NMR由Bruker DPX-400M核磁共振仪测定, 其中TMS作为内标, CDCl3或DMSO-d6作为溶剂; HR-MS由瑞士布鲁克公司高分辨率质谱测定仪: ESI源测定; 化合物熔点是由巩义市予华仪器责任有限公司的X-5显微熔点测定仪测定; TLC检测使用巩义市予华仪器责任有限公司ZF-20D暗箱式紫外分析仪; 化合物称量使用奥豪斯国际贸易有限公司电子分析天平; 溶剂旋蒸使用巩义市予华仪器责任有限公司YRE-2000旋转蒸发仪; 高分辨质谱由Bruker microToF Ⅱ高分辨率质谱仪测定.
药品和试剂见表 3.
表 3
药品和试剂
Table 3.
Starting materials and reagents
药品或试剂名称 纯度 生产厂家 苯甲酰乙酸乙酯 A.R. 上海达瑞精细化学品有限公司 对甲氧基苯甲酰乙酸乙酯 A.R. 梯希爱(上海)化成工业发展有限公司 乙酰乙酸乙酯 A.R. 上海麦克林生化科技有限公司 苯胺 A.R. 国药集团化学试剂有限公司 对溴苯胺 A.R. 上海嘉辰 对甲氧基苯胺 A.R. 阿拉丁 乙酰乙酰苯胺 A.R 青岛双桃精细化工(有限)公司 2-氯乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 4-氯乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 4-甲氧基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 2-甲氧基乙酰乙酰苯胺 A.R 青岛双桃精细化工(有限)公司 4-甲基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 2-甲基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 2, 4-甲基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 1, 2, 3-三溴丙烷 A.R. 阿拉丁 无水碳酸钾 A.R. 国药集团化学试剂有限公司 N, N-二甲基甲酰胺 A.R. 成都市科龙化工试剂厂 三氟甲磺酸 A.R. 阿拉丁 4.2 化合物2的合成
以2b为例.在25 mL圆底烧瓶中加入2-(N-邻甲氧基苯基)-3-乙酰基-5-甲基呋喃(1b) 49 mg (0.2 mmol)、三氟甲磺酸0.14 mL (1.6 mmol), 在80 ℃油浴中加热反应混合物, 直到原料1b消失, 反应过程使用TLC监测.反应结束后将反应混合物冷却至室温, 向反应混合物中加入10 mL水和1.5 mL碳酸氢钠饱和溶液, 中和反应混合物至中性, 有不溶固体析出, 抽滤, 水洗滤饼(2 mL×3), 得到的灰色固体粗产品经柱层析分离纯化[V(乙酸乙酯)/V(石油醚)=1/10], 得白色固体产物8-甲氧基-2, 4-二甲基呋喃[2, 3-b]喹啉(2b), 34 mg, 产率75%. m.p. 158~160 ℃; 1H NMR (400 MHz, CDCl3) δ:7.68 (d, J=8.4, 1H), 7.51 (s, 1H), 7.47~7.40 (m, 1H), 7.06 (d, J=7.6 Hz, 1H), 4.10 (s, 3H), 2.97 (s, 3H), 2.47 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:143.2, 126.9, 124.0, 119.9, 115.4, 115.1, 106.7, 56.0, 14.7, 11.5. HRMS (ESI) calcd for C14H13NNaO2 ([M+Na]+) 250.0838, found 250.0842.
2, 4-二甲基呋喃[2, 3-b]喹啉(2a):白色固体, 30 mg, 产率76%. m.p. 127~129 ℃; 1H NMR (600 MHz, CDCl3) δ:8.28 (d, J=8.4 Hz, 1H), 8.21 (d, J=8.4 Hz, 1H), 7.88~7.85 (m, 1H), 7.69~7.67 (m, 1H), 7.60 (s, 1H), 3.09 (s, 3H), 2.53 (s, 3H); 13C NMR (150 MHz, CDCl3) δ:158.6, 143.6, 131.4, 126.1, 125.4, 124.1, 120.9, 116.3, 15.1, 11.2. HRMS (ESI) calcd for C13H12NO2 ([M+H]+) 198.0913, found 198.0918.
2, 4, 8-三甲基呋喃[2, 3-b]喹啉(2c):白色固体, 32 mg, 产率77%. m.p. 110~112 ℃; 1H NMR (400 MHz, CDCl3) δ:7.98 (d, J=8.4 Hz, 1H), 7.57 (d, J=6.8 Hz, 1H), 7.50 (s, 1H), 7.47~7.37 (m, 1H), 2.99 (s, 3H), 2.85 (s, 3H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:161.0, 143.9, 142.6, 139.0, 136.5, 128.8, 125.8, 123.9, 121.5, 119.1, 115.3, 18.7, 14.5, 11.5. HRMS (ESI) calcd for C14H14NO ([M+H]+) 212.1070, found 212.1078.
2, 4, 6-三甲基呋喃[2, 3-b]喹啉(2d):白色固体, 30 mg, 产率72%. m.p. 135~137 ℃; 1H NMR (400 MHz, CDCl3) δ:8.00~7.98 (m, 1H), 7.79 (s, 1H), 7.56~7.48 (m, 1H), 7.44 (s, 1H), 2.89 (s, 3H), 2.56 (s, 3H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:161.0, 142.6, 142.5, 138.7, 134.1, 131.1, 128.1, 125.8, 122.5, 119.6, 115.5, 21.9, 14.3, 11.5. HRMS (ESI) calcd for C14H13NNaO ([M+Na]+) 234.0889, found 234.0890.
2, 4, 6, 8-四甲基呋喃[2, 3-b]喹啉(2e):白色固体, 36 mg, 产率77%. m.p. 102~104 ℃; 1H NMR (400 MHz, CDCl3) δ:7.68 (s, 1H), 7.45 (s, 1H), 7.39 (s, 1H), 2.90 (s, 3H), 2.79 (s, 3H), 2.52 (s, 3H), 2.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:160.4, 142.1, 137.8, 135.8, 133.0, 130.9, 125.5, 120.1, 118.7, 115.0, 99.7, 21.6, 18.4, 14.2, 11.3. HRMS (ESI) calcd for C15H16NO ([M+H]+) 226.1226, found 226.1229.
8-氯-2, 4-二甲基呋喃[2, 3-b]喹啉(2f):白色固体, 34 mg, 产率75%. m.p. 143~145 ℃; 1H NMR (600 MHz, CDCl3) δ:8.05 (d, J=9.0 Hz, 1H), 7.83 (d, J=7.2 Hz, 1H), 7.55 (s, 1H), 7.45~7.41 (m, 1H), 3.00 (s, 3H), 2.48 (s, 3H); 13C NMR (150 MHz, CDCl3) δ:161.8, 143.6, 141.2, 139.6, 132.7, 128.8, 127.2, 124.0, 122.7, 120.4, 115.3, 14.6, 11.4. HRMS (ESI) calcd for C13H11ClNO([M+H]+) 232.0524, found 232.0530.
7-氯-2, 4-二甲基呋喃[2, 3-b]喹啉(2g):白色固体, 32 mg, 产率73%. m.p. 150~153 ℃; 1H NMR (600 MHz, CDCl3) δ:8.03 (dd, J=9.6, 6.4 Hz, 1H), 7.68 (dd, J=10.4, 2.8 Hz, 1H), 7.44 (s, 1H), 7.30~7.25 (m, 1H), 2.92 (s, 3H), 2.43 (s, 3H); 13C NMR (150 MHz, CDCl3) δ:162.3, 161.4, 142.5, 139.4, 125.7, 125.6, 115.5, 114.9, 114.6, 112.3, 112.1, 14.5, 11.4. HRMS (ESI) calcd for C13H11ClNO ([M+H]+) 232.0524, found 232.0532.
6-氯-2, 4-二甲基呋喃[2, 3-b]喹啉(2h):白色固体, 36 mg, 产率79%. m.p. 175~177 ℃; 1H NMR (400 MHz, CDCl3) δ:8.10 (d, J=8.8 Hz, 1H), 8.07 (d, J=2.0 Hz, 1H), 7.64 (dd, J=8.8, 2.4 Hz, 1H), 7.53 (s, 1H), 2.95 (s, 3H), 2.47 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:161.1, 156.4, 143.6, 130.5, 129.9, 129.7, 122.7, 120.6, 116.4, 115.6, 29.7, 14.5, 11.4. HRMS (ESI) calcd for C13H11ClNO ([M+H]+) 232.0524, found 232.0524.
6-溴-2, 4-二甲基呋喃[2, 3-b]喹啉(2i):白色固体, 44 mg, 产率79%. m.p. 172~174 ℃; 1H NMR (600 MHz, CDCl3) δ:8.24 (s, 1H), 7.94 (d, J=9.0 Hz, 1H), 7.74 (d, J=9.0 Hz, 1H), 7.52 (s, 1H), 2.94 (s, 3H), 2.47 (s, 3H); 13C NMR (150 MHz, CDCl3) δ:161.9, 143.5, 143.4, 137.9, 131.9, 130.7, 127.3, 126.0, 120.4, 118.2, 115.5, 14.3, 11.5. HRMS (ESI) calcd for C13H10BrNNaO ([M+Na]+) 297.9838, found 297.9837.
2-甲基-4-苯基呋喃[2, 3-b]喹啉(2k):白色固体, 47 mg, 产率91%. m.p. 163~165 ℃; 1H NMR (400 MHz, CDCl3) δ:8.20 (d, J=8.4 Hz, 1H), 7.76~7.67 (m, 2H), 7.56 (dd, J=6.4, 3.2 Hz, 4H), 7.47~7.41 (m, 3H), 1.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:161.26, 144.28, 143.52, 143.14, 135.16, 129.91, 129.03, 128.56, 128.10, 126.03, 125.59, 124.74, 119.38, 115.67, 10.05. HRMS (ESI) calcd for C18H14NO ([M+H]+) 260.1070, found 260.1070.
辅助材料(Supporting Information)化合物的核磁谱图.这些材料可以免费从本刊网站(http://sioc-jour-nal.cn/)上下载.
-
-
[1]
(a) Abass, M. Heterocycles 2005, 65, 901.
(b) Wang, R.; Liu, Z.-Q. Med. Chem. Res. 2013, 22, 1563.
(c) Huffman, J. W.; Browder, L. E. J. Org. Chem. 1964, 29, 2598. -
[2]
(a) Grougnet, R.; Magiatis, P.; Fokialakis, N.; Mitaku, S.; Skaltsounis, A. L.; Tillequin, F.; Sevenet, T.; Litaudon, M. J. Nat. Prod. 2005, 68, 1083.
(b) Ambrozin, A. R. P.; Mafezoli, J.; Vieira, P. C.; Fernandes, J. B.; da Silva, M.; Ellena, J. A.; de Albuquerque, S. J. Braz. Chem. Soc. 2005, 16, 434.
(c) Ito, C.; Itoigawa, M.; Sato, A.; Hasan, C. M.; Rashid, M. A.; Tokuda, H.; Mukainaka, T.; Nishino, H.; Furukawa, H. J. Nat. Prod. 2004, 67, 1488.
(d) Ayafor, J. F.; Okogun, J. I. J. Chem. Soc., Perkin Trans. 11982, 909.
(e) Okogun, J. I.; Ayafor, J. F. Chem. Commun. 1977, 652. -
[3]
(a) Mabire, D.; Coupa, S.; Adelinet, C.; Poncelet, A.; Simonnet, Y.; Venet, M.; Wouters, R.; Lesage, A. S. J.; Van Beijsterveldt, L.; Bischoff, F. J. Med. Chem. 2005, 48, 2134.
(b) Michael, J. P. Nat. Prod. Rep. 2002, 19, 742.
(c) Michael, J. P. Nat. Prod. Rep. 2004, 21, 650.
(d) Michael, J. P. Nat. Prod. Rep. 2005, 22, 627.
(e) Michael, J. P. Nat. Prod. Rep. 2003, 20, 476. -
[4]
Tuppy, H.; Bohm, F. Angew. Chem., Int. Ed. 1956, 68, 388.
-
[5]
(a) Yu, L.-Z.; Hu, X.-B.; Xu, Q.; Shi, M. Chem. Commun. 2016, 52, 2701.
(b) Boyd, D. R.; Sharma, N. D.; Barr, S. A.; Carroll, J. G.; Mackerracher, D.; Malone, J. F. J. Chem. Soc., Perkin Trans. 12000, 3397. -
[6]
(a) Aillaud, I.; Bossharth, E.; Conreaux, D.; Desbordes, P.; Monteiro, N.; Balme, G. Org. Lett. 2006, 8, 1113.
(b) Sekar, M.; Prasad, K. J. R. J. Nat. Prod. 1998, 61, 294.
(c) Narasimhan, N. S.; Mali, R. S. Tetrahedron 1974, 30, 4153.
(d) Collins, J. F.; Gray, G. A.; Grundon, M. F.; Harrison, D. M.; Spyropoulos, C. G. J. Chem. Soc., Perkin Trans. 11973, 94.
(e) Narasimhan, N. S.; Paradkar, M. V.; Alurkar, R. H. Tetrahedron 1971, 27, 1351.
(f) Cooke, R. G.; Haynes, H. F. Aust. J. Chem. 1958, 11, 225.
(g) Grundon, M. F.; McCorkindale, N. J. J. Chem. Soc. 1957, 2177.
(h) Guo, R.-H.; Zhang, Q.; Ma, Y.-B.; Luo, J.; Geng, C.-A.; Wang, L.-J.; Zhang, X.-M.; Zhou, J.; Jiang, Z.-Y.; Chen, J.-J. Eur. J. Med. Chem. 2011, 46, 307. -
[7]
(a) Nagarajan, R.; Magesh, C. J.; Perumal, P. T. Synth. Stuttg. 2004, 69.
(b) Yadav, J. S.; Reddy, B. V. S.; Madhuri, C. R.; Sabitha, G. Synth. Stuttg. 2001, 1065.
(c) Yadav, J. S.; Reddy, B. V. S.; Gayathri, K. U.; Prasad, A. R. Synth. Stuttg. 2002, 2537. -
[8]
李雄武, 汪朝阳, 郑绿茵, 有机化学, 2006, 26, 1144. doi: 10.3321/j.issn:0253-2786.2006.08.024Li, X.; Wang, C; Zheng, L. Chin. J. Org. Chem. 2006, 26, 1144(in Chinese). doi: 10.3321/j.issn:0253-2786.2006.08.024
-
[9]
(a) Zhang, Z.; Zhang, Q.; Sun, S.; Xiong, T.; Liu, Q. Angew. Chem., Int. Ed. 2007, 46, 1726.
(b) Ru, T. Light Ind. Sci. Technol. 2013, 5, 58(in Chinese). (茹婷婷, 轻工科技, 2013, 5, 58.)
(c) Du, W.; Curran, D. P. Org. Lett. 2003, 5, 1765. -
[10]
Zhang, Q.; Zhang, Z.; Yan, Z.; Liu, Q.; Wang, T. Org. Lett. 2007, 9, 3651. doi: 10.1021/ol701536q
-
[11]
张志国, 博士论文, 东北师范大学, 长春, 2010.Zhang, Z. G. Ph.D. Dissertation, Northeast Normal University, Changchun, 2010(in Chinese).
-
[12]
Sai, K. K. S.; Gilbert, T. M.; Klumpp, D. A. J. Org. Chem. 2007, 72, 9761. doi: 10.1021/jo7013092
-
[13]
Heaney, H. In Comprehensive Organic Synthesis, Vol. 2, Eds.:Trost, B. M.; Fleming, I., Pergamon, Oxford, 1991, pp. 733~752.
-
[14]
(a) Zhang, Z.; Zhang, Q.; Yan, Z.; Liu, Q. J. Org. Chem. 2007, 72, 9808.
(b) Xiong, T.; Zhang, Q.; Zhang, Z.; Liu, Q. J. Org. Chem. 2007, 72, 8005.
-
[1]
-

图 1 几种代表性的含有呋喃[2, 3-b]喹啉母核结构的天然产物
Figure 1 Several representative furo[2, 3-b]quinolines-con-taining natural products

图式1 常见的呋喃[2, 3-b]喹啉类化合物的合成策略
Scheme 1 Strategies for the synthesis of furan[2, 3-b]quinolines

图式2 Ru等合成呋喃[2, 3-b]喹啉类化合物的工作
Scheme 2 Ru et al. developed the synthesis of furo[2, 3-b]-quinolines
表 1 优化条件表a
Table 1. Survey of the reaction conditions
Entry Acid (equiv.) T/℃ Time/h Yield of 2a/% 1 CF3SO3H (8) 60 14 59b 2 CF3SO3H (8) 80 10 76 3 CF3SO3H (8) 100 8 70 4 CF3SO3H (10) 80 10 67 5 PPA (8) 80 24 71 6 Con. H2SO4 (8) 80 14 68c 7 TMSOTf (8) 80 6 57d 8 p-TsOH (8) 80 10 0e 9 AcOH (8) 80 10 0f 10 PivOH (8) 80 10 0g 11 TFA (8) 80 10 0h 12 TCA (8) 80 10 0i a除非另外说明, 所有反应都是在空气氛围中进行, 反应物1a的用量为0.2 mmol, 收率为分离收率; b18%的1a被回收. c5%的1a被回收. dTMSOTf的沸点为77 ℃, 因此反应在密封管中进行. e20%的1a被回收. f16%的1a被回收. g30%的1a被回收. h5%的1a被回收. i12%的1a被回收.  下载: 导出CSV
下载: 导出CSV
表 3 药品和试剂
Table 3. Starting materials and reagents
药品或试剂名称 纯度 生产厂家 苯甲酰乙酸乙酯 A.R. 上海达瑞精细化学品有限公司 对甲氧基苯甲酰乙酸乙酯 A.R. 梯希爱(上海)化成工业发展有限公司 乙酰乙酸乙酯 A.R. 上海麦克林生化科技有限公司 苯胺 A.R. 国药集团化学试剂有限公司 对溴苯胺 A.R. 上海嘉辰 对甲氧基苯胺 A.R. 阿拉丁 乙酰乙酰苯胺 A.R 青岛双桃精细化工(有限)公司 2-氯乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 4-氯乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 4-甲氧基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 2-甲氧基乙酰乙酰苯胺 A.R 青岛双桃精细化工(有限)公司 4-甲基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 2-甲基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 2, 4-甲基乙酰乙酰苯胺 A.R. 青岛双桃精细化工(有限)公司 1, 2, 3-三溴丙烷 A.R. 阿拉丁 无水碳酸钾 A.R. 国药集团化学试剂有限公司 N, N-二甲基甲酰胺 A.R. 成都市科龙化工试剂厂 三氟甲磺酸 A.R. 阿拉丁
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 5
- 文章访问数: 1617
- HTML全文浏览量: 134
 下载:
下载:
