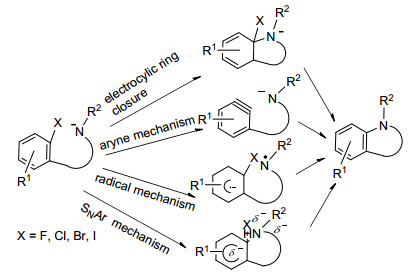
 图 1
强碱催化分子内氮芳基化C—N键耦合可能反应机理
Figure 1.
Possible reaction mechanisms of intramolecular N-arylation for the C—N bond coupling reaction by base-catal-yzed
图 1
强碱催化分子内氮芳基化C—N键耦合可能反应机理
Figure 1.
Possible reaction mechanisms of intramolecular N-arylation for the C—N bond coupling reaction by base-catal-yzed
引用本文:
李强根, 向仕凯, 毛双, 任译. 强碱催化分子内氮芳基化合成苯并咪唑反应机理及反应性研究[J]. 有机化学,
2017, 37(3): 608-616.
doi:
10.6023/cjoc201609020

Citation: Li Qianggen, Xiang Shikai, Mao Shuang, Ren Yi. Theoretical Investigations on the Intramolecular N-Arylation Mechanism and Reactivity for the Synthesis of Benzimidazoles by Base-Catalyzed[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 608-616. doi: 10.6023/cjoc201609020
Citation: Li Qianggen, Xiang Shikai, Mao Shuang, Ren Yi. Theoretical Investigations on the Intramolecular N-Arylation Mechanism and Reactivity for the Synthesis of Benzimidazoles by Base-Catalyzed[J]. Chinese Journal of Organic Chemistry, 2017, 37(3): 608-616. doi: 10.6023/cjoc201609020
强碱催化分子内氮芳基化合成苯并咪唑反应机理及反应性研究
摘要:
MP2/6-311+G**//B3LYP/6-311+G**理论水平上,对二甲基亚砜(DMSO)中强碱催化N-(2-卤基苯基)-N'-苯基乙脒分子内氮芳基化合成苯并咪唑反应机理及反应性做了理论研究.研究结果显示:标题反应的反应机理并不是Bolm等提出的自由基机理或分步的SNAr机理,而是只有一个过渡态的协同SNAr机理.几何结构宽松度分析和自然集居数分析(NPA)都不能解释标题反应的反应性大小顺序问题.多参数拟合揭示标题反应的反应能垒主要由最高占据轨道能EHOMO或亲核原子N10的区域亲核性指标ωN10-控制,另一因素为亲核原子N10所带负电荷多少.而被进攻的C2原子所带电荷以及几何结构宽松度L%对反应能垒几乎没有影响.方法对比研究发现,MP2/6-311+G**//B3LYP/6-311+G**方法所得结果与实验结果吻合较好,能更好地描述标题反应的相对能量和反应性顺序.
English
Theoretical Investigations on the Intramolecular N-Arylation Mechanism and Reactivity for the Synthesis of Benzimidazoles by Base-Catalyzed
Abstract:
Quantum chemical studies on the intramolecular N-arylation mechanism and reactivity of N-(2-halogen phenyl)-N'-phenyl ethyl amidines in dimethyl sulfoxide (DMSO) for the synthesis of benzimidazoles by base-catalyzed have been performed at MP2/6-311+G**//B3LYP/6-311+G** level of theory. The results indicate that the mechanism of the title reactions is not the radical mechanism or stepwise SNAr pathway, but the concerted SNAr pathway with a transition state, which is compared with the conclusion of Bolm et al. The reactivity of the title reactions can not be interpreted by the geometric looseness or natural population analysis. Multi parameter fitting reveals that the reactivity of the title reactions is controlled mostly by the regional nucleophilicity index ωN10- of the nucleophile N10 atom or highest occupied molecular orbital energy EHOMO of the reactant, the other factor is the charges of the nucleophilic atom N10, while the charges of the C2 atom and the geometric looseness L% have almost no effect on the reaction energy barrier. The relative energies and the reactivity of the title reactions attained at MP2/6-311+G**//B3LYP/6-311+G** level of theory are better agreement with the experimental results compared with the other methods.
-
苯并咪唑是一种拥有环化尿素骨架结构的重要杂环化合物, 其衍生物具有许多生物活性, 药物市场上广见其踪迹, 并且也是实验上的很多靶药[1~5].因此, 苯并咪唑的有效合成便是无数有机和药物化学家非常感兴趣的领域之一[6~51].传统的合成方法需要多步完成并且收益不高[6~8].近几十年发展了在高温下采用过渡金属催化合成苯并咪唑, 但是此工艺在制药工业应用中颇有争议, 因为所得产物不纯净, 要去除杂质需要多步操作, 并且需要合适的配体和造成环境污染[19~32, 49].最近几个课题组发展了在强碱催化下, 通过分子内氮芳基化来合成苯并咪唑和吲哚的新方法.此方法的优点是不需要过渡金属催化, 且反应条件温和, 产率高, 不需要多步进行, 有的甚至一锅合成[45~51].然而在反应机理上一直存在争议.由于反应在强碱中进行, 因此分子内氮芳基化C—N键耦合的初始物为N-甲苯磺酰腙阴离子[49]或N-苯基取代脒阴离子[50, 51], 目前其反应机理主要有四种假设 (图 1): (1) 电环化机理[49], (2) 苯炔机理[50], (3) 自由基机理[49, 51], (4) 芳环上亲核取代机理 (SNAr)[51]. Bolm等[49]在前期通过分子内氮芳基化合成吲哚的工作中, 提出了电环化机理, 然而他们在实验中并没有观察到具有四面体碳化合物阴离子存在, 因此排除了电环化机理, 但是他们通过电子自旋共振 (ESR) 光谱法观察到自由基中间体存在, 因此认为该反应为双自由基机理. 2013年Xiang等[50]在通过分子内氮芳基化合成苯并咪唑的工作中, 使用自由基捕获剂 (TEMPO) 后仍然发现有产物生成, 因此认为不可能是自由基机理, 同时他们提出了苯炔机理.接着Bolm等[51]在通过分子内氮芳基化合成苯并咪唑的工作中发现, 如果把反应物中的卤素取代位置从邻位换到间位, 结果没有发现产物生成, 而把邻位与间位都换成卤素取代基反而有产物生成, 并且反应活性顺序为F>I>Br>Cl, 实验结果与C—X键极性大小顺序X=F>Cl>Br>I矛盾, 因此认为不可能是苯炔机理; 另外, 由于实验中反应活性大小顺序为F>I>Br>Cl, 与经典的活化芳环SNAr反应活性顺序F>Cl>Br>I[52]不一致, 最后Bolm等[51]得出, 对于离去基团是F、Cl和Br的反应可能为SNAr亲核取代机理, 而离去基团是I的反应可能为自由基机理.
图 1
强碱催化分子内氮芳基化C—N键耦合可能反应机理
Figure 1.
Possible reaction mechanisms of intramolecular N-arylation for the C—N bond coupling reaction by base-catal-yzed
对于芳环上的SNAr反应, 通常指的是底物芳环临位或对位上存在吸电子基团, 即芳环是活化的情况, 其反应机理通常是加成-消去两步机理, 若离去基团是卤素, 其反应活性顺序通常为F>Cl>Br>I, 即加成是决速步骤[53].然而这一顺序会受亲核试剂和溶剂的影响, 例如对于一些软的, 高度可极化的亲核试剂SCN-, C6H5NHCH3或NO2−等, 反应活性顺序可能为I>Br>Cl>F[53].除了加成-消去两步机理外, 少数芳环上的SNAr反应还存在只经过一个过渡态的协同机理, 如苯酚与三嗪的反应[54], 以及甲醇阴离子与氯苯[55], 氟离子与硝基苯[56], 对氰基氯苯或对氰基硝基苯与氟离子的反应[57]等, 其反应机理都是协同的SNAr过程.
为了验证Bolm等[51]提出的反应机理的合理性, 同时解释该反应的反应性大小顺序问题 (F>I>Br>Cl), 并揭示影响反应性大小的内在因素, 本文以N-(2-卤基苯基)-N'-苯基乙脒阴离子作为反应物Re出发, 设计了四条可能的反应通道, 对二甲基亚砜 (DMSO) 中强碱催化分子内氮芳基化合成苯并咪唑反应机理做了理论探索 (图 2): Path 1为单自由基机理; Path 2为分子内电子转移的双自由基机理, 并且考察了单重态和三重态双自由基的可能性; Path 3为先加成再消去的分步SNAr机理; Path 4为协同的SNAr机理.希望我们的研究结果能为下一步的实验或理论研究提供有用的信息.
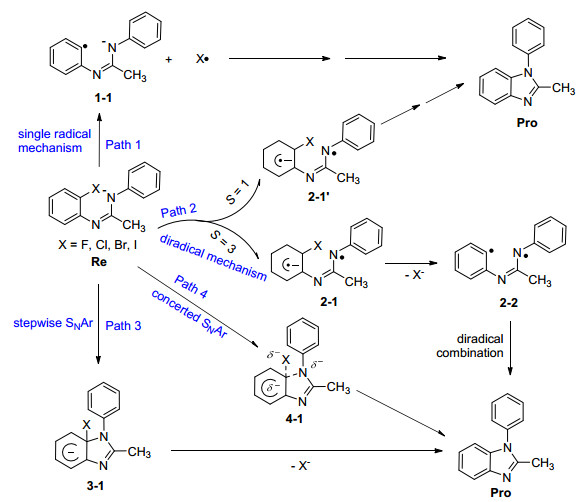 图 2
本文研究的分子内氮芳基化合成苯并咪唑可能反应通道
Figure 2.
Possible reaction pathways in this study of intramolecular N-arylations of N-(2-halogen phenyl)-N'-phenyl ethyl amidines for the synthesis of benzimidazoles
图 2
本文研究的分子内氮芳基化合成苯并咪唑可能反应通道
Figure 2.
Possible reaction pathways in this study of intramolecular N-arylations of N-(2-halogen phenyl)-N'-phenyl ethyl amidines for the synthesis of benzimidazoles
2 计算方法
采用密度泛函方法B3LYP[58~60]对所有物质几何结构在实验实际进行溶剂二甲基亚砜 (DMSO, ε=46.7) 中进行优化.选取此方法的原因是, B3LYP方法是被广泛认同并最为流行的密度泛函方法, 并且该方法在先前也被多个课题组用来研究芳环上亲核取代反应[61~68].为了得到值得信赖的计算结果, 我们先选择6-31+G*基进行优化, 接着再选择6-11+G**进行优化, 目的是考察基组的加大对几何结构与反应能垒的影响.所有优化的几何结构都用计算得到的频率来区别, 没有虚频的为全程或局域稳定点, 有且只有一个虚频的为反应的过渡态.分别在B3LYP/6-31+G*和B3LYP/6-311+G**理论水平上计算获得频率, 并且没有加任何的校正因子.对于含溴和碘的物质使用Wadt和Hay[69]的有效核电势 (ECP) 计算, 而对于其他物质, 采用全电子计算.为了评价B3LYP方法能量的精度, 我们还在MP2/6-311+G**//B3LYP/6-311+G**和MP3/6-311+G**//B3LYP/ 6-311+G**理论水平上, 采用连续介质模型 (PCM)[70]进行了液相单点能计算.各物质的原子电荷以及键极参数是基于自然键轨道 (NBO)[71]的波函数计算, 在MP2/6-311+G**//B3LYP/ 6-311+G**理论水平上计算得到.此外, 为了评价B3LYP方法的可靠性, 我们还使用密度泛函方法M06-2X[72]对所有物质几何结构在溶剂DMSO中进行优化.同样在MP2/6-311+G**//M06-2X/ 6-311+G**理论水平上进行了液相PCM单点能计算.选取该方法的原因是该方法在先前也曾被用来研究芳环上亲核取代反应[73~74].
本工作中, 所有的计算都是采用高斯09软件包[75]完成, 所有原子核间距离都用纳米 (nm) 而所有的键角都用度 (°).采用101 kPa下0 K电子能变 (ΔE, kJ·mol-1) 和298.15 K的焓变 (ΔH, kJ·mol-1) 以及吉布斯自由能变 (ΔG, kJ·mol-1) 来表示相对能量.
3 结果与讨论
3.1 自由基机理 (Path 1和Path 2)
对于自由基机理, 我们同时考察了单自由基和双自由基机理两种情况 (图 2).在单自由基路径中 (Path 1), 首先反应物Re中C—X键发生均裂形成1-1和X·两个单自由基, 然后1-1再关环形成产物. C—X键均裂能垒总结在表 1中, 从该表数据可以看出, 在M06-2X/6-311+G**理论水平上, 从F到I能垒分别为466.6 (X=F), 349.9 (X=Cl), 308.3 (X=Br) 和263.8 kJ·mol-1 (X=I); 而在MP2/6-311+G**//M06-2X/6-311+G**理论水平上却高达557.8 (X=F), 454.9 (X=Cl), 394.8 (X=Br) 和351.5 kJ·mol-1 (X=I), 因此反应物Re中C—X键发生均裂形成1-1和X·两个单自由基几乎不可能, 即标题反应单自由基机理不可能存在.对于双自由基机理, 根据双自由基反应路径 (Path 2), 反应物Re首先必须通过电子转移生成双自由基阴离子, 同时讨论了单重态的双自由基 (2-1') 和三重态的双自由基 (2-1) 的情况, 对于单重态的双自由基 (2-1'), 通过对反应物Re加关键词guess=mix进行优化, 并且对波函数的稳定性做了测试, 结果得到的结构和能量与反应物Re一样, 且<S2>=0, 说明电子总自旋为0, 为闭壳层单重态, 而非双自由基, 即不存在双自由基单线态.然而我们可以优化得到稳定的三重态双自由基, <S2>值分别为2.027 (X=F), 2.028 (X=Cl), 2.0285 (X=Br) 和2.047 (X=I).另外, 在MP2/6-311+G**//B3LYP/6-311+G**理论水平上计算得到反应物N-(2-氟基苯基)-N'-苯基乙脒阴离子及其相应的三重态双自由基的HOMO轨道 (图 3).从图 3可以看出, 单重态反应物N-(2-氟基苯基)-N'-苯基乙脒阴离子中亲核原子N的p轨道与亲核基团的苯环的π轨道存在p-π相互作用, 而在三重态的双自由基阴离子中却不存在这种作用, 因此后者能量应该比前者要高. 表 1总结了标题反应所有反应物Re(单重态) 与双自由基阴离子2-1(三重态) 的相对能量, 从该表数据可以看出, 在M06-2X/6-311+G**理论水平上, 三重态的双自由基阴离子比相应的反应物能量分别高出275.5 (X=F), 260.6 (X=Cl), 260.4 (X=Br) 和253.3 kJ·mol-1 (X=I); 而在MP2/6-311+G**//M06-2X/6-311+G**理论水平上却高达463.3 (X=F), 443.1 (X=Cl), 435.5 (X=Br) 和431.8 kJ·mol-1 (X=I), 因此要从反应物Re生成相应的三重态双自由基阴离子也是几乎不可能的.从以上分析可以得出, 标题反应的自由基机理几乎不可能存在.
 表 1
DMSO中标题反应所有反应物Re与其自由基阴离子相对能量表 (kJ·mol-1)
Table 1.
Relative energies of all the radical anions relative to reactants Re in DMSO of title reactions (kJ·mol-1)
表 1
DMSO中标题反应所有反应物Re与其自由基阴离子相对能量表 (kJ·mol-1)
Table 1.
Relative energies of all the radical anions relative to reactants Re in DMSO of title reactions (kJ·mol-1)
Single free radical Δ Ga/(kJ·mol-1) Δ Gb/(kJ·mol-1) F Cl Br I F Cl Br I M06-2X/6-311+G** 466.6 349.9 308.3 263.8 275.5 260.0 260.4 261.7 MP2/6-311+G**//M06-2X/6-311+G** 557.8 454.9 394.8 351.5 463.3 443.1 435.5 431.8 aΔG=G(1-1)+G(X·)-G(Re); bΔG=G(2-1)-G(Re). 表 1 DMSO中标题反应所有反应物Re与其自由基阴离子相对能量表 (kJ·mol-1)
Table 1. Relative energies of all the radical anions relative to reactants Re in DMSO of title reactions (kJ·mol-1)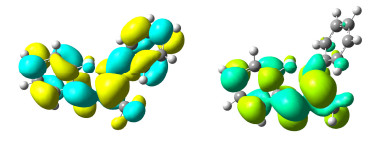 图 3
MP2/6-311+G**//B3LYP/6-311+G**理论水平上计算得到反应物N-(2-氟基苯基)-N'-苯基乙脒阴离子 (左) 及其相应的三重态双自由基阴离子 (右) HOMO轨道图
Figure 3.
HOMO orbitals of the reactant N-(2-fluorine phenyl)-N'-phenyl ethyl amidine anion (left) and its double radical anion (triplet state) (right) attained at MP2/6-311+G**//B3LYP/ 6-311+G** level
图 3
MP2/6-311+G**//B3LYP/6-311+G**理论水平上计算得到反应物N-(2-氟基苯基)-N'-苯基乙脒阴离子 (左) 及其相应的三重态双自由基阴离子 (右) HOMO轨道图
Figure 3.
HOMO orbitals of the reactant N-(2-fluorine phenyl)-N'-phenyl ethyl amidine anion (left) and its double radical anion (triplet state) (right) attained at MP2/6-311+G**//B3LYP/ 6-311+G** level
3.2 芳环上亲核取代机理 (SNAr, Path 3和Path 4)
对于芳环上的亲核取代路径, 在几何结构优化过程中, 没有得到σ络合物而只得到协同的过渡态结构, 因此标题反应不是通过先加成后消去的分步亲核取代过程 (Path 3), 而是一个协同的亲核取代机理 (Path 4).不同水平上计算所得标题反应协同SNAr路径相对能量总结在表 2中, 而B3LYP/6-311+G**理论水平上和DMSO溶剂中优化所得标题反应协同SNAr路径所有驻点几何结构总结在图 4中.从表 2能量数据可以看出, MP2/6-311+G**//B3LYP/6-311+G**方法水平上计算所得反应吉布斯自由能垒 (ΔG≠) 分别为90.5 (X=F), 109.6 (X=Cl), 102.7 (X=Br) 和100.9 kJ·mol-1 (X=I).方法对比发现, 只有这个方法和基组所得数据与Bolm等[47]在实验中观察到反应活性大小顺序F>I>Br>Cl最为接近.由于102.7 (X=Br) 和100.9 kJ·mol-1 (X=I) 数据比较接近, 怀疑可能是计算水平误差, 因此我们在MP3/6-311+G**//B3LYP/6-311+G**水平上做了较高级别的液相单点能计算, 结果发现三重微扰方法并没有拉开二者之间的距离, 因此以下的讨论均采用MP2/6-311+G**//B3LYP/6-311+G**方法水平上所得计算结果.
表 2
不同水平上计算所得标题反应SNAr路径反应能垒表
Table 2.
Reaction energy barriers for the title reactions in the SNAr pathway calculated at different level
Level Δ E≠/(kJ mol-1) Δ H≠/(kJ mol-1) Δ G≠/(kJ mol-1) F Cl Br I F Cl Br I F Cl Br I B3LYP/6-31+G* 121.6 128.4 119.3 121.4 115.8 123.5 114.9 117.0 120.8 127.9 120.3 120.2 B3LYP/6-311+G** 118.7 128.9 122.6 120.6 113.1 124.1 118.5 116.4 119.6 132.5 122.9 120.2 M062X/6-311+G** 113.2 126.0 125.2 112.1 109.1 124.1 120.7 106.2 115.5 128.5 127.2 116.1 MP2/6-311+G**//M062X/6-311+G** 94.2 102.5 96.3 82.6 87.9 97.2 91.8 78.7 94.3 101.6 98.4 88.6 MP2/6-311+G**//B3LYP/6-311+G** 89.6 106.0 102.4 101.4 84.0 101.2 98.3 97.2 90.5 109.6 102.7 100.9 MP3/6-311+G**//B3LYP/6-311+G** 126.8 146.6 141.1 140.3 121.4 141.6 136.9 136.1 127.9 150.0 141.3 139.9 表 2 不同水平上计算所得标题反应SNAr路径反应能垒表
Table 2. Reaction energy barriers for the title reactions in the SNAr pathway calculated at different level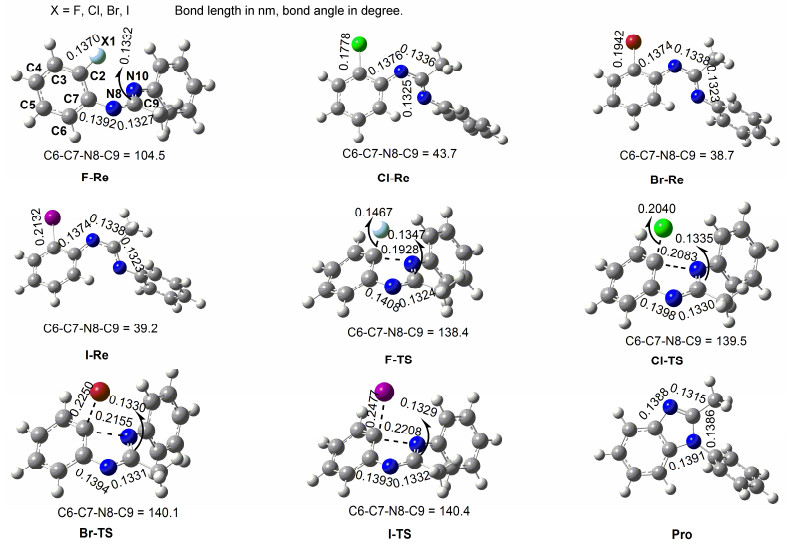 图 4
DMSO中B3LYP/6-311+G**水平上优化所得标题反应协同SNAr路径所有反应物、过渡态和产物几何结构
Figure 4.
B3LYP/6-311+G** optimized all the geometry structures of reactant, TS and product in DMSO of title reactions in the concerted SNAr pathway
图 4
DMSO中B3LYP/6-311+G**水平上优化所得标题反应协同SNAr路径所有反应物、过渡态和产物几何结构
Figure 4.
B3LYP/6-311+G** optimized all the geometry structures of reactant, TS and product in DMSO of title reactions in the concerted SNAr pathway
3.3 标题反应的反应性顺序解释
通常饱和卤代烃协同SN2亲核取代反应的能垒与C—X键极性大小顺序X=F>Cl>Br>I一致[76].最近我们研究得出, 没有活化的卤代芳烃上的SNAr亲核取代反应的能垒同样与C—X键极性大小顺序X=F>Cl>Br>I一致[77].而标题反应的反应活性顺序却是F>I>Br>Cl[51], 究竟是什么因素控制着反应的能垒?本部分将通过几何结构宽松度分析、NPA电荷分析以及反应性指标分析对标题反应的反应活性顺序作出解释.
3.3.2 NPA电荷分析
对于亲核反应来说, 如果亲核试剂所带负电荷越多而被进攻原子所带正电荷越多, 则反应的能垒就越低, 反应就越容易进行.我们在MP2/6-311+G**//B3LYP/ 6-311+G**水平上计算得到主要原子NPA电荷数据总结在表 4中.
表 4
MP2/6-311+G**//B3LYP/6-311+G**水平上计算所得主要原子NPA电荷 (Q)
Table 4.
Main atomic NPA charges (Q) calculated at MP2/6-311+G**//B3LYP/6-311+G** level
X X1 C2 C7 N8 C9 N10 Re TS Re TS Re TS Re TS Re TS Re TS F -0.4271 -0.5100 0.4021 0.5735 0.1712 0.0255 -0.8354 -0.7532 0.6142 0.5970 -0.8402 -0.7281 Cl -0.0777 -0.3445 -0.0878 0.2585 0.2243 0.0765 -0.8318 -0.7813 0.6186 0.6147 -0.8164 -0.7485 Br 0.0098 -0.3443 -0.1897 0.2228 0.2266 0.0892 -0.8321 -0.7886 0.6196 0.6186 -0.8118 -0.7577 I 0.1186 -0.3075 -0.2898 0.1807 0.2258 0.0880 -0.8355 -0.7885 0.6200 0.6200 -0.8106 -0.7682 表 4 MP2/6-311+G**//B3LYP/6-311+G**水平上计算所得主要原子NPA电荷 (Q)
Table 4. Main atomic NPA charges (Q) calculated at MP2/6-311+G**//B3LYP/6-311+G** level从表 4可以看出, 在反应物Re中, 亲核原子N10所带负电荷依次减少, 分别为-0.8402e (X=F), -0.8164e (X=Cl), -0.8118e (X=Br), -0.8106e (X=I); 而被进攻原子C2所带正电荷也是依次减少, 分别为0.4021e (X=F), -0.0878e (X=Cl), -0.1897e (X=Br) 和-0.2898e(X=I), 因此标题反应的反应性顺序理论上应该是F>Cl>Br>I, 结果与几何结构宽松度分析一致, 同样与Bolm等实验结论[51]不符.因此NPA电荷分析也不能解释标题反应反应性大小顺序问题.
3.3.3 反应性指标分析
从图 5可以看出, 标题反应电子能垒ΔE≠(kJ·mol-1) 和吉布斯自由能垒ΔG≠(kJ·mol-1) 之间呈现很好的线性关系 (R2=0.97486), 因此我们可以用ΔE≠来代替ΔG≠来分析标题反应反应性大小顺序问题.
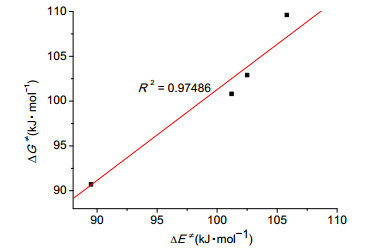 图 5
标题反应电子能垒 (ΔE≠) 和吉布斯自由能垒 (ΔG≠) 关系图
Figure 5.
Plot of electronic barriers (ΔE≠) vs the Gibbs free energy barriers (ΔG≠) of the title reactions
图 5
标题反应电子能垒 (ΔE≠) 和吉布斯自由能垒 (ΔG≠) 关系图
Figure 5.
Plot of electronic barriers (ΔE≠) vs the Gibbs free energy barriers (ΔG≠) of the title reactions
进一步研究发现, 运用反应性指标理论[80~85]或许可以很好地解释标题反应的反应性大小顺序问题.该理论曾被其他课题组成功地用来解释芳环上的亲核取代反应顺序[76, 77].根据反应性指标理论, 亲核试剂的亲核性指标可以近似用下式表示[75~77]: ω-=-I≈EHOMO, 定域亲核性指标表示为[80]: ω-(k)=ω-f-(k).其中ω-和I以及EHOMO分别为亲核试剂亲核性指标、离子势和最高占据轨道能; ω-(k) 和f-(k) 表示k原子的定域亲核性指标和k原子的定域亲核福井函数.而定域亲核福井函数表示为[83~85]: f-(k)=Pk(N)-Pk(N-1). Pk(N) 和Pk(N-1) 表示含N个电子系统中k原子的NPA电荷以及当此系统失掉一个电子后系统中k原子的NPA电荷.
根据反应性指标理论, ω-值越小或ω-(k) 值越大, 反应物的亲核性越强, 反应能垒越低, 反应性越强.我们计算得出, 各反应物最高占据轨道能 (EHOMO, eV) 和ωN10- (eV) 分别与反应电子能垒ΔE≠(kJ·mol-1) 和吉布斯自由能垒ΔG≠(kJ·mol-1) 呈现较好的线性关系 (R2=0.96811, 0.97102和0.95716, 图 6, 7), 因此可以得出标题反应的反应性主要是由最高占据轨道能 (EHOMO) 控制.
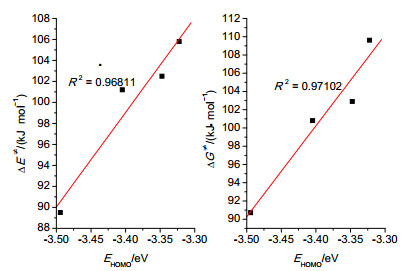 图 6
各反应物最高占据轨道能 (EHOMO) 与反应电子能垒ΔE≠和吉布斯自由能垒ΔG≠关系图
Figure 6.
Plot of the highest occupied molecular orbital energy (EHOMO) of the reactants vs electronic barriers (ΔE≠) and Gibbs free energy barriers (ΔG≠) of the title reactions
图 6
各反应物最高占据轨道能 (EHOMO) 与反应电子能垒ΔE≠和吉布斯自由能垒ΔG≠关系图
Figure 6.
Plot of the highest occupied molecular orbital energy (EHOMO) of the reactants vs electronic barriers (ΔE≠) and Gibbs free energy barriers (ΔG≠) of the title reactions
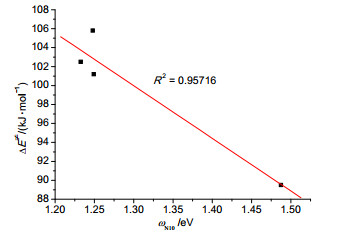 图 7
N10原子的区域亲核性指标 (ωN10-) 与反应电子能垒ΔE≠关系图
Figure 7.
Plot of the regional nucleophilicity index of N10 atom (ωN10-) vs electronic barriers (ΔE≠)
图 7
N10原子的区域亲核性指标 (ωN10-) 与反应电子能垒ΔE≠关系图
Figure 7.
Plot of the regional nucleophilicity index of N10 atom (ωN10-) vs electronic barriers (ΔE≠)
从以上分析可知, 标题反应中亲核试剂的亲核能力可以由最高占据轨道能EHOMO或者区域亲核性指标ωN10-来分析, 理论上反应性大小还与C2和N10原子所带电荷以及几何结构宽松度L%值有关.
表 5
最高占据轨道能 (EHOMO, eV)、亲核原子N10定域亲核性指标 (ωN10-, eV) 和定域亲核福井函数fN10-以及反应能垒
Table 5.
Highest occupied molecular orbital energy (EHOMO, eV), the regional nucleophilicity index (ωN10-, eV) and regional nucleophilic Fukui functionfN10-of the nucleophile N10 atom as well as the TS energy barriers in SNAr pathway of title reactions
X EHOMO ωN10- fN10- Δ E≠/(kJ·mol-1) Δ G≠/(kJ·mol-1) F -3.4929 1.4867 -0.4256 89.5 90.7 Cl -3.3212 1.2476 -0.3756 105.8 109.6 Br -3.3460 1.2321 -0.3682 102.5 102.9 I -3.4036 1.2487 -0.3669 101.2 100.8 表 5 最高占据轨道能 (EHOMO, eV)、亲核原子N10定域亲核性指标 (ωN10-, eV) 和定域亲核福井函数fN10-以及反应能垒
Table 5. Highest occupied molecular orbital energy (EHOMO, eV), the regional nucleophilicity index (ωN10-, eV) and regional nucleophilic Fukui functionfN10-of the nucleophile N10 atom as well as the TS energy barriers in SNAr pathway of title reactions通过多参数拟合得到了以上4个式子, 其R平方值都为1.从以上4个式子可以看出它们各自对反应能垒的贡献权重.很明显, 反应能垒主要由最高占据轨道能EHOMO或亲核原子N10的区域亲核性指标ωN10-决定, 另一因素为亲核原子N10电荷Q(N10) 大小, 而C2原子电荷Q(C2) 以及几何结构宽松度L%对反应能垒影响很小.
3.3.1 几何结构宽松度分析
从图 4可以看出, 标题反应协同SNAr路径 (Path 4) 过渡态中主要的几何结构变化特征为X1—C2和C2—N10单键相对于反应物与产物复合物或产物的键长变化.我们可以用Shaik等[78]提出的几何结构宽松度 (键长变化的百分数) 的概念来描述过渡态中X1—C2和C2—N10单键的变化情况, (X1—C2≠)%, (C2—N10≠)%与L%定义为:
这里R≠, RRe和RPro分别为过渡态结构TS中的键长, 反应物Re和产物复合物或产物Pro中的键长.很明显, 假如 (X1—C2≠)%, (C2—N10≠)%或L%值越大, 则反应能垒越大.计算所得几何结构宽松度 (X1—C2≠)%, (C2—N10≠)%和L%值总结在表 3中, 从表 3数据可以看出, 不管是 (X1—C2≠)%, (C2—N10≠)%还是L%值, 大小顺序为I>Br>Cl>F, 因此反应性顺序理论上应该是F>Cl>Br>I, 然而标题反应的反应活性顺序却是F>I>Br>Cl.故几何结构宽松度分析不能解释标题反应反应性大小顺序问题.
表 3
协同SNAr路径过渡态几何结构宽松 (X1—C2≠)%和 (C2—N10≠)%以及L%值与能垒
Table 3.
TS geometric looseness of the X1—C2 and C2—N10 bonds by the parameters (X1—C2≠)%, (C2—N10≠)% and L% and the TS energy barriers in SNAr pathway of title reactions
X (X1—C2≠)% (C2—N10≠)% L/% Δ E≠/(kJ·mol-1) Δ G≠/(kJ·mol-1) F 7.0 38.4 45.4 89.5 90.7 Cl 14.8 49.7 64.4 105.8 109.6 Br 15.9 54.9 70.8 102.5 102.9 I 16.2 58.7 74.9 101.2 100.8 表 3 协同SNAr路径过渡态几何结构宽松 (X1—C2≠)%和 (C2—N10≠)%以及L%值与能垒
Table 3. TS geometric looseness of the X1—C2 and C2—N10 bonds by the parameters (X1—C2≠)%, (C2—N10≠)% and L% and the TS energy barriers in SNAr pathway of title reactions4 结论
采用MP2/6-311+G**//B3LYP/6-311+G**理论方法, 对强碱催化N-(2-卤基苯基)-N'-苯基乙脒分子内氮芳基化合成苯并咪唑反应机理和反应性做了理论研究.为了评价方法、基组以及能量的可靠性, 采用密度泛函B3LYP和M06-2X理论方法以及6-31+G*和6-311+G**基组对所有物质几何结构在DMSO中进行优化, 获得了标题反应各个驻点几何结构参数, 在MP2/6-311+G**//B3LYP/6-311+G**和MP3/6-311+G**//B3LYP/ 6-311+G**以及MP2/6-311+G**//M06-2X/6-311+G**理论水平上进行了液相单点能计算, 最后得出结论如下.
反应物Re中C—X键发生均裂生成单自由基能垒太高, 因此标题反应单自由基机理不可能存在; 另外, 只存在三重态双自由基, 而单重态双自由基不存在, 并且在反应物Re中亲核原子N的p轨道与亲核基团苯环的π轨道存在p-π相互作用, 而三重态双自由基阴离子中却不存在这种作用, 导致后者能量比前者高出太多, 因此标题反应双自由基机理也不存在.
从过渡态结构以及反应能垒大小顺序可以得出, 标题反应的反应机理不是通常的分步SNAr机理, 而是只有一个过渡态的协同SNAr机理.
相比于其他方法和基组, MP2/6-311+G**//B3LYP/ 6-311+G**方法能更好地描述标题反应的相对能量和反应性顺序.
几何结构宽松度分析和NPA电荷分析都不能解释标题反应的反应性大小顺序问题.多参数拟合揭示标题反应的反应能垒主要由最高占据轨道能EHOMO或亲核原子N10的区域亲核性指标ωN10-控制, 另一因素为亲核原子N10所带负电荷的多少, 而C2原子电荷Q(C2) 以及几何结构宽松度L%对反应能垒几乎没有影响.
辅助材料 (Supporting Information) 文章中涉及的所有结构的笛卡尔坐标和能量.这些材料可以免费从本刊网站 (http://sioc-journal.cn/) 上下载
-
-
[1]
Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893. doi: 10.1021/cr020033s
-
[2]
Alamgir, M.; Black, D. St. C.; Kumar, N. Top. Heterocycl. Chem. 2007, 9, 87.
-
[3]
Kedar, M. S.; Dighe, N. S.; Pattan, S. R.; Musmade, D. S.; Thakur, D.; Bhosale, M.; Gaware, V. M. Pharma Chem. 2010, 2, 249.
-
[4]
Srikanth, L.; Varun Raj, V.; Raghunandan, N.; Venkateshwerlu, L. Pharma Chem. 2011, 3, 172.
-
[5]
Narasimhan, B.; Sharma, D.; Kumar, P. Med. Chem. Res. 2012, 21, 269. doi: 10.1007/s00044-010-9533-9
-
[6]
Lin, S. Y.; Isome, Y.; Stewart, E.; Liu, J. F.; Yohannes, D.; Yu, L. Tetrahedron Lett. 2006, 47, 2883. doi: 10.1016/j.tetlet.2006.02.127
-
[7]
Dudd, L. M.; Venardou, E.; Garcia-Verdugo, E.; Licence, P.; Blake, A. J.; Wilson, C.; Poliakoff, M. Green Chem. 2003, 5, 187. doi: 10.1039/b212394k
-
[8]
Zhang, C.; Zhang, L.; Jiao, N. Green Chem. 2012, 14, 3273. doi: 10.1039/c2gc36416f
-
[9]
Chari, M. A.; Shobha, D.; Sasaki, T. Tetrahedron Lett. 2011, 52, 5575. doi: 10.1016/j.tetlet.2011.08.047
-
[10]
Riadi, Y.; Mamouni, R.; Azzalou, R.; Haddad, M. E.; Routier, S.; Guillaumet, G.; Lazar, S. Tetrahedron Lett. 2011, 52, 3492. doi: 10.1016/j.tetlet.2011.04.121
-
[11]
Chari, M. A.; Shobha, D.; Kenawy, E. R.; Al-Deyab, S. S.; Reddy, B. V. S.; Vinu, A. Tetrahedron Lett. 2010, 51, 5195. doi: 10.1016/j.tetlet.2010.07.132
-
[12]
Bahrami, K.; Khodaei, M. M.; Nejatia, A. Green Chem. 2010, 12, 1237. doi: 10.1039/c000047g
-
[13]
Wan, J. P.; Gan, S. F.; Wu, J. M.; Pan, Y. Green Chem. 2009, 11, 1633. doi: 10.1039/b914286j
-
[14]
Saha, D.; Saha, A.; Ranu, B. C. Green Chem. 2009, 11, 733. doi: 10.1039/b823543k
-
[15]
Bahrami, K.; Khodaei, M. M.; Naali, F. Synlett 2009, 569.
-
[16]
Sharghi, H.; Aberi, M.; Doroodmand, M. M. Adv. Synth. Catal. 2008, 350, 2380. doi: 10.1002/adsc.v350:14/15
-
[17]
Mukhopadhyay, C.; Tapaswi, P. K. Tetrahedron Lett. 2008, 49, 6237. doi: 10.1016/j.tetlet.2008.08.041
-
[18]
Bahrami, K.; Khodaei, M. M.; Naali, F. J. Org. Chem. 2008, 73, 6835. doi: 10.1021/jo8010232
-
[19]
Zheng, N.; Anderson, K. W.; Huang, X.; Nguyen, H. N.; Buchwald, S. L. Angew. Chem., Int. Ed. 2007, 46, 7509. doi: 10.1002/(ISSN)1521-3773
-
[20]
Zheng, N.; Buchwald, S. L. Org. Lett. 2007, 9, 4749. doi: 10.1021/ol7020737
-
[21]
Zou, B.; Yuan, Q.; Ma, D. Angew. Chem., Int. Ed. 2007, 46, 2598. doi: 10.1002/(ISSN)1521-3773
-
[22]
Diao, X.; Wang, Y.; Jiang, Y.; Ma, D. J. Org. Chem. 2009, 74, 7974. doi: 10.1021/jo9017183
-
[23]
Kim, Y.; Kumar, M. R.; Park, N.; Heo, Y., Lee, S. J. Org. Chem. 2011, 76, 9577. doi: 10.1021/jo2019416
-
[24]
Evindar, G.; Batey, R. A. Org. Lett. 2003, 5, 133. doi: 10.1021/ol027061h
-
[25]
Brain, C. T.; Brunton, S. A. Tetrahedron Lett. 2002, 43, 1893. doi: 10.1016/S0040-4039(02)00132-6
-
[26]
Brain, C. T.; Steer, J. T. J. Org. Chem. 2003, 68, 6814. doi: 10.1021/jo034824l
-
[27]
Peng, J.; Ye, M.; Zong, C.; Hu, F.; Feng, L.; Wang, X.; Wang, Y.; Chen, C. J. Org. Chem. 2011, 76, 716. doi: 10.1021/jo1021426
-
[28]
Brasche, G.; Buchwald, S. L. Angew. Chem., Int. Ed. 2008, 47, 1932. doi: 10.1002/(ISSN)1521-3773
-
[29]
Xiao, Q.; Wang, W.; Liu, G.; Meng, F.; Chen, J.; Yang, Z.; Shi, Z. Chem. Eur. J. 2009, 15, 7292. doi: 10.1002/chem.v15:30
-
[30]
Wray, B. C.; Stambuli, J. P. Org. Lett. 2010, 12, 4576. doi: 10.1021/ol101899q
-
[31]
Deng, X.; Mani, N. S. Eur. J. Org. Chem. 2010, 4, 680.
-
[32]
Shen, M.; Driver, T. G. Org. Lett. 2008, 10, 3367. doi: 10.1021/ol801227f
-
[33]
程正, 张群峰, 许孝良, 李小年, 有机化学, 2015, 35(6), 1189. doi: 10.6023/cjoc201411031Cheng, Z.; Zhang, Q. F.; Xu, X. L.; Li, X. N. Chin. J. Org. Chem. 2015, 35(6), 1189 (in Chinese). doi: 10.6023/cjoc201411031
-
[34]
赵丹丹, 虞家涛, 王鹏程, 陆明, 有机化学, 2016, 36(1), 165. doi: 10.6023/cjoc201507010Zhao, D. D.; Yu, J. T.; Wang, P. C.; Lu, M. Chin. J. Org. Chem. 2016, 36(1), 165 (in Chinese). ( doi: 10.6023/cjoc201507010
-
[35]
余祖滔, 王泽瑜, 吴肖, 胡高云, 李乾斌, 有机化学, 2016, 36(7), 1672. doi: 10.6023/cjoc201512007Yu, Z. T.; Wang, Z. Y.; Wu, X.; Hu, G. Y.; Li, Q. B. Chin. J. Org. Chem. 2016, 36(7), 1672 (in Chinese). ( doi: 10.6023/cjoc201512007
-
[36]
蒙玉霞, 桂煜莹, 吉琼, 潘咏玲, 林志强, 吕柳, 曾向潮, 有机化学, 2016, 36(2), 384. doi: 10.6023/cjoc201507023Meng, Y. X.; Gui, Y. Y.; Ji, Q.; Pan, Y.; L.; Lin, Z. Q.; Lü, L.; Zeng, X. C. Chin. J. Org. Chem. 2016, 36(2), 384 (in Chinese). doi: 10.6023/cjoc201507023
-
[37]
Yuan, Y.; Thomé, I.; Kim, S. H.; Chen, D.; Beyer, A.; Bonnamour, J.; Zuidema, E.; Chang, S.; Bolm, C. Adv. Synth. Catal. 2010, 352, 2892. doi: 10.1002/adsc.v352.17
-
[38]
Cano, R.; Ramón, D. J.; Yus, M. J. Org. Chem. 2011, 76, 654. doi: 10.1021/jo1022052
-
[39]
Fang, Y.; Zheng, Y.; Wang, Z. Eur. J. Org. Chem. 2012, 7, 1495.
-
[40]
Zou, L. H.; Reball, J.; Mottweiler, J.; Bolm, C. Chem. Commun. 2012, 48, 11307. doi: 10.1039/c2cc36711d
-
[41]
Diness, F.; Fairlie, D. P. Angew. Chem., Int. Ed. 2012, 51, 8012. doi: 10.1002/anie.v51.32
-
[42]
Carmen Pérez-Aguilar, M.; Valdés, C. Angew. Chem., Int. Ed. 2012, 51, 5953. doi: 10.1002/anie.201200683
-
[43]
Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem.-Eur. J. 2012, 18, 14140. doi: 10.1002/chem.v18.44
-
[44]
Majumdar, K. C.; Ganai, S.; Nandi, R. K.; Ray, K. Tetrahedron Lett. 2012, 53, 1553. doi: 10.1016/j.tetlet.2012.01.015
-
[45]
Zhao, J.; Zhao, Y.; Fu, H. Angew. Chem., Int. Ed. 2011, 50, 3769. doi: 10.1002/anie.v50.16
-
[46]
Beyer, A.; Reucher, C. M. M.; Bolm, C. Org. Lett. 2011, 13, 2876. doi: 10.1021/ol2008878
-
[47]
Thomé, I.; Bolm, C. Org. Lett. 2012, 14, 1892. doi: 10.1021/ol3005134
-
[48]
Beyer, A.; Buendia, J.; Bolm, C. Org. Lett. 2012, 14, 3948. doi: 10.1021/ol301704z
-
[49]
Thom, I.; Besson, C.; Kleine, T.; Bolm, C. Angew. Chem., Int. Ed. 2013, 52, 7509. doi: 10.1002/anie.201300917
-
[50]
Xiang, S. K.; Tan, W.; Zhang, D. X.; Tian, X. L.; Feng, C.; Wang, B. Q.; Zhao, K. Q.; Hu, P.; Yang, H. Org. Biomol. Chem. 2013, 11, 7271. doi: 10.1039/c3ob41479e
-
[51]
Baars, H.; Beyer, A.; Kohlhepp, S. V.; Bolm, C. Org. Lett. 2014, 16, 536. doi: 10.1021/ol403414v
-
[52]
Bunnett, J. F.; Zahler, R. E. Chem. Rev. 1951, 49, 273. doi: 10.1021/cr60153a002
-
[53]
Terrier, F. The SNAr Reactions:Mechanistic Aspects, in Modern Nucleophilic Aromatic Substitution, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany 2013, pp. 1~84.
-
[54]
Hunter, A.; Renfrew, M.; Taylor, J. A.; Whitmore, J. M. J.; Wil-liams, A. J. Chem. Soc., Perkin Trans. 2 1993, 1703.
-
[55]
Fernandez, I.; Frenking, G.; Uggerud, E. J. Org. Chem. 2010, 75(9), 2971. doi: 10.1021/jo100195w
-
[56]
Glukhovtsev, M. N.; Bach, R. D.; Laiter, S. J. Org. Chem. 1997, 62(12), 4036. doi: 10.1021/jo962096e
-
[57]
Simkin, B. Y.; Gluz, E. B.; Glukhovtsev, M. N.; Minkin, V. I. J. Mol. Struct. (THEOCHEM) 1993, 284(1~2), 123.
-
[58]
Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
-
[59]
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785. doi: 10.1103/PhysRevB.37.785
-
[60]
Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200. doi: 10.1016/0009-2614(89)87234-3
-
[61]
Jr, J. R. P.; Veloso, D. P. Phys. Chem. Chem. Phys. 2008, 10, 1118. doi: 10.1039/B716159J
-
[62]
Cid, M. V. F.; Buijs, W.; Witkamp, G. J. Ind. Eng. Chem. Res. 2007, 46, 941.
-
[63]
Gorelsky, S. I.; Lapointe, D.; Fagnou, K. J. Am. Chem. Sos. 2008, 130, 10848. doi: 10.1021/ja802533u
-
[64]
Imoto, M.; Matsui, Y.; Takeda, M.; Tamaki, A.; Taniguchi, H.; Mizuno, K.; Ikeda, H. J. Org. Chem. 2011, 76, 6356. doi: 10.1021/jo2007219
-
[65]
Toledo, R. O.; Santos, J. G.; Ríos, P.; Castro, E. A.; Campodónico, P. R.; Contreras, R. J. Phys. Chem. B 2013, 117, 5908. doi: 10.1021/jp4005295
-
[66]
Toledo, R. O.; Contreras, R.; Tapiab, R. A.; Campodónico, P. R. Org. Biomol. Chem. 2013, 11, 2302. doi: 10.1039/c3ob27450k
-
[67]
杜丽娟, 吴彩虹, 顾红红, 李娟, 有机化学, 2015, 35(8), 1726. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344935.shtmlDu, L. J.; Wu, C. H.; Gu, H. H.; Li, J. J. Org. Chem. 2015, 35(8), 1726 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344935.shtml
-
[68]
Glukhovtsev, M. N.; Bach, R. D.; Laiter, S. J. Org. Chem. 1997, 62, 4036. doi: 10.1021/jo962096e
-
[69]
Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82, 284.
-
[70]
Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027. doi: 10.1021/cr00031a013
-
[71]
Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899. doi: 10.1021/cr00088a005
-
[72]
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215. doi: 10.1007/s00214-007-0310-x
-
[73]
Sadowsky, D.; McNeill, K.; Cramer, C. J. Environ. Sci. Technol. 2014, 48, 10904. doi: 10.1021/es5028822
-
[74]
Cairns, A. G.; Senn, H. M.; Murphy, M. P.; Hartley, R. C. Chem. Eur. J. 2014, 20, 3742. doi: 10.1002/chem.v20.13
-
[75]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. J. A; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand. J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009.
-
[76]
Glukhovtsev, M. N.; Pross, A.; Radom, L. J. Am. Chem. Soc. 1996, 118, 6273. doi: 10.1021/ja953665n
-
[77]
李强根, 毛双, 蔡皖飞, 郑妍, 刘柳斜, 化学通报, 2016, 79(5), 418.Li, Q. G.; Mao, S.; Cai, W. F.; Zheng, Y.; Liu, L. X. Chemistry 2016, 79(5), 418(in Chinese). (
-
[78]
Shaik, S. S.; Schlegel, H. B.; Wolfe, S. Theoretical Aspects of Physical Organic Chemistry. The SN2 Mechanism, Wiley, New York, 1992, pp. 181~188.
-
[79]
Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735.
-
[80]
Chattaraj, P. K.; Sarkar, U.; Roy, D. R. Chem. Rev. 2006, 106, 2065. doi: 10.1021/cr040109f
-
[81]
Ayers, P. W.; Anderson, J. S. M.; Bartolotti, L. J. Int. J. Quantum Chem. 2005, 101, 520. doi: 10.1002/(ISSN)1097-461X
-
[82]
Contreras, R.; Andres, J.; Safont, V. S.; Campodonico, P.; Santos, J. G. J. Phys. Chem. A 2003, 107(29), 5588. doi: 10.1021/jp0302865
-
[83]
Ormazábal-Toledo, R.; Contreras, R. Adv. Chem. 2014, 2014, 1.
-
[84]
Ormazábal-Toledo, R.; Contreras, R.; Campodónico, P. R. J. Org. Chem. 2013, 78, 1091. doi: 10.1021/jo3025048
-
[85]
Ormazábal-Toledo, R.; Campodónico, P. R.; Contreras, R. Org. Lett. 2011, 13, 822. doi: 10.1021/ol103033j
-
[1]
-

图 1 强碱催化分子内氮芳基化C—N键耦合可能反应机理
Figure 1 Possible reaction mechanisms of intramolecular N-arylation for the C—N bond coupling reaction by base-catal-yzed

图 2 本文研究的分子内氮芳基化合成苯并咪唑可能反应通道
Figure 2 Possible reaction pathways in this study of intramolecular N-arylations of N-(2-halogen phenyl)-N'-phenyl ethyl amidines for the synthesis of benzimidazoles

图 3 MP2/6-311+G**//B3LYP/6-311+G**理论水平上计算得到反应物N-(2-氟基苯基)-N'-苯基乙脒阴离子 (左) 及其相应的三重态双自由基阴离子 (右) HOMO轨道图
Figure 3 HOMO orbitals of the reactant N-(2-fluorine phenyl)-N'-phenyl ethyl amidine anion (left) and its double radical anion (triplet state) (right) attained at MP2/6-311+G**//B3LYP/ 6-311+G** level

图 4 DMSO中B3LYP/6-311+G**水平上优化所得标题反应协同SNAr路径所有反应物、过渡态和产物几何结构
Figure 4 B3LYP/6-311+G** optimized all the geometry structures of reactant, TS and product in DMSO of title reactions in the concerted SNAr pathway

图 5 标题反应电子能垒 (ΔE≠) 和吉布斯自由能垒 (ΔG≠) 关系图
Figure 5 Plot of electronic barriers (ΔE≠) vs the Gibbs free energy barriers (ΔG≠) of the title reactions

图 6 各反应物最高占据轨道能 (EHOMO) 与反应电子能垒ΔE≠和吉布斯自由能垒ΔG≠关系图
Figure 6 Plot of the highest occupied molecular orbital energy (EHOMO) of the reactants vs electronic barriers (ΔE≠) and Gibbs free energy barriers (ΔG≠) of the title reactions

图 7 N10原子的区域亲核性指标 (ωN10-) 与反应电子能垒ΔE≠关系图
Figure 7 Plot of the regional nucleophilicity index of N10 atom (ωN10-) vs electronic barriers (ΔE≠)
表 1 DMSO中标题反应所有反应物Re与其自由基阴离子相对能量表 (kJ·mol-1)
Table 1. Relative energies of all the radical anions relative to reactants Re in DMSO of title reactions (kJ·mol-1)
Single free radical Δ Ga/(kJ·mol-1) Δ Gb/(kJ·mol-1) F Cl Br I F Cl Br I M06-2X/6-311+G** 466.6 349.9 308.3 263.8 275.5 260.0 260.4 261.7 MP2/6-311+G**//M06-2X/6-311+G** 557.8 454.9 394.8 351.5 463.3 443.1 435.5 431.8 aΔG=G(1-1)+G(X·)-G(Re); bΔG=G(2-1)-G(Re).  下载: 导出CSV
下载: 导出CSV
表 2 不同水平上计算所得标题反应SNAr路径反应能垒表
Table 2. Reaction energy barriers for the title reactions in the SNAr pathway calculated at different level
Level Δ E≠/(kJ mol-1) Δ H≠/(kJ mol-1) Δ G≠/(kJ mol-1) F Cl Br I F Cl Br I F Cl Br I B3LYP/6-31+G* 121.6 128.4 119.3 121.4 115.8 123.5 114.9 117.0 120.8 127.9 120.3 120.2 B3LYP/6-311+G** 118.7 128.9 122.6 120.6 113.1 124.1 118.5 116.4 119.6 132.5 122.9 120.2 M062X/6-311+G** 113.2 126.0 125.2 112.1 109.1 124.1 120.7 106.2 115.5 128.5 127.2 116.1 MP2/6-311+G**//M062X/6-311+G** 94.2 102.5 96.3 82.6 87.9 97.2 91.8 78.7 94.3 101.6 98.4 88.6 MP2/6-311+G**//B3LYP/6-311+G** 89.6 106.0 102.4 101.4 84.0 101.2 98.3 97.2 90.5 109.6 102.7 100.9 MP3/6-311+G**//B3LYP/6-311+G** 126.8 146.6 141.1 140.3 121.4 141.6 136.9 136.1 127.9 150.0 141.3 139.9
下载: 导出CSV
表 3 协同SNAr路径过渡态几何结构宽松 (X1—C2≠)%和 (C2—N10≠)%以及L%值与能垒
Table 3. TS geometric looseness of the X1—C2 and C2—N10 bonds by the parameters (X1—C2≠)%, (C2—N10≠)% and L% and the TS energy barriers in SNAr pathway of title reactions
X (X1—C2≠)% (C2—N10≠)% L/% Δ E≠/(kJ·mol-1) Δ G≠/(kJ·mol-1) F 7.0 38.4 45.4 89.5 90.7 Cl 14.8 49.7 64.4 105.8 109.6 Br 15.9 54.9 70.8 102.5 102.9 I 16.2 58.7 74.9 101.2 100.8
下载: 导出CSV
表 4 MP2/6-311+G**//B3LYP/6-311+G**水平上计算所得主要原子NPA电荷 (Q)
Table 4. Main atomic NPA charges (Q) calculated at MP2/6-311+G**//B3LYP/6-311+G** level
X X1 C2 C7 N8 C9 N10 Re TS Re TS Re TS Re TS Re TS Re TS F -0.4271 -0.5100 0.4021 0.5735 0.1712 0.0255 -0.8354 -0.7532 0.6142 0.5970 -0.8402 -0.7281 Cl -0.0777 -0.3445 -0.0878 0.2585 0.2243 0.0765 -0.8318 -0.7813 0.6186 0.6147 -0.8164 -0.7485 Br 0.0098 -0.3443 -0.1897 0.2228 0.2266 0.0892 -0.8321 -0.7886 0.6196 0.6186 -0.8118 -0.7577 I 0.1186 -0.3075 -0.2898 0.1807 0.2258 0.0880 -0.8355 -0.7885 0.6200 0.6200 -0.8106 -0.7682
下载: 导出CSV
表 5 最高占据轨道能 (EHOMO, eV)、亲核原子N10定域亲核性指标 (ωN10-, eV) 和定域亲核福井函数fN10-以及反应能垒
Table 5. Highest occupied molecular orbital energy (EHOMO, eV), the regional nucleophilicity index (ωN10-, eV) and regional nucleophilic Fukui functionfN10-of the nucleophile N10 atom as well as the TS energy barriers in SNAr pathway of title reactions
X EHOMO ωN10- fN10- Δ E≠/(kJ·mol-1) Δ G≠/(kJ·mol-1) F -3.4929 1.4867 -0.4256 89.5 90.7 Cl -3.3212 1.2476 -0.3756 105.8 109.6 Br -3.3460 1.2321 -0.3682 102.5 102.9 I -3.4036 1.2487 -0.3669 101.2 100.8
下载: 导出CSV
-








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 2600
- HTML全文浏览量: 841
 下载:
下载:
