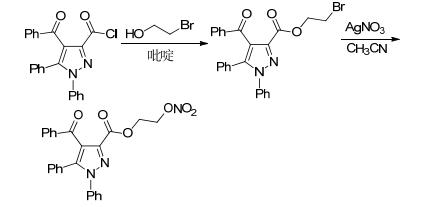
 图 图式1
带有NO供体的吡唑-3-羧酸衍生物的合成路线
Figure 图式1.
Synthetic routes of pyrazole-3-carboxylic acid derivatives bearing NO donor
图 图式1
带有NO供体的吡唑-3-羧酸衍生物的合成路线
Figure 图式1.
Synthetic routes of pyrazole-3-carboxylic acid derivatives bearing NO donor
引用本文:
王兵, 李娜, 刘腾, 王英爱, 王晓静, 孙捷. 一氧化氮供体化合物的合成方法研究进展[J]. 有机化学,
2017, 37(4): 777-797.
doi:
10.6023/cjoc201610035

Citation: Wang Bing, Li Na, Liu Teng, Wang Ying'ai, Wang Xiaojing, Sun Jie. Research Progress on Synthesis of Nitric Oxide Donor Compounds[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 777-797. doi: 10.6023/cjoc201610035
Citation: Wang Bing, Li Na, Liu Teng, Wang Ying'ai, Wang Xiaojing, Sun Jie. Research Progress on Synthesis of Nitric Oxide Donor Compounds[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 777-797. doi: 10.6023/cjoc201610035
一氧化氮供体化合物的合成方法研究进展
English
Research Progress on Synthesis of Nitric Oxide Donor Compounds
Abstract:
Nitric oxide as a biological messenger or effector molecule plays an important physiological role in the body. Owing to its various biological activities, it has received wide attention in clinical practice. Insufficient NO production in vivo is closely related with a variety of diseases. NO donor compounds can release NO in vivo to treat and prevent many diseases. With its wide application in medicine, the methods for the synthesis of NO donor compounds have attracted much attention of researchers. In this paper, the recent advances in the past 10 years in synthetic methods for NO donor compounds are reviewed.
-
Key words:
- nitric oxide donor
- / synthetic method
- / pharmacological activity
-
近几十年, NO常被人们用以阐明许多过去未能解释的生理现象, 成为近年来研究的热点. NO是一种寿命较短的自由基, 半衰期仅数秒钟, 由于其分子小且具有亲脂性, 所以很容易透过细胞膜[1].在体内由L-精氨酸和氧分子在一氧化氮合酶 (NOS) 催化下生成[2]. NO在哺乳动物生理和病理生理中发挥着非常重要的作用[3], 如参与维持微血管和大血管的动态平衡[4]、神经信号传导[5]、免疫炎症的调节[6]、肿瘤发生与转移[7]等多种生理病理过程.体内NO生成不足常与多种疾病的形成密切相关[8~11], 因此外源性NO对于这些疾病的预防和治疗有着重要意义[12].所以, NO供体药物成为了热门的新型药物研究对象. NO供体是指一类在体内经简单酶解或非酶作用后释放出NO的化合物, 是NO的储运形式, 可以克服NO本身难携带、难定量、半衰期短[13]等缺点. NO供体药物主要是指NO供体与已知功效的药物通过某些基团连接起来形成的具有协同生物活性的新化合物[14, 15].目前的研究趋势是利用前药原理, 将已知药物或已知活性化合物的结构与各类NO供体通过各种连接基团结合而制成前药.这种前药可在体内经相关酶或非酶作用释放原药和NO, 使其分别发挥各自作用.目前在研究的NO供体主要有硝酸酯类、呋咱氮氧化物类、偶氮二醇烯鎓盐类、肟类、NO-金属配合物类、S-亚硝基硫醇类、斯德酮亚胺类、胍类、羟胺类及N-羟基脲类等[16].其中硝酸酯类、呋咱氮氧化物类和偶氮二醇烯鎓盐类NO供体是近年来的研究热点.药理研究证明, 很多NO供体药物的药理活性比原药好, 而且不良反应显著低于原药[17], 同时这类药物还具有提高药物的生物利用度、减小毒副作用、增加药物稳定性、促使药物长效化[18]等优点.
NO供体类化合物多种活性的不断发现及应用范围的不断拓展已经引起了国内外很多有机合成研究人员的高度重视.通常化学方法合成NO供体类化合物主要包括两个关键点: (1) 如何构建NO供体化合物的NO释放基团; (2) 如何将NO释放基团准确地与已知功效的药物连接.本文总结了国内外学者对这方面的相关研究, 对NO供体类化合物的合成方法进行综述.
1 硝酸酯类NO供体的合成
硝酸酯类NO供体是一类经典药物, 硝酸甘油作为治疗心绞痛的药物已经应用了很多年.这类NO供体药物结构简单、易于合成、应用比较普遍[19].目前硝酸酯类NO供体药物的合成主要有以下几种合成路线.通过酯键将NO供体和药物连接, 通过醚键将NO供体和药物连接以及通过肽键连接.
1.1 酯化反应
将带有NO供体基团的分子片段与化合物通过酯键连接, 该方法条件温和, 易于操作, 适合用于多数带有羧基的化合物与NO供体基团相连.
2007年, López等[20]合成了带有NO供体基团的生育酚类似物.该反应通过成酯反应将带有NO供体基团的醇与生育酚类似物连接起来 (Eq. 1).此法适用于多数带有羧基的化合物与NO供体基团相连.
2009年, Abdel-Hafez等[21]合成了一系列具有抗菌抗炎活性的NO供体化合物.该反应首先进行酰氯的醇解合成溴代产物, 再用硝酸盐进行硝化获得有机硝酸酯类产物 (Scheme 1).药理研究表明, 该化合物比原药具有更强的抗炎活性, 而且致溃疡能力明显减弱.
图 图式1
带有NO供体的吡唑-3-羧酸衍生物的合成路线
Figure 图式1.
Synthetic routes of pyrazole-3-carboxylic acid derivatives bearing NO donor
2009年, Abdellatif等[17]报道在非甾体类抗炎药骨架上添加NO供体可以减轻非甾体类抗炎药的胃肠道副作用.该反应是先将非甾体类抗炎药的羧基酰氯化然后再通过成酯反应将NO供体与药物偶联 (Scheme 2).该合成方案通过将羧基酰氯化使整个成酯反应更加容易进行.药理研究表明, 该化合物具有与原药相当的抗炎活性, 而且可以降低非甾体类抗炎药的胃肠道不良反应.
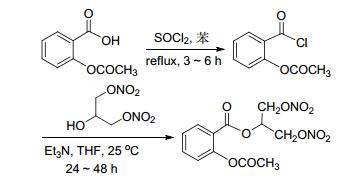 图 图式2
阿司匹林和NO供体化合物参与的偶联反应
Figure 图式2.
Cross-coupling reactions of aspirin and nitric oxide donor compound
图 图式2
阿司匹林和NO供体化合物参与的偶联反应
Figure 图式2.
Cross-coupling reactions of aspirin and nitric oxide donor compound
2012年, Rolando等[22]合成了带有NO供体基团的依达拉奉衍生物, 依达拉奉是自由基清除剂, 与NO供体基团偶联后可增强其抗氧化作用.该方法先用带有NO供体基团的羧酸进行酰氯化反应, 再和药物依达拉奉进行成酯反应获得带有NO供体基团的依达拉奉衍生物 (Scheme 3).
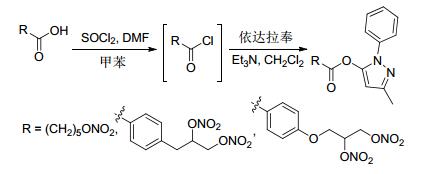 图 图式3
带有NO供体的依达拉奉衍生物的合成路线
Figure 图式3.
Synthetic routes of edaravone derivatives bearing NO donor
图 图式3
带有NO供体的依达拉奉衍生物的合成路线
Figure 图式3.
Synthetic routes of edaravone derivatives bearing NO donor
2015年, Digiacomo等[23]合成了系列带有NO供体基团的胰岛素促分泌素衍生物.该方案直接将胰岛素促分泌素药物瑞格列奈和那格列奈与带有NO供体基团的醇类化合物通过成酯反应连接起来 (Scheme 4).药理实验证明这类化合物不仅可以降低血糖还具有血管保护作用, 可以用来治疗Ⅱ型糖尿病及其引起的血管并发症.
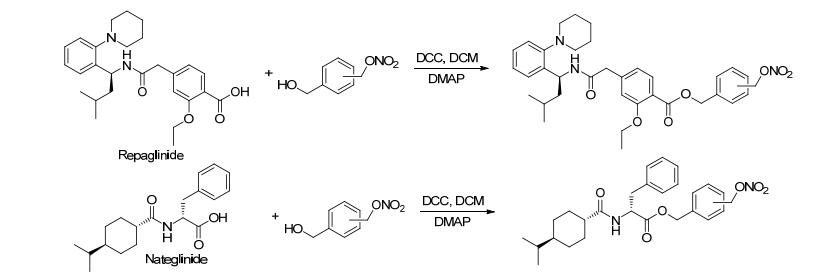 图 图式4
带有NO供体的胰岛素促分泌素衍生物的合成路线
Figure 图式4.
Synthetic routes of insulin-secretagogue derivatives bearing NO donor
图 图式4
带有NO供体的胰岛素促分泌素衍生物的合成路线
Figure 图式4.
Synthetic routes of insulin-secretagogue derivatives bearing NO donor
1.2 酰胺化反应
2008年, Konter等[24]合成了一种带有NO供体基团的抗菌药酮康唑类似物.该反应是先进行酰胺的水解反应再进行酰化反应形成带有NO供体基团的抗菌化合物 (Eq. 2).该方法可用于氨类化合物的NO供体连接.药理研究表明, 该化合物的抗真菌活性比原药酮康唑更强.
2012年, Bertinaria等[25]合成了系列具有NO释放能力的肌肽类似物NO供体 (Eq. 3).该反应可以一次引入两个NO供体基团, 能够增强NO的释放.经药理研究表明此类化合物对治疗NO缺少引起的慢性血管病和神经疾病具有较好的效果.
2014年, Biava等[26]通过酰化反应合成了一系列具有高度选择性的COX-2抑制剂 (Scheme 5).该反应通过酰胺化将羧基与带有NO供体的氨基 (Eq. 4) 直接进行偶合, 简化了反应历程.其中NO供体基团是由氨基醇经发烟硝酸处理后硝化得到.
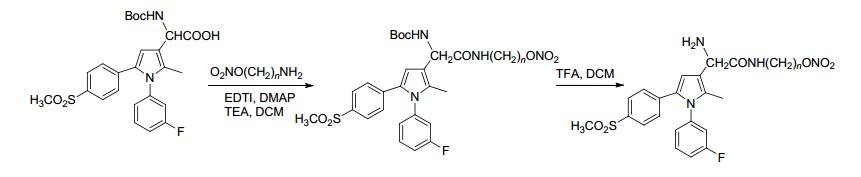 图 图式5
带有NO供体的COX-2抑制剂的合成路线
Figure 图式5.
Synthetic routes of COX-2 inhibitors bearing NO donor
图 图式5
带有NO供体的COX-2抑制剂的合成路线
Figure 图式5.
Synthetic routes of COX-2 inhibitors bearing NO donor
该方法得到的化合物通过药理实验证明具有很强的抗痛觉过敏和抗水肿作用.
1.3 取代反应
1.3.3 胺的取代
用发烟硝酸对溴代醇进行处理发生硝化反应, 得到溴代硝酸酯化合物.溴代硝酸酯与带有氨基的药物母体发生取代反应即可得到NO供体化合物 (Scheme 17).
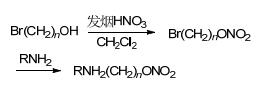 图 图式17
胺取代合成NO供体化合物
Figure 图式17.
Synthesis of NO donor compounds by substitution of amines
图 图式17
胺取代合成NO供体化合物
Figure 图式17.
Synthesis of NO donor compounds by substitution of amines
2008年, Fang等[45]合成了带有NO供体基团的抗老年痴呆药物他克林衍生物.该方法是先合成带有氨基基团的他克林衍生物, 再将用发烟硝酸硝化后的溴代烷通过取代反应连接上去.药理研究表明, 新合成的NO供体化合物与他克林相比具有较低的肝毒性.
2010年, Chowdhury等[46]合成了带有NO供体基团的塞来昔布类似物.此反应是先将塞来昔布的吡啶环进行还原, 然后再与NO供体基团通过取代反应获得具有双重效应的化合物.药理研究发现其不仅能释放NO而且具有和阿司匹林相当的抗炎效果.
2011年, Bertinaria等[47]合成了包含NO供体基团的治疗脑型疟疾的药物阿莫地喹类似物.该反应是先将中间体进行硝化, 再用苄基氯进行亲核取代反应获得带有NO供体基团的化合物.药理研究表明, 该化合物与原药相比具有更快更好的杀灭疟原虫的能力.
2016年, Zhao等[48]将硝酸酯NO供体连接到钌 (Ⅱ) 芳烃配合物.药理研究表明, 它们的组合对癌细胞具有阳性协同效应.与游离配体或非官能化复合物相比, 具有NO释放能力的4-硝基氧基甲基-吡啶配体的Ru (Ⅱ) 芳烃络合物显示出对非小细胞肺癌细胞系A549增加的细胞毒性.
综上所述, 通过酯键连接的NO供体药物可以在体内水解后发挥相连的两部分的各自功能. Zou等[30]采用的合成方案具有产率高、污染少、易于操作的优点.其反应过程是先与药物母核上的羟基成酯, 再用碱处理成酸, 得到的产物经溴代烷取代后进行硝化.此反应产率高且属于绿色化学, 是有机硝酸酯类NO供体药物合成的较好方案.另外, Tamboli等[38]采用引入溴丙烯基的合成方案实现了一次性引入两个有机硝酸酯类NO供体基团的可能, 对增强NO的体内释放有很好的效果.但此类药物也有缺点, 有机硝酸酯类药物的耐受性[49]大大限制了其在临床的应用.
1.3.2 羧酸的α-H取代
用浓硫酸和浓硝酸对溴代醇进行硝化得到溴代的NO供体基团 (Eq. 5), 再与母体化合物的羧基发生取代反应即可将母体化合物与NO供体基团连接 (Eq. 6).
2008年, Abdellatif等[41]合成了带有NO供体基团的丙烯酸衍生物.该方法是直接用2-溴乙基硝酸酯将丙烯酸衍生物的α位H取代获得带有NO供体基团的丙烯酸衍生物.药理研究表明, 该化合物不仅具有比原药更强的抗炎活性, 而且其胃肠道不良反应明显降低.
2012年, Kaur等[42]合成了带有NO供体基团的抗Ⅱ型糖尿病药物美各里替尼衍生物.该合成路线先合成了药物美各里替尼, 再与带有NO供体基团的溴代物发生羧基的α位H取代反应获得目标化合物.药理研究表明, 与原化合物相比该NO供体化合物具有更好的降血糖效果.可以用来治疗Ⅱ型糖尿病及其引起的血管并发症.
2015年, Xu等[43]合成了带有NO供体基团的乙丙昔罗衍生物.该合成路线是将溴代醇进行硝化处理, 再将NO供体基团通过羧基的α位H取代反应连接到乙丙昔罗骨架上获得NO供体化合物.该化合物具有较好的NO释放能力. Xu等还合成了以溴丙醇为起始原料的产物, NO释放研究表明其释放速率和缓释性能都会降低.
2016年, Gazzano等[44]合成了带有NO供体基团的多柔比星衍生物.该路线是将NO供体基团通过羧基的α位H取代反应连接到多柔比星骨架上获得NO供体化合物.药理研究表明, 连接两个NO供体基团的化合物比连接一个NO供体的化合物能够释放更多的NO.同时该类化合物还具有一定选择性, 只在对多柔比星敏感的肿瘤细胞中增加NO, 从而增强细胞毒性.
1.3.1 AgNO3取代
硝酸银与卤代烃亲核取代是目前NO供体制备的主要方法, 该方法是先在药物母体上创建卤代基团, 然后再与AgNO3发生亲核取代反应, 除去生成的卤化银沉淀即可得到带有硝酸酯类NO供体的药物 (Scheme 6).该方法具有广泛的应用范围.
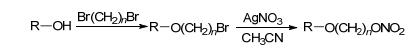 图 图式6
硝酸银法制备NO供体化合物的合成路线
Figure 图式6.
Synthetic routes of NO-donor compounds by silver nitrate process
图 图式6
硝酸银法制备NO供体化合物的合成路线
Figure 图式6.
Synthetic routes of NO-donor compounds by silver nitrate process
2009年, Liu等[27]合成了在肿瘤细胞定位释放的NO供体化合物.由于卟啉具有肿瘤细胞定位作用, 此类化合物的合成为靶向释放NO提供了重要的参考.此反应先利用1, 3-二溴丙烷将对羟基苯甲醛的羟基氢取代, 再用AgNO3处理进行亲核取代反应获得带有NO供体的中间体, 再进行卟啉的合成步骤即可合成带有NO供体的卟啉化合物 (Scheme 7).药理研究表明, 该化合物不仅具有与氟尿嘧啶类似的抗癌活性, 而且可以靶向释放.
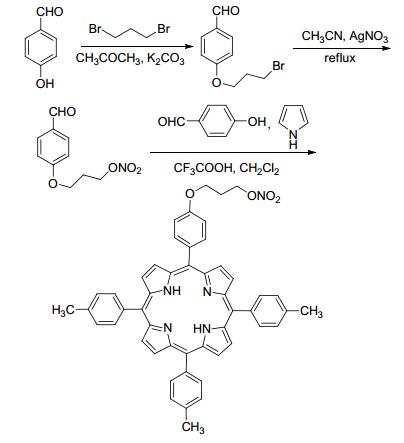 图 图式7
带有NO供体的卟啉化合物的合成路线
Figure 图式7.
Synthetic routes of porphyrins bearing NO donor
图 图式7
带有NO供体的卟啉化合物的合成路线
Figure 图式7.
Synthetic routes of porphyrins bearing NO donor
2009年, Peng等[28]合成了带有NO供体基团的白杨素衍生物.该方法先用二溴代烷进行亲核取代反应将溴代烷基连接到白杨素衍生物骨架上, 再用硝酸银进行亲核取代获得带有NO供体基团的白杨素衍生物.药理研究表明, 该化合物不仅具有促进细胞增殖的活性, 而且具有明显的促进血管再生的作用.
2010年, Dong等[29]合成了带有NO供体基团的查尔酮衍生物, 查尔酮在碳酸钾存在的丙酮溶剂中和1, 2-二溴乙烷发生亲核取代反应生成醚键, 从而使NO供体基团与查尔酮连接起来.应用此法合成NO供体化合物, 不仅操作简单且适用于大多数带有酚羟基的天然产物.应用此方法还可以在多位点同时引入多个NO供体基团.药理研究表明该化合物与原药相比具有更好的血管舒张效应, 有希望成为新的血管舒张剂.
2010年, Zou等[30]合成了系列带有NO供体基团的白杨素衍生物.用二溴代烷在7位羟基进行亲核取代反应, 再用硝酸银取代获得目标产物.药理实验表明该法合成的化合物其NO释放速率比带有羰基的产物要慢, 但其醛糖还原酶抑制性能减弱.
2012年, Bai等[31]合成了带有NO供体基团的异色满衍生物.该合成路线是将异色满的甲氧基取代物进行选择性去甲氧基反应, 再与二溴代烷进行亲核取代反应后与硝酸银发生亲核取代反应获得带有NO供体基团的异色满衍生物.药理研究表明, 与卡托普利相比该化合物具有更好的降压效果.
2013年, Wang等[32]合成了带有NO供体基团的木犀草素衍生物. NO的引入增强了原药的醛糖还原酶抑制活性, 为开发预防和治疗糖尿病并发症药物提供了参考. Wang等设计了3条合成途径.
第一种途径是先将木犀草素用二溴乙烷处理, 用二溴代烷进行亲核取代后与硝酸银发生亲核取代反应获得带有NO供体基团的木犀草素衍生物.此类产物在三条路线得到的产物中醛糖还原酶抑制活性最强.
第二条合成途径是将木犀草素先用二氯二苯基甲烷处理, 将邻二酚羟基保护起来, 然后进行二溴代烷的亲核取代反应, 再与硝酸银发生亲核取代反应, 最后再将保护基团去除.获得带有邻二酚羟基的NO供体化合物.
第三条合成途径是将木犀草素用二溴乙烷与邻二酚羟基反应成环, 再用溴乙酸乙酯进行亲核取代, 用碱进行水解处理得到带羧基的产物, 再用二溴代烷进行亲核取代, 然后与硝酸银发生亲核取代反应获得酯键连接的NO供体化合物.该方法的优点在于引入了酯键, 获得NO供体前体药物.使母体药物与NO供体基团在体内水解后可分别发挥药理作用.
2010年, Zou等[30]合成了系列带有NO供体基团的白杨素衍生物.先用溴乙酸乙酯进行亲核取代, 用碱水解成酸后与溴代烃发生酯化反应, 最后用硝酸银取代获得具有NO释放能力的白杨素衍生物 (Scheme 8).该方法运用前体药物理念, 在母体结构与NO供体基团之间引入酯键连接, 使药物进入体内水解后发挥协同作用.药理研究表明, 该化合物具有更好的降糖作用.
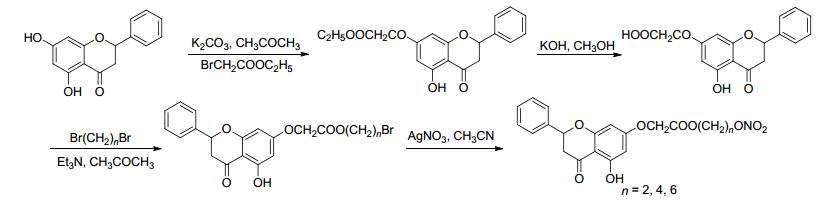 图 图式8
带有NO供体的白杨素衍生物的合成路线1
Figure 图式8.
Synthetic routes 1 of chrysin derivatives bearing NO donor
图 图式8
带有NO供体的白杨素衍生物的合成路线1
Figure 图式8.
Synthetic routes 1 of chrysin derivatives bearing NO donor
2009年, Bhandari等[33]合成了一系列具有NO释放功能的降压药.与大多数有机硝酸酯类化合物的合成一样, Bhandari等先合成了具有降血压作用的化合物骨架, 然后用AgNO3进行亲核取代反应获得有机硝酸酯类化合物 (Scheme 9).药理研究表明, 该化合物不仅具有原药的降压作用, 而且还具有抗心律失常作用.
 图 图式9
羧基连接NO供体化合物的合成路线
Figure 图式9.
Synthetic routes of antihypertensive agents bearing NO donor
图 图式9
羧基连接NO供体化合物的合成路线
Figure 图式9.
Synthetic routes of antihypertensive agents bearing NO donor
同年, Bhandari等[34]合成了具有NO供体基团的非甾体类抗炎药, 该反应是以丙二酸二乙酯为原料经过系列反应合成溴代药物母体, 再用硝酸银进行亲核取代反应获得具有NO供体基团的非甾体类抗炎药.在药理活性方面发现其具有很强的抗炎作用, 同时病理组织研究发现该化合物具有强抗溃疡作用和粘膜保护作用.这种合成途径是将来合成没有副作用的非甾体类抗炎药的重要手段.
2012年, Mourad等[35]合成了兼具抗癌活性和NO释放能力的查尔酮类化合物.该路线先通过羟醛缩合反应获得查尔酮类化合物骨架, 然后用溴乙酰溴进行亲核取代, 最后用硝酸银进行亲核取代反应获得目标化合物 (Scheme 10).药理研究表明, 该化合物具有明显的抗癌活性, 而且对结肠癌和黑素瘤具有一定的选择性.
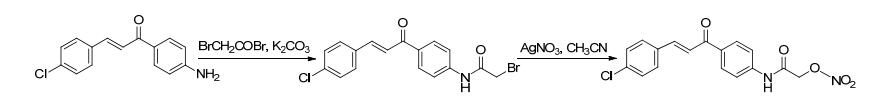 图 图式10
带有NO供体的查尔酮衍生物的合成路线
Figure 图式10.
Synthetic routes of chalcone derivatives bearing NO donor
图 图式10
带有NO供体的查尔酮衍生物的合成路线
Figure 图式10.
Synthetic routes of chalcone derivatives bearing NO donor
2012年, Abuo-Rahma等[18]合成了一系列新的带有NO供体基团的查尔酮衍生物.该方法先进行亲核取代反应, 再用硝酸银进行亲核取代反应获得NO供体化合物 (Scheme 11). NO释放研究表明该类NO供体化合物具有缓释效果.药理研究表明, 该化合物与原药相比具有更好的抗炎活性, 且明显降低了原药的副作用, 其致胃肠道溃疡作用明显降低.
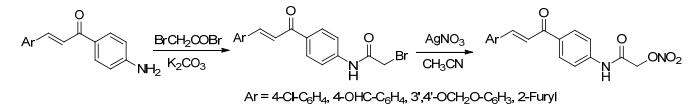 图 图式11
带有NO供体的查尔酮衍生物的合成路线
Figure 图式11.
Synthetic routes of chalcone derivatives bearing NO donor
图 图式11
带有NO供体的查尔酮衍生物的合成路线
Figure 图式11.
Synthetic routes of chalcone derivatives bearing NO donor
Abuo-Rahma等[18]还合成了一组甲基化的NO供体查尔酮衍生物 (Scheme 12).研究表明, 带有甲基的NO供体化合物的NO释放速率和持续时间都不如没有甲基取代的NO供体化合物.认为是由空间效应导致.
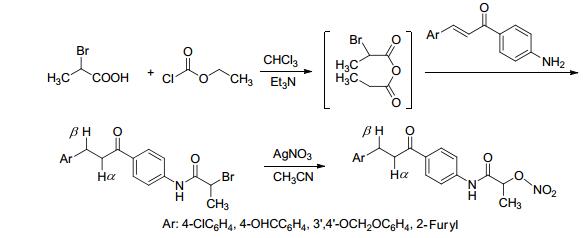 图 图式12
甲基化的NO供体查尔酮衍生物的合成路线
Figure 图式12.
Synthetic routes of methylated chalcone derivatives bearing NO donor
图 图式12
甲基化的NO供体查尔酮衍生物的合成路线
Figure 图式12.
Synthetic routes of methylated chalcone derivatives bearing NO donor
2016年, Fang等[36]制备了一种齐墩果酸-NO供体-铂 (Ⅱ) 三杂交分子.该方法先与溴代酰氯进行亲核取代反应, 再用硝酸银进行亲核取代反应得到NO供体化合物, 最后与铂进行连接获得齐墩果酸-NO供体-铂 (Ⅱ) 三杂交分子.药理研究表明, 该分子有对肝癌细胞的靶向细胞毒性, 具有协同作用模式和良好的安全性.
2009年, Zhang等[37]合成了系列带有NO供体基团的抗癌药物厄贝沙坦类似物.该反应先用二溴代烷进行亲核取代, 接着用硝酸银进行亲核取代反应而获得带有NO供体基团的厄贝沙坦类似物 (Scheme 13).药理研究表明该化合物具有较强的细胞毒性, 且NO供体部分对抗癌活性具有增强作用.
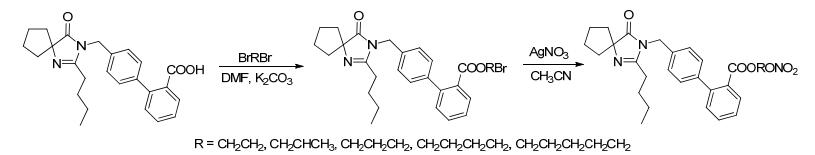 图 图式13
带有NO供体的厄贝沙坦衍生物的合成路线
Figure 图式13.
Synthetic routes of irbesartan derivatives bearing NO donor
图 图式13
带有NO供体的厄贝沙坦衍生物的合成路线
Figure 图式13.
Synthetic routes of irbesartan derivatives bearing NO donor
2012年, Tamboli等[38]在降糖药甲苯磺丁脲类似物的结构上添加NO供体获得了具有血管舒张活性和抗血栓能力的新化合物.该路线利用丙烯基的双键, 一次添加两个NO供体基团 (Scheme 14).此法不仅增加了NO的释放还节省了反应过程.药理研究表明, 该化合物具有促胰岛素释放效应、血管舒张效应和抗聚活性.
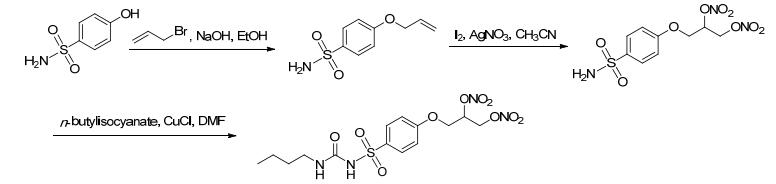 图 图式14
带有NO供体的甲苯磺丁脲类似物的合成路线
Figure 图式14.
Synthetic routes of tolbutamide analogue bearing NO donor
图 图式14
带有NO供体的甲苯磺丁脲类似物的合成路线
Figure 图式14.
Synthetic routes of tolbutamide analogue bearing NO donor
2013年, Kutty等[39]合成了带有NO供体基团的卤代呋喃酮衍生物.卤代呋喃酮类化合物是一种从海洋生物红藻中产生的天然抗菌剂.该方法是在烷烃链上用N-溴代丁二酰亚胺 (NBS) 来进行溴化, 再用硝酸银进行亲核取代反应得到带有NO供体的目标化合物 (Scheme 15).药理研究表明, NO的引入增强了其抗菌活性.
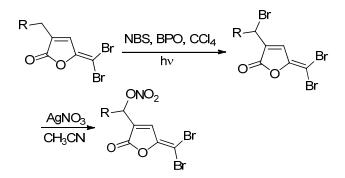 图 图式15
带有NO供体的卤代呋喃酮衍生物的合成路线
Figure 图式15.
Synthetic routes of fimbrolide derivatives bearing NO donor
图 图式15
带有NO供体的卤代呋喃酮衍生物的合成路线
Figure 图式15.
Synthetic routes of fimbrolide derivatives bearing NO donor
2015年, Huang等[40]合成了带有NO供体基团的抗青光眼药物布林佐胺衍生物.该方法先用三溴化硼进行脱甲基反应, 用氢溴酸水溶液进行亲核取代, 再与硝酸银发生亲核取代反应获得带有NO供体基团的布林佐胺衍生物 (Scheme 16).药理研究表明, 该化合物与原药布林佐胺相比具有更好降低眼内压作用.
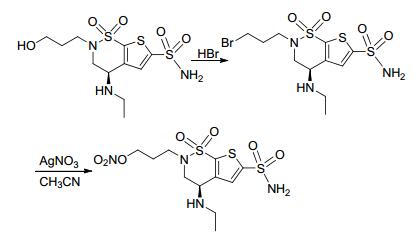 图 图式16
带有NO供体的布林佐胺衍生物的合成路线
Figure 图式16.
Synthetic routes of brinzolamide derivatives bearing NO donor
图 图式16
带有NO供体的布林佐胺衍生物的合成路线
Figure 图式16.
Synthetic routes of brinzolamide derivatives bearing NO donor
2 呋咱氮氧化物类NO供体的合成
呋咱氮氧化物类化合物对酸碱均较稳定, 环上的3、4位可连接相同或不同的取代基, 也可以与其他环骈合而形成不同的衍生物.这类化合物在进入体内后会在巯基化合物作用下释放NO.如Scheme 18所示, 呋咱氮氧化物类化合物在体内先经巯基化合物对其3或4位进行亲核加成获得加成产物 (1或4), 再经开环反应形成亚硝基衍生物 (2或5), 并进一步生成另一亚硝基衍生物 (3或6) 和亚硝酰基阴离子 (NO-).其中亚硝基衍生物可作为终产物排出体外, 也可继续水解或硫解, 还可与硫醇作用, 生成S-亚硝基硫醇, 然后裂解生成NO.而亚硝酰基阴离子经氧化形成NO释放[50].
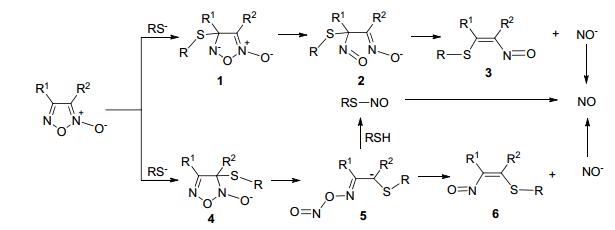 图 图式18
呋咱氮氧化物类化合物体内释放NO的机制
Figure 图式18.
Mechanism of releasing NO from furoxans in vivo
图 图式18
呋咱氮氧化物类化合物体内释放NO的机制
Figure 图式18.
Mechanism of releasing NO from furoxans in vivo
经典的有机硝酸酯类NO供体化合物需在游离巯基作用下才能释放NO, 而呋咱氮氧化物类NO供体化合物在游离巯基和巯基阴离子作用下均可释放NO.因为体内游离巯基分布很广, 所以呋咱氮氧化物类NO供体化合物可在多种组织或器官中通过非酶催化途径释放NO而产生一定的生物活性.不诱导产生耐受性也是呋咱氮氧化物类NO供体化合物的优点, 典型的呋咱氮氧化物类化合物包括苯基取代、苯磺酰基取代和甲基取代的呋咱氮氧化物等.
2.1 酯化反应
以肉桂醇为原料, 在酸性条件下加入亚硝酸钠盐反应得呋咱氮氧化物类NO供体基团3-羟甲基-4-苯基-1, 2, 5-噁二唑-2-氧化物.此化合物与带有羧基的药物分子进行酯化反应得NO供体化合物 (Scheme 19).
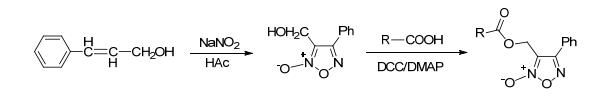 图 图式19
酯化反应制备NO供体化合物
Figure 图式19.
Synthesis of NO donor compound by esterification
图 图式19
酯化反应制备NO供体化合物
Figure 图式19.
Synthesis of NO donor compound by esterification
2007年, López等[20]合成了带有NO供体基团的α-生育酚类似物.该路线采用成酯反应将NO供体基团通过酯键连接到水溶性维生素E结构上 (Eq. 7).该方法合成的化合物在体内经酶水解后的产物可各自产生相应的活性.药理研究表明该化合物具有NO释放效应, 血管舒张效应和哺乳动物细胞毒性效应.
2008年, Chen等[51]合成了带有NO供体基团的齐墩果酸衍生物.该合成路线是将齐墩果酸与丁二酸酐进行成酯反应, 得到的产物再与带有羟基的NO供体基团进行酯化反应, 获得了带有NO供体基团的齐墩果酸衍生物.药理研究表明, 该化合物在体内能够在肝癌细胞中高水平释放, 而且对肝癌细胞具有高度选择性的细胞毒性.
2009年, Peng等[28]合成了带有NO供体基团的白杨素衍生物.该路线用白杨素衍生物对溴乙酸乙酯进行亲核取代反应, 碱处理得到带有羧基的中间产物, 再与以肉桂醇为原料合成的NO供体基团进行成酯反应获得带有NO供体基团的白杨素衍生物.鸡胚绒毛尿囊膜试验表明该化合物显著的促进血管生成作用.
2009年, Zhang等[37]合成了系列带有NO供体基团的厄贝沙坦衍生物.该方法是先以苯硫酚为原料合成带有不同取代基的NO供体基团, 再与厄贝沙坦进行偶合获得带有NO供体基团的厄贝沙坦衍生物.药理研究表明该化合物有较好的细胞毒性活性, 引入NO供体基团对抗癌活性有增强作用.
2010年, Ling等[52]合成了带有NO供体基团的法尼基硫代水杨酸 (FTA) 衍生物.该路线以苯硫酚为原料合成NO供体基团, 再与L-丙氨酸衍生物结合, 最后与苯甲酸衍生物进行偶合获得NO供体化合物.药理研究表明, 该化合物不仅在体内具有较高水平的NO释放性能, 而且还具有很好的抗癌活性.
2010年, Zou等[30]合成了系列带有呋咱氮氧化物类NO供体基团的白杨素衍生物.药理研究表明, 该化合物对醛糖还原酶有抑制作用且能够释放NO.该化合物对预防和治疗糖尿病及其引发的血管并发症有一定效果. Zou等设计了两条合成路线.
路线1是先将白杨素的7位羟基用溴乙酸乙酯进行取代, 然后用碱处理得到羧酸产物, 再跟与对羟基苯甲醇相连的NO供体基团进行成酯反应, 最后再用酸酐进行缩合反应获得带有NO供体基团的白杨素衍生物 (Scheme 20).
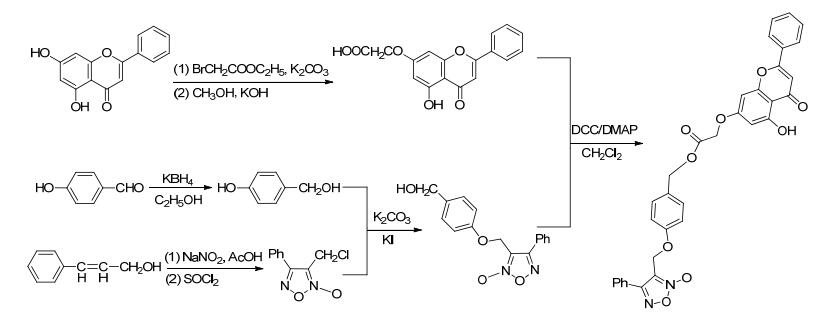 图 图式20
带有NO供体的白杨素衍生物的合成路线
Figure 图式20.
Synthetic route of chrysin derivatives bearing NO donor
图 图式20
带有NO供体的白杨素衍生物的合成路线
Figure 图式20.
Synthetic route of chrysin derivatives bearing NO donor
路线2采用的方法是用白杨素的羧酸衍生物与带有羟基的NO供体基团直接通过酯键连接起来, 然后再用酸酐进行缩合反应获得带有NO供体基团的白杨素衍生物 (Eq. 8). NO释放研究表明, NO释放均比较缓慢, 但路线2得到的化合物释放更慢.
2011年, Shi等[53]合成了系列带有NO供体基团的核苷衍生物.该方法利用成酯反应将NO供体基团与核苷衍生物连接.该化合物在体内经水解后各部分可发挥各自活性.经药理研究表明, 带有NO供体基团的核苷衍生物比药物阿昔洛韦和司他夫定有更好的抗病毒活性 (Scheme 21).
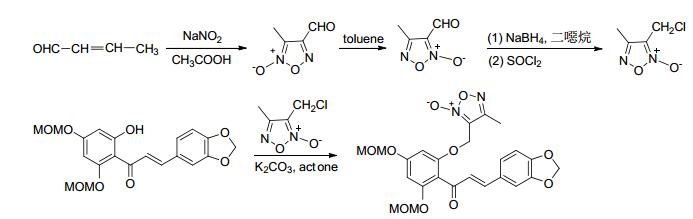 图 图式21
带有NO供体的查尔酮衍生物的合成路线
Figure 图式21.
Synthetic routes of chalcone derivative bearing NO donor
图 图式21
带有NO供体的查尔酮衍生物的合成路线
Figure 图式21.
Synthetic routes of chalcone derivative bearing NO donor
2012年, Mourad等[35]合成了系列带有NO供体基团的查尔酮衍生物.该方法是先合成查尔酮化合物骨架, 再与呋咱氮氧化物类NO供体基团经亲核取代反应连接起来.药理研究表明, 该化合物对不同的癌细胞都具有细胞毒性活性, 但其活性不如有机硝酸酯类NO供体药物显著.
2012年, Abuo-Rahma等[18]合成了带有NO供体基团的查尔酮衍生物.该路线是先用4-氯苯甲醛和对氨基苯乙酮经过羟醛缩合反应合成查尔酮化合物骨架, 再与溴乙酰溴进行偶联, 得到的产物再与NO供体基团结合获得NO供体化合物.药理研究表明, 该化合物与查尔酮相比其抗炎活性增强, 而且该化合物还降低了查尔酮的致溃疡副作用.
2014年, Tang等[54]合成了带有NO供体基团的鸦胆子苦醇衍生物.药理研究表明, 该化合物对吸烟引起的慢性肺炎具有抑制作用.该路线利用加入的丁二酸酐作为NO供体基团与鸦胆子苦醇的连接基团, 利用酯键连接可使化合物在体内水解后的产物发挥各自效应.药理研究表明, 该化合物具有较好的抗炎作用, 且由于NO供体基团的引入, 其毒性也大幅降低.
2.2 取代反应
2010年, Dong等[29]合成了系列带有NO供体基团的查尔酮衍生物.该方法是将2-丁烯醛经亚硝酸钠处理后成氧化呋咱环, 在沸腾的甲苯中加热98 h后转化成其同分异构体, 再进行还原和卤代, 然后与查尔酮化合物进行偶联, 最后进行脱二甲醚反应得到带有NO供体基团的查尔酮衍生物 (Scheme 21).该方法特点在于用甲苯对NO供体进行了同分异构转化.药理活性研究表明, 该化合物由于NO供体基团的引入使其血管舒张性能比原化合物更强, 有希望成为新的血管舒张剂.
2011年, Bertinaria等[47]合成了系列带有NO供体基团的抗脑型疟疾药物阿莫地喹类似物.此方法利用苯磺酰基在亲核取代反应中易离去的特性使NO供体基团与母体药物更易结合.该方法是将呋咱氮氧化物类NO供体基团与乙基羟乙胺进行偶联, 得到的中间产物再与阿莫地喹类似物进行偶联得到带有NO供体基团的抗脑型疟疾药物阿莫地喹类似物.药理研究表明, 得到的NO供体化合物具有阿莫地喹没有的血管舒张活性.
2012年, Tamboli等[38]合成了一系列带有NO供体基团的降糖药物甲苯磺丁脲的类似物.该方法是先将NO供体基团和4-磺酰氨基苯酚偶联, 再与异氰酸丁酯偶联获得带有NO供体的甲苯磺丁脲的类似物 (Eq. 9).药理研究表明, 与呋咱氮氧化物类NO供体基团偶合的产物比与有机硝酸酯类NO供体基团偶合产物的促胰岛素释放活性更高.
2012年, Dos Santos等[55]利用酸酐的氨解合成了带有NO供体基团的苯邻二甲酰亚胺衍生物.该反应是用邻苯二甲酸酐和呋咱氮氧化物类NO供体基团通过酰化反应得到NO供体化合物 (Eq. 10).药理研究表明, 该化合物具有多因子抗氧化活性, 且具有阻止缺血再灌注损伤的作用.
2016年, Gazzano等[44]合成了带有NO供体基团的多柔比星衍生物.该路线是将NO供体基团通过羧基的α位H取代反应连接到多柔比星骨架上获得NO供体化合物 (Eq. 11).药理研究表明, 该化合物的NO释放能力与连接两个硝酸酯N O供体基团的化合物相当.
2015年, Bertinaria等[56]合成了系列带有NO供体基团的二氢青蒿素衍生物.该路线是将NO供体基团通过取代反应连接到二氢青蒿素骨架上获得NO供体化合物 (Scheme 22).药理研究表明, 该类化合物在体外和体内对抗伯氏疟原虫实验中显示出较好活性, 而且在大鼠主动脉条的血管舒张效力中其活性比呋咱氮氧化物类NO供体强.
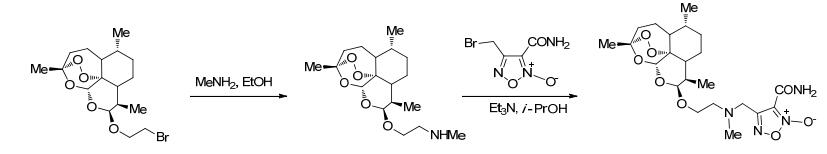 图 图式22
带有NO供体的二氢青蒿素衍生物的合成路线
Figure 图式22.
Synthetic routes of dihydroartemisinin derivative bearing NO donor
图 图式22
带有NO供体的二氢青蒿素衍生物的合成路线
Figure 图式22.
Synthetic routes of dihydroartemisinin derivative bearing NO donor
2.3 成肽反应
2011年, Bian等[57]合成了带有NO供体基团的抗白血病药物他米巴罗汀衍生物.该方法是先合成了氯取代的NO供体基团, 再通过成肽反应将NO供体和药物他米巴罗汀连接 (Eq. 12).药理研究表明, R为氢或苄基取代时, 得到的NO供体化合物抗白血病活性要比原药更强.同时NO释放研究表明, 带有氨基酸的NO供体化合物其释放率更高, 而且其抗白血病活性也是一致增高的.
2013年, Borretto等[58]合成了带有NO供体基团的恩替诺特衍生物.该合成路线是用叔丁基二甲基硅烷基保护的5-(羟甲基) 吡啶-2-甲醛由甲胺和硼氢化钠处理进行还原性胺化得到中间体, 再用4-溴甲基氧化呋咱甲酰胺处理得到氧化呋咱中间体, 经乙酸处理后与中间体8经成肽反应得到氧化呋咱羧酰胺衍生物9, 这一产物经三氟乙酸酐脱水处理后得到相关的腈, 随后用甲磺酸将其去保护, 得到目标化合物 (Eq. 13).
2013年, Lu等[59]合成了带有NO供体基团的羟基肉桂酸衍生物.该路线是先合成带有氨基的NO供体基团, 与乙酸酐保护的羟基肉桂酸衍生物通过成肽反应连接, 最后脱去羟基保护基团获得带有NO供体基团的羟基肉桂酸衍生物 (Scheme 23).药理研究发现, 该化合物具有比5-氟尿嘧啶更强的抗癌活性.
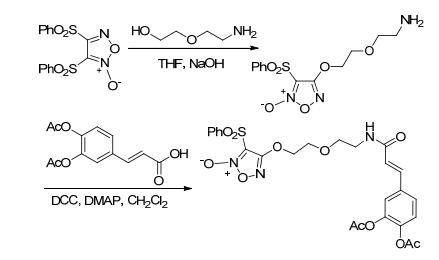 图 图式23
带有NO供体的羟基肉桂酸衍生物的合成路线
Figure 图式23.
Synthetic routes of hydroxylcinnamic acid derivative bearing NO donor
图 图式23
带有NO供体的羟基肉桂酸衍生物的合成路线
Figure 图式23.
Synthetic routes of hydroxylcinnamic acid derivative bearing NO donor
2015年, Ai等[60]合成了带有NO供体基团的齐墩果酸衍生物.该路线是先将NO供体基团与2-丁炔-1, 4-丁二醇连接, 再在碳化二亚胺等作用下形成肽键, 最后与酰氯化的齐墩果酸衍生物偶合获得NO供体基团的齐墩果酸衍生物 (Scheme 24).药理研究表明, 由于NO供体基团的加入使该化合物治疗耐药性结肠癌有较好的疗效.
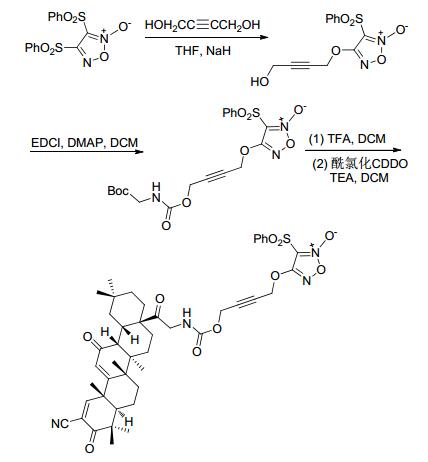 图 图式24
带有NO供体的齐墩果酸衍生物的合成路线
Figure 图式24.
Synthetic routes of oleanolic acid derivative bearing NO donor
图 图式24
带有NO供体的齐墩果酸衍生物的合成路线
Figure 图式24.
Synthetic routes of oleanolic acid derivative bearing NO donor
2015年, Duan等[61]合成了系列带有NO供体基团的组蛋白去乙酰化酶抑制剂衍生物.该路线是以2-苯硫基乙酸为原料合成NO供体基团, 再经二羟基化合物取代, 经琼斯试剂氧化成羧酸, 经成肽反应后得到带有NO供体基团的组蛋白去乙酰化酶抑制剂衍生物 (Eq. 14).药理研究表明, 该化合物具有抗白血病细胞增殖的作用, 其抗癌活性与NO的体内释放有关.
2.4 醛胺缩合
2014年, Massarico Serafim等[62]合成了带有NO供体基团的酰基腙衍生物.该方法是先用反式肉桂酸合成呋咱氮氧化物类NO供体基团, 再与酰肼类化合物进行醛胺缩合生成酰腙获得NO供体化合物 (Scheme 25).药理研究表明, 这类化合物具有很强的抗锥虫活性, 与原药相比具有更高的渗透性和更好的选择性.
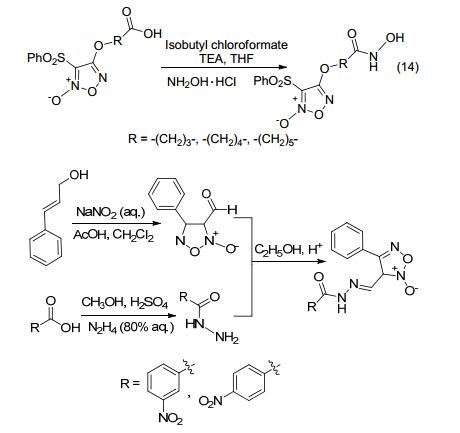 图 图式25
带有NO供体的酰基腙衍生物的合成路线
Figure 图式25.
Synthetic routes of acylhydrazone derivatives bearing NO donor
图 图式25
带有NO供体的酰基腙衍生物的合成路线
Figure 图式25.
Synthetic routes of acylhydrazone derivatives bearing NO donor
2016年, Lakshman等[63]制备了带有NO供体基团的锌 (Ⅱ)-NSAID络合物.该方法是将NO供体连接到锌 (Ⅱ) 后再与非甾体类抗炎药形成NO供体络合物.药理研究表明, 得到的络合物不仅具有较好的抗炎活性而且还具有COX-2选择抑制活性.该方法是将非选择性或COX-1选择性NSAID转化为选择性COX-2抑制剂的代表途径.
综上所述, Peng等[28]采用的合成方法具有产率高、无污染、操作简单等优点.其采用的合成路线是先以肉桂醇为原料合成了带有羟基的呋咱氮氧化物类NO供体基团, 然后再与带有羧基的查尔酮衍生物进行成酯反应得到呋咱氮氧化物类NO供体药物.该合成路线简便安全, 绿色环保, 是很好的合成NO供体药物的方法.
3 偶氮二醇烯鎓盐类NO供体的合成
偶氮二醇烯鎓盐一般可由哌嗪、吡咯、烷基胺或哌啶与NO和甲醇钠的甲醇溶液在NO吸附剂 (如纳米二氧化钛) 催化下反应生成, 代表药物有JS-K和DETANONOate.该类化合物的生物半衰期可通过引入取代基来灵活调节[64], 2个N-O上烷基化取代可使其半衰期从几分钟到几小时不等, 且某些与特定基团结合的偶氮二醇烯鎓盐化合物可在特定部位释放出定量的NO, 所以特别适合偶联特定载体制成靶向释放的NO供体药物[65].释放时, 在体内经过生物酶的催化形成氧负离子, 氧负离子在体内环境下不稳定, 极易释放出NO.
3.1 NO气体制备
2008年, Chakrapani等[66]合成了具有抗癌活性的JS-K类似物.该合成路线是用脯氨醇为原料合成偶氮二醇烯鎓盐化合物, 再用2, 4-二硝基氟苯和叔丁醇在碳酸氢钠处理下合成目标化合物10 (Scheme 26).药理研究表明, 该类NO供体基团的引入并未对此药物的抗癌活性产生影响.
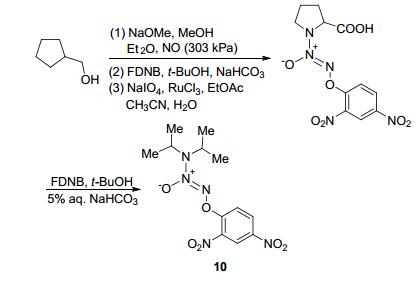 图 图式26
JS-K类似物的合成路线
Figure 图式26.
Synthetic routes of JS-K analogue
图 图式26
JS-K类似物的合成路线
Figure 图式26.
Synthetic routes of JS-K analogue
2008年, Fang等[45]合成了带有NO供体基团的抗老年痴呆药物他克林衍生物.该合成路线是先用吡咯烷与NO气体反应合成NO供体基团, 以邻氨基苯甲酸和环己酮为原料经过一系列反应合成溴代他克林衍生物, 此衍生物再与NO供体进行偶合获得带有NO供体基团的他克林衍生物 (Eq. 15).在肝中毒研究中发现, 该化合物并不会像药物他克林一样引起严重的肝中毒.
2008年, Konter等[24]合成了带有NO供体基团的酮康唑衍生物.该合成路线是将酮康唑化合物经NaOH水溶液水解后与NO气体在甲醇钠中反应生成带有偶氮二醇烯鎓盐类NO供体基团的酮康唑衍生物 (Scheme 27).药理研究发现该化合物的抗菌活性明显比原药酮康唑更强, 而单独的NO供体则没有这种效果.
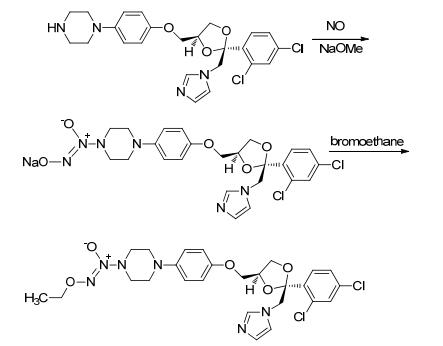 图 图式27
带有NO供体的酮康唑的合成路线
Figure 图式27.
Synthetic routes of ketoconazole bearing NO donor
图 图式27
带有NO供体的酮康唑的合成路线
Figure 图式27.
Synthetic routes of ketoconazole bearing NO donor
2008年, Chowdhury等[67]合成了带有NO供体基团的塞来昔布类似物.该合成路线是先合成了塞来昔布类似物, 再经过系列反应后与NO气体反应获得带有NO供体基团的塞来昔布类似物 (Eq. 16).药理研究表明该产物的抗炎活性比药物阿司匹林强, 但比药物塞来昔布弱.
2014年, Abdellatif等[68]合成了带有NO供体基团的塞来昔布类似物.该合成路线是以溴代塞来昔布类似物为原料与乙胺反应, 得到的产物与NO气体在四氢呋喃和甲醇钠作用下生成带有NO供体基团的塞来昔布类似物 (Scheme 28).药理研究发现与原药相比, NO供体基团的引入使其抗炎活性增强.
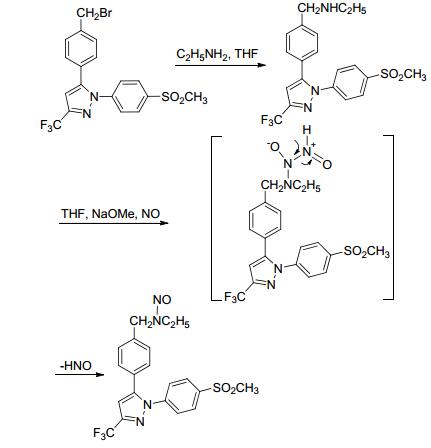 图 图式28
带有NO供体的塞来昔布类似物的合成路线
Figure 图式28.
Synthetic routes of celecoxib analogue bearing NO donor
图 图式28
带有NO供体的塞来昔布类似物的合成路线
Figure 图式28.
Synthetic routes of celecoxib analogue bearing NO donor
3.2 NH3气体制备
2010年, Abdellatif等[69]合成了带有NO供体基团的塞来昔布衍生物.该合成路线是用二酮化合物11与4-甲基磺酰基苯基肼盐酸盐 (12a) 或4-氨磺酰基苯基肼盐酸盐 (12b) 反应生成相应的吡唑产物13a或13b.将带有硝基的吡唑衍生物进行经还原得到N-羟氨基衍生物, 经NH3气体和亚硝酸丁酯处理后再与溴乙酸乙酯反应得到带有NO供体基团的塞来昔布衍生物 (Scheme 29).药理研究表明, 该化合物不仅具有抗炎活性, 而且还可以降低非甾体抗炎药引起的副作用, 如胃刺激, 高血压及血小板凝集.
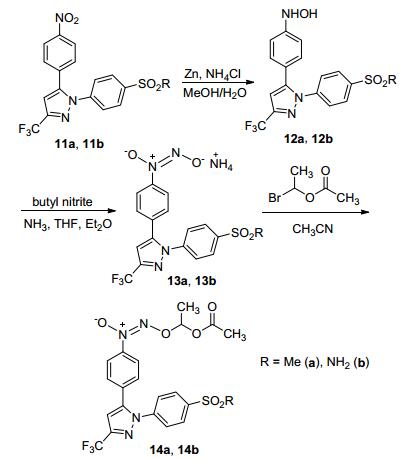 图 图式29
带有NO供体的塞来昔布衍生物的合成路线
Figure 图式29.
Synthetic routes of celecoxib derivatives bearing NO donor
图 图式29
带有NO供体的塞来昔布衍生物的合成路线
Figure 图式29.
Synthetic routes of celecoxib derivatives bearing NO donor
3.3 羧酸的α-H取代
2008年, Abdellatif等[41]合成了带有NO供体基团的丙烯酸衍生物.该方法是直接将丙烯酸衍生物与偶氮二醇烯鎓盐偶合获得带有NO供体基团的丙烯酸衍生物 (Eq. 17).药理研究表明, 该化合物不仅具有良好的稳定性, 而且其胃肠道不良反应明显降低.
Abdellatif等[70]还用同样方法合成了带有NO供体基团的塞来昔布类似物.该合成路线以苯乙酮为原料合成塞来昔布类似物, 再与偶氮二醇烯鎓盐类NO供体基团通过酯键连接.药理研究表明该化合物在鼠血清中有较好的NO释放能力, 但抗炎活性消失.
2008年, Abdellatif等[71]合成了带有NO供体基团的塞来昔布类似物.该路线是用三氟甲苯丁二酮和4-肼基苯磺酰胺合成塞来昔布, 然后进行溴代, 得到的产物再与偶氮二醇烯鎓盐类NO供体基团结合获得带有NO供体基团的塞来昔布类似物 (Eq. 18).药理研究表明, 该化合物具有较好的NO释放能力, 而且是高度选择性的COX-2抑制剂. 2011年, Abdellatif等[72]用此方法合成了带有NO供体基团的罗非昔布衍生物.该合成路线是用2-溴-1-(4-甲磺酰基) 苯乙酮与对甲基苯乙酸合成罗非昔布类似物, 产物经过溴代反应后与NO供体基团发生成酯反应获得带有NO供体基团的罗非昔布衍生物, 药理研究表明, 该化合物可以选择性地抑制COX-2从而产生抗炎作用, 而且还可以显著降低非甾体类抗炎药引起的心血管不良反应.
2008年, Velázquez等[73]合成了带有NO供体基团的阿司匹林衍生物.该合成路线是以吡咯烷-2-甲醇为原料合成偶氮二醇烯鎓盐类NO供体, 再与氯代的阿司匹林衍生物偶合获得带有NO供体基团的阿司匹林衍生物 (Eq. 19).药理研究表明, 由于NO供体基团的引入使该化合物的抗炎活性高于原药阿司匹林.
2012年, Kaur等[42]合成了带有NO供体基团的美各里替尼衍生物.该合成路线利用四氢吡咯为原料合成偶氮二醇烯鎓盐NO供体基团, 以5-氯-2-甲氧基苯甲酸为原料合成药物美各里替尼, 再通过酰胺化将两者连接获得带有NO供体基团的美各里替尼衍生物 (Scheme 30). NO释放研究表明该化合物具有较好的NO释放效应.该化合物在治疗糖尿病的同时降低了心血管发生病变的风险.
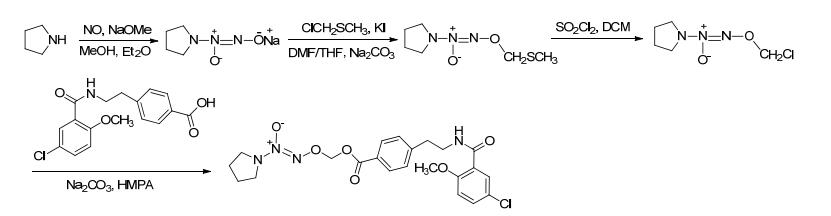 图 图式30
带有NO供体的美各里替尼衍生物的合成路线
Figure 图式30.
Synthetic routes of meglitinide derivative bearing NO donor
图 图式30
带有NO供体的美各里替尼衍生物的合成路线
Figure 图式30.
Synthetic routes of meglitinide derivative bearing NO donor
2015年, Xu等[43]合成了带有NO供体基团的乙丙昔罗衍生物.该反应是用卤代的NO供体基团和乙丙昔罗在Cs2CO3存在的DMF中发生羧基的α位H取代反应获得NO供体化合物 (Eq. 20). NO释放研究发现, 该NO供体药物在体内能够比有机硝酸酯类NO供体更有效地释放NO, 且不会产生药物耐受性.
2016年, Gazzano等[44]合成了带有NO供体基团的多柔比星衍生物.该路线是将NO供体基团通过羧基的α位H取代反应连接到多柔比星骨架上获得NO供体化合物.药理研究表明, 该化合物的NO释放能力与连接两个硝酸酯NO供体基团的化合物相当.
2016年, Xu等[74]合成了带有NO供体的卵清素衍生物.该路线是将NO供体基团通过羧基的α位H取代反应连接到卵清素骨架上获得NO供体化合物.药理研究表明, 所有目标化合物显示出有效的抗增殖活性, 而且具有卵清素不具备的诱导细胞凋亡并阻止Bel-7402细胞S期的细胞周期的活性.
3.4 酯化反应
2009年, Abdellatif等[17, 75]用酯化反应合成了一系列带有NO供体基团的非甾体类抗炎药.该合成路线是将非甾体类抗炎药 (阿司匹林、吲哚美辛、塞来昔布和布洛芬) 经过系列反应生成酰氯, 再与偶氮二醇烯鎓盐NO供体基团通过酯键连接获得带有NO供体基团的非甾体类抗炎药 (Scheme 31).药理研究表明, 这一系列化合物具有NO释放效应, 不仅增强了抗炎作用而且还可以显著降低胃肠道不良反应.
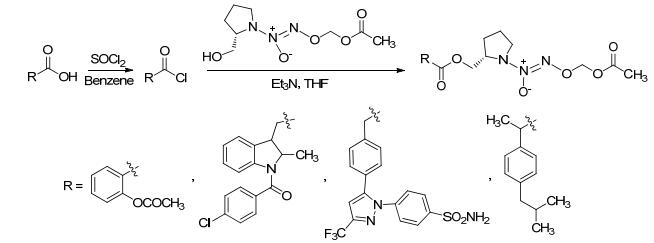 图 图式31
带有NO供体的非甾体类抗炎药衍生物的合成路线
Figure 图式31.
Synthetic routes of NSAIDs derivatives bearing NO donor
图 图式31
带有NO供体的非甾体类抗炎药衍生物的合成路线
Figure 图式31.
Synthetic routes of NSAIDs derivatives bearing NO donor
2016年, Bertinaria等[56]合成了系列带有NO供体基团的二氢青蒿素衍生物.该路线是先进行亲核取代反应将亚氨基连接到二氢青蒿素母体再与溴代的NO供体基团发生取代获得NO供体化合物 (Eq. 21).药理研究表明, 该类化合物在体外和体内对抗伯氏疟原虫实验中显示出与青蒿琥酯和蒿甲醚相当的抗疟原虫活性, 具有比原药更高的活性.
3.5 酰胺缩合
2013年, Kutty等[39]合成了带有NO供体基团的卤代呋喃酮衍生物.该合成路线是先用卤代呋喃酮衍生物与丙二酸二酰氯反应, 得到的产物再与偶氮二醇烯鎓盐类NO供体基团经酰胺缩合得到带有NO供体基团的卤代呋喃酮衍生物 (Eq. 22).药理研究表明, 该化合物具有比原药更强的抗菌活性.而且由于NO供体基团的引入, 使其具有了双效抗菌效应.
2014年, Smirnov等[76]合成了硝酸酯与偶氮二醇烯鎓盐结合的化合物.该合成路线是先用NO气体在甲醇钠存在下合成偶氮二醇烯鎓盐, 再经取代反应与不饱和烃连接得到带有双键的偶氮二醇烯鎓盐, 经加成反应加溴后与AgNO3发生取代得到目标化合物 (Scheme 32).此化合物显著地改变了NO释放速率.此合成方法可以同时引入多个NO供体基团, 改变NO在体内的释放速率, 这可以使我们在一定范围内对NO供体化合物NO释放动力学进行调节.
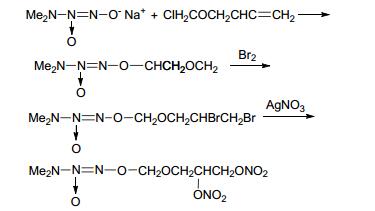 图 图式32
偶氮二醇烯鎓盐与硝酸酯杂合物的合成路线
Figure 图式32.
Synthetic routes of azo-diol enenium salts with nitrate
图 图式32
偶氮二醇烯鎓盐与硝酸酯杂合物的合成路线
Figure 图式32.
Synthetic routes of azo-diol enenium salts with nitrate
综上所述, 通过酯键将偶氮二醇烯鎓盐类NO供体与药物进行连接是偶氮二醇烯鎓盐类NO供体药物较好的合成途径, 此方法得到的目标化合物具有较高的产率, 其反应过程简单, 无污染, 易于操作, 而且药物进入体内经水解后可分别发挥各自的药理活性.
4 肟类NO供体的合成
2009年, Abdel-Hafez等[21]合成了具有NO释放能力的吡唑-3-羧酸衍生物15a.该合成路线是将吡唑-3-羧酸进行酰氯化, 与对氨基苯丙酮偶合后经NH2OH处理后获得具有NO释放能力的吡唑-3-羧酸衍生物 (Eq. 23).药理研究表明, 该化合物具有较好的NO释放能力, 同时发现其抗水肿活性比药物吲哚美辛强.
2012年, Mourad等[35]合成了带有NO供体基团的查尔酮衍生物15b.该合成路线是先合成了查尔酮的溴代衍生物, 再与对羟基苯丙酮反应, 获得的产物经NH2OH处理后得到带有肟类NO供体基团的查尔酮衍生物 (Eq. 23).药理研究发现, 该化合物的抗癌活性在加入NO供体基团后减弱.
2014年, Abuo-Rahma等[77]合成了能够释放NO的三唑类化合物15c.该合成路线是用对氨基苯丙酮和溴乙酰溴反应得到的产物与三唑化合物偶合, 然后经NH2OH处理得到能够释放NO的三唑类化合物 (Eq. 23). NO释放研究发现, 该化合物具有NO缓释作用, 同时药理研究表明该化合物具有抗炎及抗恶性细胞增殖的活性.
2011年, Shi等[53]合成了带有NO供体基团的司他夫定衍生物.该合成路线是以司他夫定为原料与丁二酸酐反应, 得到的产物再与NO供体基团偶合获得带有NO供体基团的司他夫定衍生物 (Eq. 24).药理研究表明, 该化合物不仅具有比阿昔洛韦更好的抗病毒活性, 而且其毒副作用比阿昔洛韦弱.
2016年, Cassien等[78]合成了一系列具有不同芳族取代基的新型杂化2-(二乙氧基磷酰基)-N-(亚苄基) 丙烷-2-氧化胺衍生物 (Scheme 33).该类化合物具有肟结构, 能够释放NO.药理研究表明, 该类化合物不仅具有很好的细胞毒活性, 而且对血管具有保护作用.
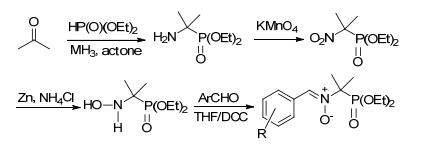 图 图式33
β-磷酸化硝酮的合成路线
Figure 图式33.
Synthetic routes of β-phosphorylated nitrone
图 图式33
β-磷酸化硝酮的合成路线
Figure 图式33.
Synthetic routes of β-phosphorylated nitrone
5 NO-金属配合物类NO供体的合成
NO-金属配合物类NO供体主要是指金属元素与具有NO释放效应的配位基形成的配合物.主要代表药物为硝普钠[79], 临床上主要用于高血压危象患者的急救, 其在体内酶或硫醇作用下释放NO而舒张血管.此类NO供体具有一些不足之处, 比如释放NO要依赖特定的酶、温度、PH值等.
2013年, Mcquilken等[80]合成了光致释放的NO-金属配合物类NO供体化合物 (Scheme 34).由于硫元素的存在, 化合物17在光致释放时可以做到完全可逆, 以氮元素连接的化合物19不可以.
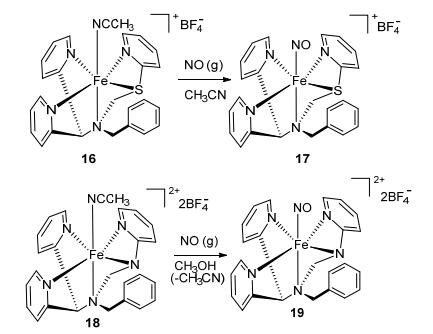 图 图式34
{FeNO}7铁配合物的合成
Figure 图式34.
Synthesis of {FeNO}7 complexes
图 图式34
{FeNO}7铁配合物的合成
Figure 图式34.
Synthesis of {FeNO}7 complexes
2014年, Sanina等[81]合成了NO-金属配合物类NO供体四亚硝基双核铁配合物 (Scheme 35).与其他NO供体化合物相比, 此化合物铁原子和NO分子的连接更加稳定.
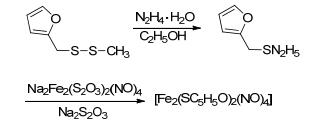 图 图式35
四亚硝基双核铁配合物的合成
Figure 图式35.
Synthesis of tetranitrosyl binuclear iron complex
图 图式35
四亚硝基双核铁配合物的合成
Figure 图式35.
Synthesis of tetranitrosyl binuclear iron complex
2016年, Monti等[82]合成了镍-哌嗪/NO供体化合物20(图 1).通过起始N-氨基乙基哌嗪与水杨醛的缩合获得该金属-壬酸盐的略微改性的形式.金属-壬酸盐基于具有壬酸基团的特定多胺, N-氨基乙基哌嗪的官能化, 并通过与金属离子络合而稳定化, 通过与水反应 (每个络合物2个NO分子) 以金属, pH和温度依赖性方式释放NO.药理研究表明, 该化合物只对高血压小鼠有很好的降压作用, 对正常小鼠血压无影响.由于其突出的血管扩张活性而被认为是抗高血压治疗的潜在候选药物.
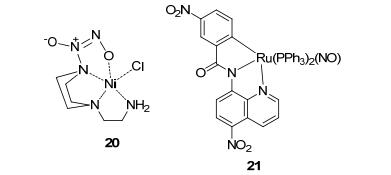 图 1
NO-金属配合物
Figure 1.
NO-metal complexes
图 1
NO-金属配合物
Figure 1.
NO-metal complexes
2016年, Sakhaei等[83]合成了NO-金属配合物类NO供体化合物NO-铜配合物 (Eq. 25).与其他NO供体化合物相比, 此化合物通过氧原子从亚硝酸盐在铜 (Ⅱ) 位点转移而释放NO, 这是从未报道过的NO释放新途径.
2016年, Kumar等[84]合成了NO-金属配合物类NO供体化合物21(图 1).该路线通过从具有酰胺键的二齿配体衍生的C—H键活化来合成新的σ-芳基钌 (Ⅲ) 络合物, 将这些有机金属钌 (Ⅲ) 络合物用NO处理, 得到亚硝酰基络合物. NO释放研究表明, 此化合物具有光致释放特性, 可以实现NO的靶向释放和定量释放, 对于NO的缓释控释具有巨大意义.
6 S-亚硝基硫醇类NO供体的合成
S-亚硝基硫醇在生物体内广泛存在, 是蛋白、多肽、硫醇等巯基亚硝基化的产物, 参与NO的储存和转运, 其可在生理条件下自发释放NO, 热、光、碱性pH以及与O2接触可促进其反应[85]. NO可以与硫醇类物质的巯基以共价键的方式结合形成RSNO, 使其性质相对稳定.外源性RSNO可通过转亚硝基反应把NO转给体内的硫醇, 最终实现内源性NO的转运. S-亚硝基硫醇是NO在体内发挥生物效应的基本形式, 主要代表性化合物为S-亚硝基谷胱甘肽[86].
2008年, Stasko等[87]合成了具有NO释放能力的S-亚硝基硫醇修饰的树状大分子.这种树状大分子设计增大了NO的储运量, 可以使NO在体内更有效地释放.药理研究表明, 该化合物具有抗菌效应和促进伤口愈合的作用. Stasko等设计了两条合成路线.
合成路线1是以N-乙酰-D/L-青霉胺为原料经系列反应与树状大分子G4-PAMAM结合后再经NaNO2处理得到具有NO释放能力的S-亚硝基硫醇修饰的树状大分子 (Scheme 36).
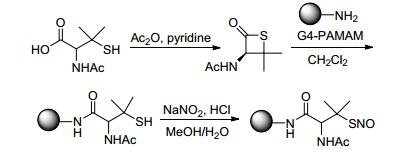 图 图式36
S-亚硝基硫醇修饰的树状大分子G4-SNAP的合成
Figure 图式36.
Synthesis of S-nitrosothiol modified generation 4 PAMAM dendrimer, G4-SNAP
图 图式36
S-亚硝基硫醇修饰的树状大分子G4-SNAP的合成
Figure 图式36.
Synthesis of S-nitrosothiol modified generation 4 PAMAM dendrimer, G4-SNAP
合成路线2是以N-乙酰半胱氨酸为原料, 经系列反应与树状大分子G4-PAMAM结合, 再经亚硝酸异戊酯处理得到具有NO释放能力的S-亚硝基硫醇修饰的树状大分子 (Scheme 37).
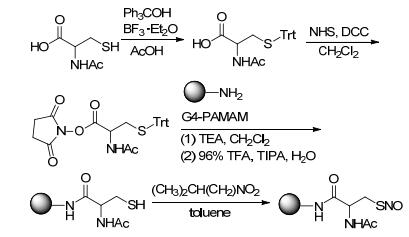 图 图式37
S-亚硝基硫醇修饰的树状大分子G4-NACysNO的合成
Figure 图式37.
Synthesis of G4-NACysNO, S-nitrosothiol modified generation 4 PAMAM dendrimer
图 图式37
S-亚硝基硫醇修饰的树状大分子G4-NACysNO的合成
Figure 图式37.
Synthesis of G4-NACysNO, S-nitrosothiol modified generation 4 PAMAM dendrimer
2009年, Llop等[88]合成了用于正电子发射断层扫描显影的NO供体化合物.该反应是以谷胱甘肽为原料合成亚硝基谷胱甘肽 (Eq. 26).该路线采用放射性亚硝基化技术, 可以在短时间内反应得到大量产物.
7 其他类NO供体
除以上所述类型的NO供体外, 还存在多种类型的NO供体[16](图 2): (1) 斯德酮亚胺类NO供体属于介离子类杂环化合物, 在生理环境下可自发降解或者经体内酶解后释放NO, 具有抗血小板凝集、抗血栓和扩张血管等生物活性, 代表性化合物为SIN-1[89]. (2) 胍类NO供体, 在体内生理环境下不能自发释放NO, 必须在体内的P450或NOS等酶的催化下经多步代谢才能生成NO, 常具有抗白血病及其他肿瘤的活性, 代表性化合物有N-hydroxydebrisoquine[90]等. (3) N-羟基-N-亚硝基胺类NO供体通常比较稳定, 需要在体内经过氧化物酶作用后释放NO, 代表性化合物有Alanosine[91]等. (4) N-羟基脲类、肽偶联NONOates类、C-亚硝基烃类等NO供体.由于近10年来这几类NO供体化合物合成方法的相关研究较少, 本文不再进行综述.
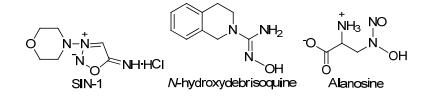 图 2
其他类NO供体药物
Figure 2.
Other types of NO donor drugs
图 2
其他类NO供体药物
Figure 2.
Other types of NO donor drugs
8 总结
综上所述, NO供体化合物研究在近几年已得到较大的发展, 已合成了多种多样的带有不同类型NO供体基团的化合物. NO供体化合物因其良好的生物活性, 较低的不良反应, 在医药卫生领域的应用越来越广泛.因此, 采用化学方法合成该类化合物具有十分重要的意义.
本文对近10年文献中报道的NO供体化合物的合成方法进行了综述, 说明了NO供体化合物的合成具有多样的反应类型和灵活性的反应体系.其中, 应用较为广泛的是制成酯类前药的设计方案.现已合成的NO供体化合物具有结构简单、易于合成、能够降低药物不良反应、提高药效等优点, 如呋咱氮氧化物类NO供体具有更广泛的生物活性, 在体内游离巯基和巯基阴离子作用下均可释放NO, 而且不易产生耐受性.偶氮二醇烯鎓盐类NO供体偶联特定的取代基团可延长其半衰期, 与某些特定载体 (如酶的底物、单克隆抗体等) 偶联可制成具有靶向缓释NO作用的NO供体化合物. NO-金属卟啉类NO供体还可以做到在肿瘤细胞定点释放NO来产生抗癌活性.但其亦具有易产生耐受性、稳定性差、无靶向性、释放不可控等缺点, 所以限制了其在临床的应用, 有进一步研究的必要.可以相信, 随着NO供体化合物研究的进一步深入, 其合成方法必将不断推新, 会开发出更多安全、高效、低毒的候选药物.
-
-
[1]
Azizzadeh, B.; Yip, H. T.; Blackwell, K. E.; Horvath, S.; Calcaterra, T. C.; Buga, G. M. Laryngoscope 2001, 111, 1896. doi: 10.1097/00005537-200111000-00004
-
[2]
Huerta, S.; Chilka, S.; Bonavida, B. Int. J. Oncol. 2008, 33, 909.
-
[3]
Agurla, S.; Gayatri, G.; Raghavendra, A. S. Nitric Oxide 2014, 43, 89. doi: 10.1016/j.niox.2014.07.004
-
[4]
Jin, R. C.; Loscalzo, J. J. Blood Med. 2010, 2010, 147.
-
[5]
Esplugues, J. V. Br. J. Pharmacol. 2002, 135, 1079. doi: 10.1038/sj.bjp.0704569
-
[6]
Wallace, J. L. Mem. Inst. Oswaldo Cruz 2005, 100(suppl. 1), 5.
-
[7]
David, H.; Tracy, R. J. Pharm. Pharmacol. 2007, 59, 3. doi: 10.1211/jpp.59.1.0002
-
[8]
Afshar, J. K.; Pluta, R. M.; Boock, R. J.; Thompson, B. G.; Oldfield, E. H. J. Neurosurg. 1995, 83, 118. doi: 10.3171/jns.1995.83.1.0118
-
[9]
Rajagopalan, S.; Harrison, D. G. Circulation 1996, 94, 240. doi: 10.1161/01.CIR.94.3.240
-
[10]
Steudel, W.; Scherrer-Crosbie, M.; Bloch, K. D.; Weimann, J.; Huang, P. L.; Jones, R. C. J. Clin. Invest. 1998, 101, 2468. doi: 10.1172/JCI2356
-
[11]
Suzuki, H.; Shimosegawa, T.; Ohara, S.; Toyota, T. J. Gastroenterol. 1999, 34, 172. doi: 10.1007/s005350050239
-
[12]
Kuo, P. C.; Schroeder, R. A. Ann. Surg. 1995, 221, 220. doi: 10.1097/00000658-199503000-00003
-
[13]
Bradley, S. A.; Steinert, J. R. J. Neurosci. Methods 2015, 245, 116. doi: 10.1016/j.jneumeth.2015.02.024
-
[14]
Serafim, R. A.; Primi, M. C.; Trossini, G. H. Curr. Med. Chem. 2012, 19, 386. doi: 10.2174/092986712803414321
-
[15]
Carradori, S.; Mollica, A.; Monte, C. Molecules 2015, 20, 5667. doi: 10.3390/molecules20045667
-
[16]
边海勇, 博士论文, 山东大学, 山东, 2011.Bian, H. Y. Ph.D. Dissertation, Shandong University, Shandong, 2011 (in Chinese).
-
[17]
Abdellatif, K. R.; Chowdhury, M. A.; Dong, Y.; Das, D.; Yu, G.; Velazquez, C. A.; Suresh, M. R.; Knaus, E. E. Bioorg. Med. Chem. Lett. 2009, 19, 3014. doi: 10.1016/j.bmcl.2009.04.059
-
[18]
Abuo-Rahma, E. D. A. A.; Abdel-Aziz, M.; Mai, A. E. M.; Farag, H. H. Bioorg. Med. Chem. 2012, 20, 195. doi: 10.1016/j.bmc.2011.11.012
-
[19]
罗刚, 陈宇瑛, 中国新药杂志, 2010, 19, 1322.Luo, G.; Chen, Y. Y. Chin. J. New Drugs 2010, 19, 1322 (in Chinese).
-
[20]
López, G. V.; Blanco, F.; Hernández, P.; Ferreira, A.; Piro, O. E.; Batthyány, C.; González, M.; Rubbo, H.; Cerecetto, H. Bioorg. Med. Chem. 2007, 15, 6262. doi: 10.1016/j.bmc.2007.06.019
-
[21]
Abdel-Hafez, E. S. M. N.; Abuo-Rahma, E. D. A. A.; Abdel-Aziz, M.; Radwan, M. F.; Farag, H. Bioorg. Med. Chem. 2009, 17, 3829. doi: 10.1016/j.bmc.2009.04.037
-
[22]
Rolando, B.; Filieri, A.; Chegaev, K.; Lazzarato, L.; Giorgis, M.; De Nardi, C.; Fruttero, R.; Martel, S.; Carrupt, P. A.; Gasco, A. Bioorg. Med. Chem. 2012, 20, 841. doi: 10.1016/j.bmc.2011.11.065
-
[23]
Digiacomo, M.; Martelli, A.; Testai, L.; Lapucci, A.; Breschi, M. C.; Calderone, V.; Rapposelli, S. Bioorg. Med. Chem. 2015, 23, 422. doi: 10.1016/j.bmc.2014.12.043
-
[24]
Konter, J.; Mollmann, U.; Lehmann, J. Bioorg. Med. Chem. 2008, 16, 8294. doi: 10.1016/j.bmc.2008.05.008
-
[25]
Bertinaria, M.; Rolando, B.; Giorgis, M.; Montanaro, G.; Marini, E.; Collino, M.; Benetti, E.; Daniele, P. G.; Fruttero, R.; Gasco, A. Eur. J. Med. Chem. 2012, 54, 103. doi: 10.1016/j.ejmech.2012.04.032
-
[26]
Biava, M.; Battilocchio, C.; Poce, G.; Alfonso, S.; Consalvi, S.; Di Capua, A.; Calderone, V.; Martelli, A.; Testai, L.; Sautebin, L.; Rossi, A.; Ghelardini, C.; Di Cesare Mannelli, L.; Giordani, A.; Persiani, S.; Colovic, M.; Dovizio, M.; Patrignani, P.; Anzini, M. Bioorg. Med. Chem. 2014, 22, 772. doi: 10.1016/j.bmc.2013.12.008
-
[27]
Liu, W.; Liu, C.; Gong, C.; Lin, W.; Guo, C. Bioorg. Med. Chem. Lett. 2009, 19, 1647. doi: 10.1016/j.bmcl.2009.02.005
-
[28]
Peng, S. M.; Zou, X. Q.; Ding, H. L.; Ding, Y. L.; Lin, Y. B. Bioorg. Med. Chem. Lett. 2009, 19, 1264. doi: 10.1016/j.bmcl.2008.12.116
-
[29]
Dong, X.; Du, L.; Pan, Z.; Liu, T.; Yang, B.; Hu, Y. Eur. J. Med. Chem. 2010, 45, 3986. doi: 10.1016/j.ejmech.2010.05.054
-
[30]
Zou, X. Q.; Peng, S. M.; Hu, C. P.; Tan, L. F.; Yuan, Q.; Deng, H. W.; Li, Y. J. Bioorg. Med. Chem. 2010, 18, 3020. doi: 10.1016/j.bmc.2010.03.056
-
[31]
Bai, R.; Yang, X.; Zhu, Y.; Zhou, Z.; Xie, W.; Yao, H.; Jiang, J.; Liu, J.; Shen, M.; Wu, X.; Xu, J. Bioorg. Med. Chem. 2012, 20, 6848. doi: 10.1016/j.bmc.2012.09.043
-
[32]
Wang, Q. Q.; Cheng, N.; Zheng, X. W.; Peng, S. M.; Zou, X. Q. Bioorg. Med. Chem. 2013, 21, 4301. doi: 10.1016/j.bmc.2013.04.066
-
[33]
Bhandari, S. V.; Bothara, K. G.; Patil, A. A.; Chitre, T. S.; Sarkate, A. P.; Gore, S. T.; Dangre, S. C.; Khachane, C. V. Bioorg. Med. Chem. 2009, 17, 390. doi: 10.1016/j.bmc.2008.10.032
-
[34]
Bhandari, S. V.; Dangre, S. C.; Bothara, K. G.; Patil, A. A.; Sarkate, A. P.; Lokwani, D. K.; Gore, S. T.; Deshmane, B. J.; Raparti, V. T.; Khachane, C. V. Eur. J. Med. Chem. 2009, 44, 4622. doi: 10.1016/j.ejmech.2009.06.035
-
[35]
Mourad, M. A.; Abdel-Aziz, M.; Abuo-Rahma Gel, D.; Farag, H. H. Eur. J. Med. Chem. 2012, 54, 907. doi: 10.1016/j.ejmech.2012.05.030
-
[36]
Fang, L.; Feng, M.; Chen, F. Bioorg. Med. Chem. 2016, 24, 4611. doi: 10.1016/j.bmc.2016.07.066
-
[37]
Zhang, Y. C.; Zhou, J. P.; Wu, X. M.; Pan, W. H. Chin. Chem. Lett. 2009, 20, 302. doi: 10.1016/j.cclet.2008.11.012
-
[38]
Tamboli, Y.; Lazzarato, L.; Marini, E.; Guglielmo, S.; Novelli, M.; Beffy, P.; Masiello, P.; Fruttero, R.; Gasco, A. Bioorg. Med. Chem. Lett. 2012, 22, 3810. doi: 10.1016/j.bmcl.2012.03.103
-
[39]
Kutty, S. K.; Barraud, N.; Pham, A.; Iskander, G.; Rice, S. A.; Black, D. S.; Kumar, N. J. Med. Chem. 2013, 56, 9517. doi: 10.1021/jm400951f
-
[40]
Huang, Q.; Rui, E. Y.; Cobbs, M.; Dinh, D. M.; Gukasyan, H. J.; Lafontaine, J. A.; Mehta, S.; Patterson, B. D.; Rewolinski, D. A.; Richardson, P. F.; Edwards, M. P. J. Med. Chem. 2015, 58, 2821. doi: 10.1021/acs.jmedchem.5b00043
-
[41]
Abdellatif, K. R.; Chowdhury, M. A.; Dong, Y.; Chen, Q. H.; Knaus, E. E. Bioorg. Med. Chem. 2008, 16, 3302. doi: 10.1016/j.bmc.2007.12.006
-
[42]
Kaur, J.; Bhardwaj, A.; Huang, Z.; Narang, D.; Chen, T. Y.; Plane, F.; Knaus, E. E. J. Med. Chem. 2012, 55, 7883. doi: 10.1021/jm300997w
-
[43]
Xu, G. G.; Deshpande, T. M.; Ghatge, M. S.; Mehta, A. Y.; Omar, A. S.; Ahmed, M. H.; Venitz, J.; Abdulmalik, O.; Zhang, Y.; Safo, M. K. Biochemistry 2015, 54, 7178. doi: 10.1021/acs.biochem.5b01074
-
[44]
Gazzano, E.; Chegaev, K.; Rolando, B. Bioorg. Med. Chem. 2016, 24, 967. doi: 10.1016/j.bmc.2016.01.021
-
[45]
Fang, L. A. D.; Decker, M.; Kiehntopf, M.; Roegler, C.; Deufel, T.; Fleck, C.; Peng, S. X.; Zhang, Y. H.; Lehmann, J. J. Med. Chem. 2008, 51, 4. doi: 10.1021/jm7009414
-
[46]
Chowdhury, M. A.; Abdellatif, K. R.; Dong, Y.; Yu, G.; Huang, Z.; Rahman, M.; Das, D.; Velazquez, C. A.; Suresh, M. R.; Knaus, E. E. Bioorg. Med. Chem. Lett. 2010, 20, 1324. doi: 10.1016/j.bmcl.2010.01.014
-
[47]
Bertinaria, M.; Guglielmo, S.; Rolando, B.; Giorgis, M.; Aragno, C.; Fruttero, R.; Gasco, A.; Parapini, S.; Taramelli, D.; Martins, Y. C.; Carvalho, L. J. Eur. J. Med. Chem. 2011, 46, 1757. doi: 10.1016/j.ejmech.2011.02.029
-
[48]
Zhao, J.; Prosser, K. E.; Chang, S. W. Dalton Trans. 2016, 45, 18079. doi: 10.1039/C6DT03661A
-
[49]
Csont, T.; Ferdinandy, P. Pharmacol. Ther. 2005, 105, 57. doi: 10.1016/j.pharmthera.2004.10.001
-
[50]
Feelisch, M.; Schonafinger, K.; Noack, E. Biochem. Pharmacol. 1992, 44, 1149. doi: 10.1016/0006-2952(92)90379-W
-
[51]
Chen, L. Z. Y.; Kong, X. W.; Lan, E. D.; Huang, Z. J.; Peng, S. X.; Kaufman, D. L.; Tian, J. J. Med. Chem. 2008, 51, 4834. doi: 10.1021/jm800167u
-
[52]
Ling, Y.; Ye, X.; Ji, H.; Zhang, Y.; Lai, Y.; Peng, S.; Tian, J. Bioorg. Med. Chem. 2010, 18, 3448. doi: 10.1016/j.bmc.2010.03.077
-
[53]
Shi, J. B.; Xu, S.; Wang, Y. P.; Li, J. J.; Yao, Q. Z. Chin. Chem. Lett. 2011, 22, 899. doi: 10.1016/j.cclet.2011.01.010
-
[54]
Tang, W.; Xie, J.; Xu, S.; Lv, H.; Lin, M.; Yuan, S.; Bai, J.; Hou, Q.; Yu, S. J. Med. Chem. 2014, 57, 7600. doi: 10.1021/jm5007534
-
[55]
Dos Santos, J. L.; Lanaro, C.; Chelucci, R. C.; Gambero, S.; Bosquesi, P. L.; Reis, J. S.; Lima, L. M.; Cerecetto, H.; Gonzalez, M.; Costa, F. F.; Chung, M. C. J. Med. Chem. 2012, 55, 7583. doi: 10.1021/jm300602n
-
[56]
Bertinaria, M.; Orjuelasanchez, P.; Marini, E. J. Med. Chem. 2015, 58, 7895. doi: 10.1021/acs.jmedchem.5b01036
-
[57]
Bian, H.; Feng, J.; Li, M.; Xu, W. Bioorg. Med. Chem. Lett. 2011, 21, 7025. doi: 10.1016/j.bmcl.2011.09.103
-
[58]
Borretto, E.; Lazzarato, L.; Spallotta, F.; Cencioni, C.; D'Alessandra, Y.; Gaetano, C.; Fruttero, R.; Gasco, A. ACS Med. Chem. Lett. 2013, 4, 994. doi: 10.1021/ml400289e
-
[59]
Lu, M.-D.; Zhou, X.; Yu, Y.-J.; Li, P.-H.; Sun, W.-J.; Zhao, C.-G.; Zheng, Z.-Q.; You, T.; Wang, F.-H. Chin. Chem. Lett. 2013, 24, 415. doi: 10.1016/j.cclet.2013.03.006
-
[60]
Ai, Y.; Kang, F.; Huang, Z.; Xue, X.; Lai, Y.; Peng, S.; Tian, J.; Zhang, Y. J. Med. Chem. 2015, 58, 2452. doi: 10.1021/jm5019302
-
[61]
Duan, W.; Li, J.; Inks, E. S.; Chou, C. J.; Jia, Y.; Chu, X.; Li, X.; Xu, W.; Zhang, Y. J. Med. Chem. 2015, 58, 4325. doi: 10.1021/acs.jmedchem.5b00317
-
[62]
Massarico Serafim, R. A.; Goncalves, J. E.; de Souza, F. P.; de Melo Loureiro, A. P.; Storpirtis, S.; Krogh, R.; Andricopulo, A. D.; Dias, L. C.; Ferreira, E. I. Eur. J. Med. Chem. 2014, 82, 418. doi: 10.1016/j.ejmech.2014.05.077
-
[63]
Lakshman, T. R.; Deb, J.; Paine, T. K. Dalton Trans. 2016, 45, 14053. doi: 10.1039/C6DT00838K
-
[64]
Davies, K. M.; Wink, D. A.; Saavedra, J. E.; Keefer, L. K. J. Am. Chem. Soc. 2001, 32, 5473.
-
[65]
Smith, D. J.; Chakravarthy, D.; Pulfer, S.; Simmons, M. L.; Hrabie, J. A.; Citro, M. L. J. Med. Chem. 1996, 39, 1148. doi: 10.1021/jm950652b
-
[66]
Chakrapani, H.; Goodblatt, M. M.; Udupi, V.; Malaviya, S.; Shami, P. J.; Keefer, L. K.; Saavedra, J. E. Bioorg. Med. Chem. Lett. 2008, 18, 950. doi: 10.1016/j.bmcl.2007.12.044
-
[67]
Chowdhury, M. A.; Abdellatif, K. R.; Dong, Y.; Knaus, E. E. Bioorg. Med. Chem. 2008, 16, 8882. doi: 10.1016/j.bmc.2008.08.059
-
[68]
Abdellatif, K. R.; Moawad, A.; Knaus, E. E. Bioorg. Med. Chem. Lett. 2014, 24, 5015. doi: 10.1016/j.bmcl.2014.09.024
-
[69]
Abdellatif, K. R.; Chowdhury, M. A.; Velazquez, C. A.; Huang, Z.; Dong, Y.; Das, D.; Yu, G.; Suresh, M. R.; Knaus, E. E. Bioorg. Med. Chem. Lett. 2010, 20, 4544. doi: 10.1016/j.bmcl.2010.06.022
-
[70]
Abdellatif, K. R.; Chowdhury, M. A.; Dong, Y.; Knaus, E. E. Bioorg. Med. Chem. 2008, 16, 6528. doi: 10.1016/j.bmc.2008.05.028
-
[71]
Abdellatif, K. R.; Chowdhury, M. A.; Dong, Y.; Velazquez, C.; Das, D.; Suresh, M. R.; Knaus, E. E. Bioorg. Med. Chem. 2008, 16, 9694. doi: 10.1016/j.bmc.2008.10.001
-
[72]
Abdellatif, K. R.; Huang, Z.; Chowdhury, M. A.; Kaufman, S.; Knaus, E. E. Bioorg. Med. Chem. Lett. 2011, 21, 3951. doi: 10.1016/j.bmcl.2011.05.017
-
[73]
Velázquez, C. A.; Chen, Q. H.; Citro, M. L.; Keefer, L. K.; Knaus, E. E. J. Med. Chem. 2008, 51, 1954. doi: 10.1021/jm701450q
-
[74]
Xu, S.; Wang, G.; Yan, L. Bioorg. Med. Chem. Lett. 2016, 26, 2795. doi: 10.1016/j.bmcl.2016.04.068
-
[75]
Abdellatif, K. R.; Chowdhury, M. A.; Dong, Y.; Das, D.; Yu, G.; Velazquez, C.; Suresh, M. R.; Knaus, E. E. Bioorg. Med. Chem. 2009, 17, 5182. doi: 10.1016/j.bmc.2009.05.046
-
[76]
Smirnov, G. A.; Gordeev, P. B.; Nikitin, S. V. Russ. Chem. Bull. 2014, 63, 487. doi: 10.1007/s11172-014-0457-2
-
[77]
Abuo-Rahma, E. D. A. A.; Abdel-Aziz, M.; Beshr, E. A.; Ali, T. F. Eur. J. Med. Chem. 2014, 71, 185. doi: 10.1016/j.ejmech.2013.11.006
-
[78]
Cassien, M.; Petrocchi, C.; Thétiot-Laurent, S. Eur. J. Med. Chem. 2016, 119, 197. doi: 10.1016/j.ejmech.2016.04.067
-
[79]
Vogt, M. A.; Vogel, A. S.; Pfeiffer, N.; Gass, P.; Inta, D. Eur. Neuropsychopharmacol. 2015, 25, 1848. doi: 10.1016/j.euroneuro.2015.06.012
-
[80]
Mcquilken, A. C.; Yang, H.; Sutherlin, K. D.; Siegler, M. A.; Hodgson, K. O.; Britt, H. J. Am. Chem. Soc. 2013, 135, 14024. doi: 10.1021/ja4064487
-
[81]
Sanina, N. A.; Kozub, G. I.; Kondratéva, T. A.; Korchagin, D. V.; Shilov, G. V.; Emelýanova, N. S. J. Mol. Struct. 2014, 1075, 159. doi: 10.1016/j.molstruc.2014.06.024
-
[82]
Monti, M.; Ciccone, V.; Pacini, A. Pharmacol. Res. 2016, 107, 352. doi: 10.1016/j.phrs.2016.03.033
-
[83]
Sakhaei, Z.; Kundu, S.; Donnelly, J. Chem. Commun. 2017, 53, 549. doi: 10.1039/C6CC08745K
-
[84]
Kumar, R.; Kumar, S.; Bala, M. RSC Adv. 2016, 6, 72096. doi: 10.1039/C6RA17223G
-
[85]
Grossi, L.; Montevecchi, P. C. J. Org. Chem. 2002, 67, 8625. doi: 10.1021/jo026154+
-
[86]
Kumari, S.; Sammut, I. A.; Giles, G. I. Eur. J. Med. Chem. 2014, 737, 168.
-
[87]
Stasko, N. A.; Fischer, T. H.; Schoenfisch, M. H. Biomacromolecules 2008, 9, 834. doi: 10.1021/bm7011746
-
[88]
Llop, J.; Gómez-Vallejo, V.; Bosque, M.; Quincoces, G.; Peñuelas, I. Appl. Radiat. Isot. 2009, 67, 95. doi: 10.1016/j.apradiso.2008.09.014
-
[89]
Priora, R.; Margaritis, A.; Frosali, S.; Coppo, L.; Summa, D.; Giuseppe, D. D. Pharmacol. Res. 2011, 64, 289. doi: 10.1016/j.phrs.2011.03.014
-
[90]
Ming, X.; Noriko, F.; Zhong, W.; Tingwei, C.; Satoshi, K.; Janczuk, A. J. Bioorg. Med. Chem. 2002, 10, 3049. doi: 10.1016/S0968-0896(02)00155-4
-
[91]
Tyagi, A. K.; Cooney, D. A. Adv. Pharmacol. Chemother. 1984, 20, 69. doi: 10.1016/S1054-3589(08)60265-3
-
[1]
-

图式1 带有NO供体的吡唑-3-羧酸衍生物的合成路线
Scheme 1 Synthetic routes of pyrazole-3-carboxylic acid derivatives bearing NO donor

图式2 阿司匹林和NO供体化合物参与的偶联反应
Scheme 2 Cross-coupling reactions of aspirin and nitric oxide donor compound

图式3 带有NO供体的依达拉奉衍生物的合成路线
Scheme 3 Synthetic routes of edaravone derivatives bearing NO donor

图式4 带有NO供体的胰岛素促分泌素衍生物的合成路线
Scheme 4 Synthetic routes of insulin-secretagogue derivatives bearing NO donor

图式5 带有NO供体的COX-2抑制剂的合成路线
Scheme 5 Synthetic routes of COX-2 inhibitors bearing NO donor

图式6 硝酸银法制备NO供体化合物的合成路线
Scheme 6 Synthetic routes of NO-donor compounds by silver nitrate process

图式8 带有NO供体的白杨素衍生物的合成路线1
Scheme 8 Synthetic routes 1 of chrysin derivatives bearing NO donor

图式9 羧基连接NO供体化合物的合成路线
Scheme 9 Synthetic routes of antihypertensive agents bearing NO donor

图式10 带有NO供体的查尔酮衍生物的合成路线
Scheme 10 Synthetic routes of chalcone derivatives bearing NO donor

图式11 带有NO供体的查尔酮衍生物的合成路线
Scheme 11 Synthetic routes of chalcone derivatives bearing NO donor

图式12 甲基化的NO供体查尔酮衍生物的合成路线
Scheme 12 Synthetic routes of methylated chalcone derivatives bearing NO donor

图式13 带有NO供体的厄贝沙坦衍生物的合成路线
Scheme 13 Synthetic routes of irbesartan derivatives bearing NO donor

图式14 带有NO供体的甲苯磺丁脲类似物的合成路线
Scheme 14 Synthetic routes of tolbutamide analogue bearing NO donor

图式15 带有NO供体的卤代呋喃酮衍生物的合成路线
Scheme 15 Synthetic routes of fimbrolide derivatives bearing NO donor

图式16 带有NO供体的布林佐胺衍生物的合成路线
Scheme 16 Synthetic routes of brinzolamide derivatives bearing NO donor

图式20 带有NO供体的白杨素衍生物的合成路线
Scheme 20 Synthetic route of chrysin derivatives bearing NO donor

图式21 带有NO供体的查尔酮衍生物的合成路线
Scheme 21 Synthetic routes of chalcone derivative bearing NO donor

图式22 带有NO供体的二氢青蒿素衍生物的合成路线
Scheme 22 Synthetic routes of dihydroartemisinin derivative bearing NO donor

图式23 带有NO供体的羟基肉桂酸衍生物的合成路线
Scheme 23 Synthetic routes of hydroxylcinnamic acid derivative bearing NO donor

图式24 带有NO供体的齐墩果酸衍生物的合成路线
Scheme 24 Synthetic routes of oleanolic acid derivative bearing NO donor

图式25 带有NO供体的酰基腙衍生物的合成路线
Scheme 25 Synthetic routes of acylhydrazone derivatives bearing NO donor

图式28 带有NO供体的塞来昔布类似物的合成路线
Scheme 28 Synthetic routes of celecoxib analogue bearing NO donor

图式29 带有NO供体的塞来昔布衍生物的合成路线
Scheme 29 Synthetic routes of celecoxib derivatives bearing NO donor

图式30 带有NO供体的美各里替尼衍生物的合成路线
Scheme 30 Synthetic routes of meglitinide derivative bearing NO donor

图式31 带有NO供体的非甾体类抗炎药衍生物的合成路线
Scheme 31 Synthetic routes of NSAIDs derivatives bearing NO donor

图式32 偶氮二醇烯鎓盐与硝酸酯杂合物的合成路线
Scheme 32 Synthetic routes of azo-diol enenium salts with nitrate

图式36 S-亚硝基硫醇修饰的树状大分子G4-SNAP的合成
Scheme 36 Synthesis of S-nitrosothiol modified generation 4 PAMAM dendrimer, G4-SNAP

图式37 S-亚硝基硫醇修饰的树状大分子G4-NACysNO的合成
Scheme 37 Synthesis of G4-NACysNO, S-nitrosothiol modified generation 4 PAMAM dendrimer
-


 下载:
下载:






































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 88
- 文章访问数: 3616
- HTML全文浏览量: 996
 下载:
下载:
