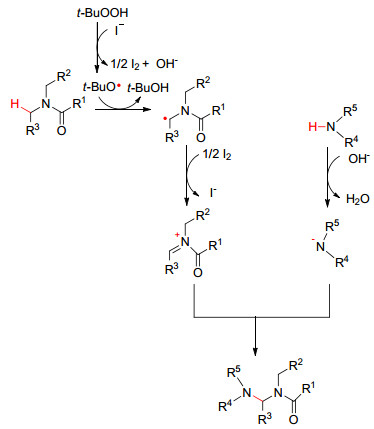
 图 图式 1
KI催化亚胺化反应机理
Figure 图式 1.
Proposed mechanism for KI-catalyzed imadation
图 图式 1
KI催化亚胺化反应机理
Figure 图式 1.
Proposed mechanism for KI-catalyzed imadation
引用本文:
袁斯甜, 王艳华, 邱观音生, 刘晋彪. 自由基引发的无过渡金属催化C-N构建研究进展[J]. 有机化学,
2017, 37(3): 566-576.
doi:
10.6023/cjoc201610011

Citation: Yuan Sitian, Wang Yanhua, Qiu Guanyinsheng, Liu Jinbiao. Recent Advances in Radical Initiated C-N Bond Formation under Transition Metal-Free Conditions[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 566-576. doi: 10.6023/cjoc201610011
Citation: Yuan Sitian, Wang Yanhua, Qiu Guanyinsheng, Liu Jinbiao. Recent Advances in Radical Initiated C-N Bond Formation under Transition Metal-Free Conditions[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 566-576. doi: 10.6023/cjoc201610011
自由基引发的无过渡金属催化C-N构建研究进展
English
Recent Advances in Radical Initiated C-N Bond Formation under Transition Metal-Free Conditions
Abstract:
Nitrogen-containing compounds are extremely important because of their abundance in synthetic organic compounds, natural products and pharmaceutical agents. Recently, transition metal-free C-N bond-formation via radical procedures has attracted wide interest. The recent advances in C-N bond formation via C (sp3, sp2 or sp)-H bond activation under transition metal-free conditions are summarized.
-
Key words:
- C-H activation
- / C-N bond
- / radical
- / dehydrogenative coupling
-
C—N键是生物活性化合物中重要的结构单元, 普遍存在药物和天然产物中, 比如胺、酰胺以及多肽等[1].因此, 快速、高效并高选择性地构建C—N键研究受到工业和学术界的极大关注, 并已取得很大进展.其中, 过渡金属催化的Ullmann反应和Buchwald-Hartwig交叉偶联反应发展迅速, 被广泛应用于天然产物、高分子以及药物合成领域的C—N键构建[2].传统的交叉偶联反应通常需要使用官能团化的底物, 例如卤代烃或者磺酸酯作为亲电偶联组分.近10年来, 过渡金属催化的C—H键直接官能化研究已成为热点, 并被有效地应用于C—N键偶联反应中[3].然而, 过渡金属催化剂比如钌、钯、铑和镱等的使用, 使得该类反应依然存在催化剂价格昂贵、金属残留等缺点, 制约了其在药物和天然产物合成中的发展.
最近几年, 在无过渡金属催化下, 自由基引发的C—H直接官能化形成新的C—N键为直接利用简单的原料进行高效、绿色的有机合成任务提供了一条新的思路和途径[4].此类方法避免了昂贵的过渡金属催化剂, 反应条件温和、原子经济性高和绿色环保, 在有机合成中具有很大的研究价值.本文将基于自由基引发的无过渡金属催化体系, 就不同杂化类型C—H键参与的C—N构建研究进展进行简要综述.
1 C (sp3)—H键官能化
1.1 直接脱氢偶联
最近, 无过渡金属催化C—H键活化的交叉脱氢偶联反应 (CDC) 引起化学家们的兴趣[5]. 2012年, Li和Meng等[6]报道了以KI为催化剂, 过氧叔丁醇 (TBHP, 70%水溶液) 为氧化剂, 氮甲基C (sp3)—H与酰亚胺直接交叉脱氢偶联生成C (sp3)—N键 (Eq. 1).该反应无需添加任何过渡金属催化剂, 化学选择性高, 产率最高可达95%.文中提出了可能的反应机理, I-诱导TBHP分解产生叔丁醇氧自由基, 该自由基夺取C (sp3)—H上的氢, 进而产生碳自由基.碳自由基随后在碘氧化下生成亚胺正离子, 最后与胺负离子结合生成终产物, 实现C (sp3)—H的直接酰亚胺化, 构建新的C—N键 (Scheme 1).
图 图式 1
KI催化亚胺化反应机理
Figure 图式 1.
Proposed mechanism for KI-catalyzed imadation
随后, Wang等[7]报道了在KI为催化剂, 过氧苯甲酸叔丁酯 (TBPB) 为氧化剂下, 吲哚和N, N-二甲基苯胺衍生物反应, 实现了吲哚C (3)-甲酰基和N-甲胺基双官能团化 (Eq. 2).有意思的是, 该反应在吲哚C (3) 位同时引入了甲酰基, 机理证实甲酰基来自胺甲基碳.反应中加入新戊酸 (PivOH) 是为了抑制吲哚在氧化剂下分解.该方法为合成官能团多样化的吲哚衍生物提供了一条高效的新途径.
除了氮邻位C (sp3)—H, 氧邻位C (sp3)—H也能够被活化, 与含氮化合物脱氢偶联构建新的C (sp3)—N键. 2014年, Sun课题组[8]报道了在无过渡金属催化下芳基甲基醚与邻磺酰苯甲酰亚胺偶联生成C—N键 (Eq. 3), 快捷地实现了糖精分子N官能团化.有意思的是, 当芳基甲基醚上含有苄基时, 此类脱氢偶联主要发生在苄基位. Du课题组[9]也报道了脂肪醚与邻苯二甲酰亚胺的自由基C—N偶联 (Eq. 4), 反应条件温和, 无需添加过渡金属催化剂, 原子经济性高.其中, 非对称烷基醚的区域选择性相对较低.
Patel课题组[10]报道的是芳醚参与的苯基四氮唑为氮源的自由基C (sp3)—N偶联反应 (Eq. 5).由于电子效应的影响, 不同碳自由基的稳定性差异导致氧原子α-位的仲碳原子反应活性优于其伯碳原子.同时, 他们也发现苄基位C—H键的反应活性高于氧原子α-位.
寻找新的亲核性含氮化合物以及碳自由基前体是拓展此类偶联反应应用范围的关键. 2014年, Zhang等[11]报道了羰基α-位C (sp3)—H活化生成C—N键.该反应中, 丙酮可以顺利地与多种酰亚胺 (如邻苯二甲酰亚胺、邻磺酰苯甲酰亚胺和琥珀酰亚胺等) 发生反应 (Eq. 6), 产率较高.他们还指出该反应可能经历了自由基胺化过程, 但具体的反应机理还在进一步研究中.
除了通过上述杂原子或者羰基稳定碳自由基, 碳碳双键或者苯环亦可以稳定碳自由基, 进而参与后续C—N键的构建. 2013年, Zhu等[12]报道了以四丁基碘化铵为催化剂, TBHP为氧化剂, 苄基位C (sp3)—H与含氮杂环偶联合成C (sp3)—N键 (Eq. 7).值得一提的是, 对二甲苯和苯并三唑反应可扩大到克量级, 产率高达88%.该反应经历了一个自由基过程, 甲苯被叔丁基氧自由基夺氢生成苄基自由基, 继而发生单电子转移生成苄基碳正离子, 最后与亲核性含氮杂环形成最终的苄基化产物.
2014年, Wang等[13]报道了在四丁基碘化铵 (TBAI)/ TBHP催化体系下, 甲苯和烷基醚均能与四唑直接脱氢偶联, 构建新的C (sp3)—N键 (Scheme 2).该方法无需过渡金属催化剂, 在80 ℃空气氛围下进行, 为四唑衍生物的合成提供了绿色高效的新途径.
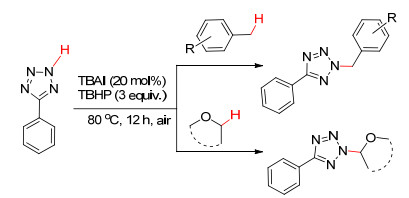 图 图式 2
无过渡金属催化四唑直接烷基化
Figure 图式 2.
Transition-metal-free direct alkylation of aryl tetrazoles
图 图式 2
无过渡金属催化四唑直接烷基化
Figure 图式 2.
Transition-metal-free direct alkylation of aryl tetrazoles
2016年, Singh等[14]报道了无过渡金属催化芳烃甲烷C (sp3)—H与N-烷基苯甲酰胺脱氢偶联生成芳烃酰亚胺 (Eq. 8).该反应是一个自由基历程, 芳甲烷在TBHP氧化下原位转化成芳甲醛后, 与N-烷基苯甲酰胺发生自由基偶联.通过氧同位素标记实验, 他们还证实了产物中新生成的羰基氧来自体系中的水.
邻位π键对于碳自由基可以起到稳定作用. 2015年, Wang等[15]报道了在TBAI/TBHP催化体系下, 烯丙基位C (sp3)—H键与环酰胺或者咪唑、三唑N—H键的直接脱氢偶联形成C—N键 (Scheme 3).该方法为含氮杂环高效地引入了烯丙基官能团, 有效地拓展了底物C (sp3)—H键活化范围.
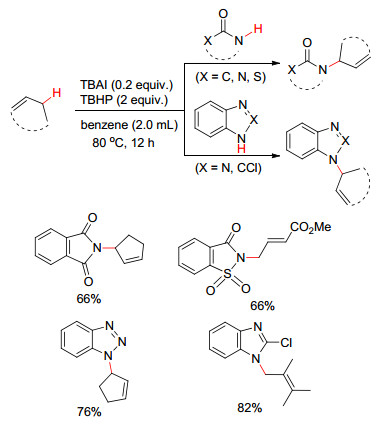 图 图式 3
无金属催化含N-杂环直接烯丙基化
Figure 图式 3.
Metal-free direct allylation of N-heterocycles
图 图式 3
无金属催化含N-杂环直接烯丙基化
Figure 图式 3.
Metal-free direct allylation of N-heterocycles
2016年Lv等[16]也报道了在TBAI/TBHP催化体系下, 烯丙基位C (sp3)—H键与系列具有生物活性的嘌呤、苯并咪唑、苯并三唑、邻磺酰苯甲酰亚胺等进行脱氢偶联反应, 构建新的C (sp3)—N键 (Eq. 9).
1.2 脱酰基偶联
除了上述直接脱氢交叉偶联方法, N-甲酰化合物也可作为氮源, 通过脱甲酰基再与C (sp3) 发生偶联. 2012年, Wang等[17]报道了以I2为催化剂, TBHP为氧化剂下, 在碱性条件, 芳基甲醇C (sp3)—H与N-烷基甲酰胺脱氢脱酰基偶联生成C—N键 (Eq. 10).该反应有着中等至良好收率, 但具有挑战性的是N上未完全烷基化的甲酰胺和苯甲醇的反应收率只有41%.文中提出的可能机理为: I-诱导TBHP分解产生叔丁醇氧自由基, 然后芳基甲醇在TBHP和碱性条件下形成芳基甲醛, 进而被叔丁基氧自由基夺走醛基碳上的氢产生芳烃甲酰基自由基, 同时N-烷基甲酰胺在叔丁基氧自由基引发下脱酰基产生氮烷基自由基.最后芳烃甲酰基自由基和氮烷基自由基发生交叉偶联生成最终的酰胺产物 (Scheme 4).
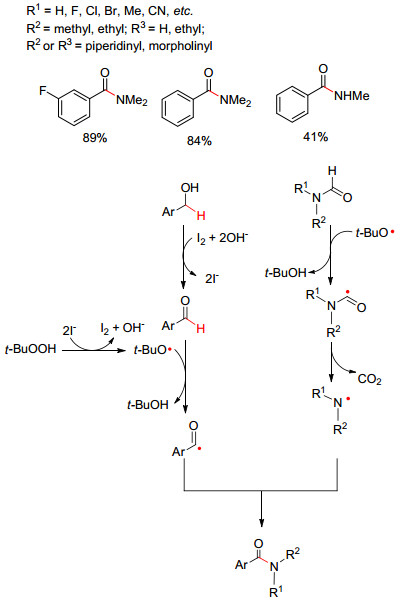 图 图式 4
芳甲醇与N-烷基甲酰胺的交叉偶联反应机理
Figure 图式 4.
Proposed mechanism for direct amidation for alcohols with N-substituted formamides
图 图式 4
芳甲醇与N-烷基甲酰胺的交叉偶联反应机理
Figure 图式 4.
Proposed mechanism for direct amidation for alcohols with N-substituted formamides
2014年, Wang等[18]报道了在I2催化, TBHP为氧化剂, 酸性条件下, 芳甲胺和甲酰胺脱氨基脱酰基偶联合成芳基酰胺, 产率中等至优秀 (Eq. 11).芳基甲胺在I2/TBHP催化体系下氧化成芳基甲亚胺, 继而水解成苯甲醛和NH3, 之后苯甲醛被叔丁氧自由基夺氢产生苯甲酰自由基, 最后与甲酰胺脱酰基产生的氮自由基发生偶联, 生成新的酰胺键.
2012年, Mai等[19]报道了在TBAI/TBHP催化体系下, 实现了芳基乙酮C (sp3)—H与N, N-二烷基甲酰胺的脱氢脱酰基偶联合成α-酮酰胺 (Eq. 12).值得一提的是, 该反应是以水作溶剂, 主要副产物是H2O和CO, 为α-酮酰胺的合成提供了绿色无污染的新方法.文中提到产生C (sp3)—N键后, 其与羰基相邻的C (sp3)—H被叔丁醇氧自由基产生碳自由基, 随后在TBHP氧化下生成亚胺正离子, 再与氢氧根负离子结合, 最后被氧化形成α-酮酰胺.同年, Prabhu等[20]报道了芳基乙酮和仲胺在N-碘代琥珀酰亚胺 (NIS)、TBHP催化体系下, 合成α-酮酰胺类衍生物.
2013年, Wang等[21]报道了芳基乙酮与甲酰胺脱氢脱酰基偶联合成α-酮酰胺类衍生物 (Eq. 13), 使用的催化体系是I2/TBHP/PhCO2H, 甲苯作溶剂.该反应经历的是自由基过程, 与前面Mai等[19]提出的机理不同, 该机理证实有α-碘代芳基乙酮中间体产生, 然后与经甲酰胺脱酰基产生的胺进行亲核取代, 最终经自由基引发和氧化生成α-酮酰, 产率中等至良好 (Scheme 5).
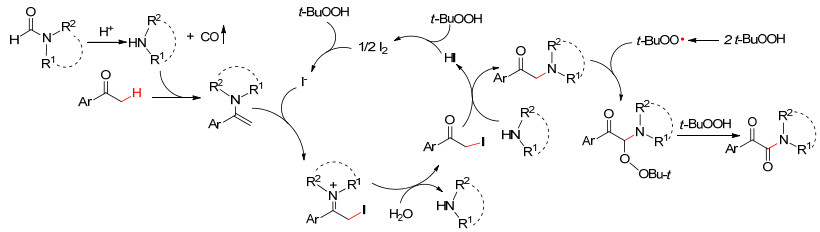 图 图式 5
芳乙酮与N-烷基甲酰胺的交叉偶联反应机理
Figure 图式 5.
Proposed mechanism for direct amidation for acetophone with N-substituted formamides
图 图式 5
芳乙酮与N-烷基甲酰胺的交叉偶联反应机理
Figure 图式 5.
Proposed mechanism for direct amidation for acetophone with N-substituted formamides
2014年, Sun等[22]报道了以n-Bu4NI为催化剂, TBHP为氧化剂的条件下, 芳基乙烷和甲酰胺生成α-酮酰胺类衍生物 (Eq. 14).值得注意的是, 通常C (sp3)—H的脱氢偶联发生在官能团的α-C (sp3)—H位, 而该反应是芳基乙烷β-C (sp3)—H键的活化.作者提出反应可能的机理为芳基乙烷在TBHP促进下, 原位生成芳基乙酮, 再发生后续脱氢脱酰基偶联 (Scheme 6).
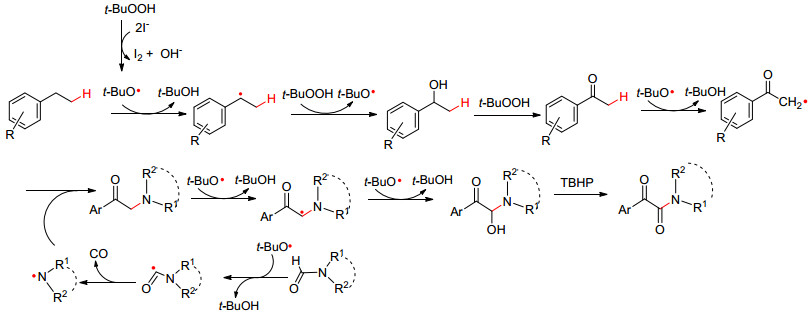 图 图式 6
芳基乙烷与N-烷基甲酰胺的交叉偶联反应机理
Figure 图式 6.
Proposed mechanism for direct amidation for ethylarenes with N-substituted formamides
图 图式 6
芳基乙烷与N-烷基甲酰胺的交叉偶联反应机理
Figure 图式 6.
Proposed mechanism for direct amidation for ethylarenes with N-substituted formamides
2 C (sp2)—H键官能化
由于sp2杂化碳自由基相对sp3杂化碳自由基更加不稳定, 引发sp2杂化碳参与自由基脱氢偶联难度较大. 2011年, DeBoef等[23]报道了在苯碘二乙酯 (PIDA) 氧化促进下, 芳烃C (sp2)—H与邻苯二甲酰亚胺直接脱氢偶联生成C (sp2)—N键 (Eq. 15), 反应无需过渡金属催化剂.富电子芳烃相比缺电子芳烃更易于与邻苯二甲酰亚胺发生反应.另外, 芳基连有取代基时, 比如甲苯, 也可以选择性地发生芳基上的C (sp2)—H活化胺化.文中提出了可能的反应机理, 即芳环被PIDA氧化为芳环碳正离子自由基, 然后邻苯二甲酰亚胺亲核进攻芳环碳正离子自由基, 最后被PIDA氧化生成目标产物.
对于烯烃sp2杂化碳参与此类自由基偶联反应时, 往往得到的是烯烃的双官能团化产物. 2013年, Zhu等[24]报道了无过渡金属催化芳基乙烯与含氮杂环发生自由基加成反应, 获得烯烃的羟基化胺化产物, 产率最高可达91% (Eq. 16).机理研究显示, TBHP分解产生叔丁氧自由基, 然后夺取胺基上的氢得到N自由基, 进而对烯烃双键进行自由基加成, 产生的苄基自由基中间体被氧化成苄基正离子, 最后水化得到羟基化胺化双取代产物 (Scheme 7).
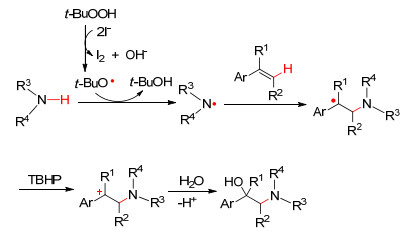 图 图式 7
烯烃的双官能团化反应机理
Figure 图式 7.
Proposed mechanism for difunctionalization of alkenes
图 图式 7
烯烃的双官能团化反应机理
Figure 图式 7.
Proposed mechanism for difunctionalization of alkenes
另外, 2014年, Du等[25]报道了以TBAI为催化剂, TBHP为氧化剂, 烯胺中C (sp2)—H与含氮化合物直接脱氢偶联生成C (sp2)—N键 (Eq. 17).文章指出烯烃双键两端的官能团对反应影响较大, 吸电子基团有利于反应的进行.该反应在2~3 h内就能转化完全, 为多取代烯烃的快速制备提供了好的选择, 产率中等至优秀.
Jiang等[26]报道了在TBAI/TBHP催化体系下, 磺酰肼对吲哚C (2) 和C (3) 位的双官能化, 同时构建C—S和C—N键.而当吲哚C (3) 位甲基取代时, 可以得到单一的C (sp2)—N偶联产物 (Scheme 8).该反应经历了自由基过程, 并且吲哚C (2)-重氮化优先于C (3)-磺酰化.
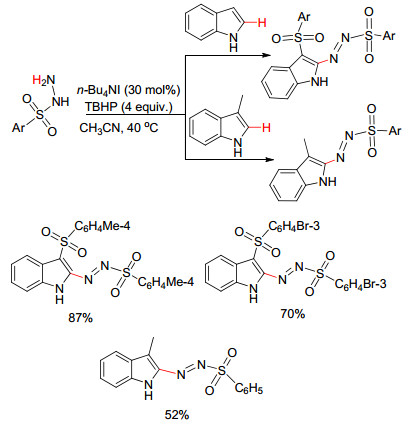 图 图式 8
吲哚的选择性磺酰化和重氮化
Figure 图式 8.
Selective sulfonylation and diazotization of indoles
图 图式 8
吲哚的选择性磺酰化和重氮化
Figure 图式 8.
Selective sulfonylation and diazotization of indoles
除了烯烃C (sp2)—H能活化脱氢, 醛氢也能被活化. 2014年, Reddy等[27]报道了在TBHP促进下, 水杨醛可与脂环胺发生氧化交叉偶联, 构建新的酰胺键 (Eq. 18).反应条件温和, 无需过渡金属催化, 产率最高可达85%.水杨醛中酚羟基与醛基的分子内氢键, 在反应中起到重要作用.
Li等[28]报道了以KI为催化剂, TBHP为氧化剂, 芳基醛和含氮杂环的直接脱氢偶联 (Eq. 19).其中苯甲醛与苯并咪唑反应可以扩大到克级别, 产率达85%, 为唑类N-酰基化 (如N-酰基吡唑、N-酰基苯并咪唑、N-酰基三氮唑、N-酰基吲唑类) 合成提供一条高效途径.机理研究揭示KI作为引诱TBHP分解产生叔丁氧自由基, 然后夺取芳基醛氢, 产生的芳基羰基自由基与氮自由基发生自由基偶联, 实现含氮杂环的酰基化.
2015年, Wang等[29]报道了在三氯异氰尿酸 (TCCA) 作用下, 室温下无金属催化醛与伯/仲胺的直接交叉脱氢偶联 (Eq. 20).有意思的是, 该反应既可以在水相中发生, 并且伯胺也能顺利参与该偶联反应.除芳香醛外, 脂肪醛亦可高效地参与该酰胺键的合成, 不足之处在于未对反应机理进行研究.
同年, Shah等[30]报道了在无金属催化下醛与胺直接脱氢偶联.在吡啶作用下苯甲醛可与仲胺反应; 而在TBAI催化下, 醛可与伯胺发生自由基偶联反应 (Scheme 9).文中指出两种反应路径不同, 在吡啶作用下存在亚胺阳离子的氧化, 而TBAI催化下经历的是自由基过程 (Scheme 10).
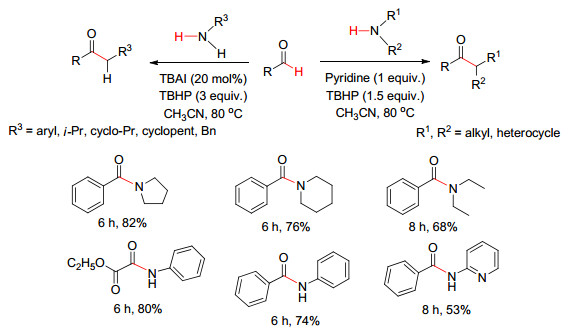 图 图式 9
醛与胺的直接交叉偶联反应
Figure 图式 9.
Dehydrogenative cross-coupling of aldehydes with amines
图 图式 9
醛与胺的直接交叉偶联反应
Figure 图式 9.
Dehydrogenative cross-coupling of aldehydes with amines
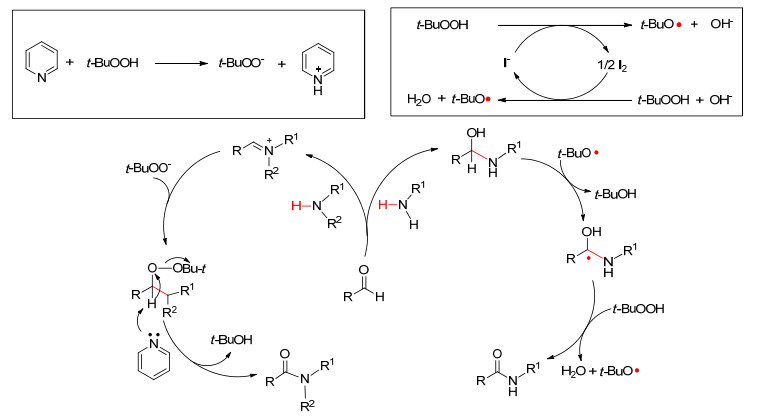 图 图式 10
醛与伯、仲胺反应的可能机理
Figure 图式 10.
Proposed mechanism for the reaction of aldehyde with primary/secondary amine
图 图式 10
醛与伯、仲胺反应的可能机理
Figure 图式 10.
Proposed mechanism for the reaction of aldehyde with primary/secondary amine
2014年, Chen和Yu等[31]报道了以碳酸氢铵为氮源, 在四乙基碘化铵 (Et4NI) 为催化剂, TBHP为氧化剂下, 醛与碳酸氢铵的氧化脱氢反应合成酰胺 (Eq. 21).另外, 醇也可以和碳酸氢铵反应, 但产率相对醛参与的反应低.碳酸氢铵受热易分解产生氨气, 和醛反应产生半缩醛胺, 然后被叔丁氧自由基夺氢, 最后与醛偶联得到酰胺.在反应中通过加入自由基抑制剂2, 2, 6, 6-四甲基哌啶氧化物 (TEMPO), 可以抑制反应发生, 证明反应可能经历了自由基路径.反应使用碳酸氢铵作为氨基源, 反应条件温和, 无需添加过渡金属, 具有较好的工业化应用前景.
2015年, Deng等[32]报道了醛在TBAI/TBHP催化体系下, 与芳基亚磺酰亚胺直接脱氢偶联构建新的C (sp2)—N键 (Eq. 22).文章指出TBAI诱导TBHP分解, 产生叔丁基氧自由基, 然后诱导醛C (sp2)—H活化, 产生酰基自由基, 最后与芳基亚磺酰亚胺偶联生成最终产物.反应同样无需过渡金属催化, 底物官能团耐受范围广, 产率最高可达90%.
2012年, Wan等[33]报道了以TBAI为催化剂, TBHP为氧化剂, 醛与甲酰胺脱氢脱酰基偶联生成C (sp2)—N键 (Eq. 23). TBAI诱导TBHP分解产生叔丁氧自由基或叔丁基过氧自由基, 进而夺取醛氢和甲酰胺的羰基, 从而产生的羰基自由基和胺自由基偶联形成新的酰胺结构.作者通过13C同位素标记实验, 证明了酰胺中羰基碳来源于醛而不是甲酰胺.
N—H和N-CO键可在上述反应条件下发生断裂, N—Me键也可在一定条件下活化, 构建新的C (sp2)—N键. 2013年, Mai等[34]报道了醛与叔胺在TBAI/TBHP催化体系下的脱氢脱甲基偶联 (Eq. 24).随后, Yu等[35]也报道了醛与二甲基胺的偶联反应, 构建新的酰胺键 (Eq. 25).不同的是, 他们以乙酸乙酯作溶剂, 催化剂的用量低至2.5 mol%, 最高产率可达99%.
2014年, Singh等[36]报道了无过渡金属催化醛与N-氯代胺脱氢脱氯偶联, 合成新的酰胺 (Eq. 26).反应以偶氮二异丁腈 (AIBN) 为自由基引发剂, TBHP为氧化剂, AIBN在加热下分解为腈基异丙基自由基, 进而夺取醛氢和N-氯分别得到酰基自由基和胺自由基, 最终交叉偶联生成酰胺.反应条件绿色温和, 底物适应性广泛.
Mal等[37]也报道了在TBAI/TBHP催化下, 醛与N-氯代胺的脱氢脱氯偶联形成C (sp2)—N键 (Eq. 27).与Sing等[36]报道的方法不同, 反应以TBAI为催化剂, 在50 ℃无需溶剂条件下或室温球磨条件即可发生.
利用可见光诱导N—Br键断裂产生氮自由基, 对芳烃和杂芳烃进行直接酰亚胺化是非常绿色高效的一种途径. 2014年, Luo等[38a]报道了N-溴代糖精分子可以在可见光促进下, 作为氮自由基前体与芳烃或杂芳烃进行交叉偶联 (Eq. 28).反应表现出优秀的化学和区域选择性, 并且无需添加任何催化剂, 常温下即可发生.机理研究揭示, 该反应经历了自由基过程, 其自由基终止阶段是由电子转移-质子转移导致的.近期该小组[38a]还报道了在类似条件下, N-溴代糖精对直链烯烃的酰亚胺化.此类方法仅需要可见光条件, 无需任何催化剂, 符合绿色化学发展的需求.
此外, Wang等[38b]还报道了无过渡金属条件下, 苯并噁唑2-位直接胺化.在TBAI/TBHP催化体系下, 苯并噁唑与甲酰胺脱氢脱酰基偶联, 合成系列2-氨基苯并噁唑衍生物 (Eq. 29).该反应需要较大量的氧化剂TBHP, 以及酸性条件.
3 C (sp)—H键官能化
sp杂化碳原子参与的自由基偶联构建C—N的例子较少, 主要以炔烃的双官能团化产物为主. 2014年, Chaskar等[39]报道了无过渡金属催化下, 以I2为催化剂, TBHP和DMSO为共氧化剂, 苯乙炔和苄脒盐酸盐反应生成α-酮酰亚胺 (Eq. 30).文中指出该反应可能经历以下历程:苯乙炔在碘和TBHP作用下产生α-碘苯乙酮, 进而在DMSO氧化下生成苯甲酰甲醛, 随后亲核性苄脒进攻醛基碳, 最终被碘氧化并水解得到α-酮酰亚胺 (Scheme 11).
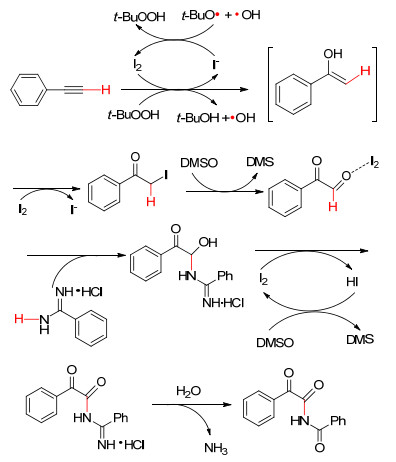 图 图式 11
苯乙炔与苄脒盐酸盐的反应机理
Figure 图式 11.
Proposed mechanism for the reaction of phenylacetylene with benzamidine hydrochloride
图 图式 11
苯乙炔与苄脒盐酸盐的反应机理
Figure 图式 11.
Proposed mechanism for the reaction of phenylacetylene with benzamidine hydrochloride
4 结论与展望
综上所述, 最近发展的无过渡金属催化下, sp3, sp2和sp C—H键断裂, 与各种胺、酰胺以及含氮杂环等进行自由基交叉偶联, 为胺、酰胺、α-酮酰胺等的合成提供了一条绿色、高效以及原子经济型途径.目前还有很多领域值得去拓展: (1) sp2-, sp-C—H键活化参与构建不饱和C—N键仍需进一步研究.尤其是碳碳重键参与反应时, 通常发生的是重键的双官能团化, 如何保留碳碳重键在无过渡金属催化下进行烯炔烃的直接胺化具有较大挑战性; (2) 借助光诱导产生自由基, 同时避免金属催化剂的使用, 是自由基诱导合成C—N键的新研究方向; (3) 无过渡金属催化的自由基偶联机理尚不明确.
-
-
[1]
(a) Humphrey, J.-M.; Chamberlin, A.-R. Chem. Rev. 1997, 97, 2243.
(b) Sheha, M.-M.; Mahfouz, N.-M.; Hassan, H.-Y.; Youssel, A.-F.; Mimoto, T.; Kiso, Y. Eur. J. Med. Chem. 2000, 35, 887. -
[2]
(a) Davies, H. M. L.; Long, M.-S. Angew. Chem., Int. Ed. 2005, 44, 3518.
(b) Naota, T.; Murahashi, S. Synlett 1991, 693. -
[3]
(a) Gunanathan, C.; Ben-David, Y.; Milstein, D. Science 2007, 317, 790.
(b) Muthaiah, S.; Ghosh, S.-C.; Jee, J.-E.; Chen, C.; Zhang, J.; Hong, S.-H. J. Org. Chem. 2010, 75, 3002.
(c) Lu, B.-L.; Li, X.-Y.; Lin, Y.-M. Chin. J. Org. Chem. 2015, 35, 2275 (in Chinese).
(卢贝丽, 李现艳, 林咏梅, 有机化学, 2015, 35, 2275.)
(d) Zhog, Y.-X.; Ren, K.; Xie, X.-M.; Zhang, Z.-G. Chin. J. Org. Chem. 2016, 36, 258 (in Chinese).
(钟业辛, 任凯, 谢小敏, 张兆国, 有机化学, 2016, 36, 258.)
(e) Qiu, D.; Qiu, M.-L.; Ma, R.; Wang, J.-B. Acta Chim. Sinica 2016, 74, 472 (in Chinese).
(邱頔, 邱孟龙, 马戎, 张艳, 王剑波, 化学学报, 2016, 74, 472.)
(f) Zhang, W.-M.; Dai, J.-J.; Xu, H.-J. Chin. J. Org. Chem. 2015, 35, 1820 (in Chinese).
(张文曼, 戴建军, 许华建, 有机化学, 2015, 35, 1820.) -
[4]
Liu, D.; Lei, A. Chem. Asian J. 2015, 10, 806. doi: 10.1002/asia.v10.4
-
[5]
Li, C.-J. Acc. Chem. Res. 2008, 42, 335.
-
[6]
Lao, Z.-Q.; Zhong, W.-H.; Lou, Q.-H.; Li, Z.-J.; Meng, X.-B. Org. Biomol. Chem. 2012, 10, 7869. doi: 10.1039/c2ob26430g
-
[7]
Li, L.-T.; Li, H.-Y.; Xing, L.-J.; Wen, L.-J.; Wang, P.; Wang, B. Org. Biomol. Chem. 2012, 10, 9519. doi: 10.1039/c2ob26636a
-
[8]
Sun, K.; Wang, X.; Li, G.; Jiang, Y.-Q.; Xiao, B.-B. Chem. Commun. 2014, 50, 12880. doi: 10.1039/C4CC06003B
-
[9]
Dian, L.; Wang, S.; Negrerie, D.-Z.; Du, Y.-F.; Zhao, K. Chem. Commun. 2014, 50, 11738. doi: 10.1039/C4CC05758A
-
[10]
Rajamanickam, S.; Majji, G.; Santra, S.-K.; Patel, B.-K. Org. Lett. 2015, 17, 5586. doi: 10.1021/acs.orglett.5b02749
-
[11]
Lv, Y.-H.; Li, Y.; Xiong, T.; Lu, Y.; Liu, Q.; Zhang, Q. Chem. Commun. 2014, 50, 2367. doi: 10.1039/c3cc48887j
-
[12]
Xue, Q.-C.; Xie, J.-X.; Li, H.-M.; Cheng, Y.-X.; Zhu, C.-J. Chem. Commun. 2013, 49, 3700. doi: 10.1039/c3cc41558a
-
[13]
Wang, L.; Zhu, K.; Chen, Q.; He, M.-Y. J. Org. Chem. 2014, 79, 11780. doi: 10.1021/jo502283h
-
[14]
Aruri, H.; Singh, U.; Kumar, S.; Kushwaha, M.; Gupta, A.-P.; Vishwakarma, R.-A.; Singh, P.-P. Org. Lett. 2016, 18, 3638 doi: 10.1021/acs.orglett.6b01684
-
[15]
Sun, J.-W.; Wang, Y.; Pan, Y. J. Org. Chem. 2015, 80, 8945. doi: 10.1021/acs.joc.5b01383
-
[16]
Lv, Y.-H.; Sun, K.; Wang, T.-T.; Wu, Y.-T.; Li, G.; Pu, W.-Y.; Mao, S.-K. Asian J. Org. Chem. 2016, 5, 325 doi: 10.1002/ajoc.v5.3
-
[17]
Xu, K.; Hu, Y.; Zhang, S.; Zhang, S.; Zha, Z.; Wang, Z.-Y. Chem.-Eur. J. 2012, 18, 9793. doi: 10.1002/chem.v18.32
-
[18]
Gao, L.-F.; Tang, H.-M.; Wang, Z.-Y. Chem. Commun. 2014, 50, 4085. doi: 10.1039/c4cc00621f
-
[19]
Mai, W.-P.; Wang, H.-H.; Li, Z.-C.; Yuan, J.-W.; Xiao, Y.-M.; Yang, L.-R.; Mao, P.; Qu, L.-B. Chem. Commun. 2012, 48, 10117. doi: 10.1039/c2cc35279f
-
[20]
Lamani, M.; Prabhu, K.-R. Chem. Eur. J. 2012, 18, 14638. doi: 10.1002/chem.201202703
-
[21]
Zhao, Q.; Miao, T.; Zhang, X.-B.; Z, W.; W, L. Org. Biomol. Chem. 2013, 11, 1867. doi: 10.1039/c3ob27433k
-
[22]
Du, B.-N.; Jin, B.; Sun, P.-P. Org. Biomol. Chem. 2014, 12, 4586. doi: 10.1039/c4ob00520a
-
[23]
Kantak, A.-A.; Potavathri, S.; Barham, R.-A.; Romano, K.-M.; DeBoef, B. J. Am. Chem. Soc. 2011, 133, 19960. doi: 10.1021/ja2087085
-
[24]
Xue, Q.; Xie, J.; Xu, P.; Hu, K.-D.; Cheng, Y.-X.; Zhu, C.-J. ACS Catal. 2013, 3, 1365. doi: 10.1021/cs400250m
-
[25]
Yuan, Y.-C.; Hou, W.-J.; Zhang-Negrerie, D.; Zhao, K.; Du, Y.-F. Org. Lett. 2014, 16, 5410. doi: 10.1021/ol5026525
-
[26]
Qiu, J.-K.; Hao, W.-J.; Wang, D.-C.; Wei, P.; Sun, J.; Jiang, B.; Tu, S.-J. Chem. Commun. 2014, 50, 14782. doi: 10.1039/C4CC06795A
-
[27]
Prasad, K.-R.; Suresh, P.; Ravikumar, B.; Reddy, N.-V.; Reddy, K.-R. Tetrahedron Lett. 2014, 55, 6307. doi: 10.1016/j.tetlet.2014.09.080
-
[28]
Zhao, J.-J.; Li, P, ; Xia, C.-G.; Li, F.-W. Chem. Commun. 2014, 50, 4751. doi: 10.1039/c4cc01587h
-
[29]
Yang, H.-Y.; Hu, W.-J.; Deng, S.-J.; Wu, T.-T.; Cen, H.-N.; Cen, Y.-P.; Zhang, D.-L.; Wang, B. New J. Chem. 2015, 39, 5912. doi: 10.1039/C5NJ01372K
-
[30]
Deshidi, R.; Rizvi, M.-A.; Shah, B.-A. RSC Adv. 2015, 5, 90521. doi: 10.1039/C5RA17425B
-
[31]
Wang, G.; Yu, Q.-Y.; Chen, S.-Y.; Yu, X.-Q. Org. Biomol. Chem. 2013, 12, 414.
-
[32]
Qin, W.-J.; Li, Y.; Yu, X.-X.; Deng, W.-P. Tetrahedron 2015, 71, 1182. doi: 10.1016/j.tet.2015.01.013
-
[33]
Liu, Z.-J.; Zhang, J.; Chen, S.-L.; Shi, E.-B.; Xu, Y.; Wan, X.-B. Angew. Chem., Int. Ed. 2012, 51, 3231. doi: 10.1002/anie.v51.13
-
[34]
Mai, W.-P.; Song, G.; Yuan, J.-W.; Yang, L.-R.; Sun, G.-C.; Xiao, Y.-M.; Mao, P.; Qu, L.-B. RSC Adv. 2013, 3, 3869. doi: 10.1039/c3ra40298c
-
[35]
Wang, S.; Wang, J.; Guo, R.; Wang, G.; Chen, S.-Y.; Yu, X.-Q. Tetrahedron Lett. 2013, 54, 6233. doi: 10.1016/j.tetlet.2013.09.018
-
[36]
Vanjari, R.; Guntreddi, T.; Singh, K.-N. Green Chem. 2014, 16, 351. doi: 10.1039/C3GC41548A
-
[37]
Achar, T.-K.; Mal, P. J. Org. Chem. 2014, 80, 666.
-
[38]
(a) Song, L.; Zhang, L.; Luo, S.; Cheng, J.-P. Chem. Eur. J. 2014, 20, 14231.
(b) Wang, R.; Liu, H.; Yue, L.; Zhang, X.-K.; Tan, Q.-Y.; Pan, R.-L. Tetrahedron Lett. 2014, 55, 2233. -
[39]
Kalmode, H.-P.; Vadagaonkar, K.-S.; Chaskar, A.-C. RSC Adv. 2014, 4, 565.
-
[1]
-

图式 2 无过渡金属催化四唑直接烷基化
Scheme 2 Transition-metal-free direct alkylation of aryl tetrazoles

图式 4 芳甲醇与N-烷基甲酰胺的交叉偶联反应机理
Scheme 4 Proposed mechanism for direct amidation for alcohols with N-substituted formamides

图式 5 芳乙酮与N-烷基甲酰胺的交叉偶联反应机理
Scheme 5 Proposed mechanism for direct amidation for acetophone with N-substituted formamides

图式 6 芳基乙烷与N-烷基甲酰胺的交叉偶联反应机理
Scheme 6 Proposed mechanism for direct amidation for ethylarenes with N-substituted formamides

图式 10 醛与伯、仲胺反应的可能机理
Scheme 10 Proposed mechanism for the reaction of aldehyde with primary/secondary amine
-


 下载:
下载:










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 40
- 文章访问数: 3060
- HTML全文浏览量: 823
 下载:
下载:
