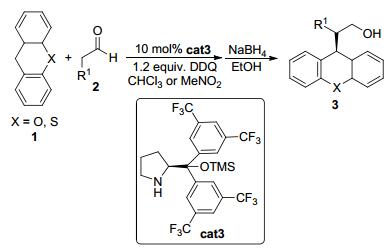
 图 图式1
有机催化的醛与苄基化合物的脱氢烷基化反应
Figure 图式1.
Organocatalytic enantioselective dehydrogenative alkylation of aldehydes with benzylic compounds
图 图式1
有机催化的醛与苄基化合物的脱氢烷基化反应
Figure 图式1.
Organocatalytic enantioselective dehydrogenative alkylation of aldehydes with benzylic compounds
引用本文:
江欣, 王斯顿, 郭贵敏, 卢贝丽. 无金属参与苄位C (sp3)—H键不对称直接官能团化研究进展[J]. 有机化学,
2017, 37(4): 841-857.
doi:
10.6023/cjoc201610010

Citation: Jiang Xin, Wang Sidun, Guo Guimin, Lu Beili. Recent Development of Metal-Free Direct Asymmetric Functionalization of Benzylic C (sp3)—H Bond[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 841-857. doi: 10.6023/cjoc201610010
Citation: Jiang Xin, Wang Sidun, Guo Guimin, Lu Beili. Recent Development of Metal-Free Direct Asymmetric Functionalization of Benzylic C (sp3)—H Bond[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 841-857. doi: 10.6023/cjoc201610010
无金属参与苄位C (sp3)—H键不对称直接官能团化研究进展
English
Recent Development of Metal-Free Direct Asymmetric Functionalization of Benzylic C (sp3)—H Bond
Abstract:
Compounds containing aryl structures are important organic synthesis intermediates, which are widely present in a large family of natural products and bioactive molecules. In recent years, direct asymmetric functionalization of benzylic C (sp3)—H bond for the efficient construction of arene and hetero-arene motifs with high stereoselectivity has drawn widespread concern from chemical community. Among the various strategies, small molecule-catalyzed metal-free functionalization of benzylic C (sp3)—H bond represents a more challenging but promising transformations. This review is intended to summarize and discuss the most recent advances in this area.
-
Key words:
- metal-free
- / benzylic position
- / C—H functionalization
- / organocatalysis
- / asymmetric synthesis
-
C—H键的直接官能团化是现代有机合成化学研究的一个重要课题.由于C—H键广泛存在于各类化合物中, 通过C—H键的直接官能团化构建C—C键、C—X键等新化学键, 无须对原料预先活化, 可有效简化操作步骤, 减少废弃物和副产物的生成, 符合“原子经济性 (atom economy)”和“路线经济性 (step economy)”等环境友好的绿色化学要求. C (sp3)—H键在有机化合物中的存在数目最多, 且其官能团化可以直接构建手性中心, 因而C (sp3)—H键的不对称直接官能团化引起了学术界的广泛关注[1].该领域目前的主要研究集中于过渡金属催化的C (sp3)—H键不对称官能团化[2].过渡金属可以在导向基团和手性配体的作用下活化C (sp3)—H键并形成新的化学键.在过去十几年里, 一系列的重要成果被发现并对该领域的进一步发展起到积极的推动作用.然而过渡金属大部分是重金属, 对环境具有一定的危害性; 并且与金属相互作用的手性配体通常需要从较为昂贵的原料出发, 经历冗长的实验步骤制备.这些缺陷迫切需要人们发展新的方法学, 例如在无金属参与的情况下实现C (sp3)—H键的不对称官能团化.
2000年以来, 由于绿色环保、高立体选择性等优点, 有机小分子催化化学得到了迅速的发展, 其重要应用之一是从简单底物出发实现复杂分子的不对称合成[3].但是有机小分子催化的不对称C (sp3)—H键官能团化仍然面临着比较大的挑战: (1) C (sp3)—H键是有机分子中最基本的化学键, 因而相比通常的不对称合成, 在众多具有类似性质的化学键中高区域和立体选择性地实现有机分子中某一特定C (sp3)—H键的不对称转化比较困难; (2) 作为键能较强的化学键之一, C—H键的键能高 (大约90~110 kcal/mol), 因而直接断裂C—H键并转化为其他有用官能团所需要的能量也较高, 这就造成反应需要在高温、化学计量的氧化剂等比较剧烈的条件下反应.这对于不对称转化是不利的, 因为要实现能量差别很小的立体异构的过渡态的有效区分, 反应必须要在比较温和的条件下进行; (3) 由于复杂的条件限制, 目前成熟的催化体系在此条件下常常失效, 必须发展新型的手性催化体系才能实现其顺利转化.
苯环、吲哚、呋喃等芳基结构广泛存在于天然产物和生物活性分子中, 含芳基的化合物是重要的有机合成中间体.苄位C (sp3)—H键由于与芳环相连, 具有一定的活性.通过合适的条件筛选, 可以有效区分苄位C (sp3)—H键与其他C—H键, 从而实现有机催化的无金属参与苄位C (sp3)—H键高区域和立体选择性的不对称官能团化, 同时构建在合成化学、生物医药、材料化工等领域具有重要作用的含芳基化合物.近年来通过有机催化的无金属参与的苄位C (sp3)—H键官能团化构建含芳基化合物的研究引起了化学家们的广泛兴趣, 许多新颖的方法学被报道, 然而目前关于这方面的综述仍然较少[4].本文将针对此方法学, 分别介绍通过交叉脱氢偶联反应、羰基化合物的β-官能团化、[1, 5]-氢迁移反应以及碱辅助的去质子化这等重要的无金属参与的苄位C (sp3)—H键的不对称直接官能团化的反应模式, 并总结该领域近年来的主要研究进展.
1 通过交叉脱氢偶联反应实现苄位C (sp3)— H键不对称官能团化
交叉脱氢偶联反应 (Cross dehydrogenative coupling reaction, CDC反应) 是由Murahashi和Li小组[5]首先发展并随后得到广泛应用的.它指的是与氮原子、氧原子或羰基相连的C (sp3)—H键与多种C (sp)—H, C (sp2)—H或C (sp3)—H键在氧化条件下直接发生偶联反应从而构建新的碳碳键.该类反应无须对底物进行预活化, 且反应选择性好、普适性广, 因而近年来该方法已经发展成为构建碳碳键的一种重要方法.采用手性催化剂来催化该类型反应且无需金属参与的的不对称转化也已经顺利实现.
2009年, Cozzi小组[6]报道了通过有机催化的C—H键活化策略实现醛α位立体选择性的烷基化反应.他们发现在2, 3-二氯-5, 6-二氰对苯醌 (DDQ) 为氧化剂的条件下, 含有苄基的化合物氧杂蒽1a的苄位C—H键可以被氧化成碳正离子 (Eq. 1).随后在MacMillan类型的手性胺催化剂cat1•TFA作用下, 烷基醛2可进攻此碳正离子得到SN1类型的加成产物3.该反应的最优反应条件是以二氯甲烷为溶剂, 在-25 ℃可以最高78% ee的立体选择性得到加成产物.
2010年, Jang小组[7]实现了有机催化的醛的α烷基化反应 (Eq. 2).该反应使用MacMillan类型的手性胺cat1作为手性催化剂, 通过环境友好的电化学方法实现氧化, 因而反应过程中无须使用化学氧化剂.对于苯丙醛底物2a而言, 反应以良好的收率和中等的立体选择性顺利实现了醛和氧杂蒽1a以及环庚三烯类底物的氧化交叉偶联.该小组通过研究发现, 该反应可能是经历了阳离子自由基烯胺中间体, 该中间体可以通过循环伏安实验证实.反应的机理也进一步地通过密度泛函理论 (DFT) 计算以及控制实验进行深入的研究.
2011年, Jiao等[8]发现采用环境友好、安全无毒害的氧气为氧化剂, 含有苄基的化合物氧杂蒽1也可以在MacMillan类型的二级胺cat2的催化下发生氧化脱氢交叉偶联反应 (Eq. 3).该反应底物氧杂蒽的苄位C—H键在实验条件下首先被氧化成碳正离子, 随后经历了形式上的苄位C (sp3)—H键官能团化从而得到最终产物3.在底物扩展中, 他们发现含有硫原子以及NMe的其他杂环化合物也能顺利反应, 以高达93% ee的手性诱导效率得到产物.
2012年, Xiao等[9]发现三甲基硅基保护的二芳基脯氨醇cat3也可以催化氧化脱氢交叉偶联反应 (Scheme 1).以DDQ作为氧化剂, 烷基醛2可与氧杂蒽或硫杂蒽1在温和的条件下发生反应.对于大多数底物, 该反应都可以大于90% ee的立体选择性得到偶联产物.另外他们还发现, 当使用1, 3, 5-环庚三烯为底物与烷基醛发生氧化偶联反应时, 也可以优秀的收率和立体选择性得到偶联产物.
图 图式1
有机催化的醛与苄基化合物的脱氢烷基化反应
Figure 图式1.
Organocatalytic enantioselective dehydrogenative alkylation of aldehydes with benzylic compounds
2011年, Tan小组[10]发现当采用催化量的玫瑰红染料 (5 mol%) 作为光敏剂, 绿光作为光源, 在空气存在下, N-芳基四氢喹啉衍生物4可与丙酮5发生氧化脱氢交叉偶联反应 (Eq. 4).通过条件筛选, 他们发现当使用L-脯胺醇甲基醚cat4作为催化剂来催化这个反应时, 可以40%的收率和15% ee的立体选择性得到苄位C (sp3)—H键官能团化产物.该反应避免了使用金属和过氧化物作为氧化剂, 空气是实际的氧化物种, 通过可见光对染料的激发来实现底物的氧化, 符合环境友好的绿色化学准则, 对后来的研究者起到了很好的启迪作用.
2013年, Toste小组[11]采用不对称氧化脱氢交叉偶联反应, 通过苄位C (sp3)—H键的直接官能团化构建C—N键 (Eq. 5).他们发现N-芳基四氢异喹啉衍生物7在以4-乙酰氨基吗啉氮氧化合物oxidant1为氧化剂时, 可发生分子内的环化反应得到多环化合物8.在该反应中, 作者发展了一系列含有三氮唑基团的手性膦酸cat5作为催化剂.原位生成手性膦酸阴离子作为抗衡离子, 从而通过手性离子对的相互作用来实现催化.这种催化剂的优点在于既可以通过位阻作用实现立体位阻的区分, 又可以通过氢键与底物互相作用诱导手性, 因而该反应可以实现很好的立体控制.
2013年, Wang等[12]报道了一例手性紧密离子对控制的三级胺N-芳基四氢异喹啉衍生物9与酮类化合物10的氧化脱氢交叉偶联反应 (Scheme 2).该反应的机理如下:在该反应中, 酮类化合物10先与手性氨基酸cat6缩合得到带有羧基的烯胺中间体.与此同时, 在氧化条件下N-芳基四氢异喹啉衍生物9会被氧化成碳正离子并进一步转化为亚胺正离子中间体.此中间体可与反应后的氧化负离子形成紧密离子对.烯胺中间体可与氧化负离子发生交换得到亚胺正离子中间体与羧基负离子烯胺中间体的紧密离子对.最后发生烯胺对亚胺正离子的亲电加成并水解就可以得到目标产物11.通过这种紧密离子对中的两种活性中间体的相互作用, 该反应可以高达81%的收率, 以13:1的非对映选择性以及94% ee的立体选择性得到带有两个相连手性中心的四氢异喹啉衍生物11.反应中没有过渡金属参与, 最终顺利实现了苄位sp3-C—H键的不对称官能团化.
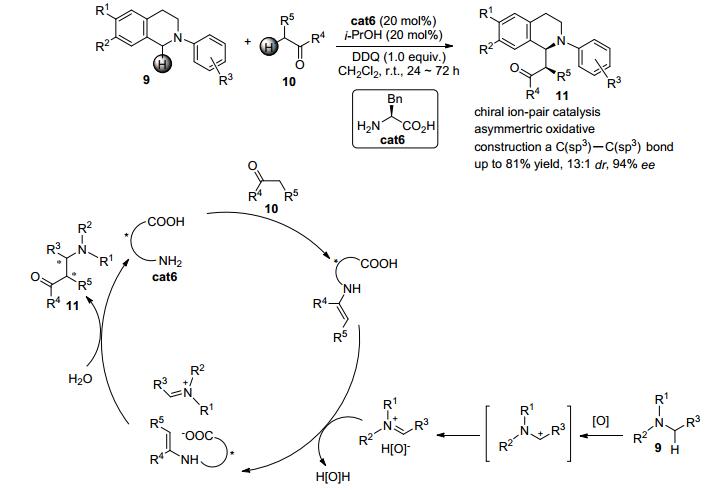 图 图式2
手性离子对控制的无金属参与的三级胺与酮的氧化脱氢偶联反应
Figure 图式2.
Chiral organic contact ion pairs in metal-free oxidative cross-dehydrogenative coupling of tertiary amines to ketones
图 图式2
手性离子对控制的无金属参与的三级胺与酮的氧化脱氢偶联反应
Figure 图式2.
Chiral organic contact ion pairs in metal-free oxidative cross-dehydrogenative coupling of tertiary amines to ketones
2014年, Liu等[13]发现醛类化合物2与苄基醚类化合物异色满12在DDQ作为氧化剂的条件下可以发生交叉脱氢偶联反应 (Eq. 6).以二级手性胺为催化剂, 异色满12可与多种醛类化合物2反应转化为含有两个连续手性中心的含氧杂环13.作者对一系列的二级手性胺进行条件筛选后发现, 当采用cat2•TFA为有机催化剂时, 该反应的立体选择性很高, 底物研究显示对于各种底物都可以90% ee以上的立体选择性得到产物13.机理研究揭示反应经历了与手性胺催化的四氢喹啉衍生物与酮的氧化脱氢偶联反应类似的反应路径.
2015年, Liu等[14]报道了第一例手性Brønsted酸催化的酰基保护的四氢β-咔啉类化合物14与芳基硼酸酯15或烯基硼酸酯17的氧化C (sp3)—H键官能团化反应 (Scheme 3).该反应采用酒石酸衍生的手性二醇为催化剂, 以二氯甲烷为溶剂、DDQ作为氧化剂, 在低温下可以实现四氢β-咔啉类化合物中吲哚单元的苄位C (sp3) —H键高区域和立体选择性的官能团化反应.需要指出的是, 在该反应中, 三氟乙醇以及六氟异丙醇作为添加剂对反应高立体选择性的实现至关重要.初步的机理研究显示, 反应中经历了氟代醇结合的四氢β-咔啉中间体, 而实际的亲核物种为二醇结合的硼酸酯化合物.
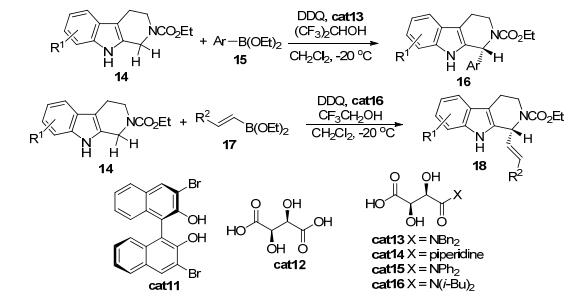 图 图式3
Brønsted酸催化的N-酰基四氢-β-咔啉类化合物的不对称C—H烯基化与芳基化反应
Figure 图式3.
Brønsted acid-catalyzed asymmetric C—H vinylation and arylation of N-carbamoyl tetrahydro-β-carbolines
图 图式3
Brønsted酸催化的N-酰基四氢-β-咔啉类化合物的不对称C—H烯基化与芳基化反应
Figure 图式3.
Brønsted acid-catalyzed asymmetric C—H vinylation and arylation of N-carbamoyl tetrahydro-β-carbolines
2015年, Liu等[15]进一步报道了手性Brønsted酸催化的酰基保护的四氢异喹啉类化合物19与烯基硼酸酯20的氧化C (sp3)—H键官能团化反应 (Eq. 7).通过仔细的条件筛选发现, 当采用酒石酸衍生酰胺cat16作为催化剂, CF3CH2OH为溶剂时, 反应结果最好.额外加入的CCl3CH2OH对反应的立体诱导起到一定的辅助作用.当反应在-20 ℃反应时, 对于大多数底物, 都可以大于90% ee的立体选择性得到氧化加成产物21.而产物中四氢异喹啉上的酰基保护基可以在温和的条件下脱去, 从而为反应的应用提供了方便, 扩大了该方法学的应用范围.
环状胺如四氢异喹啉类化合物与醛的氧化交叉偶联反应通常局限于活泼的N-芳基环状胺类化合物, 且多数情况下需要金属氧化剂参与反应, 目前无金属参与的不对称转化也有报道, 但是这些反应的底物比较局限. 2016年, Liu等[16]报道了一例无金属参与的N-酰基四氢异喹啉类化合物19与醛类化合物2的氧化交叉偶联反应, 从而实现了N-酰基四氢异喹啉类底物19中苄位C (sp3)—H键的不对称官能团化 (Eq. 8).该反应同样使用MacMillan手性二级胺催化剂cat2作为手性诱导试剂, DDQ作为氧化剂.为方便后处理, 反应得到的醛被原位还原成一级醇22.该反应可以最高97% ee的立体选择性、5:1的非对映选择性以及85%的收率得到具有连续两个手性中心的四氢异喹啉衍生物22.反应的最大特点在于使用了容易脱去的酰基保护基, 因而具有更好的底物普适性以及应用范围.
由此可见, 交叉脱氢偶联反应已经成为高区域和立体选择性地实现含芳基化合物苄位C (sp3)—H键官能团化的一种有效方法.通过该方法, 采用手性胺、手性紧密离子对以及手性Brønsted酸作为催化剂, 可以从多种底物出发在氧化条件下构建新的碳碳键.该方法也存在着明显的局限性, 如底物中C (sp3)—H键必须是两个芳基活化的氢或者既处于芳基苄位又处于杂原子的α位.总体来说, 该方法绿色高效、环境友好、可构建各种新的官能团, 因而得到了迅速的发展.
2 羰基化合物的β-官能团化实现苄位C (sp3)— H键不对称官能团化
羰基化合物的不对称α-官能团化是高立体选择性地构建C—C键的重要方法, 而关于简单的羰基化合物的不对称β-官能团化则研究得很少.二级胺与羰基缩合后生成的亚胺离子中间体可以转化为烯胺中间体, 随后通过与亲电试剂发生反应从而在醛、酮等羰基化合物的α位引入新的官能团.如果将这个过程逆转过来, 研究如何在合适的氧化剂存在下将烯胺中间体转化为亚胺离子中间体, 实现简单醛类化合物β位与亲核试剂的反应, 人们把这个过程称为“氧化烯胺催化” [17].
2011年, Hayashi等[18]报道了苯丙醛类化合物2a在氧化条件下与硝基甲烷发生不对称加成反应的例子 (Scheme 4).当采用三甲基硅基保护的二苯基脯氨醇cat10为催化剂、二氯二氰基苯醌 (DDQ) 作为氧化剂时, 苯丙醛2a中苯基的α位同时也是羰基的β位C (sp3)—H键发生形式上的C—H键活化, 与硝基甲烷高效、高立体选择性地得到产物.该反应被认为是苯丙醛 (2a) 与催化剂cat10发生缩合得到烯胺中间体, 该中间体被DDQ氧化得到烯基亚胺正离子中间体, 同时DDQ在此过程中被还原成酚式结构.在溶剂中微量水的作用下, 烯基亚胺正离子中间体会被水解为α, β-不饱和醛 (肉桂醛) 并释放出手性胺催化剂cat10, 从而实现对底物醛的氧化.而α, β-不饱和醛与手性胺在乙酸钠的作用下, 再次缩合得到烯基亚胺正离子中间体.随后硝基甲烷在碱的作用下发生去质子化并进攻烯基亚胺正离子中间体从而得到烯胺中间体, 最后水解即可得到最终的加成产物23.整体来看, 该反应具有如下特点: (1) 醛基β-H被CH2NO2取代; (2) 该反应是一个形式上的β-C (sp3)—H键官能团化; (3) 可以优秀的立体选择性得到反应产物, 且反应产物是重要的有机合成中间体; (4) 在氧化偶联过程中, 无须使用金属试剂; (5) 手性胺催化剂cat10在反应中起到了双重的作用:生成烯胺中间体及烯基亚胺正离子中间体.
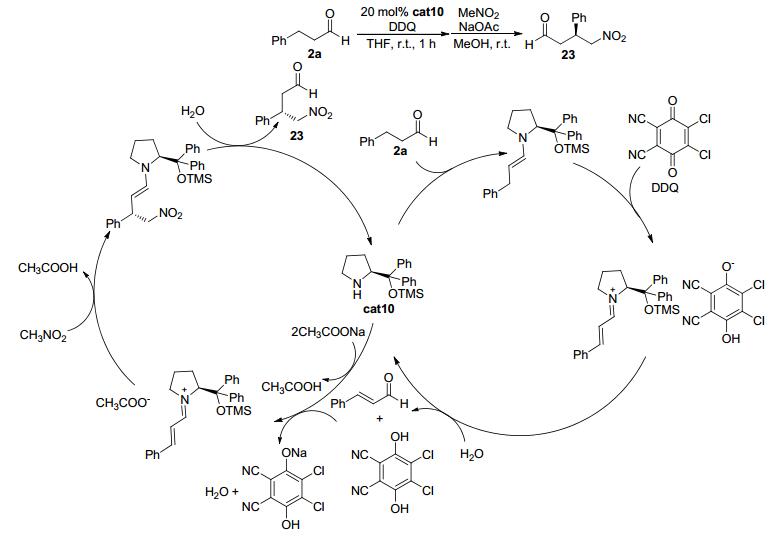 图 图式4
手性胺催化的醛与硝基甲烷的不对称氧化偶联反应
Figure 图式4.
Chiral amine-catalyzed enantioselective cross-coupling of aldehydes and nitromethane
图 图式4
手性胺催化的醛与硝基甲烷的不对称氧化偶联反应
Figure 图式4.
Chiral amine-catalyzed enantioselective cross-coupling of aldehydes and nitromethane
几乎同时, Wang课题组[19]发现使用二苯基脯胺醇硅醚cat10可以催化简单的脂肪醛2与二苯砜基氟代甲烷24 (FBSM) 的氧化交叉偶联反应, 得到β位官能团化的醛类化合物25 (Scheme 5).醛随后被原位还原成伯醇26以方便后处理.该反应使用2-碘酰基苯甲酸 (IBX) 作为氧化剂, 无须金属参与.对于大部分底物, 该反应都可以中等的收率和优秀的立体选择性得到交叉偶联产物26.另外当采用4-羟基香豆素27作为亲核试剂与苯丙醛2a反应时, 也可以良好的收率和中等的对映选择性得到多并环产物28.
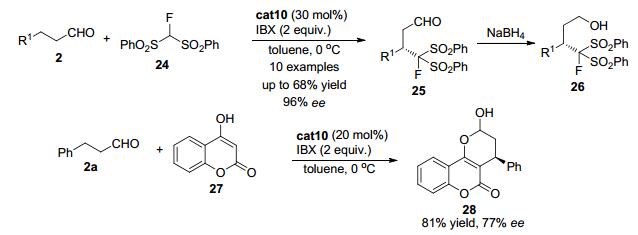 图 图式5
通过烯胺的氧化实现对映选择性的醛β-官能团化反应
Figure 图式5.
Enantioselective β-functionalization of aldehydes by oxidation of enamines
图 图式5
通过烯胺的氧化实现对映选择性的醛β-官能团化反应
Figure 图式5.
Enantioselective β-functionalization of aldehydes by oxidation of enamines
2013年, Enders等[20]发现苯丙醛2a与硝基乙烯类化合物29可以在二苯基脯胺醇硅醚cat10催化下发生串联环化反应得到五取代的环己烯类化合物30, 同时构建四个连续的手性中心 (Scheme 6).该反应可以几乎单一的非对映选择性和优秀的对映选择性得到环化产物, 且反应使用IBX作为氧化剂, 无须金属参与.另外当采用氧化吲哚31为底物与苯丙醛2a在同样条件下反应时, 可以构建含有一个螺环手性中心的化合物32.该反应的机理被认为是通过以下路径实现的:该反应过程中需要两分子的醛参与反应.两分子的醛与催化剂反应后得到两个烯胺中间体, 其中一个烯胺中间体在氧化剂的存在下被氧化为α, β-不饱和亚胺正离子中间体.另外一个烯胺中间体进攻缺电子的底物如硝基乙烯衍生物29或氧化吲哚衍生物31得到加成中间体.该中间体与α, β-不饱和亚胺正离子中间体发生加成及关环反应得到中间体, 随后发生水解从而得到目标分子.
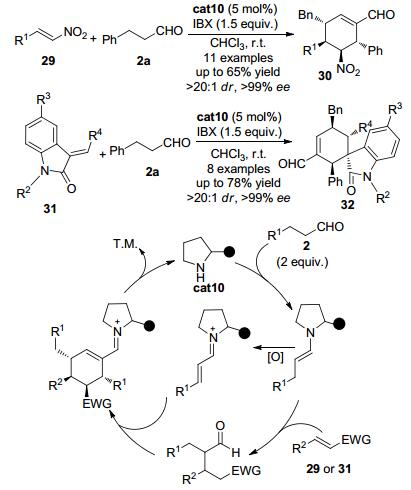 图 图式6
手性胺催化串联环化反应构建多官能团的环己烯衍生物
Figure 图式6.
Chiral amine-catalyzed domino cyclization reaction for the synthesis of polyfunctionalized cyclohexene derivatives
图 图式6
手性胺催化串联环化反应构建多官能团的环己烯衍生物
Figure 图式6.
Chiral amine-catalyzed domino cyclization reaction for the synthesis of polyfunctionalized cyclohexene derivatives
2013年, Chi等[21]报道了手性氮杂环卡宾催化剂催化的β-芳基醛类化合物2与1, 3-二羰基化合物33的氧化脱氢交叉偶联反应 (Scheme 7).使用茚胺醇衍生的三氮唑盐cat17作为氮杂环卡宾的催化剂前体, 使用1 equiv.的碳酸铯, 在醌34作为氧化剂的条件下, 该反应可以高达98%的收率和94% ee的对映选择性顺利得到环化产物.反应中化学计量的LiCl作为添加剂可以有效提高反应的手性诱导效率.该反应可能是通过以下机理来实现的:氮杂环卡宾进攻醛2中的羰基先生成Breslow中间体, 随后在氧化剂的作用下得到酰基三氮唑中间体.在碱的作用下, 该中间体可脱去一个质子得到烯醇唑类中间体.在氧化剂的作用下烯醇唑类中间体再发生一次氧化反应得到α, β-不饱和酰基唑类中间体.不饱和酰基唑类中间体最后在双亲核试剂1, 3-二羰基化合物33的作用下, 经历两次亲核进攻最终高立体选择性地得到环化产物35.
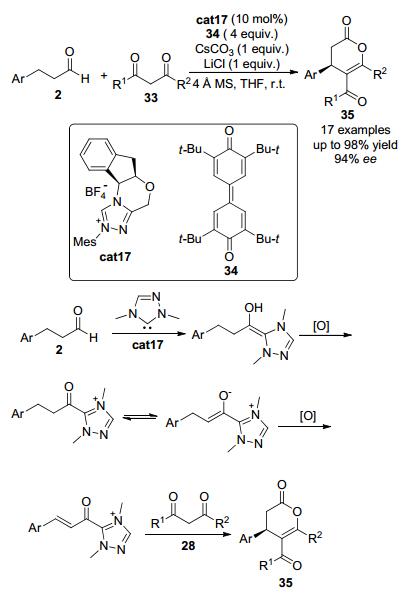 图 图式7
氧化氮杂环卡宾催化的饱和醛的β-C—H键活化
Figure 图式7.
Direct β-C—H bond activation of saturated aldehydes via oxidative NHC catalysis
图 图式7
氧化氮杂环卡宾催化的饱和醛的β-C—H键活化
Figure 图式7.
Direct β-C—H bond activation of saturated aldehydes via oxidative NHC catalysis
2013年, Chi小组[22]发现羰基β位为芳基的酯类化合物36在氮杂环卡宾催化剂作用下, 羰基β位的亚甲基可发生形式上的C (sp3)—H官能团化 (Scheme 8).以带有手性的三氮唑盐cat18作为催化剂前体, 加入化学计量的DBU为碱, 饱和酯类化合物36可与多种亲电试剂如查尔酮类化合物37, 三氟苯乙酮类化合物39以及腙类化合物41发生环化反应构建多种手性分子.该反应可以良好的收率和优秀的立体选择性得到含有两个相邻手性中心的五元环化合物, 如多取代的环戊烯类化合物38、五元内酯40以及五元内酰胺结构42.该反应被认为是通过以下反应历程来实现的:三氮唑盐cat18在碱的作用下可以原位生成氮杂环卡宾活性物种, 该活性物种可以进攻缺电子的对硝基苯酯的羰基得到酰基唑类中间体, 酰基唑类中间体在碱的作用下羰基β位可以脱去一个质子得到烯醇唑中间体.由于烯醇唑中间体中吸电子的唑类基团的存在, 羰基的β位的氢也具有一定的酸性, 可以在合适的条件下发生β-氢脱去.因而烯醇唑中间体可以在羰基的β位发生第二次去质子化作用得到类烯醇中间体 (homoenolate中间体), 并进一步发生互变异构得到β位富电子的酰基唑中间体, 该中间体具有亲核性, 可与多种亲电试剂反应, 从而实现羰基β位C (sp3)—H键的直接官能团化.以与腙类化合物41的反应为例, 酰基唑中间体由于羰基β位带有负电荷, 可以进攻缺电子的腙类底物41并进而发生环化反应得到最终产物42.
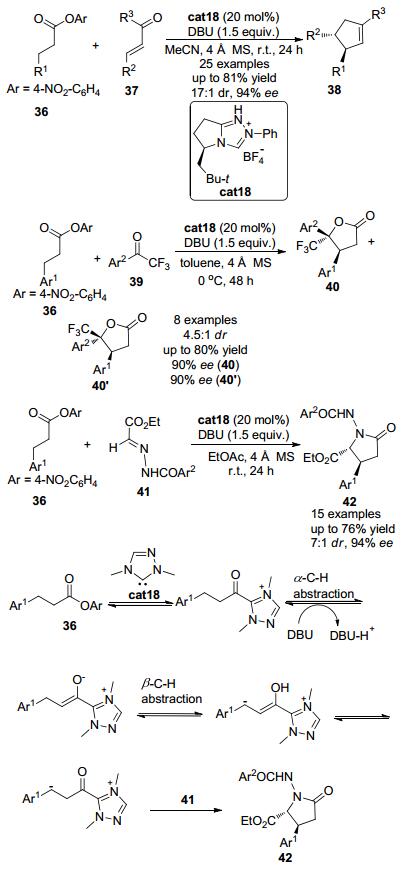 图 图式8
氮杂环卡宾催化的饱和羧酸酯的β-C—H键活化
Figure 图式8.
β-C—H bond activation of saturated carboxylic esters through N-heterocyclic carbene organocatalysis
图 图式8
氮杂环卡宾催化的饱和羧酸酯的β-C—H键活化
Figure 图式8.
β-C—H bond activation of saturated carboxylic esters through N-heterocyclic carbene organocatalysis
2014年, Chi小组[23]进一步发现以带有手性的三氮唑盐cat18作为氮杂环卡宾催化剂前体, 加入化学计量的DBU为碱, 羰基β位为芳基的对硝基苯酯类化合物36可与修饰过的查尔酮类底物43发生串联环化反应, 在此羰基β位的亚甲基也可发生形式上的C (sp3)—H官能团化 (Scheme 9).该反应可以良好的收率和优秀的立体选择性得到含有两个相邻手性中心的三并环44.该反应被认为是通过以下反应历程来实现的:三氮唑盐cat18在碱的作用下可以原位生成氮杂环卡宾活性物种 (NHC), 该活性物种可以进攻缺电子的对硝基苯酯的羰基得到酰基唑类中间体, 酰基唑类中间体在碱的作用下羰基α位C—H在外加的碱DBU的作用下脱去一个质子得到烯醇唑中间体.接着在烯醇唑中间体羰基的β位发生第二次去质子化作用得到类烯醇中间体, 并进一步与胺基烯酮类底物43发生形式上的Michael加成反应得到加成中间体.加成中间体发生氢迁移并接着发生分子内的aldol反应再发生氢迁移, 然后接着发生关环生成内酰胺并再生催化剂.内酰胺中间体发生脱水后即可得到最终产物44, 在此过程中实现羰基β位C (sp3)—H键的直接官能团化.
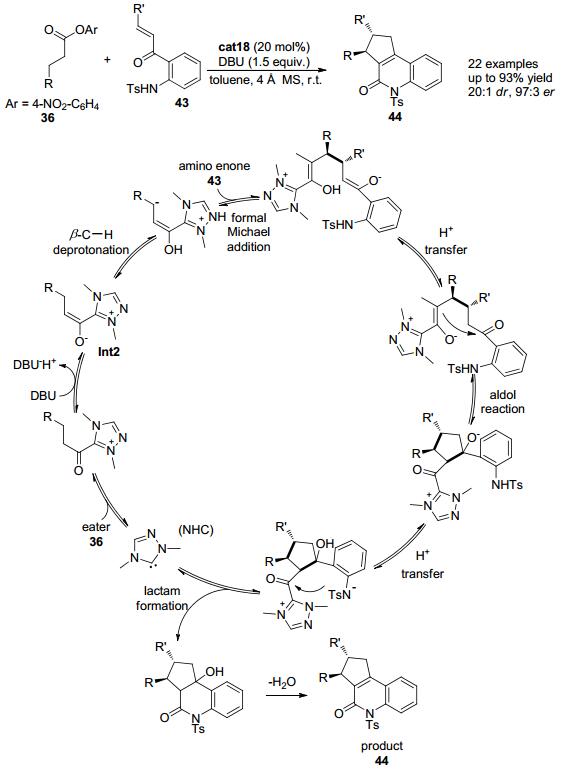 图 图式9
氮杂环卡宾催化的β-C—H键活化构建杂环化合物
Figure 图式9.
Access to heterocycles by NHC catalyzed ester β-C—H bond activation
图 图式9
氮杂环卡宾催化的β-C—H键活化构建杂环化合物
Figure 图式9.
Access to heterocycles by NHC catalyzed ester β-C—H bond activation
2014年, Chi小组[24]发现羰基β位为烷基或芳基的酸酐类化合物45也可采用手性三氮唑盐cat19作为氮杂环卡宾催化剂前体, 在化学计量的DMAP作用下与活化的α, β-不饱和酮类化合物发生串联的环化反应 (Eq. 9).在此过程中, 同样实现了芳基苄位又是羰基β位的C (sp3)—H不对称直接官能团化, 高效、高立体选择性地得到具有多个手性中心的环化产物.
2015年, Yao小组[25]使用羧酸作为底物, 通过羧酸与2-(7-偶氮苯并三氮唑)-N, N, N', N'-四甲基脲六氟磷酸酯 (HATU) 原位反应生成活化的羧酸衍生物, 中间体再与氮杂环卡宾催化剂反应, 也可以发生形式上既是苄位又是羰基β位的惰性C (sp3)—H键的直接官能团化 (Eq. 10).在该反应中, 采用氧化吲哚49作为亲电试剂, 其可与羧酸直接反应构建含有螺环及连续手性中心的多环化合物.该反应的收率和立体选择性都很好, 且以最简单的羧酸作为底物直接反应, 无需预先制备活泼的羧酸衍生物, 这也是该研究最大的特点.
综上所述, 无金属参与的羰基化合物的β-官能团化也可实现苄位C (sp3)—H键不对称官能团化.在该类反应中, 主要使用手性胺、手性氮杂环卡宾等有机小分子作为催化剂.对于手性胺催化的反应, 常通过加入合适的氧化剂实现从烯胺中间体向亚胺中间体的转化, 进而羰基β位接受亲核试剂的进攻从而构建多种官能化产物, 该反应过程被称为“氧化烯胺催化”; 对于手性氮杂环卡宾催化的不对称转化, 其特点是当采用醛类底物时, 需要加入氧化剂以使反应发生.而当采用羧酸衍生物如羧酸、羧酸酯、酸酐等底物时, 氮杂环卡宾可直接在碱性条件下实现羰基β位的官能团化构建多种化学键.
3 [1, 5]-氢迁移反应实现苄位C (sp3)— H键不对称官能团化
当苄位碳原子连有一个三级氮原子的时候, 实现苄位C (sp3)—H键官能团化可以采用另一种策略——“三级胺效应”, 利用分子内的氢迁移以及串联的成环过程从而使反应顺利进行.目前研究较多的分子内氢迁移过程为[1, 5]-氢迁移反应, 这种反应的特点在于整个反应是中性的, 不涉及氧化态的变化, 因而无需外加的氧化剂存在.通常情况下, 该反应是热驱动或Lewis酸催化的[26].最近的研究显示, 采用合适的有机催化剂, 也可以在无金属参与的温和条件下实现该反应的不对称转化.
2010年, Kim小组[27]发现邻三级胺取代的肉桂醛类化合物51在TES保护的二芳基脯胺醇cat20以及樟脑磺酸的联合作用下, 可以发生[1, 5]-氢迁移以及串联的环化反应, 高立体选择性地构建含有多个并环的四氢喹啉衍生物52 (Scheme 10).该反应被认为首先经历醛与二级胺催化剂cat20的缩合得到亚胺正离子中间体; 在正电荷的作用下, 随后发生[1, 5]-氢迁移生成带有一种新的烯胺基团的亚胺正离子中间体; 最后发生分子内的6-endo类型的烯胺对亚胺正离子的1, 2-加成反应并水解就可以得到最终产物52.对于大部分底物, 该反应都可在温和的条件下以中等的收率, 单一的非对映选择性以及优秀的立体选择性得到多并环的四氢喹啉衍生物.
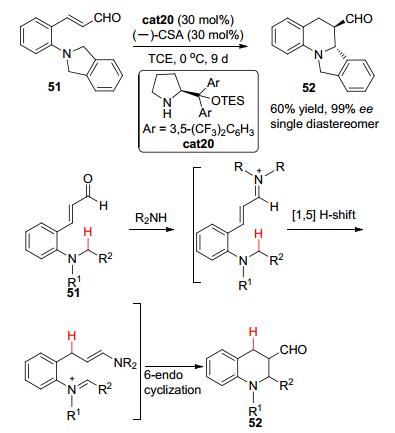 图 图式10
有机催化的通过串联[1, 5]-氢迁移/环化反应实现对映选择C—H键官能团化
Figure 图式10.
Enantioselective organocatalytic C—H bond functionalization via tandem 1, 5-hydride transfer/ring closure
图 图式10
有机催化的通过串联[1, 5]-氢迁移/环化反应实现对映选择C—H键官能团化
Figure 图式10.
Enantioselective organocatalytic C—H bond functionalization via tandem 1, 5-hydride transfer/ring closure
2011年, Akiyama等[28]发现了基于联苯基骨架的手性膦酸试剂cat21催化的[1, 5]-氢迁移反应 (Eq. 11).从与芳基相连的二苯胺底物53出发, 该反应可以在较高的温度下 (80 ℃) 以优秀的收率和立体选择性得到环化产物54.研究发现, 当底物53中与氮原子相连的芳基的邻位有取代基时, 反应活性较高.进一步的机理研究显示, 该反应不是由于一般认为的手性催化剂造成的面选择性的差异, 而在亲核试剂对亚胺正离子加成的过程中构建手性的; 该反应的手性控制很可能是由于底物与手性催化剂在反应之前发生相互作用, 苄位的两个氢原子产生立体环境的区别, 因而在随后的[1, 5]-氢迁移过程中构建了立体化学.
2014年, Kim等[29]将氧化烯胺催化的策略应用于串联[1, 5]-氢迁移/环化反应中 (Scheme 11).以二级手性胺cat22为催化剂、IBX为氧化剂, 一系列的邻位三级氮原子取代的二氢肉桂醛55首先与催化剂cat22缩合生成烯胺中间体.该烯胺中间体在氧化剂IBX的作用下可以被氧化为α, β-不饱和亚胺正离子中间体, 随后经历立体选择性的串联[1, 5]-氢迁移, 生成新的带有烯胺结构的亚胺正离子中间体.所得亚胺正离子中间体可以发生分子内的亲核加成反应从而关环得到多取代的四氢喹啉衍生物56.当R2为烷基时, 反应的立体选择性最高可以达到99% ee; 然而当R2为芳基时, 大部分底物只能以中等的立体选择性得到目标产物.
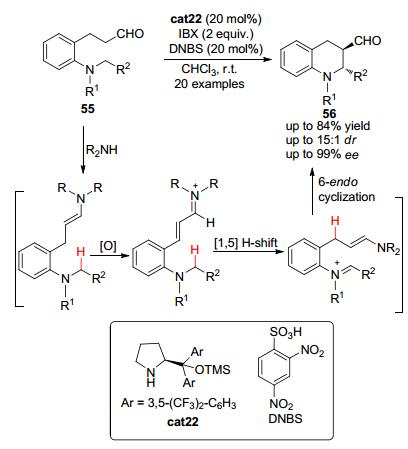 图 图式11
氧化烯胺催化-[1, 5]-氢迁移-环化反应构建手性四氢喹啉类化合物
Figure 图式11.
Oxidative enamine catalysis-1, 5-hydride transfer-cyclization sequences for the asymmetric synthesis of tetrahydroquinolines
图 图式11
氧化烯胺催化-[1, 5]-氢迁移-环化反应构建手性四氢喹啉类化合物
Figure 图式11.
Oxidative enamine catalysis-1, 5-hydride transfer-cyclization sequences for the asymmetric synthesis of tetrahydroquinolines
综上所述, 手性胺或手性膦酸等有机小分子催化的[1, 5]-氢迁移反应也可以实现苄位C (sp3)—H键高区域和立体选择性的不对称官能团化反应.该类反应无需外加氧化剂, 整个反应体系的氧化态是中性的, 不涉及氧化态的变化.通过该类型反应可以高效地构建多并环结构.
4 碱辅助的去质子化实现苄位C (sp3)— H键不对称官能团化
含有较强吸电子基团的甲苯类化合物, 其甲基可在碱性条件下脱掉一个质子生成碳负离子中间体.通常以邻芳基碳醌中间体形式存在, 该中间体随后可以进攻缺电子的烯烃、羰基等极性基团得到加成产物[30].例如Williams等[31]在2000年就发现含有多个吸电子基 (如硝基、羧酸酯基) 的甲苯化合物57, 由于多个吸电子基团的拉电子作用, 甲基上的氢原子具有一定的酸性, 从而可以在比较温和的条件下脱去质子; 另外也由于吸电子基团的存在, 可以稳定甲基去质子化后生成的碳负离子.因而底物57可以在碱性条件下脱去质子随后进攻亲电的醛类底物58并以4:1的比例得到一对非对映异构体59 (Eq. 12).他们也将这一方法学应用于合成药物活性分子FR-900482.
2011年, Melchiorre等[32]发现以手性二级胺cat10作为有机催化剂, 含有α, β-不饱和醛基的甲基吲哚、甲基吡咯或甲基呋喃类化合物可以首先与二级胺cat10缩合生成亚胺正离子中间体, 该中间体与芳环相邻的甲基可以容易地发生去质子化从而原位生成其他方法难以合成的活泼邻芳基碳醌中间体 (Scheme 12).邻芳基碳醌中间体随后可与缺电子的硝基苯乙烯类化合物61发生Diels-Alder反应从而高立体选择性地构建具有连续三个手性中心的复杂多并环杂芳香族化合物62.以甲苯为溶剂, 在70 ℃时, 对于大部分底物, 该反应都可以较好的收率和非对映选择性, 以及优秀的对映选择性得到环化产物.如果使用亚甲基吲哚酮63为底物, 则在更为温和的室温条件下, 其就可与原位生成的活泼邻芳基碳醌中间体反应, 立体选择性的构建含有一个螺环的四环结构64.
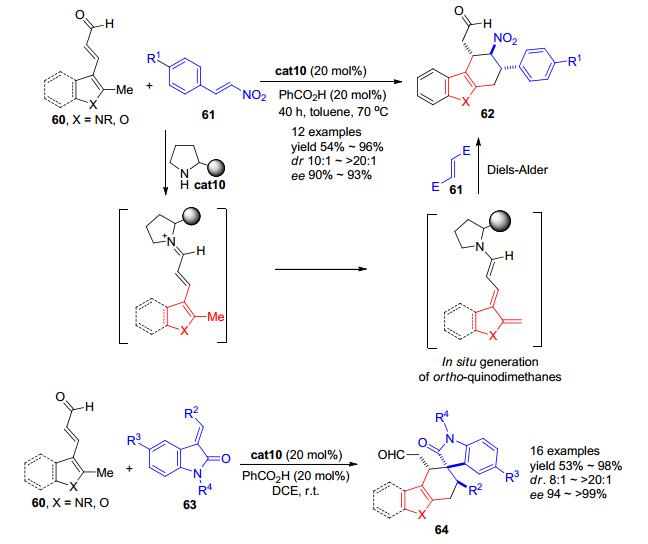 图 图式12
基于原位生成邻芳基碳醌中间体实现不对称杂Diels-Alder反应
Figure 图式12.
Asymmetric catalysis of Diels-Alder reactions with in situ generated heterocyclic ortho-quinodimethanes
图 图式12
基于原位生成邻芳基碳醌中间体实现不对称杂Diels-Alder反应
Figure 图式12.
Asymmetric catalysis of Diels-Alder reactions with in situ generated heterocyclic ortho-quinodimethanes
2012年, Melchiorre等[33]进一步扩展了这一策略.他们采用手性胺催化的三烯胺催化 (trienamine catalysis) 与氮杂环卡宾催化 (carbene catalysis) 相结合的策略来构建一些含有四个连续手性中心的四并环化合物 (Scheme 13).他们的设计合成策略为:手性二级胺cat10可在温和的条件下催化N-Boc保护的甲基吲哚类化合物60a原位生成邻芳基碳醌中间体, 该中间体可随后与α, β-不饱和酮类化合物65发生Diels-Alder环化反应而高效构建含有两个羰基的四氢咔唑类化合物66.在氮杂环卡宾催化剂cat23作用下, 四氢咔唑类化合物66可进一步发生高立体选择性和区域选择性地交叉安息香缩合反应并构建含有四个手性中心, 其中一个是季碳中心的四环结构67.无论是对于第一步三烯胺催化还是第二步卡宾催化, 反应的效率和立体选择性都很好.该方法的研究对于高效构建这种复杂的具有多个手性中心的骨架结构提供了一种新的合成思路.
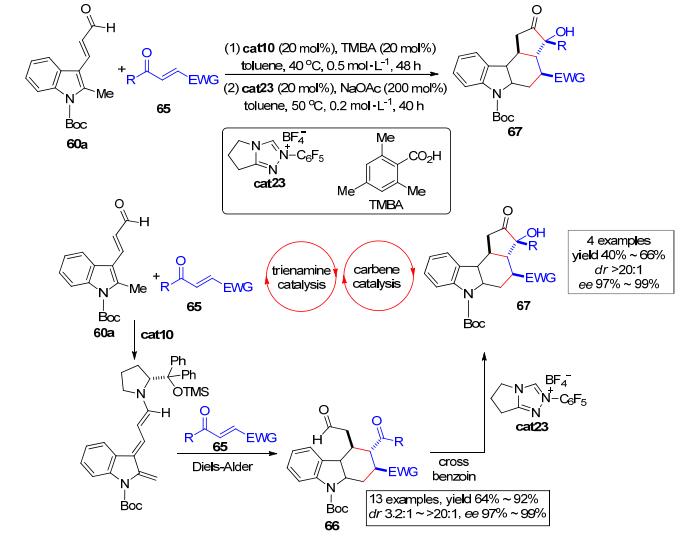 图 图式13
多种催化剂下通过Diels-Alder/安息香缩合不对称构建四氢咔唑衍生物
Figure 图式13.
Multicatalytic asymmetric synthesis of tetrahydrocarbazoles via a Diels–Alder/Benzoin reaction sequence
图 图式13
多种催化剂下通过Diels-Alder/安息香缩合不对称构建四氢咔唑衍生物
Figure 图式13.
Multicatalytic asymmetric synthesis of tetrahydrocarbazoles via a Diels–Alder/Benzoin reaction sequence
2012年, Chen小组[34]发现二级手性胺催化剂cat10可在弱酸性条件下催化2-甲基-3-吲哚甲醇类化合物68和α, β-不饱和醛69的环化反应, 高效、立体专一地构建含有多个手性中心的四氢咔唑类化合物70 (Scheme 14) 为方便分离, 醛基随后在NaBH4的作用下还原为含有一级醇的四氢咔唑衍生物71.该反应被认为2-甲基-3-吲哚甲醇68在弱酸性条件下先发生脱水生成烯基亚胺中间体, 随后发生氢迁移及异构化反应得到邻吲哚基碳醌中间体.二级手性胺cat10可通过与α, β-不饱和醛类化合物形成亚胺正离子从而活化双键, 随后可与邻吲哚基碳醌中间体发生分子间的不对称Diels-Alder反应得到四氢咔唑类化合物70, 并最终被还原为一级醇71.
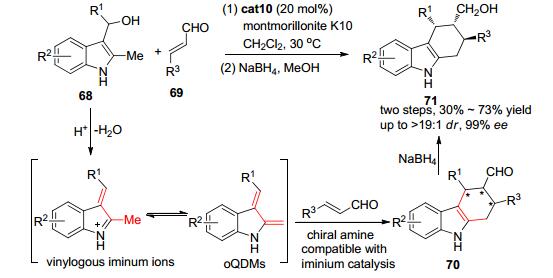 图 图式14
基于原位生成邻芳基碳醌中间体的2-甲基-3-吲哚甲醇的不对称Diels-Alder反应
Figure 图式14.
Asymmetric Diels-Alder reaction of 2-methyl-3-indolylmethanols via in situ generation of o-quinodimethanes
图 图式14
基于原位生成邻芳基碳醌中间体的2-甲基-3-吲哚甲醇的不对称Diels-Alder反应
Figure 图式14.
Asymmetric Diels-Alder reaction of 2-methyl-3-indolylmethanols via in situ generation of o-quinodimethanes
2013年, Wang等[35]报道了含有硝基的甲苯类化合物72在二级手性胺cat24催化下可与α, β-不饱和醛69发生1, 4-加成反应 (Scheme 15).在该反应中, 通过在芳香环上引入强拉电子基团, 硝基邻位的甲基可以在比较温和的条件下发生去质子化, 随后可与手性胺催化剂和醛缩合所得的含有亚胺正离子的烯烃反应, 立体专一地得到羰基β位带有手性中心的化合物73.产物中芳环上的硝基可通过简单的化学转化为其他有用的官能团, 从而得到新的官能化产物74, 因而该反应在构建开链的β-苄基取代的醛类化合物方面具有一定的应用性.
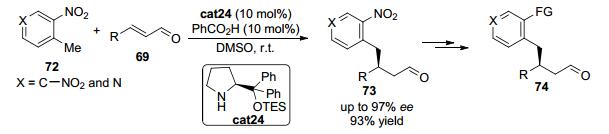 图 图式15
手性胺催化的惰性芳基甲烷作为亲核试剂的对映选择性共轭加成反应
Figure 图式15.
Enantioselective direct conjugate addition of inert aryl methane nucleophiles to enals with a chiral amine catalyst
图 图式15
手性胺催化的惰性芳基甲烷作为亲核试剂的对映选择性共轭加成反应
Figure 图式15.
Enantioselective direct conjugate addition of inert aryl methane nucleophiles to enals with a chiral amine catalyst
2013年, Jørgensen等[36]几乎同时报道了在手性二苯基脯胺醇硅醚cat25催化下, 含有强吸电子基团如硝基的甲苯衍生物72与α, β-不饱和醛69的不对称1, 4-共轭加成反应, 以优秀的收率和高达99% ee的立体选择性得到具有生物活性的β-苄基取代的醛类加成产物73 (Eq. 13).该反应是通过手性胺与醛缩合得到亚胺正离子从而活化双键, 以及强吸电子基团活化与芳环相连的甲基这两种效应共同作用而实现的.
2014年, Lee等[37]报道二苯基脯胺醇硅醚cat10可催化含有多个硝基取代的甲苯类化合物75与两分子α, β-不饱和醛69a发生串联环化反应, 高效、立体专一地构建含有多达五个手性中心的三并环结构76 (Scheme 16).该反应被认为首先α, β-不饱和醛69a与手性胺缩合得到α, β-不饱和亚胺中间体; 硝基活化的甲苯衍生物75的甲基在碱的作用下脱掉一个质子得到比较稳定的亲核性碳负离子, 亲核性碳负离子随后与α, β-不饱和亚胺中间体发生Michael加成得到1, 4-加成产物; 1, 4-加成产物可发生分子内的Michael加成, 接着进攻另一分子α, β-不饱和亚胺中间体; 接着发生手性胺催化的分子内的aldol反应并脱去一分子水最终得到目标产物76.在该反应过程中共发生了三次Michael反应和一次Aldol反应, 最终以单一的非对映选择性和>99% ee的对映选择性高效地制备复杂的化合物骨架, 其结构和绝对构型可通过产物的衍生物单晶衍射进行确认.
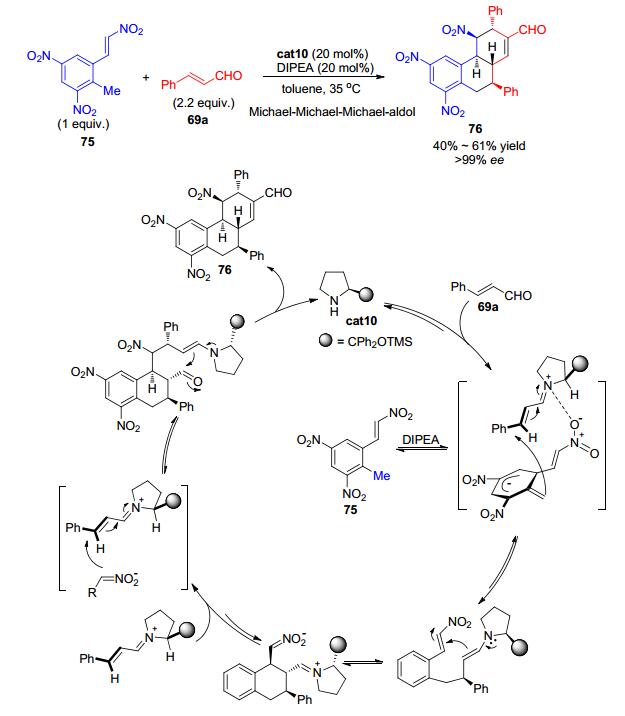 图 图式16
有机催化的四重串联反应不对称构建多官能团化合物
Figure 图式16.
Organocatalytic quadruple-cascade reactions for the asymmetric synthesis of highly functionalized compounds
图 图式16
有机催化的四重串联反应不对称构建多官能团化合物
Figure 图式16.
Organocatalytic quadruple-cascade reactions for the asymmetric synthesis of highly functionalized compounds
2015年, Albrecht等[38]报道了一种手性胺催化呋喃衍生物的立体远程烷基化的新方法 (Scheme 17). 5-烷基呋喃醛77与含有氢键结构的二级胺催化剂cat26发生缩合、去质子化、去芳构化得到一种新型的三烯胺中间体.该中间体随后与硝基烯烃61发生不对称共轭加成, 水解后可以较高的非对映和良好到优秀的对映选择性构建含有两个相邻手性中心的开链呋喃衍生物78.该反应的手性控制被认为是通过含有氢键的手性胺cat26以及呋喃底物与硝基烯烃的π-π堆积作用来实现的.
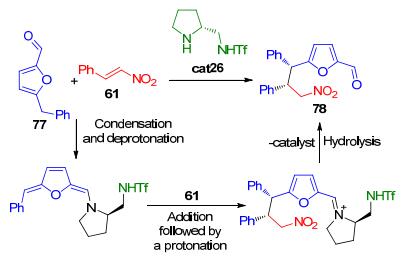 图 图式17
通过非典型三烯胺活化实现呋喃衍生物的远程烷基化
Figure 图式17.
Organocatalytic nonclassical trienamine activation in the remote alkylation of furan derivatives
图 图式17
通过非典型三烯胺活化实现呋喃衍生物的远程烷基化
Figure 图式17.
Organocatalytic nonclassical trienamine activation in the remote alkylation of furan derivatives
2013年, Chi等[39]发现含有醛基的甲基吲哚、甲基苯并呋喃以及甲基苯并噻吩等杂芳基醛类化合物79在手性三氮唑盐cat17作为氮杂环卡宾前体以及醌类氧化剂34的存在下, 可与三氟苯乙酮类化合物39以及氧化吲哚类化合物81发生环化反应得到含有一个季碳中心或螺环的三并环结构80或82 (Scheme 18).该反应被认为是通过以下机理来实现的:杂芳基醛79与原位生成的氮杂环卡宾活性物种先发生反应得到富电子的Breslow中间体; 接着在氧化剂34的作用下, Breslow中间体转化为与NHC连接的酰基唑类中间体; 由于三氮唑基团上正电荷的存在, 杂芳基醛的甲基被活化因而酸性增加, 在碱的作用下得到关键的邻芳基碳醌中间体; 邻芳基碳醌中间体可与三氟苯乙酮类化合物39以及氧化吲哚类化合物81发生形式上的[4+2]环加成反应最终得到潜在的具有药物活性的目标分子.
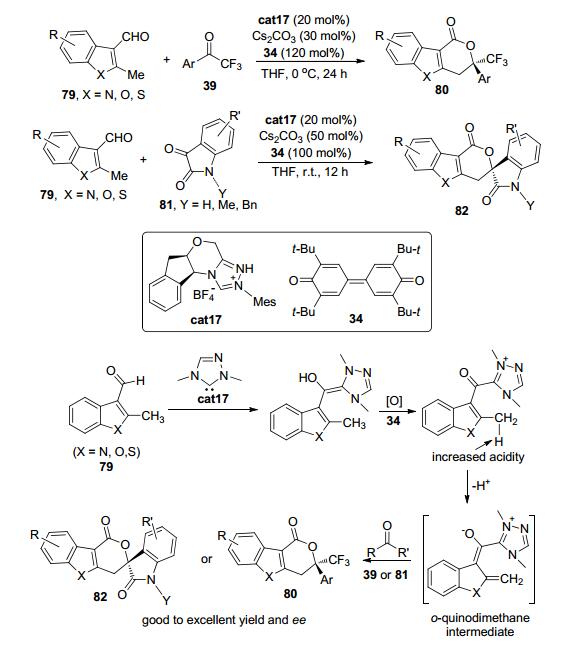 图 图式18
有机催化的吲哚醛的C (sp3)—H键官能团化反应
Figure 图式18.
Organocatalytic C (sp3)—H bond functionalization of indole aryl aldehydes
图 图式18
有机催化的吲哚醛的C (sp3)—H键官能团化反应
Figure 图式18.
Organocatalytic C (sp3)—H bond functionalization of indole aryl aldehydes
2016年, Xu等[40]报道了手性三氮唑盐cat17作为氮杂环卡宾催化剂前体也可以催化2-甲基杂芳基-3-羧酸酯83与氧化吲哚衍生的酮亚胺类化合物84的[4+2]环化反应, 高效、高立体选择性地构建杂芳基并六元内酯类结构85, 在此过程中同时构建了一个季碳手性中心 (Eq. 14).该反应也是通过氮杂环卡宾与羧酸酯底物83反应原位生成邻芳基碳醌中间体, 采用羧酸酯作为底物避免了氧化剂的使用, 更加绿色、环境友好.该反应的特色在于从稳定、易制备的简单底物出发, 在温和的条件下高选择性地实现了2-甲基杂环中甲基的C (sp3)—H键的直接官能团化, 并构建了在药物化学中具有重要作用的复杂骨架.
由此可见, 碱辅助的去质子化实现苄位C (sp3)—H键不对称官能团化, 通常需要预先将含有较强吸电子能力的基团引入到芳环上, 随后在碱性条件下脱掉一个质子生成碳负离子中间体.该中间体常以邻芳基碳醌中间体形式存在, 随后进攻缺电子的烯烃、羰基等极性基团得到加成产物.该反应可使用手性胺以及手性氮杂环卡宾等有机小分子来催化该反应的发生, 可高立体选择性地构建复杂的多环、螺环骨架结构.
5 结论与展望
芳环化合物广泛应用于生物医药、材料化工等领域, 也是重要的有机合成中间体.苄位C (sp3)—H键的直接不对称官能团化可以构建多种新型的多并环结构、螺环结构以及含有多个连续手性中心的开链结构等复杂骨架, 因而不断开发高效高选择性的苄位C (sp3)—H键不对称官能团化新方法仍然是一项富有意义的研究工作.本文主要从通过交叉脱氢偶联反应、羰基化合物的β-官能团化、[1, 5]-氢迁移反应以及碱辅助的去质子化实现苄位C (sp3)—H键不对称官能团化这四类不同的反应模式对其中比较有代表性的合成方法进行概括.由此可以看出近年来通过有机催化的无金属参与的苄位C (sp3)—H键官能团化构建芳环化合物的研究确实推动了该领域的发展.但是目前的苄位C (sp3)—H键不对称官能团化也存在着反应温度较高、氧化剂的使用、底物局限性、官能团容忍度不强等缺点.继续发展条件温和、操作简单、绿色高效、副产物少的有机催化方法学并将其应用于天然产物和生物活性分子的全合成中仍然是该领域发展需要解决的一些问题.相信在不久的将来会有更多突破性的进展, 为构建多种类型的芳环化合物提供有效手段.
-
-
[1]
(a) Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. Eur. J. 2010, 16, 2654.
(b) Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem., Int. Ed. 2012, 51, 8960.
(c) Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624.
(d) Davies, H. M. L.; Morton, D. J. Org. Chem. 2016, 81, 343.
(e) Rouquet, G.; Chatani, N. Angew. Chem., Int. Ed. 2013, 52, 11726.
(f) Wang, M.; Wang, Z.; Shang, M.; Dai, H. Chin. J. Org. Chem. 2015, 35, 570 (in Chinese). (王明明, 王子潇, 商明, 戴辉雄, 有机化学, 2015, 35, 570.)
(g) Tan, M.; Gu, Y.; Luo, X.; Zhang, P. Chin. J. Org. Chem. 2015, 35, 781 (in Chinese). (谭明雄, 顾运琼, 罗旭健, 张培, 有机化学, 2015, 35, 781.)
(h) Chen, T.; Zhang, M. Chin. J. Org. Chem. 2015, 35, 813 (in Chinese). (陈天保, 章明, 有机化学, 2015, 35, 813.)
(i) Zhao, J.; Zhang, Q. Acta Chim. Sinica 2015, 73, 1235 (in Chinese). (赵金钵, 张前, 化学学报, 2015, 73, 1235.)
(j) Shang, X.; Liu, Z. Acta Chim. Sinica 2015, 73, 1275 (in Chinese). (尚筱洁, 柳忠全, 化学学报, 2015, 73, 1275.) -
[2]
(a) Davies, H. M. L.; Lian, Y. Acc. Chem. Res. 2012, 45, 923.
(b) Motevalli, S.; Sokeirik, Y.; Ghanem, A. Eur. J. Org. Chem. 2016, 1459.
(c) Ye, B.; Cramer, N. Acc. Chem. Res. 2015, 48, 1308.
(d) He, G.; Wang, B.; Nack, W. A.; Chen, G. Acc. Chem. Res. 2016, 49, 635. -
[3]
(a) Retamosa, M. G.; Matador, E.; Monge, D.; Lassaletta, J. M.; Fernandez, R. Chem. Eur. J. 2016, 22, 13430.
(b) Bulfield, D.; Huber, S. M. Chem. Eur. J. 2016, 22, 14434.
(c) Tanriver, G.; Dedeoglu, B.; Catak, S.; Aviyente, V. Acc. Chem. Res. 2016, 49, 1250.
(d) Donslund, B. S.; Johansen, T. K.; Poulsen, P. H.; Halskov, K. S.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2015, 54, 13860. -
[4]
(a) Zhao, Y.-L.; Wang, Y.; Luo, Y.-C.; Fu, X.-Z.; Xu, P.-F. Tetrahedron Lett. 2015, 56, 3703.
(b) Zheng, C.; You, S.-L. RSC Adv. 2014, 4, 6173
(c) Wei, G.; Basheer, C.; Tan, C.-H.; Jiang, Z. Tetrahedron Lett. 2016, 57, 3801. -
[5]
(a) Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74.
(b) Stockerla, S.; Mancheño, O. G. Org. Chem. Front. 2016, 3, 277.
(c) Murahashi, S.-I.; Komiya, N.; Terai, H.; Nakae, T. J. Chem. Soc. 2003, 125, 15312. -
[6]
Benfatti, F.; Capdevila, M. G.; Zoli, L.; Benedetto, E.; Cozzi, P. G. Chem. Commun. 2009, 5919.
-
[7]
Ho, X.-H.; Mho, S.; Kang, H.; Jang, H.-Y. Eur. J. Org. Chem. 2010, 4436.
-
[8]
Zhang, B.; Xiang, S.-K.; Zhang, L.-H.; Cui, Y.; Jiao, N. Org. Lett. 2011, 13, 5212. doi: 10.1021/ol202090a
-
[9]
Huang, F.; Xu, L.; Xiao, J. Chin. J. Chem. 2012, 30, 2721. doi: 10.1002/cjoc.v30.11
-
[10]
Pan, Y.; Kee, C. W.; Chen, L.; Tan, C.-H. Green Chem. 2011, 13, 2682. doi: 10.1039/c1gc15489c
-
[11]
Neel, A. J.; Hehn, J. P.; Tripet, P. F.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 14044. doi: 10.1021/ja407410b
-
[12]
Zhang, G.; Ma, Y.; Wang, S.; Kong, W.; Wang, R. Chem. Sci. 2013, 4, 2645. doi: 10.1039/c3sc50604e
-
[13]
Meng, Z.; Sun, S.; Yuan, H.; Lou, H.; Liu, L. Angew. Chem., Int. Ed. 2014, 53, 543. doi: 10.1002/anie.v53.2
-
[14]
Liu, X.; Meng, Z.; Li, C.; Lou, H.; Liu, L. Angew. Chem., Int. Ed. 2015, 54, 6012. doi: 10.1002/anie.201500703
-
[15]
Liu, X.; Sun, S.; Meng, Z.; Lou, H.; Liu, L. Org. Lett. 2015, 17, 2396. doi: 10.1021/acs.orglett.5b00909
-
[16]
Xie, Z.; Zan, X.; Sun, S.; Pan, X.; Liu, L. Org. Lett. 2016, 18, 3944. doi: 10.1021/acs.orglett.6b01625
-
[17]
(a) Xiao, J. ChemCatChem 2012, 4, 612.
(b) Nielsen, M.; Worgull, D.; Zweifel, T.; Gschwend, B.; Bertelsen, S.; Jørgensen, K. A. Chem. Commun. 2011, 47, 632. -
[18]
Hayashi, Y.; Itoh, T.; Ishikawa, H. Angew. Chem., Int. Ed. 2011, 50, 3920. doi: 10.1002/anie.201006885
-
[19]
Zhang, S.-L.; Xie, H.-X.; Zhu, J.; Li, H.; Zhang, X.-S.; Li, J.; Wang, W. Nat. Commun. 2011, 2, 211. doi: 10.1038/ncomms1214
-
[20]
Zeng, X.; Ni, Q.; Raabe, G.; Enders, D. Angew. Chem., Int. Ed. 2013, 52, 2977. doi: 10.1002/anie.201209581
-
[21]
Mo, J.; Shen, L.; Chi, Y. R. Angew. Chem., Int. Ed. 2013, 52, 8588. doi: 10.1002/anie.201302152
-
[22]
Fu, Z.; Xu, J.; Zhu, T.; Leong, W. W. Y.; Chi, Y. R. Nat. Chem. 2013, 5, 835. doi: 10.1038/nchem.1710
-
[23]
Fu, Z.; Jiang, K.; Zhu, T.; Torres, J.; Chi, Y. R. Angew. Chem., Int. Ed. 2014, 53, 6506. doi: 10.1002/anie.201402620
-
[24]
Jin, Z.; Chen, S.; Wang, Y.; Zheng, P.; Yang, S.; Chi, Y. R. Angew. Chem., Int. Ed. 2014, 53, 13506. doi: 10.1002/anie.201408604
-
[25]
Xie, Y.; Yu, C.; Li, T.; Tu, S.; Yao, C. Chem. Eur. J. 2015, 21, 5355. doi: 10.1002/chem.201500345
-
[26]
Wang, M. ChemCatChem 2013, 5, 1291. doi: 10.1002/cctc.v5.6
-
[27]
Kang, Y. K.; Kim, S. M.; Kim, D. Y. J. Am. Chem. Soc. 2010, 132, 11847. doi: 10.1021/ja103786c
-
[28]
Mori, K.; Ehara, K.; Kurihara, K.; Akiyama, T. J. Am. Chem. Soc. 2011, 133, 6166. doi: 10.1021/ja2014955
-
[29]
Kang, Y. K.; Kim, D. Y. Chem. Commun. 2014, 50, 222. doi: 10.1039/C3CC46710D
-
[30]
Jaworski, A. A.; Scheidt, K. A. J. Org. Chem. 2016, 81, 10145. doi: 10.1021/acs.joc.6b01367
-
[31]
Williams, R. M.; Rollins, S. B.; Judd, T. C. Tetrahedron 2000, 56, 521. doi: 10.1016/S0040-4020(99)01046-7
-
[32]
Liu, Y.; Nappi, M.; Arceo, E.; Vera, S.; Melchiorre, P. J. Am. Chem. Soc. 2011, 133, 15212. doi: 10.1021/ja206517s
-
[33]
Liu, Y.; Nappi, M.; Escudero-Adán, E. C.; Melchiorre, P. Org. Lett. 2012, 14, 1310. doi: 10.1021/ol300192p
-
[34]
Xiao, Y.-C.; Zhou, Q.-Q.; Dong, L.; Liu, T.-Y.; Chen, Y.-C. Org. Lett. 2012, 14, 5940. doi: 10.1021/ol302853m
-
[35]
Li, T.; Zhu, J.; Wu, D.; Li, X.; Wang, S.; Li, H.; Li, J.; Wang, W. Chem. Eur. J. 2013, 19, 9147. doi: 10.1002/chem.v19.28
-
[36]
Dell'Amico, L.; Companyó, X.; Naicker, T.; Bräuer, T. M.; Jørgensen, K. A. Eur. J. Org. Chem. 2013, 5262.
-
[37]
Raja, A.; Hong, B.-C.; Lee, G.-H. Org. Lett. 2014, 16, 5756. doi: 10.1021/ol502821e
-
[38]
Skrzyńska, A.; Przydacz, A.; Albrecht, Ł. Org. Lett. 2015, 17, 5682. doi: 10.1021/acs.orglett.5b02979
-
[39]
Chen, X.; Yang, S.; Song, B.-A.; Chi, Y. R. Angew. Chem., Int. Ed. 2013, 52, 11134. doi: 10.1002/anie.201305861
-
[40]
Xu, J.; Yuan, S.; Miao, M. Org. Lett. 2016, 18, 3822. doi: 10.1021/acs.orglett.6b01831
-
[1]
-

图式1 有机催化的醛与苄基化合物的脱氢烷基化反应
Scheme 1 Organocatalytic enantioselective dehydrogenative alkylation of aldehydes with benzylic compounds

图式2 手性离子对控制的无金属参与的三级胺与酮的氧化脱氢偶联反应
Scheme 2 Chiral organic contact ion pairs in metal-free oxidative cross-dehydrogenative coupling of tertiary amines to ketones

图式3 Brønsted酸催化的N-酰基四氢-β-咔啉类化合物的不对称C—H烯基化与芳基化反应
Scheme 3 Brønsted acid-catalyzed asymmetric C—H vinylation and arylation of N-carbamoyl tetrahydro-β-carbolines

图式4 手性胺催化的醛与硝基甲烷的不对称氧化偶联反应
Scheme 4 Chiral amine-catalyzed enantioselective cross-coupling of aldehydes and nitromethane

图式5 通过烯胺的氧化实现对映选择性的醛β-官能团化反应
Scheme 5 Enantioselective β-functionalization of aldehydes by oxidation of enamines

图式6 手性胺催化串联环化反应构建多官能团的环己烯衍生物
Scheme 6 Chiral amine-catalyzed domino cyclization reaction for the synthesis of polyfunctionalized cyclohexene derivatives

图式7 氧化氮杂环卡宾催化的饱和醛的β-C—H键活化
Scheme 7 Direct β-C—H bond activation of saturated aldehydes via oxidative NHC catalysis

图式8 氮杂环卡宾催化的饱和羧酸酯的β-C—H键活化
Scheme 8 β-C—H bond activation of saturated carboxylic esters through N-heterocyclic carbene organocatalysis

图式9 氮杂环卡宾催化的β-C—H键活化构建杂环化合物
Scheme 9 Access to heterocycles by NHC catalyzed ester β-C—H bond activation

图式10 有机催化的通过串联[1, 5]-氢迁移/环化反应实现对映选择C—H键官能团化
Scheme 10 Enantioselective organocatalytic C—H bond functionalization via tandem 1, 5-hydride transfer/ring closure

图式11 氧化烯胺催化-[1, 5]-氢迁移-环化反应构建手性四氢喹啉类化合物
Scheme 11 Oxidative enamine catalysis-1, 5-hydride transfer-cyclization sequences for the asymmetric synthesis of tetrahydroquinolines

图式12 基于原位生成邻芳基碳醌中间体实现不对称杂Diels-Alder反应
Scheme 12 Asymmetric catalysis of Diels-Alder reactions with in situ generated heterocyclic ortho-quinodimethanes

图式13 多种催化剂下通过Diels-Alder/安息香缩合不对称构建四氢咔唑衍生物
Scheme 13 Multicatalytic asymmetric synthesis of tetrahydrocarbazoles via a Diels–Alder/Benzoin reaction sequence

图式14 基于原位生成邻芳基碳醌中间体的2-甲基-3-吲哚甲醇的不对称Diels-Alder反应
Scheme 14 Asymmetric Diels-Alder reaction of 2-methyl-3-indolylmethanols via in situ generation of o-quinodimethanes

图式15 手性胺催化的惰性芳基甲烷作为亲核试剂的对映选择性共轭加成反应
Scheme 15 Enantioselective direct conjugate addition of inert aryl methane nucleophiles to enals with a chiral amine catalyst

图式16 有机催化的四重串联反应不对称构建多官能团化合物
Scheme 16 Organocatalytic quadruple-cascade reactions for the asymmetric synthesis of highly functionalized compounds

图式17 通过非典型三烯胺活化实现呋喃衍生物的远程烷基化
Scheme 17 Organocatalytic nonclassical trienamine activation in the remote alkylation of furan derivatives
-


 下载:
下载:

















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 51
- 文章访问数: 3117
- HTML全文浏览量: 563
 下载:
下载:
