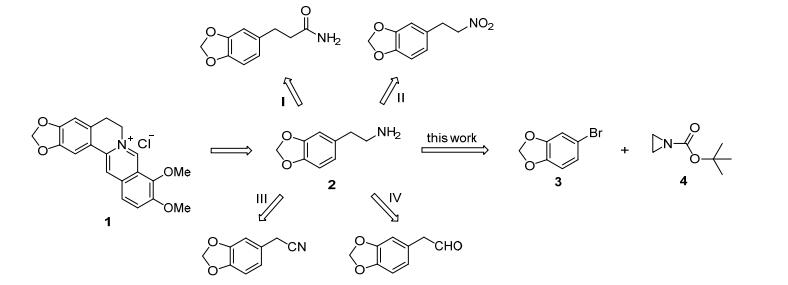
 图 1
盐酸黄连素的逆合成分析
Figure 1.
Retrosynthesis analysis of berberine chloride
图 1
盐酸黄连素的逆合成分析
Figure 1.
Retrosynthesis analysis of berberine chloride
引用本文:
陈程, 徐蒙蒙, 赵青, 刘承秀, 杨鸿均, 冯豫川. 盐酸黄连素的汇聚式合成研究[J]. 有机化学,
2017, 37(2): 503-507.
doi:
10.6023/cjoc201612005

Citation: Chen Cheng, Xu Mengmeng, Zhao Qing, Liu Chengxiu, Yang Hongjun, Feng Yuchuan. A Concisely Convergent Synthesis of Berberine Chloride[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 503-507. doi: 10.6023/cjoc201612005
Citation: Chen Cheng, Xu Mengmeng, Zhao Qing, Liu Chengxiu, Yang Hongjun, Feng Yuchuan. A Concisely Convergent Synthesis of Berberine Chloride[J]. Chinese Journal of Organic Chemistry, 2017, 37(2): 503-507. doi: 10.6023/cjoc201612005
盐酸黄连素的汇聚式合成研究
摘要:
报道了以1,2-亚甲二氧基苯为起始原料合成盐酸黄连素的汇聚式路线,该路线包括格氏试剂亲核开环、脱保护、环合等5步反应,总收率为33%.该路线反应条件温和、操作简单,且所有中间体及产物都经过1H NMR、13C NMR、MS鉴定.
-
关键词:
- 盐酸黄连素
- / 汇聚式合成
- / 1, 2-亚甲二氧基苯
- / 格氏试剂
English
A Concisely Convergent Synthesis of Berberine Chloride
Abstract:
The berberine chloride was synthesized starting from benzo[d][1, 3]dioxole with the mild condition and the simple operation in overall 33% yield. The route includes the five steps of nucleophilic ring opening, deprotection and cyclization and so on. All intermediates were determined by 1H NMR, 13C NMR and MS techniques.
-
Key words:
- berberine chloride
- / convergent synthesis
- / benzo[d][1, 3]dioxole
- / grignard regent
-
盐酸黄连素是存在于植物中的异喹啉类生物碱, 可从多种植物中提取, 如黄连、黄柏、三颗针、伏牛花、白屈菜、南天竹、小檗、铁皮莲等[1a].大量临床研究表明, 盐酸黄连素具有抗癌[1]、抗炎[2]、抗微生物[3]、抗病毒[5]、抗利什曼原虫[6]、抗寄生虫[7]、抗结核[8, 9]等多种药理活性, 而且其毒性低、副作用小, 市场需求大.但可供提取的天然植物资源有限.因此, 人们致力于研究盐酸黄连素及其类似物的化学合成方法[8].
据文献报道, 盐酸黄连素的合成大多是以3, 4-亚甲二氧基苯乙胺 (2) 作关键中间体, 且主要有四种合成策略 (图 1). 1913年, Decker等课题组[8a]通过霍夫曼反应合成化合物2, 该路线具有原料不易得、合成收率低等不足 (路线I); 南宁制药厂[8c]以胡椒醛为起始原料, 采用Henry反应、催化加氢等合成化合物2; 杭州制药厂[8e]通过胡椒苄氯与氰化钠亲核取代反应, 再经过还原而得化合物2(路线Ⅱ), 尽管收率较高但需使用氰化钠 (路线Ⅲ); 东北制药厂[8f]以苯酚为起始原料, 经过亚甲基化, 水解等8步合成盐酸黄连素 (路线Ⅳ).尽管盐酸黄连素已经工业化生产, 但现有的路线仍然需要深入地研究和改进, 从而开发出一条反应条件温和、路线简短的新路线.
图 1
盐酸黄连素的逆合成分析
Figure 1.
Retrosynthesis analysis of berberine chloride
受到以上研究的启发, 2016年我们课题组[8n]对霍夫曼路线进行了改进, 开发了盐酸黄连素新的合成路线.同时我们也发现, 合成盐酸黄连素都是采用直线式逐步合成的策略, 效率较低、路线长.因此, 我们设想开发效率更高, 路线更短的汇聚式合成路线 (Scheme 1).通过对盐酸黄连素的逆合成分析, 盐酸黄连素可通过化合物2合成得到, 化合物2可通过3和4反应得到.基于逆合成分析, 本文以1, 2-亚甲二氧基苯为起始原料, 经溴化反应得化合物4, 化合物5经二碳酸二叔丁酯保护得化合物3.在化合物4的格氏试剂作用下化合物3亲核开环得7, 再脱叔丁氧羰基即得关键中间体2.中间体2再经缩合、环化得盐酸黄连素, 该汇聚式合成路线仅需5步, 收率高达33%.
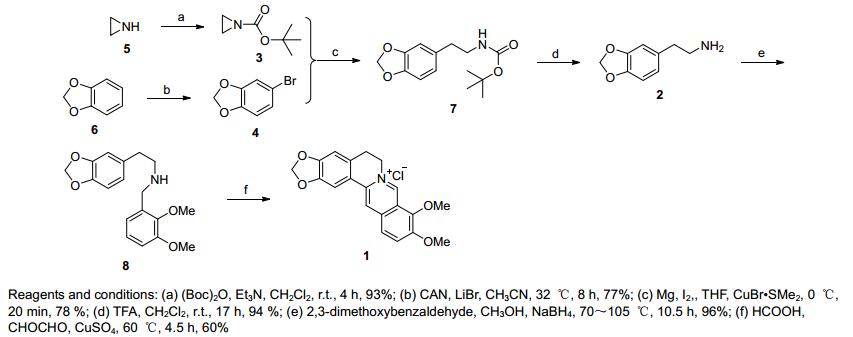 图 图式1
盐酸黄连素的汇聚式合成
Figure 图式1.
Convergent synthesis of berberine chloride
图 图式1
盐酸黄连素的汇聚式合成
Figure 图式1.
Convergent synthesis of berberine chloride
1 结果与讨论
1.1 化合物4的合成
在化合物6的溴代反应中, 最初我们尝试用溴化氢-双氧水作为溴化试剂, 但副产物多, 收率仅为45%.以硝酸铈铵 (CAN)-溴化锂作为溴化试剂, 收率可提高至77%, 反应条件温和, 因而选CAN-溴化锂为溴化试剂合成化合物4. CAN作为单电子氧化剂, 将Br-氧化为Br+, 进而Br+进攻富电子的芳环发生亲电取代, 得到化合物4[9] (Scheme 2).
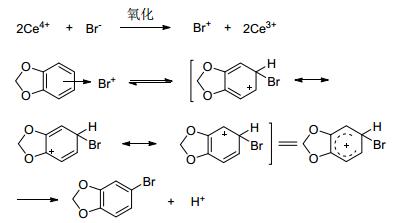 图 图式2
合成化合物4的反应机理
Figure 图式2.
Reaction mechanism on the preparation of 4
图 图式2
合成化合物4的反应机理
Figure 图式2.
Reaction mechanism on the preparation of 4
1.2 化合物7的合成
在格氏试剂的作用下, 化合物3可开环得化合物7.为了提高收率, 我们对这一步反应进行条件研究, 考察了催化剂、温度、溶剂对收率的影响 (表 1).实验结果表明, -40 ℃时, 以四氢呋喃 (THF) 作溶剂, 格氏试剂直接与3反应主要生成化合物9, 并没有得到中间体7(表 1, Entry 1);当以CuI作催化剂, 尽管能得到中间体7, 但收率仅为7%(表 1, Entry 2);改用CuBr作为催化剂时, 收率略微提高 (表 1, Entry 3);以CuBr•SMe2作催化剂, 收率可提高至34 %(表 1, Entry 4).为了进一步提高收率, 探讨温度对反应的影响, 随着温度的提高, 收率逐渐增加, 在0 ℃时收率可达78 %, 但继续升高温度, 收率急剧下降 (表 1, Entry 7).在0 ℃, 以乙醚作溶剂时, 格氏试剂很难生成 (表 1, Entry 8).因此, 合成化合物7的最佳反应条件为:干燥THF为溶剂, CuBr•SMe2为反应催化剂, 反应温度为0 ℃, 在该反应条件下, 合成化合物7的收率为78%(表 1, Entry 7).
 表 1
合成化合物7的反应条件优化
Table 1.
Optimization study on the preparation of 7
表 1
合成化合物7的反应条件优化
Table 1.
Optimization study on the preparation of 7
Entry Catalyst t/℃ Solvent/mL Yielda/% 1 — -40 THF — 2 CuI -40 THF 7 3 CuBr -40 THF 23 4 CuBr•SMe2 -40 THF 34 5 CuBr•SMe2 -20 THF 43 6 CuBr•SMe2 0 THF 78 7 CuBr•SMe2 30 THF 28 8 CuBr•SMe2 0 Et2O — a纯化的收率. 表 1 合成化合物7的反应条件优化
Table 1. Optimization study on the preparation of 71.3 化合物7的合成反应机理推测
根据实验反应的现象和结果, 对合成化合物7的反应机理进行预测 (Scheme 3).当反应中无催化剂时, 格氏试剂直接进攻羰基碳原子而生成化合物9; 当加入催化剂CuBr•SMe2时, 首先CuBr•SMe2与格氏试剂形成芳基铜络合物, 可能由于铜络合物位阻较大, 选择性地进攻位阻较小的氮杂环丙烷碳原子, 开环得到目标化合物7.
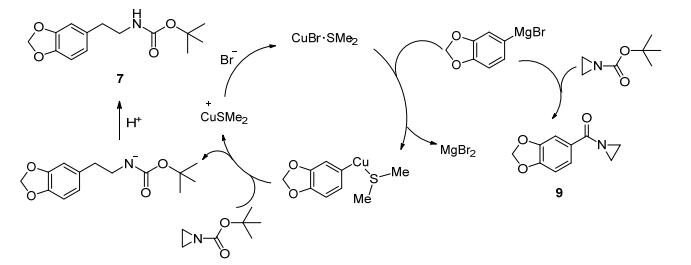 图 图式3
化合物7的合成反应机理推测
Figure 图式3.
Proposed reaction mechanism of 7
图 图式3
化合物7的合成反应机理推测
Figure 图式3.
Proposed reaction mechanism of 7
2 结论
本文开发了盐酸黄连素的汇聚式合成路线, 以1, 2-亚甲二氧基苯为起始原料, 经格氏试剂的亲核开环作为本路线的关键步骤, 仅需5步反应合成盐酸黄连素, 收率为33 %.所有中间体都经核磁氢谱、碳谱、质谱, 红外和熔点鉴定.该路线具有路线短、收率高、反应条件温和等特点, 为合成盐酸黄连素提供一种新思路新途径, 具有潜在的工业应用价值.
3 实验部分
3.1 仪器与试剂
1H NMR、13C NMR用Agilent-400 (400 MHz) 型核磁共振仪测定, TMS作内标, CDCl3或d6-DMSO作溶剂; 质谱用LCQ ADVANTAGE MAX测定; 熔点用WRS-1B数字熔点仪测定; 红外用FTS 3000傅里叶变换红外光谱仪 (美国DIGILAB公司) 测定.其余试剂均为国产分析纯试剂, 四氢呋喃和二氯甲烷干燥后使用, 其余试剂未经纯化直接使用.
3.2 实验方法
3.2.2 叔丁基氮丙啶-1-羧酸酯 (3) 的合成
将氮丙啶 (1.00 g, 23.3 mmol)、20 mL二氯甲烷和9.6 mL三乙胺依次加入100 mL三口瓶中, 于0 ℃下, 搅拌下滴加二碳酸二叔丁酯 (5.03 g, 23.3 mmol) 的二氯甲烷 (20 mL) 溶液, 室温下搅拌反应, TLC跟踪反应完全, 冷却至室温, 在旋转蒸发仪上蒸去溶剂, 得到的残余液用中性Al2O3(石油醚) 纯化, 得3.10 g无色油状液体3, 收率为93%. 1H NMR (400 MHz, CDCl3) δ: 1.42 (s, 9H), 2.10 (s, 4H); 13C NMR (100 MHz, CDCl3) δ: 25.71, 27.84, 81.11, 162.76; IR (KBr) v: 2992, 2934, 1747, 1396, 1343, 1230, 1176, 1052 cm-1; ESI-MS m/z (%): 144.01 [M+ H]+.
3.2.6 5, 6-二氢-9, 10-二甲氧基苯并 (g)1, 3-苯并二恶茂 (5, 6-a) 喹嗪盐酸盐 (1) 的合成
将6.6 mL 88 %甲酸、硫酸铜 (1.23 g, 7.71 mmol) 依次加入反应瓶中, 升温至55 ℃搅拌反应, 保温脱水0.5 h, 再加入化合物8 (1.32 g, 4.19 mmol)、1.1 mL 40 %乙二醛溶液, 升温至100 ℃反应4 h.在反应过程中按下列比例加入浓盐酸, 保温后50 min, 加入0.1 mL浓盐酸, 隔50 min后, 加入0.1 mL浓盐酸, 再隔1 h后, 加入0.1 mL浓盐酸, 再隔1 h, 加入0.1 mL浓盐酸, 再保温反应20 min.搅拌冷却至0 ℃以下, 过滤, 用5%食盐水洗至pH=2.0, 过滤抽干得盐酸黄连素粗品.将盐酸黄连素粗品先加部分水调成浆状, 然后加水至足量, 升温至75~80 ℃, 保温1 h, 然后加入氧化钙调至pH=8.0~8.5趁热过滤, 滤饼用热水洗涤, 洗液与滤液合并后用10%盐酸调至pH=2.0, 室温放置8 h, 过滤, 滤饼用常水洗涤, 抽干、干燥, 得一次精制盐酸黄连素, 得839 mg黄色固体1[11], 收率60%. m.p. 195~197 ℃(文献值[8n]195~197 ℃); 1H NMR (400 MHz, DMSO-d6) δ: 3.19~3.22 (t, J=5.8 Hz, 2H), 4.07 (s, 3H), 4.10 (s, 3H), 4.88~4.91 (t, J=6 Hz, 2H), 6.18 (s, 2H), 7.09 (s, 1H), 7.80 (s, 1H), 7.99~8.01 (d, J=8.8 Hz, 1H), 8.20~8.22 (d, J=8.8 Hz, 1H), 8.94 (s, 1H), 9.89 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 26.7, 55.5, 57.4, 62.3, 102.5, 105.8, 108.8, 120.6, 120.8, 121.8, 123.9, 127.0, 131.0, 133.3, 137.8, 144.0, 145.8, 148.0, 150.1, 150.8; IR (KBr) v: 3424, 3071, 2925, 1612, 1510, 1393, 1343, 1278, 1232, 1110, 1043 cm-1; ESI-MS m/z (%): 337.18 [M+H]+.
辅助材料 (Supporting Information)产物和中间体的核磁共振和质谱图谱.这些材料可以免费从本刊网站 (http://sioc-journal.cn/) 上下载.
3.2.3 [2-(苯并[d][1, 3]二氧杂环戊烯-5-基) 乙基]氨基甲酸叔丁酯 (7) 的合成
在N2保护下, 将镁条 (134 mg, 2.82 mmol) 和I2依次加入25 mL三口瓶, 往反应瓶中先加入一部分化合物4(1.02 g, 5.12 mmol) 的无水四氢呋喃 (THF) (8 mL) 溶液, 格氏反应引发完毕后缓慢加入剩余溶液, 制成格氏试剂, 以备使用.将CuBr•SMe2 (0.0421 mg, 0.205 mmol) 用4 mL的THF溶解, 在0 ℃下, 用注射器将制备好的格氏试剂加入到CuBr•SMe2的反应液中, 搅拌反应5 min, 后滴加化合物3 (400 mg, 2.80 mmol) 的无水THF (4 mL) 溶液, 搅拌反应20 min, 停止反应.加入20 mL的饱和NH4Cl溶液, 二氯甲烷萃取 (50 mL×3), 合并有机相, 饱和食盐水洗涤, 最后经无水Na2SO4干燥得初产品, 经柱层析[V(石油醚):V(二氧甲烷)=1:10]纯化后, 得576 mg白色固体结晶7, 收率为78%. m.p. 59~61 ℃(文献值[10]: 60 ℃); 1H NMR (400 MHz, CDCl3) δ: 1.42 (s, 9H), 2.69 (t, J=6.6 Hz, 2H), 3.30 (s, 2H), 4.56 (s, 1H), 5.91 (s, 2H), 6.66~6.61 (m, 2H), 6.73 (dd, 1H); 13C NMR (100 MHz, CDCl3)δ: 28.37, 35.86, 41.93, 79.19, 100.82, 108.28, 109.08, 121.62, 132.72, 146.05, 147.69, 155.81; IR (KBr) v: 3375, 2991, 2901, 1689, 1535, 1500, 1451, 1374, 1254, 1181, 1044, 936, 876, 811, 629 cm-1; ESI-MS m/z(%): 266.21 [M+ H]+.
3.2.5 N-(2, 3-二甲氧基苄基)-3, 4-亚甲二氧基苯乙胺 (8) 的合成
将化合物2 (3.00 g, 18.2 mmol)、2, 3-二甲氧基苯甲醛 (3.01 g, 18.1 mmol) 依次加入三口瓶中, 在105 ℃条件下反应0.5 h, 随后冷却至室温, 加入24 mL甲醇, 分多次加入NaBH4 (381 mg, 10.0 mmol), 在70 ℃的条件下反应10 h.室温下旋转蒸除去多余的甲醇, 残留物用20 mL水溶解后用乙醚萃取 (50 mL×3), 合并有机层, 用20 mL水洗、饱和食盐水洗 (20 mL×2), 有机相用无水Na2SO4干燥, 过滤洗涤, 合并滤液, 旋干, 得5.48 g淡黄色油状液体8[8e], 收率96%. 1H NMR (400 MHz, CDCl3)δ: 2.52 (s, 2H), 2.75 (t, J=7.2 Hz, 2H), 2.84 (t, J=6.8 Hz, 2H), 3.80 (s, 3H), 3.85 (s, 3H), 5.90 (s, 2H), 6.62~6.72 (m, 3H), 6.83~6.86 (m, 2H), 7.00 (t, J=8.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ: 13C NMR (100 MHz, CDCl3) δ: 35.8, 48.4, 50.4, 55.6, 60.6, 100.7, 108.1, 109.0, 111.4, 121.5, 121.7, 123.8, 133.2, 133.6, 145.8, 147.2, 147.5, 152.5; IR (KBr) v: 3341, 2947, 2851, 1597, 1494, 1453, 1360, 1248, 1049, 934, 865, 808 cm-1; ESI-MS m/z: 316.21 ([M+H]+).
3.2.1 5-溴苯并[d][1, 3]二氧杂环戊烯 (4) 的合成
在N2保护下, 将化合物6 (20.0 g, 164 mmol)、无水溴化锂 (15.7 g, 181 mmol) 和100 mL乙腈依次加入1000 mL三口瓶中.将反应液温度升至32 ℃, 搅拌下滴加硝酸铈铵 (99.0 g, 181 mmol) 的乙腈 (400 mL) 溶液, 薄层色谱 (TLC) 跟踪反应完全, 冷却至室温, 加入100 mL H2O, 乙醚萃取 (300 mL×3), 合并有机相, 再依次用饱和NaHCO3 (80 mL×3), H2O (80 mL×3), 饱和食盐水洗涤, 最后经无水Na2SO4干燥得初产品, 经减压蒸馏收集120~128 ℃ (0.095 MPa) 的馏分, 得25.3 g浅黄色油状液体4[9a], 收率为77%. 1H NMR (400 MHz, CDCl3)δ: 5.96 (s, 2H), 6.68 (d, J=4.0 Hz, 1H), 6.94 (dd, J=4.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 101.5, 109.5, 112.2, 113.0, 124.2, 146.9, 148.5; IR (KBr) v: 2907, 1615, 1483, 1239, 1158, 1109, 1043, 937, 866 cm-1; ESI-MS m/z(%): 200.71 [M+H]+.
3.2.4 3, 4-亚甲二氧基苯乙胺 (2) 的合成
将化合物7 (200 mg, 0.755 mmol) 和7 mL二氯甲烷依次加入25 mL的三口瓶中, 0 ℃下滴加三氟乙酸 (TFA) (714 mg, 6.27 mmol) 的二氯甲烷 (2.5 mL) 溶液, 在室温条件下反应17 h, 停止反应, 加入15 mL 2 mol/L HCl, 分出水相, 调pH=10.0, 用二氯甲烷萃取 (30 mL×3), 合并有机相, 饱和食盐水洗涤, 最后经无水Na2SO4干燥, 得116 mg浅黄色油状液体2, 收率为94%. 1H NMR (400 MHz, CDCl3) δ: 2.66 (t, J=6.8 Hz, 2H), 2.90 (t, J=6.8 Hz, 2H), 5.92 (s, 2H), 6.64 (d, J=7.6 Hz, 1H), 6.69 (s, 1H), 6.74 (d, J=8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 39.6, 43.6, 100.8, 108.2, 109.1, 121.6, 133.5, 145.9, 147.6; IR (KBr) v: 3382, 2936, 1604, 1499, 1366, 1250, 1199, 1106, 1043, 933, 866, 813 cm-1; ESI-MS m/z (%): 166.16 [M+H]+.
-
-
[1]
(a) Lo, C. Y.; Hsu, L. C.; Chen, M. S.; Lin, Y. J.; Chen, L. G.; Kuo, C. D.; Wu., J. Y. Bioorg. Med. Chem. Lett. 2013, 23, 305.
(b) Li, Q.; Xiang, J. F.; Tang, Y. L. Chin. J. Chem. 2015, 33, 1041. -
[2]
Zhang, S. L.; Chang, J. J.; Damu, G. V.; Fang, B.; Zhou, X. D.; Geng, R. X.; Zhou, C. H. Bioorg. Med. Chem. Lett. 2013, 23, 1008. doi: 10.1016/j.bmcl.2012.12.036
-
[3]
(a) Samosorn, S.; Tanwirat, B.; Muhamad, N.; Casadei, G.; Tomkiewicz, D.; Lewis, K.; Suksamrarn, A.; Prammananan, T.; Gornall, K. C.; Beck, J. L.; Bremner, J. B. Bioorg. Med. Chem. 2009, 17, 3866.
(b) Zhang, S. L.; Damu, G. L.; Zhang, L.; Geng, R. X.; Zhou, C. H. Eur. J. Med. Chem. 2012, 55, 164. -
[4]
Bodiwala, H. S.; Sabde, S.; Mitra, D.; Bhutani, K. K.; Singh, I. P. Eur. J. Med. Chem. 2011, 46, 1045. doi: 10.1016/j.ejmech.2011.01.016
-
[5]
Vennerstrom, J. L.; Lovelace, J. K.; Wsits, V. B.; Hanson, W. L.; Klayman, D. L. Antimicrob. Agents Chemother. 1990, 34, 918. doi: 10.1128/AAC.34.5.918
-
[6]
Bahar, M.; Deng, Y.; Zhu, X.; He, S.; Pandharkar, T.; Drew, M. E.; Navarro-Vazquez, A.; Anklin, C.; Gil, R. R.; Dos Kotch, R. W.; Werbovetz, K. A.; Kinghorn, A. D. Bioorg. Med. Chem. Lett. 2011, 21, 2606. doi: 10.1016/j.bmcl.2011.01.101
-
[7]
(a) Letasiova, S.; Jantova, S.; Cipak, L.; Muckova, M. Cancer Lett. 2006, 239, 254.
(b) Ma, Y.; Ou, T. M.; Tan, J. H.; Hou, J. Q.; Huang, S. L.; Gu, L. Q.; Huang, Z. S. Bioorg. Med. Chem. Lett. 2009, 19, 3414. -
[8]
(a) Decker, V. I. Justus Liebigs Ann. Chem. 1913, 395, 295.
(b) Kametani, T.; Noguchi, l. J. Chem. Soc. (C) 1969, 2036.
(c) Nanning Pharmaceutical Chin. J. Pharm. 1973, (7), 1 (in Chinese).(广西南宁制药厂, 医药工业, 1973, (7), 1.)
(d) Masayuki, O.; Kumiko, Y.; Junko, O. Chem. Pharm. Bull. 1974, 22, 2365.
(e) Hangzhou First Pharmaceutical Reagent Chamber. Chin. J. Pharm. 1974, (8), 6 (in Chinese).(杭州第一制药厂试剂室, 医药工业, 1974, (8), 6.)
(f) North-east Pharmaceutical Factory Chin. J. Pharm. 1975, (4), 12 (in Chinese).(东北制药总厂, 医药工业, 1975, (4), 12.)
(g) Vinogradova, V. I.; Yunusov, M. S.; Kuchin, A. V.; Tolstikov, G. A.; Sagandykov, R. T.; Khaimuratov, K. A.; Ali mov, A. Chem. Nat. Compd. 1990, 26, 54.
(h) Hisashi, I.; Mayumi, O.; Shuji, O.; Takashi, H.; Tsutomu, I. Heterocycles 1994, 37, 897.
(i) Matulenko, M. A.; Meyers, A. I. J. Org. Chem. 1996, 61, 573.
(j) Yang, P.; Song, D. Q.; Li, Y. H.; Kong, W. J.; Wang, Y. X.; Gao, L. M.; Liu, S. Y.; Cao, R. Q.; Jiang, J. D. Bioorg. Med. Chem. Lett. 2008, 18, 4675.
(k) Gatland, A. E.; Pilgrim, B. S.; Procopiou, P. A.; Donohoe, T. J. Angew. Chem. Int. Ed. 2014, 53, 14555.
(l) He, Y.; Zheng, Y.; Hai, L.; Wu, Y. Chin. J. Chem. 2014, 32, 1121.
(m) Reddy, V.; Jadhav, A. S.; Vijaya Anand, R. Org. Biomol. Chem. 2015, 13, 3732.
(n) Chen, C.; Luo, Z. M.; Yang, H. J.; Feng, Y. C. Chin. J. Org. Chem. 2016, 36, 1426 (in Chinese).(陈程, 罗卓玛, 杨鸿均, 冯豫川, 有机化学, 2016, 36, 1426.) -
[9]
(a) Roy, S. C.; Guin, C.; Rana, K. K.; Maiti, G. Tetrahedron Lett. 2001, 42, 6941.
(b) Roy, S. C.; Guin, C.; Maiti, G. Tetrahedron Lett. 2001, 42, 9253. -
[10]
Tietze, L. F., Schirok, H. J. Am. Chem. Soc. 1999, 121, 10264. doi: 10.1021/ja991650+
-
[11]
Pan, J. F.; Yu, C.; Zhu, D. Y.; Zhang, H.; Ren, J. Y. CN 1314347, 2001 [Chem. Abstr. 2002, 137, 370266].
-
[1]
-

图式1 盐酸黄连素的汇聚式合成
Scheme 1 Convergent synthesis of berberine chloride
Reagents and conditions: (a) (Boc)2O, Et3N, CH2Cl2, r.t., 4 h, 93%; (b) CAN, LiBr, CH3CN, 32 ℃, 8 h, 77%; (c) Mg, I2, , THF, CuBr•SMe2, 0 ℃, 20 min, 78 %; (d) TFA, CH2Cl2, r.t., 17 h, 94 %; (e) 2, 3-dimethoxybenzaldehyde, CH3OH, NaBH4, 70~105 ℃, 10.5 h, 96%; (f) HCOOH, CHOCHO, CuSO4, 60 ℃, 4.5 h, 60%
表 1 合成化合物7的反应条件优化
Table 1. Optimization study on the preparation of 7
Entry Catalyst t/℃ Solvent/mL Yielda/% 1 — -40 THF — 2 CuI -40 THF 7 3 CuBr -40 THF 23 4 CuBr•SMe2 -40 THF 34 5 CuBr•SMe2 -20 THF 43 6 CuBr•SMe2 0 THF 78 7 CuBr•SMe2 30 THF 28 8 CuBr•SMe2 0 Et2O — a纯化的收率.  下载: 导出CSV
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 28
- 文章访问数: 2248
- HTML全文浏览量: 635
 下载:
下载:
