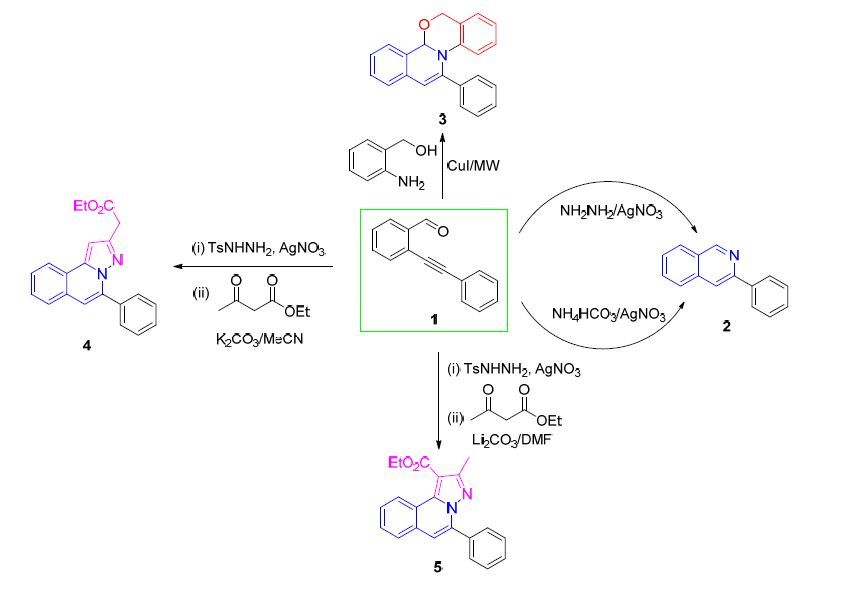
 图 图式 1
基于邻炔基醛的异喹啉合成研究
Figure 图式 1.
Synthesis of isoquinolines from o-alkynyl aldehydes
图 图式 1
基于邻炔基醛的异喹啉合成研究
Figure 图式 1.
Synthesis of isoquinolines from o-alkynyl aldehydes
引用本文:
李余波, 赵云辉, 罗明检, 唐子龙, 曹朝暾, 邓克勤. 基于邻炔基醛的异喹啉及其衍生物的合成研究[J]. 有机化学,
2016, 36(10): 2504-2509.
doi:
10.6023/cjoc201604031

Citation: Li Yubo, Zhao Yunhui, Luo Mingjian, Tang Zilong, Cao Chaotun, Deng Keqin. Synthesis of Isoquinolines Derivatives from o-Alkynyl Aldehydes[J]. Chinese Journal of Organic Chemistry, 2016, 36(10): 2504-2509. doi: 10.6023/cjoc201604031
Citation: Li Yubo, Zhao Yunhui, Luo Mingjian, Tang Zilong, Cao Chaotun, Deng Keqin. Synthesis of Isoquinolines Derivatives from o-Alkynyl Aldehydes[J]. Chinese Journal of Organic Chemistry, 2016, 36(10): 2504-2509. doi: 10.6023/cjoc201604031
基于邻炔基醛的异喹啉及其衍生物的合成研究
English
Synthesis of Isoquinolines Derivatives from o-Alkynyl Aldehydes
Abstract:
The isoquinoline and dihydroisoquinoline ring systems are found as the core nucleus in a wide variety of natural products and pharmaceutical agents with good biologically activity. Isoquinoline, benzoxazine-fused isoquinlines and pyrazo-lo[5,1-a]isoquinolines are prepared from o-alkynyl aldehydes and nitrogen-containing compounds via tandem reactions catalyzed by CuI or AgNO3. The reactions have some advantages, such as mild reaction conditions, simple operation and easily available catalysts.
-
异喹啉、二氢异喹啉及其衍生物是一类特殊的骨架结构,广泛存在于天然产物、药物分子以及生物碱中[1].含有这类特殊结构的化合物大部分具有较好的生物活性,如抗肿瘤、抑制小肠收缩、抗心血管等活性,使得这些小分子被作为合成砌块而大量地应用于有机合 成[2]. 异喹啉化合物同时还可以作为过渡金属离子的手性配体而应用于发光材料领域[3].
因其重要的应用价值,有机化学家已经研发多种方法来合成异喹啉类化合物[4]. 早期用于合成异喹啉的有 Bischler-Napieralski[5]、Pomeranz-Fritsch[6]和Pictet- Spengler[7]等反应,但这些反应操作复杂、条件苛刻而没有得到推广. 随后,Pfeffer[8]、Heck[9]和Widdowson[10]等报道了使用钯作为催化剂来合成多取代的异喹啉化合物,这种方法具有反应条件温和、高效、选择性好等优点,开创了过渡金属催化合成异喹啉化合物的先河.现在,过渡金属催化的分子间或分子内环化反应已经成为合成异喹啉最重要的合成方法. 在这些过渡金属中,如Pd、Ag、Cu、Pt、Au、Ni、Co、Rh等[11],Ag因其在成环过程中对炔键良好的活化作用而得到了广泛地应用. 吴劼小组[12]报道了多种利用AgOTf催化合成异喹啉及其衍生物的方法,梁永民等[13]实现了AgSbF6催化分子内叠氮成环构筑异喹啉的方法,Shin等[14]证明邻炔基芳香肟醚在AgOTf与TfOH的共催化下,能断裂N—O键与炔形成异喹啉环. 本文将基于邻炔基醛发展几种构筑异喹啉及其衍生物的合成方法(Scheme 1).
图 图式 1
基于邻炔基醛的异喹啉合成研究
Figure 图式 1.
Synthesis of isoquinolines from o-alkynyl aldehydes
1 结果与讨论
1.1 硝酸银促进的N—N键断裂合成异喹啉
邻炔基醛与单盐酸肼在乙醇中先形成腙,然后除去溶剂,得到的腙无需纯化即进行下一步反应. 首先以1,2-二氯乙烷(DCE)为溶剂,10 mol%硝酸银为催化剂,在80 ℃反应0.5 h,可以36%的产率得到异喹啉. 继续考察其它溶剂对反应产率的影响,发现在非极性溶剂(CCl4、CHCl3、PhMe)中,反应产率普遍偏低,如在PhMe中,只能以26%的产率得到异喹啉; 而极性溶剂有助于产物的生成,如在MeOH、EtOH、i-PrOH或DMSO中,产率均在40%以上. 其中以DMSO为溶剂,反应产率最高(44%),反应时间也最短,只需10 min左右. 增加硝酸银至1 equiv.,产率增加至57%,继续增加催化剂的使用量,对反应产率提升没有帮助. 由此可以得出,本方法中最佳的反应条件是: DMSO为溶剂,1 equiv.硝酸银为催化剂,反应温度为100 ℃. 因肼具有较强还原银离子的能力,在本实验中也观察到反应瓶壁上会有单质银产生,需要消耗一部分的硝酸银,故而,反应需要1 equiv.硝酸银作为催化剂.
1.2 硝酸银促进的NH4HCO3法合成异喹啉
NH4HCO3在受热的条件下容易产生NH3,NH3在密闭的反应体系中与邻炔基醛形成亚胺,然后在AgNO3的催化下发生环化反应得到异喹啉. 温度对反应产率有重要的影响,当反应在80 ℃、以乙腈为溶剂时,产率为65%,升温至100 ℃,产率可以提升到79%,继续升温至120 ℃,可以86%的收率得到异喹啉,而在室温条件下,反应几乎不进行(图 1). 使用其它溶剂如EtOH或DCE时,反应产率会有明显的下降. 降低催化剂AgNO3的使用量,产物的收率也会有一定程度的降低,这可能与反应体系中形成部分的银氨溶液有关.
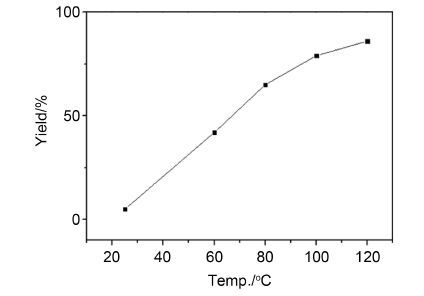 图 1
温度对产率的影响关系曲线图
Figure 1.
Influence of temperature on yields
图 1
温度对产率的影响关系曲线图
Figure 1.
Influence of temperature on yields
1.3 CuI催化的苯并噁嗪并异喹啉的微波合成
以邻氨基苄醇为原料,在10 mol% Ni(AcO)2催化下与邻炔基醛在乙醇中、微波100 W、70 ℃反应,以45%的产率得到目标产物苯并恶嗪并异喹啉3,使用NiCl2与CoCl2催化,反应效率没有提升. 采用Zn(NO3)2与PdCl2作催化剂,反应收率可以达到50%以上,改用AgNO3,反应效率进一步提高. 选用CuI催化该反应,收率则可以达到81%. 继续考察溶剂对反应收率的影响. 使用极性溶剂如N,N'-二甲基甲酰胺(DMF)、四氢呋喃(THF)、H2O等,反应产率有明显的下降; 采用非极性溶剂如二氯甲烷(DCM)、PhMe,反应产率则急剧下降. 故在微波辐照条件下合成苯并噁嗪并异喹啉的最佳催化剂为CuI、最适宜的溶剂为EtOH.
1.4 [3+2]环加成合成吡唑并异喹啉化合物
邻炔基醛1与对甲苯磺酰肼在乙醇中形成腙I,然后在AgNO3的催化下得到1,3-偶极中间体Ⅱ (Scheme 2)[15]. 1 equiv.中间体Ⅱ与3 equiv.乙酰乙酸乙酯溶于MeCN中,体系在K2CO3作用下80 ℃反应10 h,以46%的产率得到吡唑并异喹啉乙酸乙酯(4)和17%的产率得到吡唑并异喹啉甲酸乙酯(5). 增加乙酰乙酸乙至5 equiv.,化合物4的产率提升至57%,而化合物5的收率变化不大. 使用其它无机碱,如Na2CO3、Li2CO3、t-BuOK、K3PO4、Cs2CO3等,化合物4的产率均有不同程度的下降,使用较弱的碱Na2CO3,化合物4的收率只有12%,但化合物5的收率迅速提升到50%. 有趣的是使用Li2CO3为碱,以45%的收率只得到单一产物5,但反应时间需延长到120 h. 为了进一步提高反应效率,继续考察溶剂对反应的影响,使用THF、DMSO、EtOH、PhMe、DCE等为溶剂,产物4或5的收率均未有明显的提升. 但是研究中发现,使用Li2CO3为碱、DMF为溶剂,可以高达72%的产率得到单一的化合物5,并且反应时间也缩短至10 h. 研究得出,合成吡唑并异喹啉乙酸乙酯(4)的最佳条件为: 原料比1:5,MeCN为溶剂,K2CO3为碱,80 ℃反应10 h. 而制备吡唑并异喹啉甲酸乙酯(5)的最优条件为: DMF为溶剂,Li2CO3为碱.
根据上述实验现象,我们提出了可能的反应机理(Scheme 2). 1,3-偶极中间体Ⅱ与乙酰乙酸乙酯反应可能经历两条不同的路径分别得到产物4和5. 路径a: 乙酰乙酸乙酯在K2CO3的作用下,γ-位质子被夺去形成端位的烯醇中间体Ⅲ,Ⅲ与中间体Ⅱ发生1,3-偶极环加成,再经芳构化反应得到产物4. 路径b: 乙酰乙酸乙酯在Li2CO3的作用下,α-位质子被夺去形成烯醇中间体V,然后进攻中间体Ⅱ发生1,3-偶极环加成,再经系列转化得到产物5. 在此过程中,V由于Li离子的良好络合能力可能形成了比较稳定的六元环过渡态,因而反应体系中只得到了单一产物5.
该1,3-偶极环加成反应的优势在于,采用相同的底物,通过简单改变反应条件就可以得到不同的产物4和5,而且化合物4是三组分“一锅法”所不能提供的[16]. 新化合物4的构型通过含有取代基衍生物6的单晶衍射得到确定(图 2).
2 结论
以邻炔基苯甲醛为原料,在硝酸银、碘化亚铜等过渡金属化合物的催化下,合成了4种不同类型的异喹啉及其衍生物,反应条件温和,操作简便,产率中等到优秀,为含氮杂环化合物的合成提供了新的方法与策略.
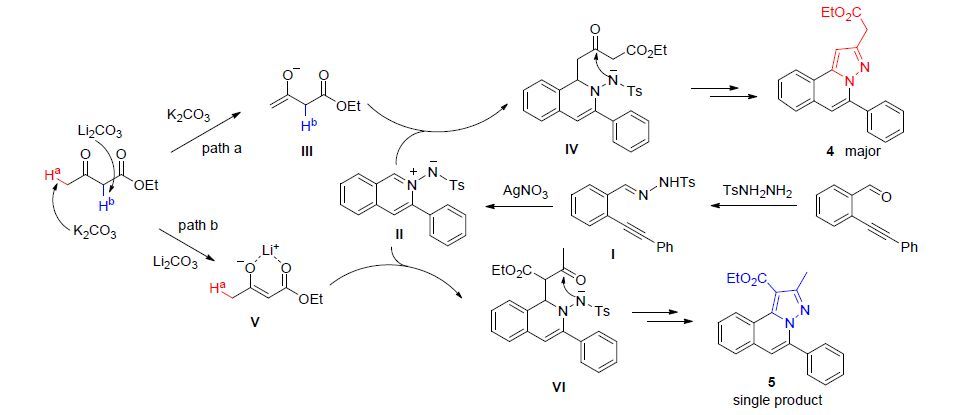 图 图式 2
吡唑并异喹啉化合物的合成机理推测
Figure 图式 2.
Probable mechanism of synthesizing pyrazolo[5,1-a]isoquinolines
图 图式 2
吡唑并异喹啉化合物的合成机理推测
Figure 图式 2.
Probable mechanism of synthesizing pyrazolo[5,1-a]isoquinolines
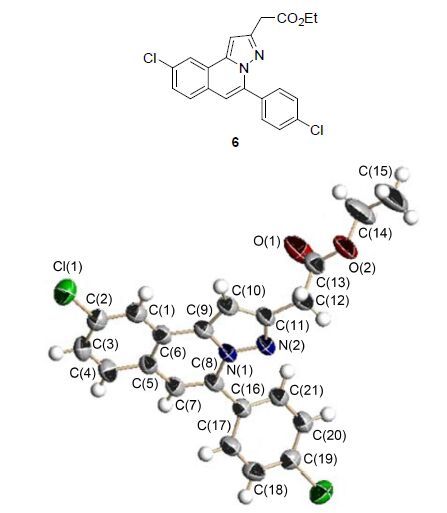 图 2
化合物6的单晶衍射
Figure 2.
X-ray crystal structure of 6
图 2
化合物6的单晶衍射
Figure 2.
X-ray crystal structure of 6
3 实验部分
3.1 仪器与试剂
熔点用Yanaco MP500型显微熔点测定仪(温度未校正); 高分辨质谱在液相色谱/质谱联用仪Xevo Q-Tof MS上测定; IR 光谱在Nicolet 5-DX 型FT-IR红外光谱仪上测定(KBr 压片); 核磁共振仪: Bruker AV-Ⅱ 500 MHz NMR,以TMS 为内标,氘代氯仿为溶剂. 邻炔基醛根据文献[15]制备,其它所用的药品均为市售的分析纯或化学纯,除特别注明外,未经进一步处理.
3.2 实验方法
3.2.3 吡唑并异喹啉基乙酸酯(4)的合成
将邻苯乙炔基苯甲醛(1) (62.0 mg,0.3 mmol)和对甲苯磺酰肼(56 mg,0.3 mmol)溶于10 mL无水乙醇中,加入1滴浓盐酸,室温下搅拌,反应完毕后待固体析出,抽滤得到中间体产物I. 然后将所得到的固体溶于1,2-二氯乙烷中,加入硝酸银(10 mg,0.06 mmol),60 ℃下搅拌反应2 h,除去溶剂,得中间体产物Ⅱ.
将中间体产物Ⅱ (112.2 mg,0.3 mmol)溶于3 mL乙腈中,加入乙酰乙酸乙酯 (195.1 mg,1.5 mmol)和K2CO3 (82.9 mg,0.6 mmol),反应体系在70 ℃反应12 h,冷却至室温,溶剂减压旋蒸除去,粗产物柱层析[V(石油醚):V(乙酸乙酯)=5:1]分别得到纯净的吡唑并异喹啉化合物4 (产率57%)和5 (产率20%).
2-(5-苯基吡唑[5,1-a]异喹啉)乙酸乙酯(4),浅黄色液体. 1H NMR (500 MHz,CDCl3) δ: 8.09~8.04 (m,1H),7.94~7.85 (m,2H),7.72~7.66 (m,1H),7.57~7.43 (m,5H),7.09 (s,1H),6.99 (s,1H),4.21 (q,J=7.0 Hz,2H),3.92 (s,2H),1.29 (t,J=7.0 Hz,3H); 13C NMR (125 MHz,CDCl3) δ: 170.9,147.2,140.4,138.2,133.8,129.5,129.3,129.2,128.3,127.9,127.3,127.1,123.8,123.6,112.3,97.9,61.0,35.0,14.3; IR (KBr) v: 2979,2927,1735,1549,1466,1323,1251,1159,1031,760,695 cm-1. HRMS calcd for C21H19N2O2 [M+H]+ 331.14410,found 331.14407.
2-[9-氯-5-(4-氯苯基)吡唑[5,1-a]异喹啉]乙酸乙酯(6),产率41%. 浅黄色固体,m.p. 77~78 ℃; 1H NMR (500 MHz,CDCl3) δ: 8.02 (d,J=2.0 Hz,1H),7.86~7.82 (m,2H),7.61 (d,J=8.5 Hz,1H),7.49~7.43 (m,3H),7.06 (s,1H),6.92 (s,1H),4.22 (q,J=7.0 Hz,2H),3.90 (s,2H),1.30 (t,J=7.0 Hz,3H); 13C NMR (125 MHz,CDCl3) δ: 170.7,147.5,139.3,137.2,135.5,133.2,131.7,130.9,130.8,128.9,128.6,128.5,128.4,127.3,124.8,123.1,111.6,98.6,61.1,34.8,14.3; IR (KBr) v: 974,2921,1737,1549,1491,1332,1217,1193,1088,832,748 cm-1. HRMS calcd for C21H17Cl2N2O2 [M+H]+ 399.06616,found 399.06616.
3.2.4 吡唑并异喹啉基甲酸酯(5)的合成
取上述中间体产物Ⅱ (112.2 mg,0.3 mmol)溶于3 mL N,N'-二甲基甲酰胺中,加入乙酰乙酸乙酯(195.1 mg,1.5 mmol)和Li2CO3 (44.3 mg,0.6 mmol). 反应体系在70 ℃反应10 h,冷却至室温,溶剂减压旋蒸除去,粗产物柱层析[V(石油醚):V(乙酸乙酯)=5:1]得到纯净的2-5-苯基吡唑[5,1-a]异喹啉-1-甲酸乙酯(5),产率72%. 白色固体,m.p. 131~132 ℃; 1H NMR (500 MHz,CDCl3) δ: 9.56~9.54 (m,1H),7.87~7.81 (m,2H),7.75~7.71 (m,1H),7.63~7.57 (m,2H),7.54~7.47 (m,3H),7.13 (s,1H),4.47 (q,J=7.0 Hz,2H),2.64 (s,3H),1.47 (t,J=7.0 Hz,3H); 13C NMR (125 MHz,CDCl3) δ: 165.3,153.5,140.3,137.9,133.63,130.9,129.8,129.4,129.1,128.3,127.3,127.1,127.0,123.5,114.7,106.4,60.5,15.9,14.5; IR (KBr) v: 2973,2930,1708,1527,1453,1337,1271,1179,1124,1087,758,687 cm-1. HRMS calcd for C21H19N2O2 [M+H]+ 331.14410,found 331.14407.
辅助材料(Supporting Information) 所有新化合物的1H NMR和13C NMR图谱. 这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
3.2.1 异喹啉的合成
AgNO3催化的NH4HCO3法合成异喹啉: 将邻苯乙炔基苯甲醛(1) (62.0 mg,0.3 mmol)溶于3 mL乙腈中,然后加入NH4HCO3 (79 mg,1 mmol)和 AgNO3 (10 mg,0.06 mmol). 反应体系在120 ℃搅拌12 h,冷却至室温,向反应体系中加入饱和食盐水,用乙酸乙酯萃取3次,合并有机相,使用无水Na2SO4干燥1 h. 将固体过滤,减压旋蒸除去溶剂,粗产物柱层析[V(石油醚):V(乙酸乙酯)=10:1]得到纯净的异喹啉2,产率86%.
AgNO3催化的N—N键断裂法合成异喹啉: 将邻苯乙炔基苯甲醛(1) (62.0 mg,0.3 mmol)溶于5 mL乙醇中,然后加入单盐酸肼(30.6 mg,0.45 mmol),反应体系在室温搅拌3 h. 将反应体系中的固体过滤得到中间体,然后将中间体溶于3 mL的二甲亚砜中,再加入AgNO3 (51.0 mg,0.3 mmol). 反应体系在100 ℃搅拌15 min原料即消失. 冷却至室温,向反应体系中加入饱和食盐水,用乙酸乙酯萃取3次,合并有机相,使用无水Na2SO4干燥1 h. 将固体过滤,减压旋蒸除去溶剂,粗产物柱层析[V(石油醚):V(乙酸乙酯)=10:1]得到纯净的异喹啉2,产率57%.
3-苯基异喹啉(2)[13]: 白色固体,m.p. 97~98 ℃; 1H NMR (500 MHz,CDCl3) δ: 9.37 (s,1H),8.16 (d,J=7.5 Hz,2H),8.10 (s,1H),8.02 (d,J=8.0 Hz,1H),7.91~7.89 (d,J=8.0 Hz,1H),7.75~7.70 (m,1H),7.63~7.60 (m,1H),7.56~7.53 (m,2H),7.47~7.44 (m,1H); 13C NMR (125 MHz,CDCl3) δ: 152.5 ,151.3,139.6,136.7,130.6,128.8,128.6,127.8,127.6,127.1,127.0,126.9,116.6.
3.2.2 苯并噁嗪并异喹啉(3)的合成
邻苯乙炔基苯甲醛(1) (62.0 mg,0.3 mmol)溶于3 mL乙醇中,加入邻氨基苯乙醇(92 mg,0.75 mmol)和CuI (9.5 mg,0.05 mmol),反应体系在70 ℃微波加热条件下反应10 min,原料消失. 溶剂减压旋蒸除去,粗产物柱层析[V(石油醚):V(乙酸乙酯)=5:1]得到纯净的苯并噁嗪并异喹啉(3)[17],产率81%. 浅黄色固体,m.p. 164~165 ℃; 1H NMR (500 MHz,CDCl3) δ: 7.42 (d,J=7.5 Hz,1H),7.32 (t,J=7.5 Hz,1H),7.23~7.17 (m,7H),7.05 (d,J=7.5 Hz,1H),6.92 (t,J=7.5 Hz,1H),6.78 (t,J=7.5 Hz,1H),6.22 (d,J=8.0 Hz,1H),6.09 (s,1H),5.96 (s,1H),5.26 (d,J=14.5 Hz,1H),5.09 (d,J=14.5 Hz,1H); 13C NMR (125 MHz,CDCl3) δ: 140.8,139.9,136.9,132.4,129.0,128.6,128.5,128.0,127.9,126.8,126.0,125.8,124.72,124.4,123.8,122.6,105.7,85.0,68.0.
-
-
[1]
Scott, J. D.; Williams, R. M. Chem. Rev. 2002, 102, 1669. (b) Chrzanowska, M.; Rozwadowska, M. D. Chem. Rev. 2004, 104, 3341. (c) Inoue, K.; Kulsum, U.; Chowdhury, S. A.; Fujisawa, S. I.; Ishihara, M.; Yokoe I.; Sakagami, H. Anticancer Res. 2005, 25, 4053. (d) Su, S.; Porco Jr., J. A. Org. Lett. 2007, 9, 4983. doi: 10.1021/cr010212u
-
[2]
Wada, Y.; Nishida, N.; Kurono, N.; Ohkuma, T.; Orito, K. Eur. J. Org. Chem. 2007, 4320. (b) Sun, Y.; Xun, K.; Wang Y.; Chen, X. Anti-Cancer Drugs 2009, 20, 757. (c) Wong, N. C. W.; Tucker, J. E. L.; Hansen, H. C.; Chiacchia, F. S.; McCaffrey, D. US 2008188467, 2008,[Chem. Abstr. 2008, 149, 246409].
-
[3]
Liu, S.-J.; Zhao, Q.; Chen, R.-F.; Deng, Y.; Fan, Q.-L.; Li, F.-Y.; Wang, L.-H.; Huang, C.-H.; Huang, W. Chem. Eur. J. 2006, 12, 4351. (b) Zhao, Q.; Liu, S.; Shi, M.; Wang, C.; Yu, M.; Li, L.; Li, F.; Yi, T.; Huang, C. Inorg. Chem. 2006, 45, 6152. (c) Tsuboyama, A.; Iwawaki, H.; Furugori, M.; Mukaide, T.; Kamatani, J.; Igawa, S.; Moriyama, T.; Miura, S.; Takiguchi, T.; Okada, S.; Hoshino, M.; Ueno, K. J. Am. Chem. Soc. 2003, 125, 12971. doi: 10.1002/(ISSN)1521-3765
-
[4]
Tiwari, V. K.; Pawar, G. G.; Jena, H. S.; Kapur, M. Chem. Commun. 2014, 50, 7322. (b) Liu, Y.; Zeng, R.; Pan, J.; Zou, J. Chin. J. Chem, 2014, 32, 883. (c) Shi, L.; Ji, Y.; Huang, W.; Zhou, Y. Acta Chim. Sinica 2014, 72, 820(in Chinese). (时磊, 姬悦, 黄文学, 周永贵, 化学学报, 2014, 72, 820.) (d) Ma, J.; Zhai, Y.; Zhu, L.; Zhao, Y. Chin. J. Org. Chem. 2015, 35, 2366(in Chinese). (马金龙, 翟艳光, 朱俐, 赵育, 有机化学, 2015, 35, 2366.)
-
[5]
Sotomayor, N.; Dominguez, E. J. Org. Chem. 1996, 61, 4062. (b) Ishikawa, T.; Shimooka, K.; Narioka, T.; Noguchi, S.; Saito, T.; Ishikawa, A.; Yamazaki, E.; Harayama, T.; Seki, H.; Yamaguchi, K. J. Org. Chem. 2000, 65, 9143. doi: 10.1021/jo960007s
-
[6]
Boudou, M.; Enders, D. J. Org. Chem. 2005, 70, 9486. (b) Walker, E. R.; Leung, S. Y.; Barrett, A. G. M. Tetrahedron Lett. 2005, 46, 6537. doi: 10.1021/jo051554t
-
[7]
Cox, E. D.; Cook, J. M. Chem. Rev. 1995, 95, 1797. (b) Chrzanowska, M.; Rozwadowska, M. D. Chem. Rev. 2004, 104, 341. (c) Youn, S. W. J. Org. Chem. 2006, 71, 2521. doi: 10.1021/cr00038a004
-
[8]
Maassarani, F.; Pfeffer, M.; Le Borgne, G. J. Chem. Soc., Chem. Commun. 1987, 8, 565.
-
[9]
Wu, G.; Geib, S. J.; Rheingold, A. L.; Heck, R. F. J. Org. Chem. 1988, 53, 3238. doi: 10.1021/jo00249a018
-
[10]
Girling, I. R.; Widdowson, D. A. Tetrahedron Lett. 1982, 23, 4281. doi: 10.1016/S0040-4039(00)88725-0
-
[11]
Dai, G.; Larock, R. C. J. Org. Chem. 2002, 67, 7042. (b) Huang, Q.; Larock, R. C. J. Org. Chem. 2003, 68, 980. (c) Ramakrishna, T. V. V.; Sharp, P. R. Org. Lett. 2003, 5, 877. (d) Lim, S.-G.; Lee, J. H.; Moon, C. W.; Hong, J.-B.; Jun, C.-H. Org. Lett. 2003, 5, 2759. (e) Korivi, R. P.; Cheng, C.-H. Org. Lett. 2005, 7, 5179. (f) Wang, B.; Lu, B.; Jiang, Y.; Zhang, Y.; Ma, D. Org. Lett. 2008, 10, 2761. (g) Chiba, S.; Xu, Y.-J.; Wang, Y.-F. J. Am. Chem. Soc. 2009, 131, 12886. (h) Florentino, L.; Aznar, F.; Valdés, C. Org. Lett. 2012, 14, 2323. (i) Li, C.; An, S.; Zhu, Y.; Zhang, J.; Kang, Y.; Liu, P.; Wang, Y.; Li, J. RSC Adv. 2014, 4, 49888. doi: 10.1021/jo026016k
-
[12]
Chen, Z.; Yang, X.; Wu, J. Chem. Commun. 2009, 3469. (b) Chen, Z.; Ding, Q.; Yu, X.; Wu, J. Adv. Synth. Catal. 2009, 351, 1692. (c) Liu, G.; Liu, H.; Qiu, G.; Pu, S.; Wu, J. Chem. Commun. 2012, 7049.
-
[13]
Niu, Y.-N.; Yan, Z.-Y.; Gao, G.-L.; Wang, H.-L.; Shu, X.-Z.; Ji, K.-G.; Liang, Y.-M. J. Org. Chem. 2009, 74, 2893.
-
[14]
Hwang S.; Lee, Y.; Lee, P.; Shin, S. Tetrahedron Lett. 2009, 50, 2305.
-
[15]
Kobayashi, M.; Kondo, K.; Aoyama, T. Tetrahedron Lett. 2007, 48, 7019.
-
[16]
Yu, X.; Yang, Q.; Lou, H.; Peng, Y.; Wu, J. Org. Biomol. Chem. 2011, 9, 7033. doi: 10.1039/c1ob05917c
-
[17]
Zhao, Y.-H.; Li, Y.; Guo, T.; Tang, Z.; Deng, K.; Zhao, G. Synth. Commun. 2016, 46, 355. (b) Zhao, Y.-H.; Li, Y.; Guo, T.; Tang, Z.; Xie, W.; Zhao, G. Tetrahedron Lett. 2016, 57, 2257.
-
[1]
-

图式 2 吡唑并异喹啉化合物的合成机理推测
Scheme 2 Probable mechanism of synthesizing pyrazolo[5,1-a]isoquinolines
-


 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 820
- HTML全文浏览量: 155
 下载:
下载:
