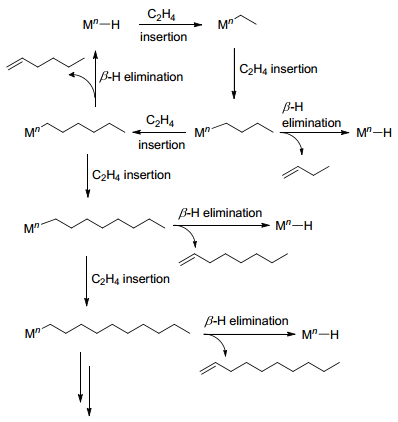
 图 图式1
乙烯二聚Cossee-Alrman机理
Figure 图式1.
Cossee-Alrma mechanism of ethylene dimerization
图 图式1
乙烯二聚Cossee-Alrman机理
Figure 图式1.
Cossee-Alrma mechanism of ethylene dimerization
引用本文:
宋闯, 毛国梁, 刘振华, 宁英男, 姜涛. 均相Cr系催化剂催化乙烯选择性齐聚反应机理研究进展[J]. 有机化学,
2016, 36(9): 2105-2120.
doi:
10.6023/cjoc201602034

Citation: Song Chuang, Mao Guoliang, Liu Zhenhua, Ning Yingnan, Jiang Tao. Advances in Mechanistic Research of Ethylene Selective Oligomeri-zation Catalyzed by Homogeneous Chromium-Based Catalysts[J]. Chinese Journal of Organic Chemistry, 2016, 36(9): 2105-2120. doi: 10.6023/cjoc201602034
Citation: Song Chuang, Mao Guoliang, Liu Zhenhua, Ning Yingnan, Jiang Tao. Advances in Mechanistic Research of Ethylene Selective Oligomeri-zation Catalyzed by Homogeneous Chromium-Based Catalysts[J]. Chinese Journal of Organic Chemistry, 2016, 36(9): 2105-2120. doi: 10.6023/cjoc201602034
均相Cr系催化剂催化乙烯选择性齐聚反应机理研究进展
English
Advances in Mechanistic Research of Ethylene Selective Oligomeri-zation Catalyzed by Homogeneous Chromium-Based Catalysts
Abstract:
Linear α-olefin is an important feed stock for chemical industry. Production of specific α-olefin through selective ethylene oligomerization catalyzed by homogeneous chromium-based catalysts is an important research on orientation under rapid development in recent years. The research of catalytic mechanism plays an important role in guiding the exploration of novel high-efficient catalysts. The application of methods such as labelled atom, organometallic precursor, EPR-XAS, density functional theory (DFT) calculation etc. in the research of reaction path and the metal oxidation state is introduced, with the latest achievement in the mechanistic research summarized. Research works carried out by various researchers are compared and discussed from methodology. New perspective on binuclear Cr metal ring mechanistic research is proposed and an outlook on the further research orientation is presented.
-
线性α-烯烃是一类重要的化工原料, 主要用于制备增塑剂、润滑剂、表面活性剂、环氧化合物等.其中1-己烯和1-辛烯是合成线性低密度聚乙烯(LLDPE)重要的共聚单体[1~3].传统的生产方法中, 乙烯非选择性齐聚工艺中特定组分选择性差, 得到的是符合Schulz-Flory分布或Poisson分布的混合物, 需要通过高耗能的分离过程得到较高纯度的线性α烯烃, 这无法满足工业生产对高纯度α烯烃的需求[4, 5].近些年乙烯选择性齐聚技术的研究取得许多重要进展.在1987年, IFP-SABIC公司研发的Alphabutol乙烯二聚工艺实现1-丁烯工业化生产[6~9]. 2003年, Phillips公司在卡塔尔首次实现乙烯三聚工业化生产[10]. 2014年, 南非Sasol公司在美国路易斯安那州建造世界上第一个工业化乙烯四聚装置[11].
目前乙烯选择性齐聚催化剂中, 钛系、钽系、铬系等催化剂都表现了良好的催化性能, 其中Cr系催化剂具有优越的综合性能(活性和选择性), 所以备受关注[12~16].这些催化剂的性能表现主要受配体结构(空间位阻效应、给电子效应)的影响, 其配体上配位原子大多为P、N、O、S等, 近几年研究热点主要集中在多齿的PP、PNN、PNP、PNNP、SNS、NNZ、NZN (Z为P、N、O、S原子)等类型的配体.
催化机理方面的研究对于新型催化剂的研发具有重要的指导作用, 鉴于Cr系催化剂在乙烯选择性三聚和四聚过程中表现出优越性能, 人们从反应路径、活性结构金属中心价态两方面采用不同的研究方法对其催化机理进行了深入的研究并取得了一些重要的进展.
1 乙烯选择性齐聚反应路径研究
在乙烯选择性齐聚催化体系中, 主要以制备高纯度1-丁烯、1-己烯、1-辛烯为主.起初高选择性Ti系乙烯二聚催化体系的研究使人们认为反应路径是遵循Cossee-Arlman机理.随着高选择性乙烯三聚催化体系的诞生, 人们对其反应路径的认识由最初的Cossee-Arlman机理转向单核金属环机理.新型乙烯四聚催化体系的研究引发了单核和双核金属环机理之间的争论.
1.1 基于乙烯二聚的Cossee-Arlman机理
研究初期, 由于乙烯齐聚产物中α-烯烃符合Schulz-Flory或者正态分布, 乙烯齐聚反应被认为是Cossee-Arlman机理[15]作用的结果, 如Scheme 1所示, 因β氢转移而发生的还原消去反应与乙烯的插入链增长反应形成竞争.
图 图式1
乙烯二聚Cossee-Alrman机理
Figure 图式1.
Cossee-Alrma mechanism of ethylene dimerization
当用Cossee-Arlman机理解释乙烯二聚生成1-丁烯的反应过程, 并得到实验的间接证明. Suttil等[17]基于Ziegler等[18]发现的Ti(OR')4/AlR3乙烯二聚催化体系, 对采用不同的C2H4/C2D4配比的乙烯二聚产物进行GC-MS分析.当使用Ti(OBu)4/AlMe3作为催化剂, C2H4/C2D4的配比为1:1时, 齐聚产物中H/D分布不规则[19], 但是1-丁烯中离子碎片峰丰度分布与按Cossee-Arlman机理预测的理论分布基本一致, 1-丁烯与乙烯共聚按照Cossee-Arlman机理预测的共聚产物离子碎片峰丰度的理论分布与C2H4/C2D4齐聚产物中C6烯烃实际分布基本一致(如图 1所示).分子计算方面Robinson等[20]利用密度泛函理论(DFT)基于Ti(OR')4/AlR3乙烯二聚催化体系, 对Al-Sa'doun等[21]用ESR推测的Al/Ti比为2时三核活性结构进行了优化, 并按Cossee-Arlman机理对乙烯齐聚过渡态进行了计算.研究表明, 在1-丁烯生成前乙烯的插入的速率比链转移速率快, 这为1-丁烯的生成提供了可能.然而, 由于LnTi-Et (Ln=[(μ-OMe)2AlMe2]2)的乙烯插入活化能与LnBu的乙烯插入活化能相当, 并且LnHex中β氢转移的活化能比LnBu的乙烯插入活化能高, 故此机理不能解释高选择性生成1-己烯、1-辛烯以及更高碳数产物的Schulz-Flory分布等问题.
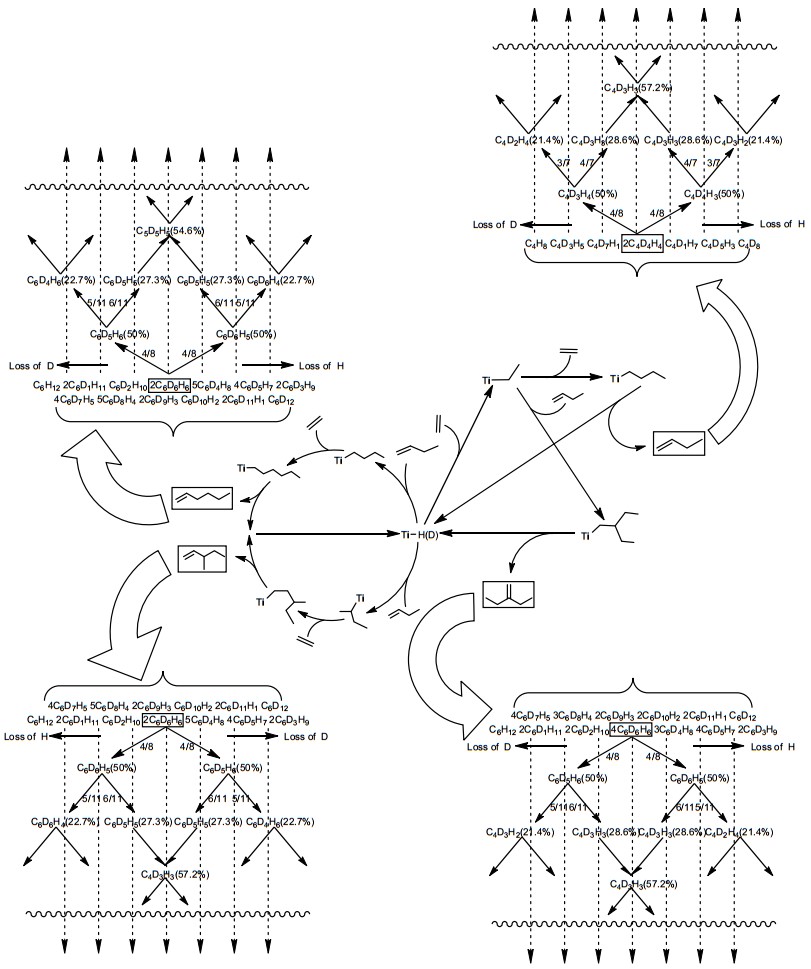 图 1
Cossee-Alrman机理下C2H4/C2D4产物异构体质谱(EI)裂解过程
Figure 1.
Process according to Cossee-Alrman mechanism on relative intensities of C2H4/C2D4 isotopomer fragmentation pattern as a result of electron impact ionization
图 1
Cossee-Alrman机理下C2H4/C2D4产物异构体质谱(EI)裂解过程
Figure 1.
Process according to Cossee-Alrman mechanism on relative intensities of C2H4/C2D4 isotopomer fragmentation pattern as a result of electron impact ionization
1.2 基于乙烯三聚的金属环机理
1973年, McDermott等[22]首次合成了Pt(Ⅱ)的金属环状化合物, 并认为在热分解中得到的烯烃产物和金属环上β氢转移有关, 如Scheme 2所示.这为Manyik等[23]基于他们发现的乙烯齐聚Cr(Ⅲ)-2-ethylhexanoate/PIBAO (PIBAO为部分水解的三异丁基铝氧烷)催化体系提出乙烯三聚金属环机理提供了重要的现实依据.
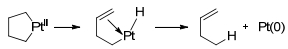 图 图式2
McDermott等提出的Pt(Ⅱ)金属环热分解机理
Figure 图式2.
Pt(Ⅱ)
metal ring thermal decomposition mechanism suggested by McDermott et al.
图 图式2
McDermott等提出的Pt(Ⅱ)金属环热分解机理
Figure 图式2.
Pt(Ⅱ)
metal ring thermal decomposition mechanism suggested by McDermott et al.
Manyik等认为首先两个乙烯分子通过π电子与金属中心空位配位, 经过氧化耦合形成Cr的金属五元环, 接着第三个乙烯分子与金属中心配位, 通过β氢转移形成烯烃烷基Cr结构或者直接还原消去释放1-己烯, 如Scheme 3所示.
然而, 随着Phillip公司高选择性乙烯三聚催化体系的诞生[24~26], Manyik等提出的金属五元环机理仍不能解释高选择性1-己烯的生成和没有其他C6烯烃生成的原因.
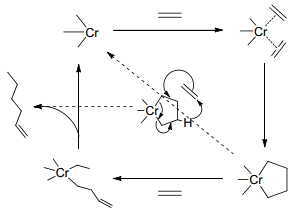 图 图式3
Manyik等提出的乙烯三聚金属环状机理
Figure 图式3.
Metal ring mechanism for ethylene trimerization supposed by Manyik et al.
图 图式3
Manyik等提出的乙烯三聚金属环状机理
Figure 图式3.
Metal ring mechanism for ethylene trimerization supposed by Manyik et al.
1989年, Briggs[27]基于他们发现的Cr(Ⅲ)/2-ethylhexanoate/PIBAO/DME (DME为乙二醇二甲醚)乙烯三聚催化体系对此金属五元环机理做了改进, 如Scheme 4所示.当第三个乙烯分子与金属中心配位后, 迅速插入五元环中使其扩大成为Cr的金属七元环, 再通过β氢的转移释放1-己烯.当乙烯插入Cr金属五元环的速率大于其五元环β氢还原消去的速率, 并且金属七元环还原消去1-己烯的速率大于乙烯进一步插入成环的速率时, 这就得到1-己烯的高选择性.
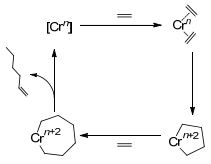 图 图式 4
Briggs 等改进的乙烯三聚金属环机理
Figure 图式 4.
Metal ring mechanism for ethylene trimerization modified by Briggs et al.
图 图式 4
Briggs 等改进的乙烯三聚金属环机理
Figure 图式 4.
Metal ring mechanism for ethylene trimerization modified by Briggs et al.
很多实验证明了Briggs等改进的乙烯三聚金属环机理的合理性. Emrich等[28]于1997年首次合成了Cr杂金属五元环和七元环化合物, 这为其金属环中间体的存在提供了证据, 相对于Cr杂金属五元环, Cr杂金属七元环更容易热分解产生1-己烯, 所以1-己烯被认为是金属七元环经β氢转移开环还原消去形成的.而1-丁烯没有生成则是由于Cr杂金属五元环实际上基本没有发生热分解.此外, Ti系金属五元环化合物未见报道, 刘睿等[29]认为基于乙烯二聚的钛系催化剂不会发生类似的反应过程.
Agapie等[30, 31]基于Cr(Ⅲ)/PNPOMe/MAO (MAO为甲基铝氧烷)乙烯三聚催化体系进行了氘代实验, 使用[((o-MeO-Phenyl)2P)2NCH3]CrPh3/H(Et2O)2B[C6H3-(CF3)2]4及((o-MeO-Phenyl)2P)2NCH3]CrBr-(o, o'-biphenyldiyl)/NaB(C6H3(CF3)2)4作为催化剂, MAO作为助催化剂, C2H4/C2D4配比为1时, 三聚产物的异构体只有C6D12、C6D8H4、C6D4H8和C6H12, 且其比例为1:3:3:1, 这一现象支持了乙烯三聚金属环机理, 如Scheme 5所示.
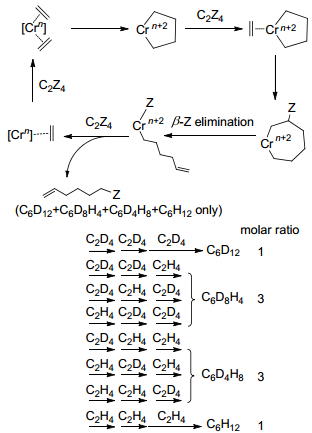 图 图式5
Agapie等提出的金属环机理下乙烯三聚产物异构体的理论分布
Figure 图式5.
Theoretical distribution of isomers in ethylene trimerization product under metal ring mechanism proposed by Agapie et al.
图 图式5
Agapie等提出的金属环机理下乙烯三聚产物异构体的理论分布
Figure 图式5.
Theoretical distribution of isomers in ethylene trimerization product under metal ring mechanism proposed by Agapie et al.
基于单核Cr系催化剂乙烯三聚金属环机理的计算表明[32, 33], 相比于乙烯的进一步插入成环, Cr杂七元环更容易还原消去释放1-己烯.然而能量的计算表明金属环稳定性是与金属中心的环境和配体结构有关的, 这意味着Cr杂九元环乃至更大的Cr杂十一元环是有可能形成的.
在乙烯三聚金属环机理中, Cr杂七元环开环过程存在两种方式(Scheme 6):一是β氢转移至金属中心, 形成烷基氢化物, 之后氢转移至α'碳上还原消去释放1-己烯.二是β氢直接转移至α'碳上.目前很多理论计算表明乙烯三聚金属环机理中β氢转移可能是由后者完成的.
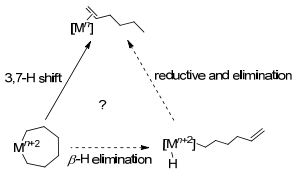 图 图式6
β-H转移的两种可能方式
Figure 图式6.
Two possible ways of β-H transfer
图 图式6
β-H转移的两种可能方式
Figure 图式6.
Two possible ways of β-H transfer
2003年, 余志祥等[34]基于Ta系乙烯三聚催化剂在探索乙烯三聚金属环机理时, 首次提出金属七元环开环释放1-己烯过程是由β氢直接转移至α'碳上导致的.
刘柏平等[35]利用DFT研究DME对Cr(EH)3/PIBAO (EH=2-ethylhexanoate)催化体系催化行为转换机制中, 对乙烯选择性齐聚金属环机理进行了过渡态计算, 基于Cr(Ⅰ)/Cr(Ⅲ)、Cr(Ⅱ)/Cr(Ⅳ)建立了10种初始模型.经研究发现, 只有Cr(Ⅰ)+/DME和Cr(Ⅰ)OR两种模型能建立β氢转移两步法反应路径, 其余八种模型β氢均是通过Agostic-Assisted作用直接转移至α'碳上, 所以作者认为在乙烯选择性三聚过程中, Cr金属七元环开环过程中β氢转移是通过3, 7位转移一步完成的.
Budzelaar[36]基于乙烯三聚Cr(Ⅰ)催化剂[37]建立了C(Ⅰ)和Cr(Ⅰ)+的四种模型, 以Cr(Ⅰ)/Cr(Ⅲ)循环为基础利用DFT对乙烯选择性齐聚金属环机理进行了过渡态计算, β氢移过程如Scheme 7所示.研究发现, 能垒TS(D-G) < TS(D-E) < TS(G-L) < TS(F-E), 由此得出此体系中Cr七元环开环过程β氢同样是直接转移至α'碳上, 且此β氢转移过程是乙烯选择性齐聚过程的决速步, 作者推测以Cr(Ⅱ)/Cr(Ⅳ)循环的乙烯三聚机理中Cr金属七元环β氢同样也是直接转移至α'碳上.
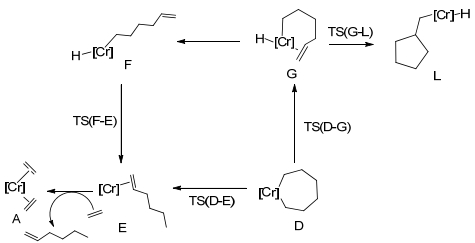 图 图式7
Budzelaar提出的金属环机理β-H转移过渡态
Figure 图式7.
β-H transfer transition state of metal ring mechanism proposed by Budzelaar
图 图式7
Budzelaar提出的金属环机理β-H转移过渡态
Figure 图式7.
β-H transfer transition state of metal ring mechanism proposed by Budzelaar
1.3 乙烯四聚金属环机理探索
2004年, Bollmann等[38]首次发现了Cr/PNP/MAO乙烯选择性四聚催化体系, 研究人员对其机理开展了大量的研究工作, Overett等[39]利用不同配比的C2H4/C2D4基于Cr(acac)3/PNP(PNP=Ph2PN(iPr)PPh2)/MMAO乙烯四聚催化体系对其主要产物及副产物(1-己烯、1-辛烯、甲基环戊烷、亚甲基环戊烷及C10以上异构体)形成机理(如Scheme 8所示)采用GC-MS进行了分析.研究表明, Cr金属环机理相比于Cossee-Alrman机理更适合解释乙烯四聚催化体系产物形成过程.目前已报道的乙烯四聚金属环机理主要有Cr单核及双核两种.
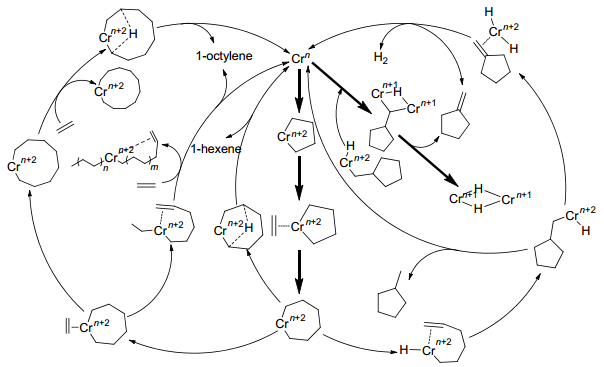 图 图式8
Overett等基于Cr(acac)3/PNP (PNP=Ph2PN(iPr)PPh2)/MMAO乙烯四聚催化体系提出的产物形成机理
Figure 图式8.
Product formation mechanism based on ethylene tetramerization catalyst of Cr(acac)3/PNP (PNP=Ph2PN(iPr)PPh2)/MMAO proposed by Overett et al.
图 图式8
Overett等基于Cr(acac)3/PNP (PNP=Ph2PN(iPr)PPh2)/MMAO乙烯四聚催化体系提出的产物形成机理
Figure 图式8.
Product formation mechanism based on ethylene tetramerization catalyst of Cr(acac)3/PNP (PNP=Ph2PN(iPr)PPh2)/MMAO proposed by Overett et al.
1.3.1 Cr系催化剂乙烯四聚单核金属环机理
基于乙烯三聚的机理可以推测出乙烯四聚的金属环机理, 如Scheme 9所示.即在Cr金属七元环的基础上进一步插入一个乙烯分子使其生成更大的Cr金属九元环, 接着再通过β氢转移和还原消去释放1-辛烯.然而, 这很难解释当形成Cr杂金属九元环后却不能被乙烯分子继续插入形成更大的金属环, 进而生成一系列按一定规律分布的α烯烃产物的原因, 且目前为止乙烯四聚并没有得到和三聚一样高的选择性( > 95%).余志祥和Blok等[34, 40]分别基于Ta系和Ti系乙烯三聚催化体系利用DFT方法证明了金属七元环在被乙烯插入进一步形成更大的环的不稳定性.最近Britovsek等[41]利用DFT方法基于Cr/PNP/MAO乙烯选择性齐聚催化体系按照Cr(Ⅰ)/Cr(Ⅲ)单核金属环机理对齐聚产物的形成进行了全分析计算, 研究表明, 采用阳离子的Cr(Ⅰ)(P、N上取代基均为甲基)模型中Cr杂金属九元环再次插入乙烯的能垒比其通过β氢直接转移至α'碳上释放1-辛烯的能垒高, 然而Cr杂金属五元环与乙烯分子单配位经过氧化耦合形成Cr杂金属七元环和双配位形成Cr杂金属九元环形成竞争关系, 在较为合适的动力学与热力学条件下, 后者成为高选择性1-辛烯生成的必要条件.
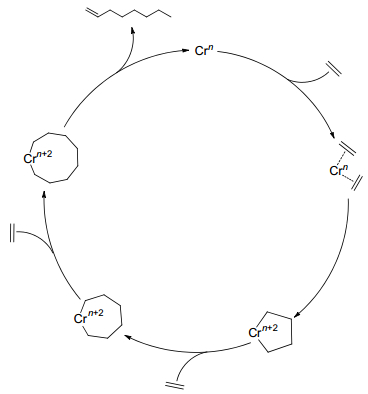 图 图式9
单核Cr乙烯四聚金属环机理
Figure 图式9.
Mononuclear
Cr ethylene tetramerization metal ring mechanism
图 图式9
单核Cr乙烯四聚金属环机理
Figure 图式9.
Mononuclear
Cr ethylene tetramerization metal ring mechanism
1.3.2 Cr系催化剂乙烯四聚双核金属环机理
针对Cr系催化剂单核金属九元环的不稳定性, 很多学者提出具有双核活性结构的乙烯四聚反应机理.
Young等[42]针对乙烯四聚催化体系1-辛烯的高选择性(70%)提出了一种具有双金属中心活性结构的乙烯四聚反应机理, 如Scheme 10所示.
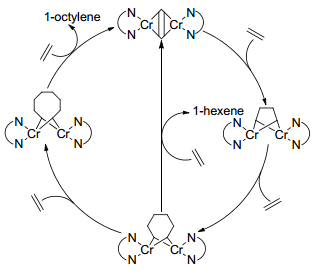 图 图式10
Young等提出的双核Cr乙烯四聚金属环机理
Figure 图式10.
Binuclear Cr ethylene tetramerization metal ring mechanism proposed by Young et al.
图 图式10
Young等提出的双核Cr乙烯四聚金属环机理
Figure 图式10.
Binuclear Cr ethylene tetramerization metal ring mechanism proposed by Young et al.
Peitz等[43]提出了一种新的乙烯四聚反应机理, 如Scheme 11所示.这是一个双核低价态Cr的结构, 乙烯分子分别和两个金属中心配位形成两个Cr金属五元环, 通过C—C偶联形成双金属十元环, 接着再通过β氢的还原消去释放1-辛烯.此机理避开了金属七元环的形成, 但还需经过进一步的验证.
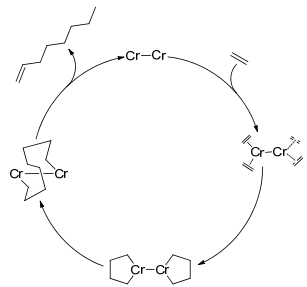 图 图式11
Peitz等提出的乙烯四聚双核Cr金属环机理
Figure 图式11.
Binuclear Cr ethylene tetramerization metal ring mechanism proposed by Peitz et al.
图 图式11
Peitz等提出的乙烯四聚双核Cr金属环机理
Figure 图式11.
Binuclear Cr ethylene tetramerization metal ring mechanism proposed by Peitz et al.
2 基于单核Cr系催化剂金属环机理金属中心价态研究
金属中心价态的确定对于乙烯选择性齐聚机理的研究可以起到很重要的作用.目前普遍认为助催化剂的引入可能改变了中心金属的价态, 伴随着可能发生的烷基转移、去质子化、阳离子化、配体半配位化等作用使其成为活性物种, 接着乙烯分子的氧化加成使金属中心价态升高, 还原消除过程使金属中心价态还原为初始价态, 完成催化循环.然而, 人们对金属中心价态的变化情况尚存在争议.对于乙烯选择性齐聚单核Cr系金属环机理而言, Cr(Ⅰ)/Cr(Ⅲ)、Cr(Ⅱ)/Cr(Ⅳ)被认为是可能的金属耦合价态.
2.1 基于预制催化剂前驱体对金属中心价态研究
1959年, Breslow等[44~48]基于他们发现的Cp2TiCl2/Et2AlCl催化体系首次提出了活性物种金属烷基化的概念, 并通过不同配比的Cp2TiCl2/Et2AlCl与Cp2CH2TiCl/AlCl3在紫外可见分光光度计下吸光度值进行了论证. 1961年Zefirova等[49]在此基础上提出了阳离子型活性物种Cp2TiMe+. 1967年, Dyachkovskii等[50]基于电化学方法提出乙烯聚合发生在阳离子活性中心Cp2Ti+-R (Scheme 12).这为人们后续研究乙烯选择性齐聚机理提供了重要基础.
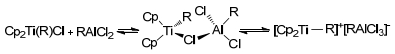 图 图式12
Dyachkovskii等提出的Ti系催化剂金属烷基化与离子化过程
Figure 图式12.
Ti metal alkylation and ionization process proposed by Dyachkovskii et al.
图 图式12
Dyachkovskii等提出的Ti系催化剂金属烷基化与离子化过程
Figure 图式12.
Ti metal alkylation and ionization process proposed by Dyachkovskii et al.
一般认为, 作为助催化剂的铝试剂在与主催化剂的作用中可以使金属中心烷基化, 金属中心价态经氧化还原可能被改变, 并形成离子产物[15]. Cr-Al双核化合物被认为是可能的乙烯选择性齐聚活性前驱体.
Jabri等[51]基于CrCl3/PNP (PNP=Ar2PN(R)PAr2, R=Cy, Ar=Ph)催化体系在AlMe3的作用下合成了阳离子型Cr(Ⅱ)—Al双金属化合物(Eq. 1), 它在MAO的作用下具有乙烯齐聚催化活性.
McGuinness等[52, 53]基于他们发现的CrCl3/PNP (PNP=NH[(CH2)2PPh2]2)乙烯三聚催化体系在DABCO {1, 4-diazabicyclo[2.2.2]octane}的作用下合成了NH去质子化的Cr(Ⅱ)双核配合物(Eq. 2), 他们认为DABCO与烷基铝有同样的去质子化作用, 此Cr(Ⅱ)配合物在MAO或者B(C6F5)3作用下具有乙烯齐聚活性.此外他们[54]基于CrCl2(THF)/SNS [SNS=CySCH2CH2N(H)CH2CH2SCy]催化体系在AlMe3的作用下得到Cr(Ⅲ)和Cr(0)物质(Eq. 3), 并在EPR下确定了Cr(Ⅲ)配合物结构.此Cr(Ⅲ)配合物具有乙烯三聚活性, 由此他们认为烷基铝不仅可以使高价的Cr(Ⅲ)还原, 也可以使低价态Cr(Ⅱ)再次氧化.
Zhang等[55]基于Yoshida等[56]发现的CrCl3/NNN (NNN为三齿的吡唑基配体)乙烯三聚催化体系在AlMe3的作用下合成了一系列Cr(Ⅲ)—Al双金属化合物(Eqs. 4~6), 研究发现在此过程中主要有自由基加成、甲基化、还原、配合物阳离子化、卤化、N—H去质子化和苯环上C—H键活化作用, 此系列配合物在MAO作用下可催化乙烯三聚反应.
在乙烯选择性齐聚催化体系中, 除使用烷基铝试剂作为助催化剂外, 还使用诸如B[3, 5-(CF3)2C6H3]4-、B(C6F5)4-、{Al[OC(CF3)3]4}−等弱配位或者非配位的阴离子作为助催化剂.它们在乙烯聚合中过量的配比会使配体分解, 并且在外加不同铝试剂时会表现出不同催化行为[57, 58].
在2008年, Gambarotta等[59, 60]首次对Cr系乙烯选择性金属环机理中的金属中心价态的变化进行了研究, 他们用2, 3, 4, 5-tetrahydro-1H-carbazole/Cr(octanoate)3/AlEt2Cl合成的Cr(Ⅰ)配合物在甲基环己烷中和Phillips催化剂[61]具有类似的乙烯三聚催化行为, 但在甲苯中失去了催化活性, 作者认为这可以解释Phillips乙烯三聚催化剂在甲苯中活性不高的原因:这是由于配合物其中一个配体解离使得空位被甲苯占据, 阻止了乙烯与其配位.而用2, 3, 4, 5-tetrahydro-1H-carbazole/CrCl3/AlEt3合成的Cr(Ⅱ)配合物不具有乙烯三聚活性.接着他们用tetramethylpyrrole/CrCl2(thf)2/AlMe3合成的双核Cr(Ⅱ)配合物具有乙烯三聚催化行为, 他们利用DFT对此双核配合物可能的解离过程的ΔG进行了计算, 认为此Cr(Ⅱ)双核配合物在乙烯齐聚过程中经歧化过程产生一个Cr(Ⅰ)活性物种{η5-2, 3, 4, 5-Me4C4N(AlClMe2)Cr}和Cr(Ⅲ)物种{η5-2, 3, 4, 5-Me4C4N(AlClMe2)CrMe2}.由此他们认为Cr(Ⅰ)/Cr(Ⅲ)参与了乙烯三聚过程, 如Scheme 13所示.
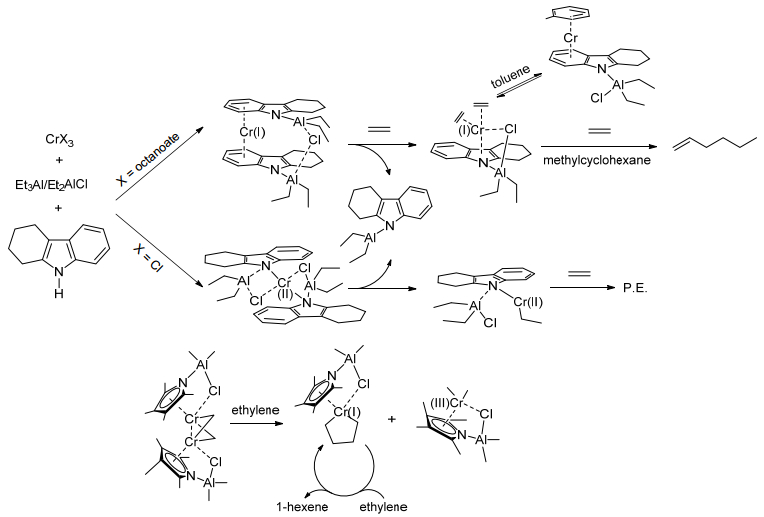 图 图式13
Gambarotta等分别基于单核和双核Cr前驱体提出的活性物种齐聚过程
Figure 图式13.
Oligomerization of reactive species based on
mononuclear and binuclear Cr precursors proposed by Gambarotta et al
图 图式13
Gambarotta等分别基于单核和双核Cr前驱体提出的活性物种齐聚过程
Figure 图式13.
Oligomerization of reactive species based on
mononuclear and binuclear Cr precursors proposed by Gambarotta et al
然而, McGuinness等[53]基于PNP & SNS/Cr/MAO乙烯三聚催化体系研究去质子化作用时, 发现合成的双核Cr(SNS)配合物在AlR3/B(C6F5)3的作用下对乙烯三聚具有高选择性, 利用伊万斯核磁共振法[62]提出在乙烯聚合过程中产生了一个配体去质子化的Cr(Ⅱ)离子型活性物种[当CrCl3(C2H5SCH2CH2)2NH (μeff=4.05μB)与25 equiv.的MAO混合后μeff=2.95μB, 这与低自旋C(Ⅱ)或Cr(Ⅳ)金属中心磁矩一致], 并认为对于Cr—SNS乙烯三聚催化过程是由离子型的活性物种完成Cr(Ⅱ)/Cr(Ⅳ)循环或者由非离子型活性物种完成Cr(Ⅰ)/Cr(Ⅲ)循环, 如Scheme 14所示.
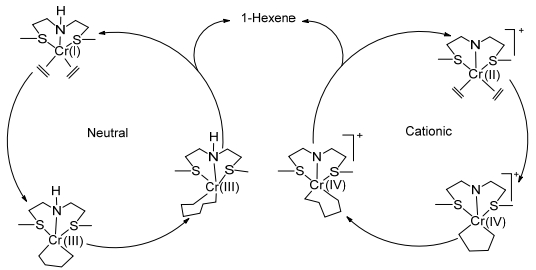 图 图式14
McGuinness等分别基于中性Cr(Ⅰ)活性物种和Cr(Ⅱ)阳离子活性物种提出的乙烯三聚机理
Figure 图式14.
Ethylene trimerization mechanism based on neutral Cr(Ⅰ) reactive species and Cr(Ⅱ) cation reactive species proposed by McGuinness et al.
图 图式14
McGuinness等分别基于中性Cr(Ⅰ)活性物种和Cr(Ⅱ)阳离子活性物种提出的乙烯三聚机理
Figure 图式14.
Ethylene trimerization mechanism based on neutral Cr(Ⅰ) reactive species and Cr(Ⅱ) cation reactive species proposed by McGuinness et al.
Rucklidge等[63]基于Cr/PNP/MAO [PNP=Ph2PN(i-Pr)PPh2]乙烯四聚催化体系首次合成了一种Cr(Ⅰ)的离子型配合物(Eq. 7), 在AlEt3的作用下具有较高的乙烯四聚活性.由此他们认为乙烯四聚过程是由单核Cr(Ⅰ)/Cr(Ⅲ)完成的, 如Scheme 15所示.
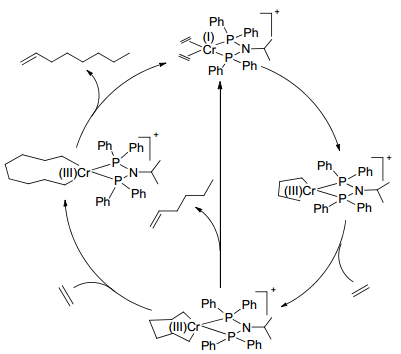 图 图式15
Rucklidge等基于阳离子活性物种提出的乙烯四聚机理
Figure 图式15.
tetramerization mechanism based on cation reactive species proposed by Rucklidge et al.
图 图式15
Rucklidge等基于阳离子活性物种提出的乙烯四聚机理
Figure 图式15.
tetramerization mechanism based on cation reactive species proposed by Rucklidge et al.
通过大量研究发现, 基于不同助催化剂, 不同配体合成的Cr催化剂前驱体不能体现乙烯选择性齐聚中Cr中心价态对催化行为的影响, 这可能由于合成的前驱体并不是真正的活性结构.
2.2 基于EPR检测技术对金属价态的研究
由于乙烯选择性齐聚催化剂中过渡金属具有未成对电子, 这使得催化体系具有顺磁性.电子顺磁共振(electron paramagnetic resonance, EPR)可以通过检测金属中心未成对电子进而判断金属化合价, 为其催化机理的推断提供重要支撑, 这使得EPR相对于其它表征手段具有独特的优势.
2008年Brückner等[64]首次利用EPR检测技术对基于Cr(acac)3/PNP/MMAO (MMAO为改性的甲基铝氧烷)和[(PNP)CrCl2(μ-Cl)]2/MMAO [PNP=PPh2PN(i-Pr)PPh2]催化体系的乙烯齐聚过程进行了监测.前者在反应过程中Cr(Ⅲ)物种转化为Cr(Ⅰ)物种, 然而Cr(Ⅲ)还原速度比Cr(Ⅰ)生成速度快, 作者认为这可能是Cr(Ⅲ)还原生成了对EPR不响应的Cr(Ⅰ)二聚物或者Cr(Ⅱ)物种.后者在齐聚反应中活性较低, 其甲苯溶液中EPR谱图超精细结构发现giso和Aiso值以及53Cr同位素的电子自旋与核自旋的超精细耦合常数与CH3CN溶液中的[Cr(η6-CH3C6H5)2]+的giso和Aiso理论计算值惊人的一致, 由此作者推测[(PNP)CrCl2(μ-Cl)]2在反应过程中配体被解离后生成[Cr(η6-CH3C6H5)2]+.
Skobelev等[65]基于Phillips乙烯三聚催化体系[66]利用EPR检测技术对Cr(acac)3 & Cr(EH)3/HPyr/AlEt3体系中Cr(Ⅰ)和Cr(Ⅲ)组分变化与乙烯齐聚活性的关系进行了分析.研究发现Cr(acac)3/HPyr/AlEt3 (1:3:30)相比于Cr(acac)3/AlEt3 (1:30), Cr(Ⅰ)物种在EPR中更快得到响应, Cr(Ⅰ)物种信号强度随温度升高和时间的延长变大, 且g=1.989处信号峰出现了细微变化, 作者认为这是由于产生了Cr(Ⅰ)-Pyr物种, 所以乙烯三聚活性升高.此外, Cr(EH)3:HPyr:AlEt3与Cr(EH)3:AlEt3通过EPR检测得到了类似的结果.作者认为Cr(Ⅰ)/Cr(Ⅲ)可能对乙烯三聚过程提供了支撑, 然而也不能排除Cr(Ⅱ)/Cr(Ⅳ)的作用, 因为它们对EPR不响应.
在2012年, Rabeah等[67]在Brückner等工作的基础上基于Cr(acac)3/PNP/MMAO [PNP=PPh2PN(i-Pr)PPh2]催化体系对乙烯齐聚反应的真实条件进行了模拟, 根据PNP—Cr(Ⅰ)与体系中配体解离的Cr(Ⅰ)物种占比随乙烯聚合压力变化情况发现, Cr(Ⅰ)物种与乙烯选择性齐聚活性相关性不大.通过EPR-XAS分析, 作者认为对EPR未响应的Cr(Ⅱ)物种参与了乙烯选择性齐聚[68], 推测在MMAO作用下其结构为(PNP)Cr(Ⅱ)(CH3)2非离子型四配位中间体, 据此作者认为乙烯选择性四聚过程是由Cr(Ⅱ)/Cr(Ⅳ)或Cr(Ⅱ)/Cr(Ⅲ)完成的, 如Scheme 16所示.
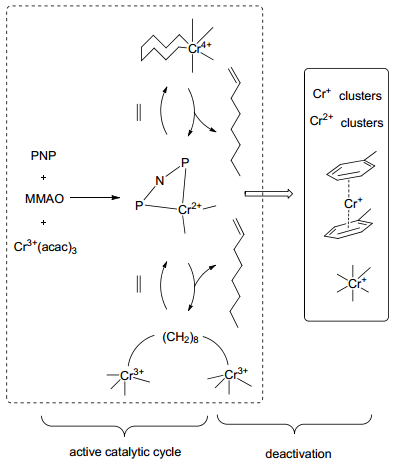 图 图式16
Rabeah等基于Cr(Ⅱ)活性物种提出的乙烯四聚机理
Figure 图式16.
Ethylene tetramerization mechanism based on Cr(Ⅱ) reactive species proposed by Rabeah et al.
图 图式16
Rabeah等基于Cr(Ⅱ)活性物种提出的乙烯四聚机理
Figure 图式16.
Ethylene tetramerization mechanism based on Cr(Ⅱ) reactive species proposed by Rabeah et al.
然而由于助催化剂的参与导致不同化合价的Cr物种存在于体系中, 有的Cr活性物种不对EPR响应且配位环境复杂, 这使得EPR确定结构时遇到很多困难.
2.3 基于DFT方法对金属价态的研究
实验数据表明配体结构会影响金属中心氧化态Cr(Ⅰ)/Cr(Ⅲ)、Cr(Ⅱ)/Cr(Ⅳ), 而金属中心的配位环境由自旋多重度决定, 所以金属中心自旋多重度对于乙烯选择性齐聚机理中金属氧化态的确定是尤为重要的.在Cr双核结构研究中[69], 根据Cr烷基氢化物C—H键还原反应的不同可以分为两类同分异构体, 一种是以Cr(Ⅱ)2(μ-Ph)(μ-H)为主的双核配合物, 另外一种是以Cr(Ⅰ)2(μ-η6:η6-C6H6)为主的双核配合物.这两种化合物的自旋多重度是不同的, 其中Cr(Ⅱ)化合物的自旋态是S=0, Cr(Ⅰ)化合物的自旋态是S=3.当Cr(Ⅱ)化合物加热至120 ℃时, 结构仍然保持稳定, 因为“自旋禁阻[70]”存在于上述两类自旋态不同的双核配合物中, 所以Cr(Ⅱ)2(μ-Ph)(μ-H)配合物不能通过C—H键还原消除形成Cr(Ⅰ)2(μ-η6:η6-C6H6)配合物. K hn[71]认为这可以说明Cr系催化剂催化乙烯四聚金属环机理[39]中Crn+2甲基环戊烷基金属氢化物中间体经歧化作用产生的双核Crn+1烷基金属氢化物不会通过C—H还原消除生成甲基环戊烷的原因(如Scheme 17所示).初步认为由于化合物的不同自旋态会对反应动力学造成影响.但是在含有顺磁性铬化合物的催化反应中, 其中间体的自旋态及它们对反应的影响需要进一步研究.在实验室中不同氧化态的化合物分离是非常困难的, 得益于计算化学的支持, 其分子计算结果表明, 对于Cr(Ⅰ)/Cr(Ⅲ)氧化还原过程, 四重态是最稳定的, 而Cr(Ⅱ)/Cr(Ⅳ)氧化还原过程, 三重态被认为是最稳定的.
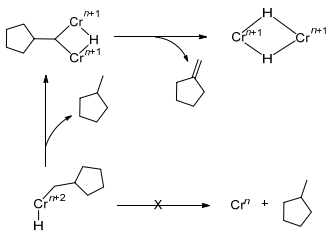 图 图式17
Cr系催化剂乙烯四聚中C6副产物(甲基环戊烷、亚甲基环戊烷)形成机理
Figure 图式17.
C6 product formation (methylcyclopentane and methylenecyclopentane) mechanism based on ethylene tetramerization catalyst of Cr
图 图式17
Cr系催化剂乙烯四聚中C6副产物(甲基环戊烷、亚甲基环戊烷)形成机理
Figure 图式17.
C6 product formation (methylcyclopentane and methylenecyclopentane) mechanism based on ethylene tetramerization catalyst of Cr
大量基于DFT方法关于Cr金属中心价态的计算表明, 配体结构的不同导致在乙烯齐聚过程中金属氧化态也不同, Cr(Ⅱ)/Cr(Ⅳ)氧化还原过程适用于Cr/pyrrolide和仅Cl原子为配体的催化体系, Cr(Ⅰ)/Cr(Ⅲ)氧化还原过程适用于Cr/PNP催化体系, Cr(Ⅱ)/Cr(Ⅳ)和Cr(Ⅰ)/Cr(Ⅲ)氧化还原过程适用于Cr/DME/carboxylate催化体系[32, 33, 41, 52, 72~74].唐思扬等[75]基于DFT-QSAR计算表明乙烯四聚过程可能是由单核Cr(Ⅱ)/Cr(Ⅳ)完成的, 然而乙烯四聚Cr金属中心价态尚不是很明朗.
3 方法学评价
3.1 示踪原子
以McGuinness和Bercaw等为代表开展的氘代乙烯交叉实验为Cr系催化剂催化乙烯选择性齐聚机理的揭示提供了重要支撑:利用SPSS软件对基于Suttil等[17]进行的氘代交叉实验中的1-丁烯、1-己烯同位素异构体实验与理论预测结果(图 1质谱裂解后的相对丰度, 如图 2所示)进行了方差分析, 如表 1所示, 基于Ti系金属环机理下形成的产物(1-丁烯和1-己烯)其实验与理论计算结果的组间差异与组内差异的比值[F1-丁烯: 17.492 (Metallacycle) > 2.388 (Cossee-Alrman), F1-己烯: 12.873 (Metallacycle) > 0.015 (Cossee-Alrman)]明显大于Ti系Cossee-Alrman链增长机理组间差异与组内差异的比值, 且后者拟合度较高(R1-2丁烯=0.979, R1-2己烯=0.952), 这表明Ti系金属环机理并不适用于解释Ti(OR')4/AlR3催化体系催化乙烯高选择性生成1-丁烯和少量1-己烯的过程.在Cr系催化剂催化乙烯选择性齐聚(三聚)的研究中[30, 31], 基于考虑到上述Suttil等进行的氘代交叉实验中1-己烯的生成符合Cossee-Alrman链增长机理, 通过Cr系金属环机理与Cossee-Alrman链增长机理氘代交叉实验下理论同位素异构体含量对比(如图 3所示), 可以看出两种机理1-己烯同位素异构体分布形式和氘原子数量有显著不同(在Cosse-Alrman链增长机理中1-己烯氘原子出现了奇数分布), 研究表明Cr系金属环机理相对于Cosse-Alrman机理更可靠.然而Cr杂五元环配合物和Cr系乙烯二聚氘代实验机理研究尚未见报道, 笔者认为Cr系乙烯二聚并不是按照金属环机理发生的.
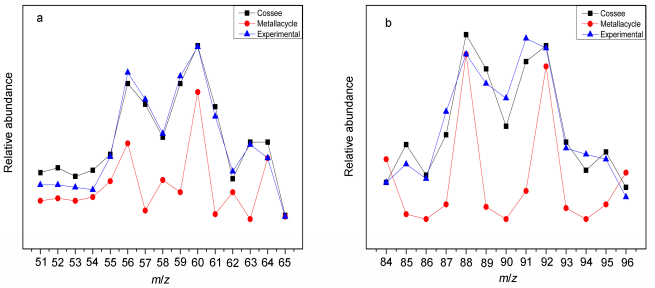 图 2
Suttil等进行的氘代交叉实验中1-丁烯(a)、1-己烯(b)同位素异构体实验与理论预测结果
Figure 2.
Results of experiment and theoretical prediction on 1-butene (a) and 1-hexene(b) isotope isomer in deuterium cross-over experiment conducted by Suttil et al.
图 2
Suttil等进行的氘代交叉实验中1-丁烯(a)、1-己烯(b)同位素异构体实验与理论预测结果
Figure 2.
Results of experiment and theoretical prediction on 1-butene (a) and 1-hexene(b) isotope isomer in deuterium cross-over experiment conducted by Suttil et al.
 表 1
Suttil等进行的氘代交叉实验中1-丁烯、1-己烯同位素异构体实验与理论预测结果方差分析
Table 1.
Variance analysis about the results of experiment and theoretical prediction on 1-butene and 1-hexene isotope isomer in deuterium cross-over experiment conducted by Suttil et al.
表 1
Suttil等进行的氘代交叉实验中1-丁烯、1-己烯同位素异构体实验与理论预测结果方差分析
Table 1.
Variance analysis about the results of experiment and theoretical prediction on 1-butene and 1-hexene isotope isomer in deuterium cross-over experiment conducted by Suttil et al.
Alkene Mechanism F Sig. Adjusted R2 1-Butene Cossee-Alrman 2.388 0.145 0.979 Metallacycle 17.492 0.001 0.637 1-Hexene Cossee-Alrman 0.015 0.904 0.952 Metallacycle 12.873 0.004 0.589 表 1 Suttil等进行的氘代交叉实验中1-丁烯、1-己烯同位素异构体实验与理论预测结果方差分析
Table 1. Variance analysis about the results of experiment and theoretical prediction on 1-butene and 1-hexene isotope isomer in deuterium cross-over experiment conducted by Suttil et al. 图 3
氘代交叉实验C6生成原理示意图
Figure 3.
Schematic diagram of the principle for the formation of C6 in labelled experiment
图 3
氘代交叉实验C6生成原理示意图
Figure 3.
Schematic diagram of the principle for the formation of C6 in labelled experiment
在Cr系催化剂催化乙烯三聚反应中, 氘代交叉实验不能揭示β氢的两种转移方式(3, 7位转移和β氢转移至金属中心再转移至α'碳上), 因为同位素异构体均来自相同的β位氢转移(如图 3a所示).
此外在Cr系催化剂催化乙烯四聚反应中, 其金属环反应路径目前尚存争议(单核、双核). McGuinness等[39]利用氘代交叉实验采用不同配比的C2H4/C2D4对乙烯插入成环结构和1-己烯、1-辛烯等同位素异构体含量进行了预测, 但其预测模型是基于概率性的乙烯单体依次插入和β位氢转移, 笔者对此模型中C8同位素异构体分布量进行了计算(如图 4所示), 此概率模型对双核金属环机理具有一定普适性且Cr杂九元环配合物至今尚未报道.
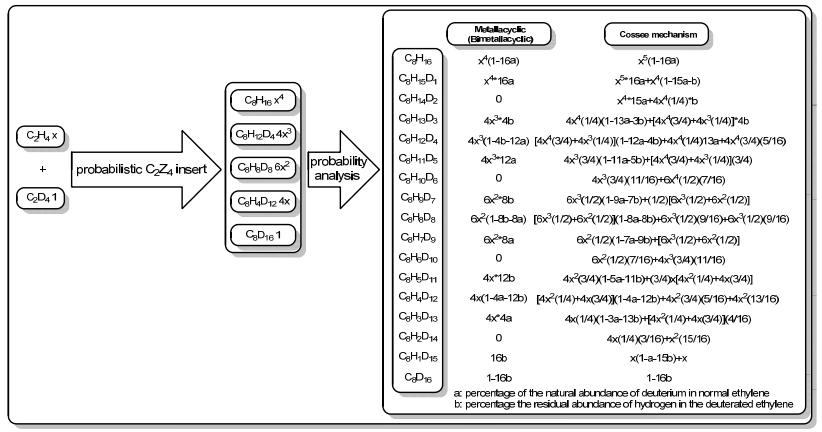 图 4
氘代交叉实验C8生成原理示意图
Figure 4.
Schematic diagram of the principle for the formation of C8 in labelled experiment
图 4
氘代交叉实验C8生成原理示意图
Figure 4.
Schematic diagram of the principle for the formation of C8 in labelled experiment
3.2 预制催化剂前驱体
预制催化剂前驱体揭示了主催化剂与助催化剂之间的相互作用, 包括金属中心烷基化、去质子化、卤化、配合物离子化、C—H键活化等[51~58].这为探索不同助催化剂参与的Cr系催化剂催化乙烯选择性齐聚反应机理提供了极其重要的指导.特别是Gambarotta等[59, 60]基于Phillips乙烯三聚催化体系预制的催化剂前驱体, 在无助催化剂的条件下即可催化乙烯三聚, 这为研究Cr系催化剂催化乙烯选择性齐聚反应机理中金属中心价态的变化奠定了非常重要的基础.
在乙烯选择性齐聚工业化生产的今天, 乙烯四聚技术仍然是发展的重点与难点, MAO是乙烯四聚的高效助催化剂, 一些学者对其分子结构进行了研究[76~79].目前普遍认为, 其有效成分是一种具有笼式单元结构的聚合物.主催化剂与它的作用十分复杂, 通过与其它助催化剂预制催化剂前驱体类比齐聚行为间接揭示MAO与主催化剂的作用对探索其反应机理并找到能替代MAO的高效廉价助催化剂是十分重要的.
双核金属环机理最大的难点在于揭示Cr—Cr间的相互作用, 合成Cr—Cr多核配合物并探索Cr—Cr间可能存在的化学键作用及金属中心价态对乙烯四聚机理研究有积极作用.此外, 合成Cr金属氘代氢化物可以探索Cr系催化剂在乙烯选择性齐聚金属环机理中β氢转移方式.
3.3 EPR检测技术
利用EPR对乙烯三聚、四聚催化体系乙烯齐聚过程进行的实时监测对揭示催化反应机理中金属中心价态变化有积极意义, 它解决了传统核磁方法难以表征具有顺磁性物质的问题, 特别是Rabeah等[67]进行了乙烯齐聚高压环境对活性组分中不同价态的金属中心的含量变化研究, 这对揭示乙烯齐聚反应条件的内在本质影响具有促进作用.
然而, 由于一些不具有顺磁性的物质如Cr(Ⅰ)的二聚物和Cr(Ⅱ)配合物等对EPR信号不响应, 这增加了EPR鉴定活性物种结构和金属中心价态的难度.此外, 由于助催化剂的参与, 导致活性组分复杂化及金属中心价态多样化, 从而提高了EPR检测的数据分析的难度.
合成无助催化剂的Cr系乙烯齐聚催化剂并应用EPR技术对其齐聚过程中金属中心价态变化研究可以消除助催化剂对机理研究的影响, 均相乙烯齐聚反应实时在线波谱检测技术的发展将为机理研究提供便利.
3.4 DFT方法
由于在实验室中很难分离和确定活性中间体, 很多学者利用DFT方法基于不同催化体系对Cr系催化剂催化乙烯选择性齐聚机理中金属中心价态[Cr(Ⅰ)/Cr(Ⅳ)、Cr(Ⅱ)/Cr(Ⅳ)等]、活性中间体结构以及乙烯插入过程中的过渡态进行计算与讨论, 为乙烯选择性齐聚反应机理的研究提供了重要的依据.然而目前该方法仍然有很多问题.
基于演绎法下不同催化体系提出的活性中间体结构优化模型在解决乙烯插入成环机理研究中起到了决定作用, 但计算结果中乙烯选择性齐聚Cr系催化剂金属中心价态变化不同且存在争议.
由于MAO在乙烯三聚、四聚中具有卓越的助催化剂活性, 所以MAO作用下乙烯齐聚反应机理方面理论认识是人们一直追求的, van Rensburg等[73]利用DFT方法基于(η5-C5H4CMe2C6H5)-TiCl3/MAO乙烯三聚催化体系分别在(η5-C5H4CMe2C6H5)-Ti+和(η5-C5H4CMe2C6H5)-Ti+[MAO-Cl]-活性结构模型基础上对乙烯插入成环过渡态进行了计算.针对均相Cr系乙烯选择性齐聚催化剂在MAO作用下的活性结构模型, Pasha等[80]基于Cr/PNP/MAO [PNP=Me2PN(Me)PMe2、Ph2PN(iPr)PPh2]乙烯齐聚催化体系对MAO笼式结构模型与Cr杂金属五元环下乙烯分子的进一步插入释放1-己烯过程, 利用DFT方法进行了计算.然而由于基于不同配体的Cr系乙烯选择性齐聚催化体系与MAO作用复杂(离子化、去质子化、烷基化等), 目前此方面计算报道较少尚未形成有统计意义的结论.
DFT方法取决于人们对反应机理合理的认识和推测(如Cosse-Alram机理、单核金属环机理、抓氢键[81]等), 然而现目前人们对双核乙烯四聚反应路径的认识尚不清楚.
由于DFT方法中泛函的特点、适用场合不同, 对于单核的过渡金属配合物, 人们常采用BP86和B3LYP等泛函. McGuinness等[74]针对Cr/PNP/MAO乙烯选择性齐聚催化体系基于简单阳离子模型(P、N上均为甲基, 经MAO活化的金属中心为甲基化Cr)利用不同DFT方法对金属环机理中活性中间体自旋态、金属中心价态[Cr(Ⅰ)/Cr(Ⅲ)、Cr(Ⅱ)/Cr(Ⅳ)]进行基准性研究中发现, 使用BP86、BPW91和M06L定域泛函在金属中心价态的稳定性、活性物种几何结构优化以及乙烯插入成环过程势能面上能垒计算方面获得较好的结果, 但计算速度较慢.此外, 由于一些Cr系催化体系活性金属中心可能存在电子自旋交叉现象[82](Minimum Energy Crossing Points, MECPs), 这会导致B3LYP等泛函会高估一些过渡金属配合物高自旋态的稳定性[83, 84], 提高计算精度和速度是有待解决的一对矛盾.
基于上述方法学的讨论, 在笔者看来, 均相Cr系催化剂催化乙烯选择性齐聚反应机理的研究需将上述方法合理有效结合起来:氘代交叉实验在解决反应路径上具有极其重要的作用, EPR技术可以建立反应条件、反应活性以及反应活性组分之间的定量关系, DFT方法可以为反应机理做理论支撑, 然而这三者均离不开催化剂前驱体的设计与合成.预制催化剂前驱体不仅可以为新型配体设计做指导、揭示助催化剂与主催化剂的作用、为DFT理论计算提供理论模型结构, 还可以为EPR研究反应机理中金属中心价态变化提供理想的标本.
配体结构对乙烯齐聚行为影响是极大的, 目前国内还没有建立乙烯齐聚配体方面数据库.对现有配体结构数据分析有利于揭示反应机理和设计合成新型配体.通过聚类算法、多因素方差分析等一系列统计学方法可以将含配体的多流形结构数据在不同水平下分类(乙烯三聚、乙烯四聚以及非选择性乙烯齐聚), 并基于Tomov等[85, 86]对Cr系(配体分别为NNN和NNO)乙烯齐聚催化剂氘代交叉实验的研究以及人们对双核金属环机理的猜测, 笔者认为Cr系乙烯四聚双核反应机理中金属中心可能由于活性物种空间几何结构特点使其金属中心具有两个以上位点, 导致成环具有交替性(如图 5所示).基于对Cr系催化剂乙烯齐聚配体统计学的研究, 对乙烯四聚金属环机理中的交替性进行探索, 乙烯四聚双核机理的研究为设计新型配体及催化剂提供理论指导.
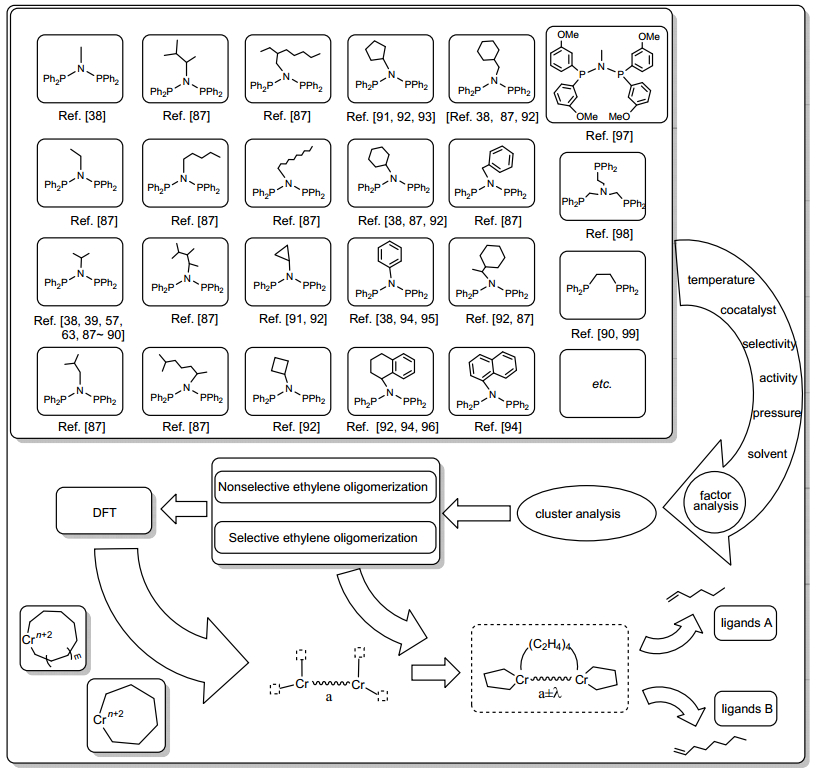 图 5
基于统计学分析的乙烯选择性齐聚双核金属环机理探索示意图
Figure 5.
Schematic diagram of the exploration on selective ethylene oligomerization binuclear metal ring mechanism based on statistic analysis
图 5
基于统计学分析的乙烯选择性齐聚双核金属环机理探索示意图
Figure 5.
Schematic diagram of the exploration on selective ethylene oligomerization binuclear metal ring mechanism based on statistic analysis
4 总结与展望
对均相Cr系催化剂催化乙烯选择性齐聚的研究迄今已有四十多年, 取得了大量的研究成果, 完善了乙烯三聚催化体系, 并且开创了乙烯四聚催化技术, 为满足市场对特定α-烯烃的需求提供了技术支撑.进一步提高催化剂的活性及对目标产物的选择性是学界和业界永恒的追求目标, 乙烯选择性齐聚反应机理的研究可以为新型催化剂的研发提供指导.
氘代实验结果证明了在Cr系乙烯选择性齐聚中金属环机理相对于Cossee-Arlman机理更可靠.然而目前机理中的很多方面在细节上仍然存在争议, 如β氢转移过程、金属中心价态、金属环扩张趋势等.基于大量已发表的实验结果与方法学讨论, 笔者认为, 均相Cr系催化剂催化乙烯选择性齐聚相对于非选择性乙烯齐聚而言金属环机理本身具有各自特点, 进一步的氘代实验与统计学方法研究可以揭示这一内在本质.
活性中间体的确定与捕获对解决反应机理研究中的细节问题是至关重要的, 随着EPR、XAS等表征手段的引入, 实验和分子计算的结合可以不断地推进这一领域的研究进程.以配体设计为主导的新型催化体系的研发与反应机理的研究相结合将有利于建立聚烯烃催化剂构效关系数据库, 进而促进乙烯选择性齐聚技术的发展.
-
-
[1]
Skupinska, J.Chem.Rev.1991, 91, 613. doi: 10.1021/cr00004a007
-
[2]
Forestière A.; Olivier-Bourbigou H.; Saussine L.Oil Gas Sci.Technol.2009, 64, 649. doi: 10.2516/ogst/2009027
-
[3]
钱伯章, 石化技术, 2011, 18, 58.Qian, B.Z.Petrochem.Ind.Technol.2011, 18, 58(in Chinese).
-
[4]
金文, 石油知识, 2014, 4, 24.Jin, W.Petroleum.Knowl.2014, 4, 24(in Chinese).
-
[5]
Carter, A.; Cohen, S.A.; Cooley, N.A.; Murphy, A.; Scutt, J.; Wass, D.F.Chem.Commun.2002, 8, 858.
-
[6]
Commereuc, D.; Chauvin, Y.; Gaillard, J.; Leonard, J.; Andrews, J.Hydrocarbon Process.1984, 63, 118.
-
[7]
Al-Jarallah, A.M.; Anabtawi, J.A.; Siddiqui, M.A.B.; Aitani, A.M.; Al-Sa'doun, A.W.Catal.Today 1992, 14, 1. doi: 10.1016/0920-5861(92)80128-A
-
[8]
Pillai, S.M.; Ravindranathan, M.; Sivaram, S.Chem.Rev.1986, 86, 353. doi: 10.1021/cr00072a004
-
[9]
Al-Sherehy, F.A.Stud.Surf.Sci.Catal.1996, 100, 515. doi: 10.1016/S0167-2991(96)80052-8
-
[10]
Van Leeuwen, P.W.; Clément, N.D.; Tschan, M.J.L.Coord.Chem.Rev.2011, 255, 1499. doi: 10.1016/j.ccr.2010.10.009
-
[11]
许建耘, 石油炼制与化工, 2014, 45, 67. http://www.cnki.com.cn/Article/CJFDTotal-SYLH201304025.htmXu, J.Y.Pet.Process.Petrochem.2014, 45, 67(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-SYLH201304025.htm
-
[12]
McGuinness, D.S.Chem.Rev.2010, 111, 2321.
-
[13]
Agapie, T.Coord.Chem.Rev.2011, 255, 861. doi: 10.1016/j.ccr.2010.11.035
-
[14]
Bryliakov, K.P.; Talsi, E.P.Coord.Chem.Rev.2012, 256, 2994. doi: 10.1016/j.ccr.2012.06.023
-
[15]
Dixon, J.T.; Green, M.J.; Hess, F.M.; Morgan, D.H.J.Organomet.Chem.2004, 689, 3641. doi: 10.1016/j.jorganchem.2004.06.008
-
[16]
Belov, G.P.Pet.Chem.2012, 52, 139. doi: 10.1134/S0965544112030036
-
[17]
Suttil, J.A.; McGuinness, D.S.Organometallics 2012, 31, 7004. doi: 10.1021/om3008508
-
[18]
Ziegler, K.; Martin, H.US 2943125, 1960[Chem.Abstr.1983, 54, 134637].
-
[19]
Belov, G.P.; Dzhabiev, T.S.; Kolesnikov, I.M.J.Mol.Catal.1982, 14, 105. doi: 10.1016/0304-5102(82)80053-9
-
[20]
Robinson Jr, R.; McGuinness, D.S.; Yates, B.F.ACS Catal.2013, 3, 3006. doi: 10.1021/cs4006875
-
[21]
Al-Sa'doun, A.W.Appl.Catal., A 1993, 105, 1. doi: 10.1016/0926-860X(93)85131-8
-
[22]
McDermott, J.X.; White, J.F.; Whitesides, G.M.J.Am.Chem.Soc.1973, 95, 4451. doi: 10.1021/ja00794a068
-
[23]
Manyik, R.M.; Walker, W.E.; Wilson, T.P.J.Catal.1977, 47, 197. doi: 10.1016/0021-9517(77)90167-1
-
[24]
Pettijohn, T.M.; Reagen, W.K.; Martin, S.J.US 5331070, 1994[Chem.Abstr.1994, 122, 56808].
-
[25]
Reagen, W.K.; Pettijohn, T.M.; Freeman, J.W.; Benham, E.A.US 5786431, 1998[Chem.Abstr.1998, 115, 93172].
-
[26]
Freeman, J.W.; Buster, J.L.; Knudsen, R.D.US 5919996, 1999[Chem.Abstr.1999, 130, 95984].
-
[27]
Briggs, J.R.J.Chem.Soc., Chem.Commun.1989, 11, 674.
-
[28]
Emrich, R.; Heinemann, O.; Jolly, P.W., Krüger, C.; Verhovnik, G.P.Organometallics 1997, 16, 1511. doi: 10.1021/om961044c
-
[29]
刘睿, 肖树萌, 钟向宏, 曹育才, 梁胜彪, 刘振宇, 叶晓峰, 沈安, 朱红平, 有机化学, 2015, 35, 1861. doi: 10.6023/cjoc201504009Liu, R.; Xiao, S.M.; Zhong, X.H.; Cao, Y.C.; Liang, S.B.; Liu, Z.Y.; Ye, X.F.; Shen, A.; Zhu, H.P.Chin.J.Org.Chem.2015, 35, 1861(in Chinese). doi: 10.6023/cjoc201504009
-
[30]
Agapie, T.; Schofer, S.J.; Labinger, J.A.; Bercaw, J.E.J.Am.Chem.Soc.2004, 126, 1304. doi: 10.1021/ja038968t
-
[31]
Agapie, T.; Labinger, J.A.; Bercaw, J.E.J.Am.Chem.Soc.2007, 129, 14281. doi: 10.1021/ja073493h
-
[32]
Klemps, C.; Payet, E.; Magna, L.; Saussine, L.; Le Goff, X.F.; Le Floch, P.Chem.Eur.J.2009, 15, 8259. doi: 10.1002/chem.v15:33
-
[33]
Yang, Y.; Liu, Z.; Zhong, L.; Qiu, P.Y.; Dong, Q.; Cheng, R.H.; Vanderbilt, J.; Liu, B.P.Organometallics 2011, 30, 5297. doi: 10.1021/om200722r
-
[34]
Yu, Z.X.; Houk, K.N.Angew.Chem., Int.Ed.2003, 42, 808. doi: 10.1002/anie.200390215
-
[35]
Qi, Y.; Dong, Q.; Zhong, L.; Liu, Z.; Qiu, P.; Cheng, R.H.; He, X.L.; Vanderbilt, J.; Liu, B.P.Organometallics 2010, 29, 1588. doi: 10.1021/om900917k
-
[36]
Budzelaar, P.H.Can.J.Chem.2009, 87, 832.
-
[37]
Albahily, K.; Koç, E.; Al-Baldawi, D.; Savard, D.; Gambarotta, S.; Burchell, T.J.; Duchateau, R.Angew.Chem., Int.Ed.2008, 47, 5816. doi: 10.1002/anie.v47:31
-
[38]
Bollmann, A.; Blann, K.; Dixon, J.T.; Hess, F.M.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H.; Neveling, A.; Otto, S.; Overett, M.; Slawin, A.M.Z.; Wasserscheid, P.; Kuhlmann, S.J.Am.Chem.Soc.2004, 126, 14712. doi: 10.1021/ja045602n
-
[39]
Overett, M.J.; Blann, K.; Bollmann, A.; Dixon, J.T.; Haasbroek, D.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H.J.Am.Chem.Soc.2005, 127, 10723. doi: 10.1021/ja052327b
-
[40]
Blok, A.N.; Budzelaar, P.H.; Gal, A.W.Organometallics 2003, 22, 2564. doi: 10.1021/om030049o
-
[41]
Britovsek, G.J.; McGuinness, D.S.; Wierenga, T.S.; Young, C.T.ACS Catal.2015, 5, 4152. doi: 10.1021/acscatal.5b00989
-
[42]
Young, J.F.; Yap, G.Theopold, K H.In INOR 554-Reactivity of Chromium (Ⅱ) Bimetallocycles and Their Role in Ethylene Trimerization, Abstracts of Papers of the 234th ACS National Meeting, American Chemical Society, America, 2007, p.234.
-
[43]
Peitz, S.; Aluri, B.R.; Peulecke, N.; Müller, B.H.; Wöhl, A.; Müller, W.; Al-Hazmi, M.H.; Mosa, F.M.; Rosenthal, U.Chem.Eur.J.2010, 16, 7670. doi: 10.1002/chem.201000750
-
[44]
Breslow, D.S.; Newburg, N.R.J.Am.Chem.Soc.1957, 79, 5072.
-
[45]
Long, W.P.; Breslow, D.S.J.Am.Chem.Soc.1960, 82, 1953. doi: 10.1021/ja01493a029
-
[46]
Lee, P.Y.; Liang, L.C.Inorg.Chem.2009, 48, 5480. doi: 10.1021/ic802030d
-
[47]
Long, W.P.J.Am.Chem.Soc.1959, 81, 5312. doi: 10.1021/ja01529a017
-
[48]
Chien, J.C.J.Am.Chem.Soc.1959, 81, 86. doi: 10.1021/ja01510a019
-
[49]
Zefirova, A.K.; Shilov, A.E.Dokl.Akad.Nauk SSSR 1961, 136, 599.
-
[50]
Dyachkovskii, F.S.; Shilova, A.K.; Shilov, A.E.J.Polym.Sci., Part C:Polym.Symp.1967, 16, 2333.
-
[51]
Jabri, A.; Crewdson, P.; Gambarotta, S.; Korobkov, I.; Duchateau, R.Organometallics 2006, 25, 715. doi: 10.1021/om050886l
-
[52]
McGuinness, D.S.; Wasserscheid, P.; Keim, W.; Morgan, D.; Dixon, J.T.; Bollmann, A.; Maumela, H.; Hess, F.; Englert, U.J.Am.Chem.Soc.2003, 125, 5272. doi: 10.1021/ja034752f
-
[53]
McGuinness, D.S.; Brown, D.B.; Tooze, R.P.; Hess, F.M.; Dixon, J.T.; Slawin, A.M.Organometallics 2006, 25, 3605. doi: 10.1021/om0601091
-
[54]
Temple, C.; Jabri, A.; Crewdson, P.; Gambarotta, S.; Korobkov, I.; Duchateau, R.Angew.Chem.2006, 118, 7208. doi: 10.1002/(ISSN)1521-3757
-
[55]
Zhang, J.; Li, A.; Hor, T.A.Organometallics 2009, 28, 2935. doi: 10.1021/om9002347
-
[56]
Yoshida, T.; Yamamoto, T.; Okada, H.; Murakita, H.US 0035029, 2002[Chem.Abstr.2002, 136, 249387].
-
[57]
McGuinness, D.S.; Overett, M.; Tooze, R.P.; Blann, K.; Dixon, J.T.; Slawin, A.M.Organometallics 2007, 26, 1108. doi: 10.1021/om060906z
-
[58]
Deckers, P.J.; van der Linden, A.J.; Meetsma, A.; Hessen, B.Eur.J.Inorg.Chem.2000, 5, 929.
-
[59]
Jabri, A.; Mason, C.B.; Sim, Y.; Gambarotta, S.; Burchell, T.J.; Duchateau, R.Angew.Chem., Int.Ed.2008, 47, 9717. doi: 10.1002/anie.v47:50
-
[60]
Vidyaratne, I.; Nikiforov, G.B.; Gorelsky, S.I.; Gambarotta, S.; Duchateau, R.; Korobkov, I.Angew.Chem., Int.Ed.2009, 48, 6552. doi: 10.1002/anie.v48:35
-
[61]
Reagan, W.K.EP 0417477, 1991[Chem.Abstr.1991, 115, 93172].
-
[62]
Evans, D.F.J.Chem.Soc.1959, 2003. doi: 10.1039/jr9590002003
-
[63]
Rucklidge, A.J.; McGuinness, D.S.; Tooze, R.P.; Slawin, A.M.; Pelletier, J.D.; Hanton, M.J.; Webb, P.B.Organometallics 2007, 26, 2782. doi: 10.1021/om0701975
-
[64]
Brückner, A.; Jabor, J.K.; McConnell, A.E.; Webb, P.B.Organometallics 2008, 27, 3849. doi: 10.1021/om800316m
-
[65]
Skobelev, I.Y.; Panchenko, V.N.; Lyakin, O.Y.; Bryliakov, K.P.; Zakharov, V.A.; Talsi, E.P.Organometallics 2010, 29, 2943. doi: 10.1021/om100215t
-
[66]
Freeman, J.W.; Buster, J.L.; Knudsen, R.D.US 5856257, 1999[Chem.Abstr.1999, 130, 95984].
-
[67]
Rabeah, J.; Bauer, M.; Baumann, W.; McConnell, A.E.; Gabrielli, W.F.; Webb, P.B.; Selent, D.; Bruckner, A.ACS Catal.2012, 3, 95.
-
[68]
Weckhuysen, B.M.; Schoonheydt, R.A.; Jehng, J.M.; Wachs, I.E.; Cho, S.J.; Ryoo, R.; Kijlstra, S.; Poels, E.J.Chem.Soc., Faraday Trans.1995, 91, 3245. doi: 10.1039/FT9959103245
-
[69]
Monillas, W.H.; Yap, G.; Theopold, K.H.Angew.Chem., Int.Ed.2007, 46, 6692. doi: 10.1002/(ISSN)1521-3773
-
[70]
Poli, R.; Harvey, J.N.Chem.Soc.Rev.2003, 32, 1. doi: 10.1039/b200675h
-
[71]
Köhn, R.D.Angew.Chem., Int.Ed.2008, 47, 245. doi: 10.1002/(ISSN)1521-3773
-
[72]
Bhaduri, S.; Mukhopadhyay, S.; Kulkarni, S.A.J.Organomet.Chem.2009, 694, 1297. doi: 10.1016/j.jorganchem.2008.12.012
-
[73]
Janse van Rensburg, W.; van den Berg, J.A.; Steynberg, P.J.Organometallics 2007, 26, 1000. doi: 10.1021/om060890c
-
[74]
McGuinness, D.S.; Chan, B.; Britovsek, G.J.; Yates, B.F.Aust.J.Chem.2014, 67, 1481. doi: 10.1071/CH14436
-
[75]
唐思扬, 刘振, 占兴稳, 程瑞华, 何雪莲, 刘柏平, 化工学报, 2014, 65, 131.Tang, S.Y.; Liu, Z,; Zhan, X.W.; Cheng, R.H.; He, X.L.; Liu, B.P.J.Chem.Eng.2014, 65, 131(in Chinese).
-
[76]
Mason, M.R.; Smith, J.M.; Bott, S.G.; Barron, A.R.J.Am.Chem.Soc.1993, 115, 4971. doi: 10.1021/ja00065a005
-
[77]
Harlan, C.J.; Mason, M.R.; Barron, A.R.Organometallics 1994, 13, 2957. doi: 10.1021/om00020a011
-
[78]
Babushkin, D.E.; Semikolenova, N.V.; Panchenko, V.N.; Sobolev, A.P.; Zakharov, V.A.; Talsi, E.P.Macromol.Chem.Phys.1997, 198, 3845. doi: 10.1002/macp.1997.021981206
-
[79]
Zakharov, I.I.; Zakharov, V.A.Macromol.Theory Simul.2001, 10, 108. doi: 10.1002/(ISSN)1521-3919
-
[80]
Pasha, F.A.; Basset, J.M.; Toulhoat, H.; de Bruin, T.Organometallics 2015, 34, 426. doi: 10.1021/om5008874
-
[81]
Grubbs, R.H.; Coates, G.W.Acc.Chem.Res.1996, 29, 85. doi: 10.1021/ar9501683
-
[82]
Harvey, J.N.; Poli, R.; Smith, K.M.Coord.Chem.Rev.2003, 238, 347.
-
[83]
Salomon, O.; Reiher, M.; Hess, B.A.J.Chem.Phys.2002, 117, 4729. doi: 10.1063/1.1493179
-
[84]
Reiher, M.; Salomon, O.; Hess, B.A.Theor.Chim.Acta 2001, 107, 48. doi: 10.1007/s00214-001-0300-3
-
[85]
Tomov, A.K.; Chirinos, J.J.; Jones, D.J.; Long, R.J.; Gibson, V.C.J.Am.Chem.Soc.2005, 127, 10166. doi: 10.1021/ja051523f
-
[86]
Tomov, A.K.; Chirinos, J.J.; Long, R.J.; Gibson, V.C.; Elsegood, M.R.J.Am.Chem.Soc.2006, 128, 7704. doi: 10.1021/ja0615369
-
[87]
Blann, K.; Bollmann, A.; de Bod, H.; Dixon, J.T.; Killian, E.; Nongodlwana, P.; Maumela, M.C.; Maumela, H.; McConnell, A.E.; Morgan, D.H.; Overett, M.J.; Prétoriusa, M.; Kuhlmannb, S.; Wasserscheidb, P.J.Catal.2007, 249, 244. doi: 10.1016/j.jcat.2007.04.009
-
[88]
Overett, M.J.; Blann, K.; Bollmann, A.; de Villiers, R.; Dixon, J.T.; Killian, E.; Maumela, M.C.; Maumela, H.McGuineess, D.S.; Morgan, D.H.; Rucklidge, A.; Slawin, A.M.Z.J.Mol.Catal.A:Chem.2008, 283, 114. doi: 10.1016/j.molcata.2007.11.036
-
[89]
Jiang, T.; Liu, X.Y.; Ning, Y.N.; Chen, H.X.; Luo, M.J.; Wang, L.B.; Huang, Z.J.Catal.Commun.2007, 8, 1145. doi: 10.1016/j.catcom.2006.10.032
-
[90]
McGuinness, D.S.; Rucklidge, A.J.; Tooze, R.P.; Slawin, A.M.Organometallics 2007, 26, 2561. doi: 10.1021/om070029c
-
[91]
Chen, H.X.; Liu, X.Y.; Hu, W.B.; Ning, Y.N.; Jiang, T.J.Mol.Catal.A:Chem.2007, 270, 273. doi: 10.1016/j.molcata.2007.02.013
-
[92]
Kuhlmann, S.; Blann, K.; Bollmann, A.; Dixon, J.T.; Killian, E.; Maumela, M.C.; Maumela, H.; Morgan, D.H.; Prétoriusb, M.; Taccardia, N.; Wasserscheid, P.J.Catal.2007, 245, 279. doi: 10.1016/j.jcat.2006.10.020
-
[93]
Jiang, T.; Ning, Y.N.; Zhang, B.J.; Li, J.Z.; Wang, G.; Yi, J.J.; Huang, Q.J.Mol.Catal.A:Chem.2006, 259, 161. doi: 10.1016/j.molcata.2006.06.026
-
[94]
Killian, E.; Blann, K.; Bollmann, A.; Dixon, J.T.; Kuhlmann, S.; Maumela, M.C.; Maumela, H.; Morgan, D.H.; Nongodlwanaa, P.; Overetta, M.J.; Pretoriusa, M.; Höfenerb, K.; Wasserscheidb, P.J.Mol.Catal.A:Chem.2007, 270, 214. doi: 10.1016/j.molcata.2007.01.046
-
[95]
Jiang, T.; Zhang, S.; Jiang, X.L.; Yang, C.F.; Niu, B.; Ning, Y.N.J.Mol.Catal.A:Chem.2008, 279, 90. doi: 10.1016/j.molcata.2007.10.009
-
[96]
Kuhlmann, S.; Dixon, J.T.; Haumann, M.; Morgan, D.H.; Ofili, J.; Spuhl, O.; Taccardi, N.; Wasserscheid, P.Adv.Synth.Catal.2006, 348, 1200. doi: 10.1002/(ISSN)1615-4169
-
[97]
Overett, M.J.; Blann, K.; Bollmann, A.; Dixon, J.T.; Hess, F.; Killian, E.; Maumela, H.; Morgan, D.H.; Neveling, A.; Otto S.Chem.Commun.2005, 5, 622.
-
[98]
Mao, G.L.; Ning, Y.N.; Hu, W.B.; Li, S.M.; Song, X.F.; Niu, B.; Jiang, T.Chin.Sci.Bull.2008, 53, 3511.
-
[99]
Aluri, B.R.; Peulecke, N.; Müller, B.H.; Peitz, S.; Spannenberg, A.; Hapke, M.; Rosenthal, U.Organometallics 2010, 29, 226. doi: 10.1021/om900925b
-
[1]
-

图 1 Cossee-Alrman机理下C2H4/C2D4产物异构体质谱(EI)裂解过程
Figure 1 Process according to Cossee-Alrman mechanism on relative intensities of C2H4/C2D4 isotopomer fragmentation pattern as a result of electron impact ionization

图式2 McDermott等提出的Pt(Ⅱ)金属环热分解机理
Scheme 2 Pt(Ⅱ) metal ring thermal decomposition mechanism suggested by McDermott et al.

图式3 Manyik等提出的乙烯三聚金属环状机理
Scheme 3 Metal ring mechanism for ethylene trimerization supposed by Manyik et al.

图式 4 Briggs 等改进的乙烯三聚金属环机理
Scheme 4 Metal ring mechanism for ethylene trimerization modified by Briggs et al.

图式5 Agapie等提出的金属环机理下乙烯三聚产物异构体的理论分布
Scheme 5 Theoretical distribution of isomers in ethylene trimerization product under metal ring mechanism proposed by Agapie et al.

图式7 Budzelaar提出的金属环机理β-H转移过渡态
Scheme 7 β-H transfer transition state of metal ring mechanism proposed by Budzelaar

图式8 Overett等基于Cr(acac)3/PNP (PNP=Ph2PN(iPr)PPh2)/MMAO乙烯四聚催化体系提出的产物形成机理
Scheme 8 Product formation mechanism based on ethylene tetramerization catalyst of Cr(acac)3/PNP (PNP=Ph2PN(iPr)PPh2)/MMAO proposed by Overett et al.

图式9 单核Cr乙烯四聚金属环机理
Scheme 9 Mononuclear Cr ethylene tetramerization metal ring mechanism

图式10 Young等提出的双核Cr乙烯四聚金属环机理
Scheme 10 Binuclear Cr ethylene tetramerization metal ring mechanism proposed by Young et al.

图式11 Peitz等提出的乙烯四聚双核Cr金属环机理
Scheme 11 Binuclear Cr ethylene tetramerization metal ring mechanism proposed by Peitz et al.

图式12 Dyachkovskii等提出的Ti系催化剂金属烷基化与离子化过程
Scheme 12 Ti metal alkylation and ionization process proposed by Dyachkovskii et al.

图式13 Gambarotta等分别基于单核和双核Cr前驱体提出的活性物种齐聚过程
Scheme 13 Oligomerization of reactive species based on mononuclear and binuclear Cr precursors proposed by Gambarotta et al

图式14 McGuinness等分别基于中性Cr(Ⅰ)活性物种和Cr(Ⅱ)阳离子活性物种提出的乙烯三聚机理
Scheme 14 Ethylene trimerization mechanism based on neutral Cr(Ⅰ) reactive species and Cr(Ⅱ) cation reactive species proposed by McGuinness et al.

图式15 Rucklidge等基于阳离子活性物种提出的乙烯四聚机理
Scheme 15 tetramerization mechanism based on cation reactive species proposed by Rucklidge et al.

图式16 Rabeah等基于Cr(Ⅱ)活性物种提出的乙烯四聚机理
Scheme 16 Ethylene tetramerization mechanism based on Cr(Ⅱ) reactive species proposed by Rabeah et al.

图式17 Cr系催化剂乙烯四聚中C6副产物(甲基环戊烷、亚甲基环戊烷)形成机理
Scheme 17 C6 product formation (methylcyclopentane and methylenecyclopentane) mechanism based on ethylene tetramerization catalyst of Cr

图 2 Suttil等进行的氘代交叉实验中1-丁烯(a)、1-己烯(b)同位素异构体实验与理论预测结果
Figure 2 Results of experiment and theoretical prediction on 1-butene (a) and 1-hexene(b) isotope isomer in deuterium cross-over experiment conducted by Suttil et al.

图 3 氘代交叉实验C6生成原理示意图
Figure 3 Schematic diagram of the principle for the formation of C6 in labelled experiment

图 4 氘代交叉实验C8生成原理示意图
Figure 4 Schematic diagram of the principle for the formation of C8 in labelled experiment

图 5 基于统计学分析的乙烯选择性齐聚双核金属环机理探索示意图
Figure 5 Schematic diagram of the exploration on selective ethylene oligomerization binuclear metal ring mechanism based on statistic analysis
表 1 Suttil等进行的氘代交叉实验中1-丁烯、1-己烯同位素异构体实验与理论预测结果方差分析
Table 1. Variance analysis about the results of experiment and theoretical prediction on 1-butene and 1-hexene isotope isomer in deuterium cross-over experiment conducted by Suttil et al.
Alkene Mechanism F Sig. Adjusted R2 1-Butene Cossee-Alrman 2.388 0.145 0.979 Metallacycle 17.492 0.001 0.637 1-Hexene Cossee-Alrman 0.015 0.904 0.952 Metallacycle 12.873 0.004 0.589  下载: 导出CSV
下载: 导出CSV
-























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 4108
- HTML全文浏览量: 905
 下载:
下载:
