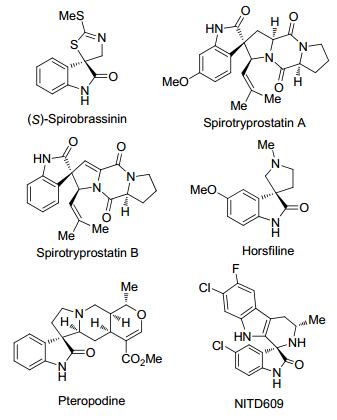
 图 1
代表性的含有螺环氧化吲哚骨架的天然产物或药物分子
Figure 1.
Representative examples of natural products and pharmaceutical molecules containing a spirooxindole core structure
图 1
代表性的含有螺环氧化吲哚骨架的天然产物或药物分子
Figure 1.
Representative examples of natural products and pharmaceutical molecules containing a spirooxindole core structure
引用本文:
谭芬, 肖文精, 曾国平. 3-异硫氰酸酯氧化吲哚参与的不对称串联反应研究进展[J]. 有机化学,
2017, 37(4): 824-840.
doi:
10.6023/cjoc201611017

Citation: Tan Fen, Xiao Wenjing, Zeng Guoping. Recent Advances in 3-Isothiocyanato Oxindoles Engaged Asymmetric Cascade Reactions[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 824-840. doi: 10.6023/cjoc201611017
Citation: Tan Fen, Xiao Wenjing, Zeng Guoping. Recent Advances in 3-Isothiocyanato Oxindoles Engaged Asymmetric Cascade Reactions[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 824-840. doi: 10.6023/cjoc201611017
3-异硫氰酸酯氧化吲哚参与的不对称串联反应研究进展
摘要:
3-异硫氰酸酯氧化吲哚是一类高活性的新型反应试剂,已经被广泛地应用于串联反应中,并用于结构多样的手性螺环氧化吲哚骨架的构建.简单综述了近六年来3-异硫氰酸酯氧化吲哚参与的几类串联环化反应的最新研究进展,主要介绍各反应的特点、活化模式及合成应用,并展望它的发展前景.
-
关键词:
- 3-异硫氰酸酯氧化吲哚
- / 螺环氧化吲哚
- / 不对称合成
- / 串联反应
English
Recent Advances in 3-Isothiocyanato Oxindoles Engaged Asymmetric Cascade Reactions
Abstract:
3-Isothiocyanato oxindoles have been widely employed as a class of highly reactive and novel reagents in the enantioselective synthesis of diverse spirooxindoles. This review summarizes the recent advances of 3-isothiocyanato oxindoles mediated some types of cascade process in the past six years, including properties of reaction, activation models and synthetic applications. Furthermore, the prospects of this concept are also discussed.
-
Key words:
- 3-isothiocyanato oxindole
- / spirooxindole
- / asymmetric synthesis
- / cascade reaction
-
螺环氧化吲哚骨架广泛存在于天然产物和药物分子中, 由于其含有电负性比较强的氧和氮等杂原子, 可以与其它生物大分子之间有较强的分子间作用力, 是一类重要的类药性骨架.在已发现的手性螺环氧化吲哚类化合物中, 很多都具有较高的生物活性, 诸如抗病毒、抗细胞毒素、抗氧化性等[1, 2].由于螺环结构在创新药物领域越来越受到关注, 作为优势骨架的氧化吲哚也逐渐受到有机化学和药学工作者的青睐.近年来, 人们十分重视该领域的研究, 分离并合成了成千上万种螺环氧化吲哚衍生物[3].如图 1所示, 化合物 (S)-Spirobrassinin[4]是一类从十字花科植物中分离出来的植物抗毒素, 具有杀菌和抗肿瘤活性. Spirotryprostatins A和B[5]是从菌类Aspergillus fumigatus的发酵液中分离得到的一种吲哚类生物碱, 具有良好的抗肿瘤生物活性. Horsfiline[6]是从马来西亚一种用来制作鼻烟的植物Horsfieldia superba中首次分离得到的, 这类化合物具有镇痛作用. Pteropodine[7]是从常绿藤本植物钩藤Uncaria tomentosa的茎皮中分离所得, 是一类重要的药用植物. NITD609[8]则是最初从化合物库中筛选出来并加以人工合成改造的一种螺环氧化吲哚化合物, 研究表明它具有良好的抗疟疾活性, 目前已经作为候选药物在临床前研究中.鉴于螺环氧化吲哚化合物广谱的生物活性及其潜在的生理用途, 探索新颖的反应试剂、发展新型高效的合成方法用于构建这类骨架将具有重要的理论意义和应用前景.
图 1
代表性的含有螺环氧化吲哚骨架的天然产物或药物分子
Figure 1.
Representative examples of natural products and pharmaceutical molecules containing a spirooxindole core structure
天然活性异硫氰酸酯类化合物是十字花科植物中重要的化学组分, 也是一种潜在的癌症化学预防化合物.该类化合物在生物体内可以诱导二型解毒酶的生成、抑制癌细胞的增殖、诱导癌细胞凋亡等.此外, 异硫氰酸酯还具有抗氧化、抗炎和抗菌的功效.异硫氰酸酯可分为脂肪族和芳香族两类, 它们的母体结构主要存在于十字花科蔬菜中, 例如西兰花、荠菜、山葵、萝卜、芝麻菜、辣根、独行菜、西洋菜等.异硫氰酸酯被广泛应用于有机合成中, 其中以烯丙基异硫氰酸酯、苯乙基异硫氰酸酯的研究最为广泛[9].最近, 由袁伟成教授课题组[10]率先发展的新型3-异硫氰酸酯氧化吲哚作为一种高活性的反应试剂, 因其独特的骨架结构而受到化学家们的青睐.另一方面, 新颖高效的串联反应策略在构建复杂螺环分子中起到了非常重要的作用[11].因此, 本文将对近六年来3-异硫氰酸酯氧化吲哚参与的串联环化反应在不对称合成螺环氧化吲哚衍生物方面的研究进展进行简单的综述.如图 2所示, 根据反应类型的不同主要分为以下五类进行综述: (1) 与醛、酮及其衍生物的串联Aldol/Cyclization反应; (2) 与亚胺的串联Mannich/Cyclization反应; (3) 与各种不同的缺电子烯烃或炔烃的串联Michael/Cyclization反应; (4) 与偶氮化合物的串联环化反应; (5) 与氮杂环丙烷的串联开环/关环反应.
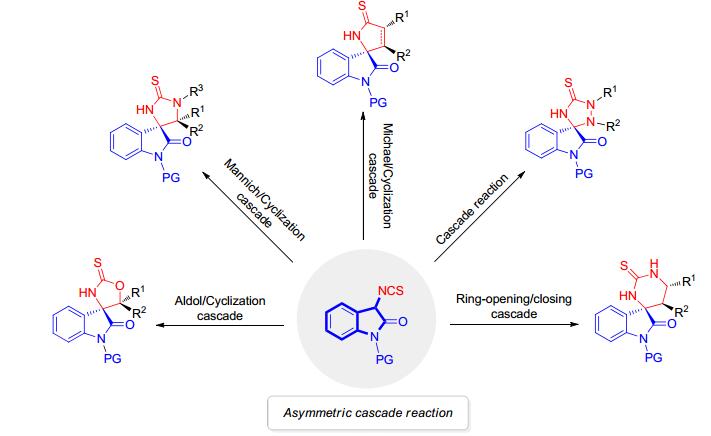 图 2
3-异硫氰酸酯氧化吲哚参与的五类不同类型的串联反应
Figure 2.
3-Isothiocyanato oxindoles engaged five different types of cascade reactions
图 2
3-异硫氰酸酯氧化吲哚参与的五类不同类型的串联反应
Figure 2.
3-Isothiocyanato oxindoles engaged five different types of cascade reactions
1 与醛、酮及其衍生物的串联Aldol/Cyclization反应
1.1 3-异硫氰酸酯氧化吲哚与酮的不对称串联Aldol/ Cyclization反应
2011年, Yuan课题组[10a]率先设计并合成了新型的3-异硫氰酸酯氧化吲哚1, 首次成功报道了该合成子与酮2在双功能有机催化剂3作用下的不对称串联Aldol/ Cyclization反应, 高效、高对映选择性地合成了含有两个连续四取代手性碳中心的螺环氧化吲哚衍生物4 (Eq. 1).这一开创性的工作为后续3-异硫氰酸酯氧化吲哚参与其它类型的串联反应的发展奠定了基础.
通过对产物4a进行简单的合成转换 (如取代反应、缩合反应、氧化反应等), 可以实现更多结构多样的螺环氧化吲哚化合物6~9的合成, 而产物的立体选择性在反应过程中几乎不受到任何影响 (Scheme 1).值得注意的是, 利用碱性条件下的取代反应, 可以实现含有噁唑啉片段的 (S)-Spirobrassinin同系物7和8的生成.
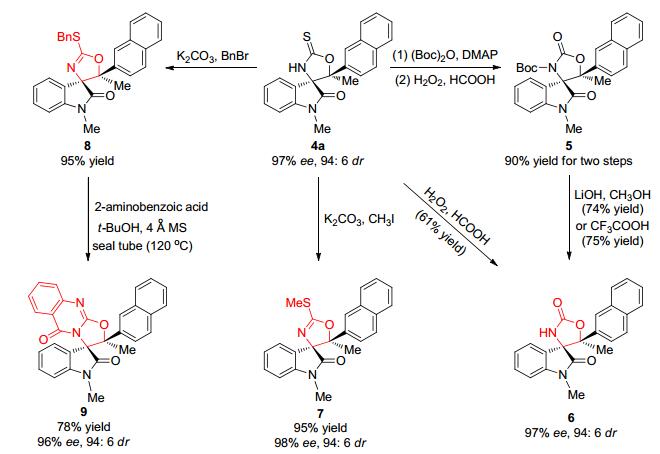 图 图式1
合成应用
Figure 图式1.
Synthetic applications
图 图式1
合成应用
Figure 图式1.
Synthetic applications
3-异硫氰酸酯氧化吲哚能与简单的酮及靛红[12]反应, 此外也可以和酮的衍生物作用. 2015年, Mukherjee小组[13]成功实现了奎宁衍生的三级胺硫脲催化剂11促进的3-异硫氰酸酯氧化吲哚1与α-酮膦酸酯10的串联Aldol/Cyclization反应, 高立体选择性地合成了含有两个连续季碳中心的氧化吲哚螺β-氨基-α-羟基膦酸酯衍生物12 (Eq. 2).该反应可以在很短的时间内快速完成, 作者发现当使用2-甲基四氢呋喃作为溶剂时, 在-95 ℃的条件下, 缓慢加入3-异硫氰酸酯氧化吲哚约30 min (反应条件A), 反应即可完成; 当在甲苯及-78 ℃时 (反应条件B), 反应在5 min内即可迅速完成.在这两种反应情况下, 串联环化反应的效率和对映选择性基本上相似, 不同之处在于前者获得的非对映选择性要优于后者.同时底物具有很好的官能团兼容性, 两种反应条件都适用于脂肪族α-酮膦酸酯, 对于芳香族α-酮膦酸酯而言, 反应条件A获得的效果要更好.最后作者也对反应产物进行了简单的转换研究 (取代反应、氧化反应等), 可以实现不同官能化的螺环氧化吲哚衍生物的合成.
1.2 3-异硫氰酸酯氧化吲哚与醛的不对称串联Aldol/ Cyclization反应
除了各种酮, 3-异硫氰酸酯氧化吲哚同样可以和醛作用, 并用于氧化吲哚螺噁唑啉骨架的构建. Yuan小组[14]在2013年成功报道了双功能有机催化剂3催化的3-异硫氰酸酯氧化吲哚1与各种简单醛13的串联Aldol/ Cyclization反应, 高效实现了螺环氧化吲哚衍生物14的立体选择性合成 (Eq. 3).反应的普适性比较宽广, 芳醛、杂芳醛及脂肪醛都能较好的适用于该串联过程.值得一提的是, 该反应在1 min内即可快速完成.此外, 串联反应产物在碳酸钾作碱的条件下与碘甲烷作用, 可以实现含有噁唑啉片段的 (S)-Spirobrassinin同系物的生成.
通过单晶衍射及核磁二维谱实验, 作者推测了反应可能的过渡态来解释立体选择性的产生.如图 3所示, 手性的硫脲-三级胺催化剂具有双功能的性质, 可以同时活化亲核和亲电试剂.其中, 硫脲片段作为氢键给体与醛羰基片段相互作用, 而三级胺部分作为碱促使3-异硫氰酸酯氧化吲哚烯醇化, 通过氢键作用实现立体控制.
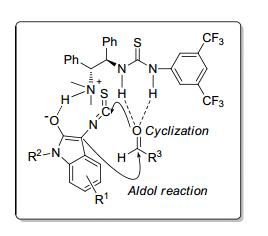 图 3
可能的过渡态
Figure 3.
Proposed transition state
图 3
可能的过渡态
Figure 3.
Proposed transition state
2013年, Kanai和Matsunaga小组[15]也发展了3-异硫氰酸酯氧化吲哚1与脂肪醛15的串联Aldol/ Cyclization反应, 成功实现了氧化吲哚螺噁唑啉衍生物17的高效、高立体选择性合成 (Eq. 4).与Yuan小组工作不同的是, 该串联反应是在双核Ni/席夫碱16复合物的催化作用下顺利完成的.对于各种支链、直链及环状的烷基醛, 反应的效率和立体选择性都比较好, 而芳香醛在该反应条件下的效果并不十分理想.
2 与亚胺的串联Mannich/Cyclization反应
Kanai和Matsunaga课题组[16]在2012年首次发展了双功能的金属Sr (OiPr)2/席夫碱19复合物催化的3-异硫氰酸酯氧化吲哚1与磷酸酯保护的醛亚胺18的不对称串联Mannich/Cyclization反应, 高对映选择性地合成了结构多样的螺环氧化吲哚衍生物20 (Eq. 5).作者对席夫碱进行了细致的研究, 发现当苯环上缺少邻位甲氧基取代时, 反应的立体选择性很差, 由此可以说明甲氧基在决定选择性方面的重要性.
为了拓展该方法学的重要性, 作者对氧化吲哚螺咪唑啉产物20a的合成转换进行了研究 (Scheme 2).对20a进行简单的苯甲酰化, 能以94%的收率得到21.通过进一步钯催化的脱硫交叉偶联反应, 可以实现新型咪唑啉螺环氧化吲哚22的构建.值得一提的是, 由于产物22中两个芳基处于顺式位置, 因此22可以作为以下两种物质的合成前体: (1) Nutlin[17], 一种基于咪唑骨架的可以有效抑制p53/E3泛素连接酶Mdm2之间相互作用的拮抗剂; (2) MI-219[18], 基于吡咯啉螺环氧化吲哚的p53/ Mdm2抑制剂.鉴于此, 该方法将为药物化学领域中设计和合成新的潜在抗肿瘤试剂提供一条非常重要的途径.
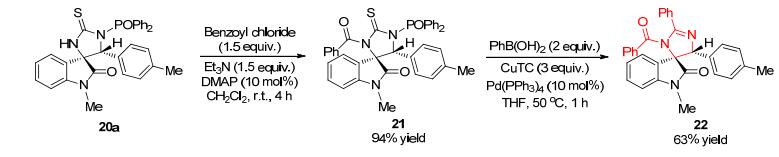 图 图式2
合成转换
Figure 图式2.
Synthetic transformations
图 图式2
合成转换
Figure 图式2.
Synthetic transformations
2014年, Liu和Xu等[19]实现了双功能Takemoto催化剂24促进的3-异硫氰酸酯氧化吲哚1和磺酰亚胺23的串联Mannich/Cyclization反应, 快速、高效地构建了咪唑烷类螺环氧化吲哚骨架25 (Eq. 6).反应在两种条件下都可以顺利完成, 在方法A中, 对氰基苯甲酸的加入可以提高反应的对映选择性, 而非对映选择性略有降低.
为了验证该方法学的有用性, 作者对串联反应产物咪唑烷类螺环氧化吲哚25a进行了转化研究 (Scheme 3).在碳酸钾的存在下与碘甲烷作用, 能以95%的收率得到2-甲基硫咪唑烷螺环氧化吲哚26.通过进一步萘钠促进的去磺酰化脱保护过程, 可以实现Spirobrassinin同系物27的合成.
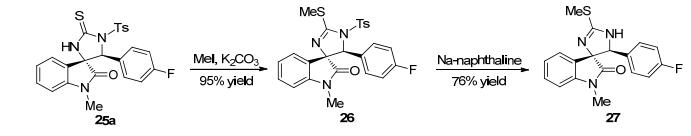 图 图式3
合成转换
Figure 图式3.
Synthetic transformations
图 图式3
合成转换
Figure 图式3.
Synthetic transformations
随后在2015年, Yuan小组[20]也探讨了奎宁28调节的咪唑烷类螺环氧化吲哚衍生物25的快速合成.当使用1 mol%催化剂时, 反应在10 min之内即可快速、高效完成.与Liu小组工作的不同之处在于, 在本文中作者不仅验证了对甲苯磺酰基保护的醛亚胺的适用范围, 同时还尝试使用了其他保护基取代的醛、酮亚胺29~ 32, 得到了一系列结构多样的螺环氧化吲哚衍生物33~36 (Scheme 4).虽然产物的立体选择性不是十分理想, 但是反应底物的适用范围得到了进一步拓展.
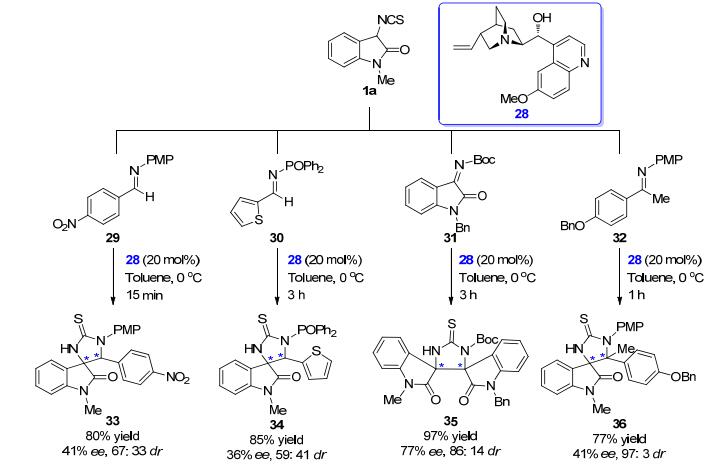 图 图式4
3-异硫氰酸酯氧化吲哚与其它亚胺的串联Mannich/Cyclization反应
Figure 图式4.
Mannich/Cyclization cascade reaction of 3-isothiocyanato oxindoles with some other imines
图 图式4
3-异硫氰酸酯氧化吲哚与其它亚胺的串联Mannich/Cyclization反应
Figure 图式4.
Mannich/Cyclization cascade reaction of 3-isothiocyanato oxindoles with some other imines
在实验结果及其他课题组相关工作的基础上, 作者提出了可能的过渡态来解释串联Mannich/Cyclization反应的立体选择性.如图 4所示, 喹啉28中与C (9) 相连的羟基氢原子通过氢键作用活化对甲苯磺酰基保护的亚胺氮原子; 另一方面, 3-异硫氰酸酯氧化吲哚在三级胺的作用下通过去质子后形成其烯醇式, 紧接着从其Si面亲核进攻亚胺完成分子间的Mannich反应, 最后通过分子内的环化过程获得光学纯的螺环氧化吲哚.
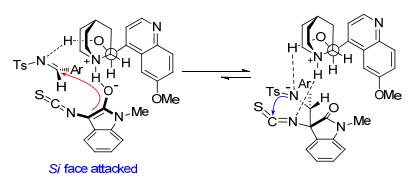 图 4
可能的过渡态
Figure 4.
Proposed transition state
图 4
可能的过渡态
Figure 4.
Proposed transition state
2016年, Shi小组[21]成功发展了金鸡纳碱衍生的硫脲催化剂11促进的3-异硫氰酸酯氧化吲哚1与α, β-不饱和亚胺37的串联Mannich/Cyclization反应, 简单、高效地得到了一系列咪唑烷类螺环氧化吲哚38 (Eq. 7).此外, 通过串联反应得到的手性螺环氧化吲哚产物在mCPBA的氧化作用下, 能够有效转变成相应的γ内酰胺类化合物.
3 与缺电子烯烃或炔烃的串联Michael/ Cyclization反应
3.1 3-异硫氰酸酯氧化吲哚与3-烯基氧化吲哚的不对称串联Michael/Cyclization反应
2013年, 兰州大学的Wang小组[22], 南开大学的Huang和Wang等[23]以及我们课题组[24]成功实现了3-异硫氰酸酯氧化吲哚1与3-烯基氧化吲哚39的串联Michael/Cyclization反应, 非常简便、高效、高立体选择性地合成了一系列具有潜在生理活性的手性双螺环氧化吲哚衍生物43(Eq. 8).通过该反应可以一步构建两个螺环和三个连续的手性中心, 其中包含两个季碳中心.同时该反应条件温和, 反应时间短, 底物适用范围宽广, 具有很好的官能团兼容性.三份工作的不同之处主要在于所使用的双功能有机催化剂的种类不一样, 当使用催化剂40~42时, 在不同的用量下, 反应的最终效果有所差别.同年, Jing等[25]也探讨了3-异硫氰酸酯氧化吲哚与3-烯基氧化吲哚的串联环化反应, 用于非对映选择性的构建双螺环氧化吲哚.在文中作者只尝试了一例不对称催化, 当使用10 mol% DHQ时, 能够以98%的收率, 98%的对映选择性以及>95:5的非对映选择性获得手性双螺环氧化吲哚.
3.2 3-异硫氰酸酯氧化吲哚与硝基烯烃的不对称串联Michael/Cyclization反应
3.3 3-异硫氰酸酯氧化吲哚与3-硝基吲哚的不对称串联Michael/Cyclization反应
3-硝基吲哚是一类特殊的反应试剂, 当N (1) 和C (3) 位都是吸电子取代基时, 该合成子可以被看作是缺电子的烯烃.通过文献调研发现该试剂很少被用于催化不对称合成中. 2015年, Yuan小组[29a]首次成功发展了双功能三级胺-硫代氨基甲酸酯55促进的3-异硫氰酸酯氧化吲哚1与3-硝基吲哚54的串联Michael/Cyclization反应, 通过一步反应过程可以快速构建含有三个连续手性中心, 其中有两个是季碳中心的多环螺环氧化吲哚衍生物56 (Eq. 12).该反应条件温和, 官能团兼容性较好, 同时产物的收率和立体选择性都很理想.随后, 该小组[29b]也成功实现了手性Zn (OTf)2/ent-45金属复合物促进的3-异硫氰酸酯氧化吲哚1与3-硝基吲哚54的串联环化反应 (Eq. 13).相较于此前的有机催化策略, 作者发现手性Lewis酸作用效果更好, 能以几乎定量的收率及非常好的立体选择性得到多环螺环氧化吲哚产物56.
为了进一步拓展该方法学的实用性, 作者进行了一系列合成转换实验, 得到了结构多样的新型螺环氧化吲哚衍生物57~62 (Scheme 5).根据已知文献, 串联环化产物56a在碳酸钾的条件下分别与碘甲烷和溴化苄作用, 能以几乎定量的收率得到螺环噁唑啉化合物57和58.在双氧水和甲酸作用下, 56a能够很容易被氧化成γ内酰胺类化合物59 (90% yield).有锌粉和三甲基氯硅烷存在时, 螺环氧化吲哚56a的硝基片段很容易被还原成氨基, 能以61%的收率得到相应的含有氨基的螺环氧化吲哚衍生物60.在这四种反应过程中, 产物的立体选择性几乎不受影响.重要的是, 化合物57中的硝基在强酸性和镍硼试剂还原的作用下可以被有效脱除分别得到结构新颖的螺环氧化吲哚61 (67% yield, 87% ee) 和62 (67% yield, 97% ee, >99:1 dr).
 图 图式5
合成应用
Figure 图式5.
Synthetic applications
图 图式5
合成应用
Figure 图式5.
Synthetic applications
3.4 3-异硫氰酸酯氧化吲哚与联烯酯、丁炔二酸二酯或炔酮的不对称串联Michael/Cyclization反应
2013年, Shi和Xu等[30]成功报道了3-异硫氰酸酯氧化吲哚1与联烯酯63或丁炔二酸二酯64的串联Michael/Cyclization反应, 在金鸡纳碱衍生的四方酰胺双功能催化剂42的作用下, 高效、高对映选择性实现了新型螺环氧化吲哚骨架65与66、67的构建 (Scheme 6).值得注意的是, 在不同的反应情况下, 可以得到不同的螺环氧化吲哚衍生物66或67.当使用大大过量 (10 equiv.) 的丁炔二酸二酯64时, 加入30 mg分子筛, 在室温条件下可以有效构建结构新颖的螺环氧化吲哚67.作者在本文工作中对串联环化产物进行了转换研究, 通过简单的取代、偶联和氧化反应可以实现结构多样的氧化吲哚螺环化合物的合成.
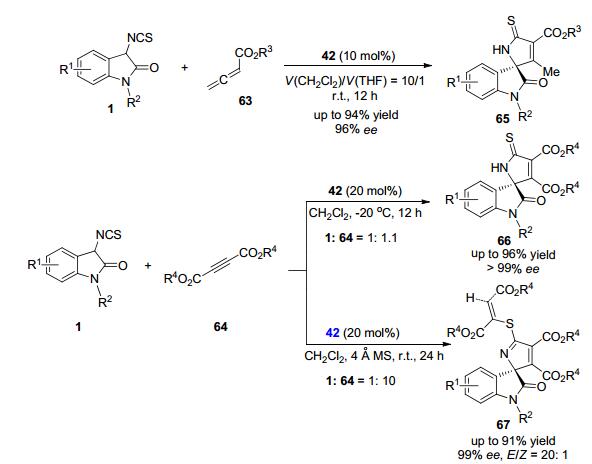 图 图式6
3-异硫氰酸酯氧化吲哚与联烯酯和丁炔二酸二酯的串联Michael/Cyclization反应
Figure 图式6.
Michael/Cyclization cascade reactions of 3-isothiocyanato oxindoles with allenic esters and 2-butynedioic acid diesters
图 图式6
3-异硫氰酸酯氧化吲哚与联烯酯和丁炔二酸二酯的串联Michael/Cyclization反应
Figure 图式6.
Michael/Cyclization cascade reactions of 3-isothiocyanato oxindoles with allenic esters and 2-butynedioic acid diesters
2015年, Wang小组[31]从o-羟基苯乙酸出发合成了噁唑啉羟基型手性配体69, 并成功发展了Mg/69原位生成的金属复合物促进的3-异硫氰酸酯氧化吲哚1与炔酮68的串联Michael/Cyclization反应, 以较好的化学收率和对映选择性得到了一系列手性螺环氧化吲哚衍生物70 (Eq. 14).该反应的发现进一步拓展了炔基化合物的适用范围.
作者推测了反应可能的机理, 如图 7所示.金属Mg盐与手性配体原位生成金属络合物A, 主要以其二聚体形式B存在.烯醇化的3-异硫氰酸酯氧化吲哚与活性物种B作用, 形成中间体C, 进一步与炔酮化合物68a反应得到中间体D, 其中Mg作为Lewis酸活化羰基.通过中间体E中的反应模式实现螺环氧化吲哚70a的立体选择性构建.
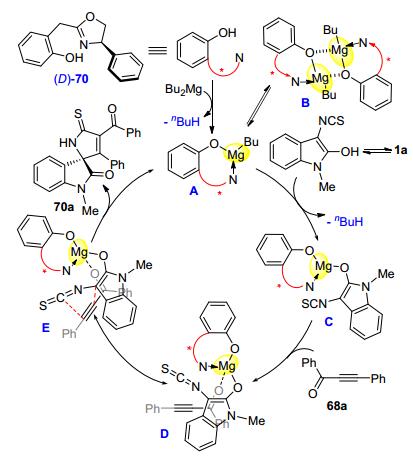 图 7
可能的反应机理
Figure 7.
Proposed mechanism
图 7
可能的反应机理
Figure 7.
Proposed mechanism
3.5 3-异硫氰酸酯氧化吲哚与其它缺电子烯烃的不对称串联Michael/Cyclization反应
在构建Spirobrassinin核心结构及类似物方面, Shi和Xu小组也做出了突出的贡献. 2015年, 该课题组[32]成功发展了3-异硫氰酸酯氧化吲哚1与三氟甲基-2-丁烯酸二酯 (71) 在有机催化剂72作用下的不对称串联Michael/Cyclization反应, 高效、高立体选择性地构建了一系列含三氟甲基的螺环氧化吲哚化合物73 (Eq. 15).
作者进行了一系列合成转换实验来说明此方法学的重要性 (Scheme 7).通过简单的兰尼镍氢化反应, 可以高效构建吡咯烷螺环氧化吲哚74 (84% yield, 94% ee).串联环化产物73a在HgCl2的作用下可以将硫原子移除, 同时生成金属复合物75 (90% yield).在碱性条件下与碘甲烷作用后得到的产物76, 在mCPBA的进一步氧化下可以获得新型螺环氧化吲哚化合物77.此物质后续可以与TMSN3、烯丙醇反应或在氢化锂铝的还原下, 分别得到结构新颖的螺环氧化吲哚衍生物78~81.
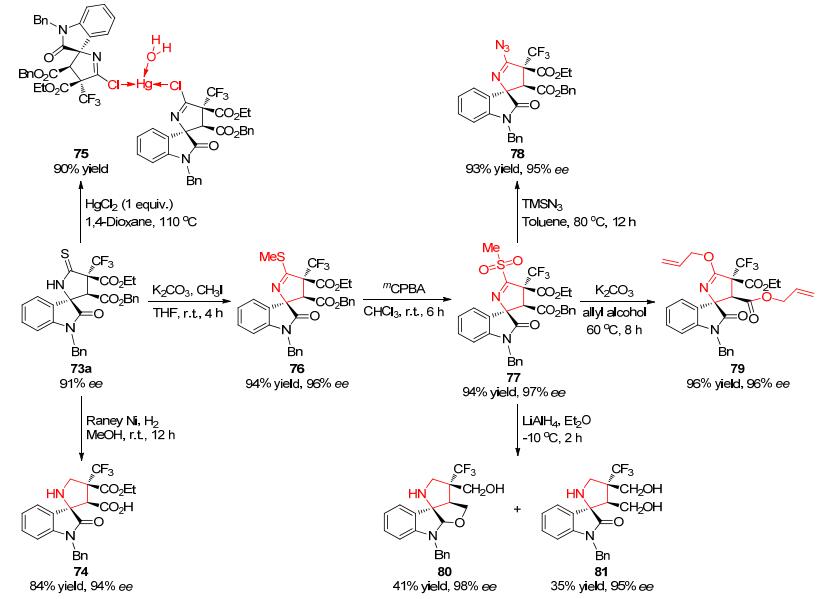 图 图式7
合成转换
Figure 图式7.
Synthetic transformations
图 图式7
合成转换
Figure 图式7.
Synthetic transformations
2016年, Shi小组[21]成功发展了金鸡纳碱衍生的硫脲催化剂11促进的3-异硫氰酸酯氧化吲哚1与α, β-不饱和亚胺的串联环化反应 (Eq. 16).当使用不饱和酮亚胺82时, 得到的主要是串联Michael/Cyclization环化反应产物—吡咯啉类螺环氧化吲哚83.值得一提的是, 当使用含有三个缺电子不饱和键的化合物84时, 在双功能机催化剂85的作用下, 通过串联[3+2]/[4+2]反应能够简便、高效、高立体选择性地得到一系列结构新颖的多环螺环氧化吲哚衍生物86 (Eq. 17).在该方法学中, 只需要简单地改变反应底物, 就可以有效地控制反应的区域选择性.此外, 通过不同的串联反应得到的手性螺环氧化吲哚产物中的C=S和C=N在mCPBA的氧化作用下, 能够有效地转变成C=O, 极大地丰富了螺环氧化吲哚的应用范围.
最近, Ghosh和Chowdhury等[33]实现了金鸡纳碱衍生的双功能硫脲催化剂11促进的3-异硫氰酸酯氧化吲哚1与亚芳基丙二酸酯87的不对称串联Michael/Cyclization反应, 高效、高立体选择性地构建了高度官能化的3, 2'-吡咯啉螺环氧化吲哚衍生物88 (Eq. 18).
Yuan小组在构建3, 2'-吡咯啉螺环氧化吲哚骨架方面也做出了相应的工作. 2013年, 该课题组[34]通过引入3-甲基-4-硝基-5-烯基异噁唑底物89, 成功地实现了奎宁28作用下该合成子与3-异硫氰酸酯氧化吲哚1的串联Michael/Cyclization反应, 在温和的反应条件下得到了一系列具有三个连续手性中心的氧化吲哚螺环化合物90 (Eq. 19).当催化剂用量仅为1 mol%时, 反应的效率和立体选择性仍然很好.
此外, 3-异硫氰酸酯氧化吲哚也能与其它缺电子杂环化合物发生反应.同年, 该小组[35]实现了双功能三级胺—硫代氨基甲酸酯催化剂55促进的3-异硫氰酸酯氧化吲哚1与亚烷基吖内酯91的串联Michael/Cyclization反应, 在温和的反应条件下, 快速、高效地构筑了含有三个连续手性中心的双螺环硫代吡咯啉氧化吲哚骨架92 (Eq. 20).同时作者在本文工作中也完成了3-异硫氰酸酯氧化吲哚与3-烯基氧化吲哚的串联环化反应, 高立体选择性地得到了一系列结构多样的双螺环氧化吲哚衍生物 (up to 99% yield, >99% ee, >99:1 dr).与前面3-异硫氰酸酯氧化吲哚1与3-烯基氧化吲哚39串联反应相关工作的不同之处在于3-烯基氧化吲哚底物取代基的不同.在此文中作者主要使用的是3位苯基取代, 对于乙酯官能团, 在标准反应条件下, 同样可以得到很好的效果 (98% yield, 98% ee, >99:1 dr).
不饱和吡唑啉酮也是一类有效的反应试剂. 2013年, Wang小组[36]成功地发展了3-异硫氰酸酯氧化吲哚1与该合成子93在新型三级胺硫脲催化剂94作用下的串联Michael/Cyclization反应, 高效、高立体选择性地合成了含有三个连续手性中心, 其中两个是季碳中心的螺环氧化吲哚衍生物95 (Eq. 21). 2014年, Yuan等[37]也成功地实现了3-异硫氰酸酯氧化吲哚1与不饱和吡唑啉酮93或不饱和异噁唑酮96的串联Michael/Cyclization反应.与Wang小组的不同之处在于所使用的催化剂不一样, 在奎宁作用下, 该过程能顺利进行, 并且能以非常好的收率和较好的立体选择性得到两类不同的螺环氧化吲哚衍生物95和97 (Scheme 8).
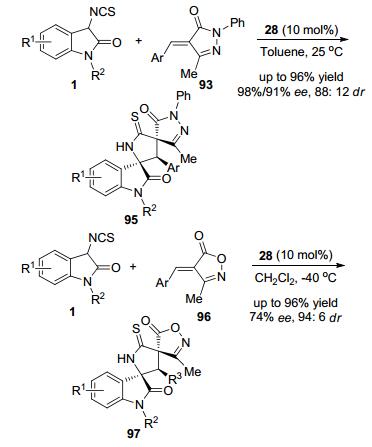 图 图式8
3-异硫氰酸酯氧化吲哚与不饱和吡唑啉酮和不饱和异噁唑酮的串联Michael/Cyclization反应
Figure 图式8.
Michael/Cyclization cascade reactions of 3-isothiocyanato oxindoles with unsaturated pyrazolones and unsaturated isoxazolones
图 图式8
3-异硫氰酸酯氧化吲哚与不饱和吡唑啉酮和不饱和异噁唑酮的串联Michael/Cyclization反应
Figure 图式8.
Michael/Cyclization cascade reactions of 3-isothiocyanato oxindoles with unsaturated pyrazolones and unsaturated isoxazolones
巴比妥酸盐的螺环衍生物是一类结构独特的螺杂环化合物, 具有重要的生物活性.迄今为止, 已有很多方法来构建消旋的螺环巴比妥酸骨架, 但是其不对称合成的例子却很少. 2016年, Zhao小组[38]发展了金鸡纳碱衍生的催化剂11调节的3-异硫氰酸酯氧化吲哚1与巴比妥酸烯烃98的串联Michael/Cyclization反应, 成功实现了手性螺环巴比妥酸化合物99的快速、高效合成 (Eq. 22).作者发现4-甲基苯甲酸是最有效的添加剂, 可以提高反应的对映选择性.
根据文献调研及实验结果, 作者对该串联环化反应的机理进行了研究, 并提出了可能的过渡态.如图 8所示, 在4-甲基苯甲酸的作用下, 3-异硫氰酸酯氧化吲哚1d异构化为其烯醇式.金鸡纳生物碱衍生的硫脲催化剂有双功能性质, 通过氢键作用同时活化巴比妥酸烯烃和烯醇化的3-异硫氰酸酯氧化吲哚.在催化剂构成的手性环境下, 3-异硫氰酸酯氧化吲哚1d的烯醇式从其Si面进攻巴比妥酸烯烃98a的Si面, 继而完成串联环化反应.
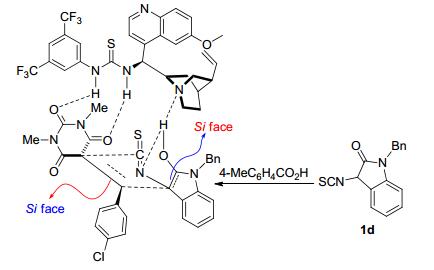 图 8
可能的过渡态
Figure 8.
Proposed transition state
图 8
可能的过渡态
Figure 8.
Proposed transition state
2016年, Mukherjee小组[39]发展了3-异硫氰酸酯氧化吲哚1与α, β-不饱和环酮100的不对称串联Michael/ Cyclization反应, 使用的是金鸡纳碱衍生的四方酰胺双功能有机催化剂42.通过该过程可以有效构建含有三个连续手性中心, 其中两个是季碳中心的3, 2'-吡咯啉双螺环氧化吲哚衍生物101 (Eq. 23).作者对产物进行了官能化转换, 通过简单的取代和氧化反应, 能以较高的化学收率得到相应的产物, 反应的立体选择性不受任何影响.值得注意的是, 在四氢锂铝作为还原剂的条件下, 很容易发生脱硫和羰基还原成醇的反应.
拥有环外双键的巴比妥酸烯烃能与3-异硫氰酸酯氧化吲哚有效作用, 而当双键在环内时, 该串联环化反应也能顺利完成.同年, Du小组[40]实现了金鸡纳碱衍生的四方酰胺双功能有机催化剂42促进的3-异硫氰酸酯氧化吲哚1与马来酰亚胺102的串联Michael/Cyclization反应, 高效、高立体选择性构建了含有三个连续手性中心的吡咯啉螺环氧化吲哚核心骨架103 (Eq. 24).该反应条件温和, 底物普适性相对宽广, 但是作者在实验中发现马来酰亚胺上与N相连的芳环上取代基的电子和位置效应对反应的效率有较大的影响.当芳环上取代基是邻位取代或者芳基被烷基取代时, 反应的效率很低, 甚至反应有可能不发生.
3.2.2 3-异硫氰酸酯氧化吲哚与β-硝基烯烃的串联环化反应
硝基烯烃是一类重要的合成子, 而硝基具有易转化性, 因此硝基烯烃已经被广泛应用于含氮类化合物的合成中. 2014年, Mukherjee等[28]发展了3-异硫氰酸酯氧化吲哚1与β-硝基烯烃51的串联Michael/Cyclization反应, 在辛可尼定衍生的三级胺硫脲催化剂52的调节下, 成功获得了一系列螺环氧化吲哚衍生物53 (Eq. 11).通过该过程能以较好的收率和中等的立体选择性得到含有三个连续手性中心的高度官能化的3, 2'-吡咯啉螺环氧化吲哚.值得注意的是, 该串联环化反应的产物并不稳定, 很容易分解, 甚至在-20 ℃条件下也不能长时间保存.
在辛可尼定衍生的手性三级胺硫脲催化剂52的作用下, 作者推测了该串联Michael/Cyclization反应的机理, 认为可能是通过双活化的模式来实现手性诱导.如图 6所示, 在反应过程中, 催化剂52的三级胺片段作为碱活化3-异硫氰酸酯氧化吲哚1, 硫脲部分通过双氢键作用活化β-硝基烯烃51的硝基部位.按照下图的反应模式, 烯醇化的3-异硫氰酸酯氧化吲哚从β-硝基烯烃的Re面亲核进攻其双键, 快速实现螺环氧化吲哚衍生物53的立体选择性合成.
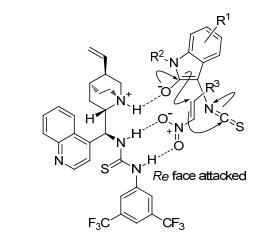 图 6
可能的立体化学模式
Figure 6.
Proposed stereochemical model
图 6
可能的立体化学模式
Figure 6.
Proposed stereochemical model
3.2.1 3-异硫氰酸酯氧化吲哚与环状硝基烯烃的串联环化反应
3-硝基-2-氢-色烯是一类非常有用的环状硝基烯烃, 被广泛应用于合成多种多样具有显著生物活性的色满衍生物. 2014年, 我们小组[26]成功发展了3-异硫氰酸酯氧化吲哚1与3-硝基-2-氢-色烯44在手性Zn (OTf)2/45金属复合物作用下的串联Michael/Cyclization反应, 高效、高立体选择性地合成了一系列多环螺环氧化吲哚衍生物46 (Eq. 9).值得一提的是, 在反应过程中我们发现了一个有趣的现象, 随着反应的进行, 体系中会有大量的黄色固体析出.当反应完成后, 通过对反应体系进行简单的抽滤, 即能以较好的收率和非常好的立体选择性得到纯净的目标产物.这一操作非常简便, 具有很好的工业应用前景.重要的是, 该反应不仅适用于环状硝基烯烃, 对于非环状硝基烯烃, 例如β-甲基-β-硝基烯烃, 能以82%的分离收率, 99.5%的对映选择性, 6:1的非对映选择性成功得到相应的反应产物.然而遗憾的是, 对于最简单的β-硝基苯乙烯, 虽然也能顺利参与该串联环化反应并获得相应的螺环产物, 但是在手性金属复合物催化体系下得到的效果并不理想, 我们仅以29%的分离收率, 4%的对映选择性和3:1的非对映选择性得到相应的串联环化过程产物.
为了解释串联Michael/Cyclization反应的立体选择性, 我们提出了可能的过渡态.如图 5所示, 依据相关文献中金属/双噁唑啉配体催化反应提出的活化模式, 我们建立了与文献报道完全不同的Lewis酸/Lewis碱协同催化的模型.手性的Zn (Ⅱ) 复合物有两方面的作用, 其中Zn (Ⅱ) 作为Lewis酸活化缺电子烯烃中的硝基片段, 配体中NH基团的N原子作为Lewis碱与底物1a通过氢键作用, 使得亲核进攻从3-硝基-2-氢-色烯44a的Re面完成.
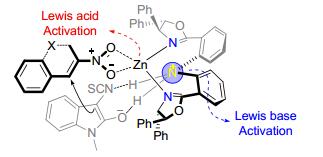 图 5
可能的过渡态
Figure 5.
Proposed transition state
图 5
可能的过渡态
Figure 5.
Proposed transition state
除了手性Lewis酸可以催化3-异硫氰酸酯氧化吲哚与环状硝基烯烃的串联环化反应, 有机催化剂同样能够有效作用于该过程.同年, Xie小组[27]实现了双功能硫脲催化剂48促进的3-异硫氰酸酯氧化吲哚1与3-硝基-2-氢-色烯衍生物47的串联Michael/Cyclization反应, 快速合成了结构多样的多环螺环氧化吲哚衍生物49及其异构体50 (Eq. 10).在催化量的无机碱作用下, 串联环化产物49能以几乎定量的收率转换成50, 同时对映选择性会提高.
4 与偶氮二羧酸酯的不对称串联环化反应
2013年, Shi小组[41]成功发展了 (DHQD)2PHAL催化的3-异硫氰酸酯氧化吲哚1与偶氮二羧酸酯104的形式上[3+2]环加成反应, 快速地得到了在C (3') 位含有两个杂环的螺环氧化吲哚衍生物105 (Eq. 25).该反应条件温和, 底物范围宽广, 而且反应效率很高, 在5 min内即能完成, 同时可以获得较好的收率和对映选择性.
有趣的是, 作者在实验过程中发现, 当偶氮二异丙酯104a增加到2 equiv.时, 正常的环加成产物105a会与过量的104a作用, 生成新产物106a.通过一系列的条件筛选, 作者观察到产物106a的对映选择性与105a的几乎一样, 由此说明新产物的对映选择性是由第一步产生105a的反应决定的, 而进一步的控制实验也证明了第二步反应过程并不会影响最终产物的对映选择性 (Scheme 9).更为重要的是, 作者对螺环氧化吲哚衍生物105a和106a的生物活性进行了评估.通过对抑制人类宫颈癌细胞扩散的细胞毒性实验研究发现, 当浓度为1 mg/mL时, 这两个化合物表现出很强的抑制效率, 细胞存活率基本为零, 由此可以说明该类螺环氧化吲哚骨架具有重要的生理活性.
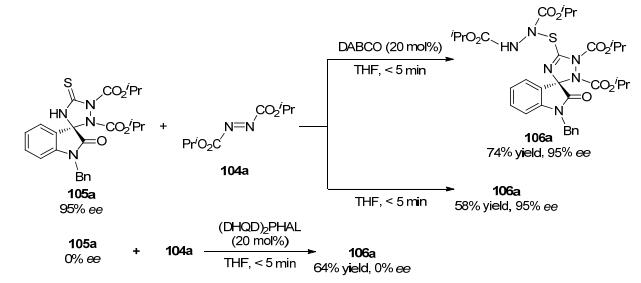 图 图式9
控制实验
Figure 图式9.
The control experiments
图 图式9
控制实验
Figure 图式9.
The control experiments
5 与氮杂环丙烷的串联开环/关环反应
作为NITD609类似物, 含有嘧啶结构的螺环氧化吲哚也是药物化学家青睐的合成目标.基于此, 2015年, Wang小组[42]成功实现了Bu2Mg/108复合物催化的3-异硫氰酸酯氧化吲哚1与N-(2-吡啶甲酰基) 氮杂环丙烷107的不对称开环反应, 而后加入叔丁醇钾和碘甲烷完成关环反应, 高效、高立体选择性地合成了一系列嘧啶螺氧化吲哚化合物109 (Eq. 26).而早在2013年, 该小组[43]就完成了3-异硫氰酸酯氧化吲哚与1, 3-偶极偶氮甲碱亚胺之间非对映选择性的形式上的[3+3]环加成反应, 在三乙胺的作用下可以高效、高非对映选择性地合成结构多样的3, 3'-三嗪基螺环氧化吲哚衍生物.遗憾的是, 该反应的对映选择性控制并没有成功实现.
6 结论与展望
本文主要论述了3-异硫氰酸酯这样一个高活性的物种参与的不同类型的不对称串联环化反应, 通过该方法学可以得到许多结构多样并且具有重要生物活性的螺环氧化吲哚核心骨架.近些年, 在这一研究领域, 有机化学家们取得了一定的成绩.但是纵观之前的工作, 我们认为该方向还存在很多值得深入研究和发展的空间.从已有的研究成果中不难分析得出以下几点结论: (1) 反应类型, 主要是串联Aldol/Mannich/Michael环化反应, 反应种类不够丰富, 需要进一步探索新型反应; (2) 催化剂方面, 主要使用的是有机小分子催化剂或手性金属复合物, 种类比较少, 而且反应选择性比较单一, 因此仍需要发展高效、高选择性、广普适性的催化体系; (3) 底物方面, 主要是缺电子双键 (C=O、C=N、C=C) 或叁键, 需要进一步拓展底物的适用范围; (4) 产物方面, 多数合成五元或六元环结构的化合物, 七元或更大环结构仍然难以构建.我们相信, 通过在上述四个方面继续拓展3-异硫氰酸酯氧化吲哚参与的反应类型, 寻找合适的催化体系, 发展新型底物, 并由此构建以螺环氧化吲哚为基本骨架的新化合物, 将会被广泛地应用于药物化学的研究中, 同时也将为药物研发中新的先导化合物发现提供一定数量的化合物库.
-
-
[1]
Lin, H.; Danishefsky, S. J. Angew. Chem., Int. Ed. 2003, 42, 36. doi: 10.1002/(ISSN)1521-3773
-
[2]
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209.
-
[3]
(a) Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381.
(b) Yu, J.; Shi, F.; Gong, L.-Z. Acc. Chem. Res. 2011, 44, 1156.
(c) Rios, R. Chem. Soc. Rev. 2012, 41, 1060.
(d) Cheng, D.-J.; Ishihara, Y.; Tan, B.; Barbas Ⅲ, C. F. ACS Catal. 2014, 4, 743.
(e) Xiao, Y.-L.; Zhou, Y.; Wang, J.; Wang, J.-X.; Liu, H. Chin. J. Org. Chem. 2015, 35, 2035 (in Chinese). (肖永龙, 周宇, 王江, 王进欣, 柳红, 有机化学, 2015, 35, 2035. -
[4]
Suchy, M.; Kutschy, P.; Monde, K. J. Org. Chem. 2001, 66, 3940. doi: 10.1021/jo0155052
-
[5]
(a) Cui, C. B.; Kakeya, H.; Osada, H. J. Antibiot. 1996, 49, 832.
(b) Edmondson, S.; Danishefsky, S.-J.; Sepp-Lorenzino, L.; Rosen, N. J. Am. Chem. Soc. 1999, 121, 2147.
(c) Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Tomita, Y.; Parrish, D. A.; Deschamps, J. R.; Wang, S. J. Am. Chem. Soc. 2005, 127, 10130.
(d) Cheng, M.-N.; Wang, H.; Gong, L.-Z. Org. Lett. 2011, 13, 2418. -
[6]
Jossang, A.; Jossang, P.; Hadi, H. A.; Sevenet, T.; Bodo, B. J. Org. Chem. 1991, 56, 6527. doi: 10.1021/jo00023a016
-
[7]
(a) Potawel, S. E.; Mehta, U. K.; Waseem, S.; Dhalawat, H. J.; Lunya, K. P.; Mantri, R. A.; Vetol, Y. D. Pharmacology 2008, 2, 197.
(b) Litvinov, Y. M.; Mortikov, V. Y.; Shestopalov, A. M. J. Comb. Chem. 2008, 10, 741. -
[8]
Rottmann, M.; McNamara, C.; Yeung, B. K. S.; Lee, M. C. S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D. M.; Dharia, N. V.; Tan, J.; Cohen, S. B.; Spencer, K. R.; González-Páez, G. E.; Lakshminarayana, S. B.; Goh, A.; Suwanarusk, R.; Jegla, T.; Schmitt, E. K.; Beck, H. P.; Brun, R.; Nosten, F.; Renia, L.; Dartois, V.; Keller, T. H.; Fidock, D. A.; Winzeler, E. A.; Diagana, T. T. Science 2010, 329, 1175. doi: 10.1126/science.1193225
-
[9]
梁浩, 李瑞敏, 袁其朋, 北京化工大学学报 (自然科学版), 2015, 42, 1.Liang, H.; Li, R.-M.; Yuan, Q.-P. J. Beijing Univ. Chem. Technol. (Nat. Sci.) 2015, 42, 1 (in Chinese).
-
[10]
(a) Chen, W.-B.; Wu, Z.-J.; Hu, J.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2011, 13, 2472.
(b) Han, W.-Y.; Zhao, J.-Q.; Zuo, J.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Adv. Synth. Catal. 2015, 357, 3007. -
[11]
Jiang, K.; Jia, Z.-J.; Yin, X.; Wu, L.; Chen, Y.-C. Org. Lett. 2010, 12, 2766. doi: 10.1021/ol100857s
-
[12]
Han, Y.-Y.; Chen, W.-B.; Han, W.-Y.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2012, 14, 490. doi: 10.1021/ol203081x
-
[13]
Kayal, S.; Mukherjee, S. Org. Lett. 2015, 17, 5508. doi: 10.1021/acs.orglett.5b02929
-
[14]
Chen, W.-B.; Han, W.-Y.; Han, Y.-Y.; Zhang, X.-M.; Yuan, W.-C. Tetrahedron 2013, 69, 5281. doi: 10.1016/j.tet.2013.05.002
-
[15]
(a) Kato, S.; Kanai, M.; Matsunaga, S. Chem. Asian J. 2013, 8, 1768.
(b) Kato, S.; Kanai, M.; Matsunaga, S. Heterocycles 2014, 88, 475. -
[16]
Kato, S.; Yoshino, T.; Shibasaki, M.; Kanai, M.; Matsunaga, S. Angew. Chem., Int. Ed. 2012, 51, 7007. doi: 10.1002/anie.201203005
-
[17]
(a) Vassilev, L. T.; Vu, B. T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E. A. Science 2004, 303, 844.
(b) Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B. T.; Qing, W.; Packman, K.; Myklebost, O.; Heimbrook, D. C.; Vassilev, L. T. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1888. -
[18]
Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R. S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; Bernard, D.; Zhang, J.; Lu, Y.; Gu, Q.; Shah, R. B.; Pienta, K. J.; Ling, X.; Kang, S.; Guo, M.; Sun, Y.; Yang, D.; Wang, S. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3933. doi: 10.1073/pnas.0708917105
-
[19]
Cai, H.; Zhou, Y.; Zhang, D.; Xu, J.-Y.; Liu, H. Chem. Commun. 2014, 50, 14771. doi: 10.1039/C4CC06000H
-
[20]
Bai, M.; Cui, B.-D.; Zuo, J.; Zhao, J.-Q.; You, Y.; Chen, Y.-Z.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Tetrahedron 2015, 71, 949. doi: 10.1016/j.tet.2014.12.074
-
[21]
Du, D.; Xu, Q.; Li, X.-G.; Shi, M. Chem. Eur. J. 2016, 22, 4733. doi: 10.1002/chem.v22.14
-
[22]
Cao, Y.-M.; Shen, F.-F.; Zhang, F.-T.; Wang, R. Chem. Eur. J. 2013, 19, 1184. doi: 10.1002/chem.201204114
-
[23]
Wu, H.; Zhang, L.-L.; Tian, Z.-Q.; Huang, Y.-D.; Wang, Y.-M. Chem. Eur. J. 2013, 19, 1747. doi: 10.1002/chem.201203221
-
[24]
Tan, F.; Cheng, H.-G.; Feng, B.; Zou, Y.-Q.; Duan, S.-W.; Chen, J.-R.; Xiao, W.-J. Eur. J. Org. Chem. 2013, 2071.
-
[25]
Wu, S.; Zhu, X.-L.; He, W.-J.; Wang, R.-M.; Xie, X.-H.; Qin, D.-B.; Jing, L.-H.; Chen, Z.-Q. Tetrahedron 2013, 69, 11084. doi: 10.1016/j.tet.2013.11.016
-
[26]
Tan, F.; Lu, L.-Q.; Yang, Q.-Q.; Guo, W.; Bian, Q.; Chen, J.-R.; Xiao, W.-J. Chem. Eur. J. 2014, 20, 3415. doi: 10.1002/chem.v20.12
-
[27]
Fu, Z.-K.; Pan, J.-Y.; Xu, D.-C.; Xie, J.-W. RSC Adv. 2014, 4, 51548. doi: 10.1039/C4RA07860H
-
[28]
Kayal, S.; Mukherjee, S. Eur. J. Org. Chem. 2014, 6696.
-
[29]
(a) Zhao, J.-Q.; Zhou, M.-Q.; Wu, Z.-J.; Wang, Z.-H.; Yue, D.-F.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2015, 17, 2238.
(b) Zhao, J.-Q.; Wu, Z.-J.; Zhou, M.-Q.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2015, 17, 5020. -
[30]
Du, D.; Jiang, Y.; Xu, Q.; Shi, M. Adv. Synth. Catal. 2013, 355, 2249. doi: 10.1002/adsc.201300460
-
[31]
Wang, L.-Q.; Yang, D.-X.; Li, D.; Liu, X.-H.; Zhao, Q.; Zhu, R.-R.; Zhang, B.-Z.; Wang, R. Org. Lett. 2015, 17, 4260. doi: 10.1021/acs.orglett.5b02052
-
[32]
Du, D.; Jiang, Y.; Xu, Q.; Tang, X.-Y.; Shi, M. ChemCatChem 2015, 7, 1366. doi: 10.1002/cctc.201500141
-
[33]
Chowdhury, R.; Kumar, M.; Ghosh, S. K. Org. Biomol. Chem. 2016, 14, 11250. doi: 10.1039/C6OB02104B
-
[34]
Liu, X.-L.; Han, W.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2013, 15, 1246. doi: 10.1021/ol400183k
-
[35]
Han, W.-Y.; Li, S.-W.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Chem. Eur. J. 2013, 19, 5551. doi: 10.1002/chem.v19.18
-
[36]
Chen, Q.; Liang, J.-Y.; Wang, S.-L.; Wang, D.; Wang, R. Chem. Commun. 2013, 49, 1657. doi: 10.1039/c3cc38386e
-
[37]
Cui, B.-D.; Li, S.-W.; Zuo, J.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Tetrahedron 2014, 70, 1895. doi: 10.1016/j.tet.2014.01.036
-
[38]
Zhao, H.-W.; Tian, T.; Pang, H.-L.; Li, B.; Chen, X.-Q.; Yang, Z.; Meng, W.; Song, X.-Q.; Zhao, Y.-D.; Liu, Y.-Y. Adv. Synth. Catal. 2016, 358, 2619. doi: 10.1002/adsc.v358.16
-
[39]
Kayal, S.; Mukherjee, S. Org. Biomol. Chem. 2016, 14, 10175. doi: 10.1039/C6OB02187E
-
[40]
Liu, L.; Zhao, B.-L.; Du, D.-M. Eur. J. Org. Chem. 2016, 4711.
-
[41]
Jiang, Y.; Pei, C.-K.; Du, D.; Li, X.-G.; He, Y.-N.; Xu, Q.; Shi, M. Eur. J. Org. Chem. 2013, 7895.
-
[42]
Wang, L.-Q.; Yang, D.-X.; Li, D.; Wang, R. Org. Lett. 2015, 17, 3004. doi: 10.1021/acs.orglett.5b01291
-
[43]
Zhu, G.-M.; Sun, W.-S.; Wu, C.-Y.; Li, G.-F.; Hong, L.; Wang, R. Org. Lett. 2013, 15, 4988. doi: 10.1021/ol402295m
-
[1]
-

图 1 代表性的含有螺环氧化吲哚骨架的天然产物或药物分子
Figure 1 Representative examples of natural products and pharmaceutical molecules containing a spirooxindole core structure

图 2 3-异硫氰酸酯氧化吲哚参与的五类不同类型的串联反应
Figure 2 3-Isothiocyanato oxindoles engaged five different types of cascade reactions

图式4 3-异硫氰酸酯氧化吲哚与其它亚胺的串联Mannich/Cyclization反应
Scheme 4 Mannich/Cyclization cascade reaction of 3-isothiocyanato oxindoles with some other imines

图式6 3-异硫氰酸酯氧化吲哚与联烯酯和丁炔二酸二酯的串联Michael/Cyclization反应
Scheme 6 Michael/Cyclization cascade reactions of 3-isothiocyanato oxindoles with allenic esters and 2-butynedioic acid diesters

图式8 3-异硫氰酸酯氧化吲哚与不饱和吡唑啉酮和不饱和异噁唑酮的串联Michael/Cyclization反应
Scheme 8 Michael/Cyclization cascade reactions of 3-isothiocyanato oxindoles with unsaturated pyrazolones and unsaturated isoxazolones
-


 下载:
下载:
















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 2037
- HTML全文浏览量: 368
 下载:
下载:
