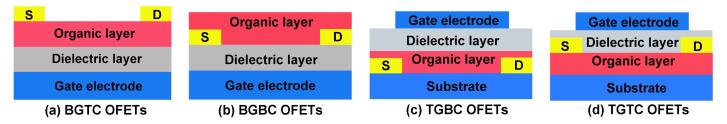
 图 1
常见的OFET器件结构平面示意图
Figure 1.
OFETs structures in this perspective
图 1
常见的OFET器件结构平面示意图
Figure 1.
OFETs structures in this perspective
引用本文:
陈华杰. 高迁移率聚合物半导体材料最新进展[J]. 有机化学,
2016, 36(3): 460-479.
doi:
10.6023/cjoc201511026

Citation: Chen Huajie. Recent Advances in High-Mobility Polymeric Semiconductor Materials[J]. Chinese Journal of Organic Chemistry, 2016, 36(3): 460-479. doi: 10.6023/cjoc201511026
Citation: Chen Huajie. Recent Advances in High-Mobility Polymeric Semiconductor Materials[J]. Chinese Journal of Organic Chemistry, 2016, 36(3): 460-479. doi: 10.6023/cjoc201511026
高迁移率聚合物半导体材料最新进展
摘要:
自20世纪80年代以来, 聚合物半导体材料及其薄膜场效应晶体管器件(OFETs)已取得系列突破性进展.目前, 已有数百种聚合物半导体材料被成功应用于OFETs中, 空穴迁移率值最高已达36.3 cm2·V-1·s-1, 可与有机小分子半导体材料甚至可同无定形硅相媲美.综述了近年来国内外高迁移率聚合物半导体的最新进展.分类对比总结和评述了空穴传输型(p-型)、电子传输型(n-型)和双极传输型聚合物半导体材料, 并对聚合物半导体材料分子设计思路、薄膜OFETs器件制备及其性能参数进行了重点阐述.同时, 总结了聚合物半导体材料的分子结构、聚集态结构与OFETs器件性能之间的内在关系, 为今后设计与合成综合性能优异的聚合物半导体材料提供一定理论指导.
English
Recent Advances in High-Mobility Polymeric Semiconductor Materials
Abstract:
Significant progress has been made in polymeric semiconductors and their organic field-effect transistors (OFETs) since 1980s. To date, hundreds of polymeric semiconductors have been reported and used for OFETs. The hole mobility above 36.3 cm2·V-1·s-1 has been achieved, which can be competitive with organic small semiconductors and even amorphous silicon. In this review, the recent progress in high-mobility polymeric semiconductor materials has been summarized from the perspective of design, synthesis, and OFET devices performance. Moreover, the recent developments are systematically summarized and analyzed according to different types of polymeric semiconductors, including p-type, n-type, and ambipolar polymeric semiconductors. The analysis about the relationship among the molecular structure-aggregation structure-OFET devices performance may guide the rational molecule design in the polymeric semiconductor materials with excellently comprehensive performance in the future.
-
Key words:
- organic field-effect transistors
- / polymer semiconductors
- / high mobility
- / air stability
-
有机共轭半导体是一类新型的半导体材料, 它独特的光、电、磁等特性吸引了全球科学家的关注[1~4].早在20世纪初, Mccoy和Moore就预言: “人们有望合成出不含金属元素的有机金属导体”. 1970年, Wudl等成功合成了四硫富瓦烯(TTF)类有机共轭电子给体. 1973年, Ferraris等发现TTF给体与有机共轭受体7, 7, 8, 8-四氰基对亚甲基苯醌(TCNQ)之间能够形成高导电性的有机电荷转移复合物, 从此开创了有机导体和超导体研究的新纪元. 1986年, Tsumura等[5]报道了第一个以聚噻吩为活性层的有机场效应晶体管(Organic field-effect transistors, OFETs).同年, Tang[6]首次报道了基于酞青电子给体和苝酰亚胺衍生物电子受体的双层异质结有机太阳能电池(Organic photovoltaics, OPVs). 1987年, Tang等[7]报道了第一个基于8-羟基喹啉衍生物活性层的电致发光二极管(Organic light-emmiting diodes, OLEDs). 2000年, Heeger等因发现导电高分子被授予诺贝尔化学奖, 开拓了有机电子学研究的新时代.作为有机电子学领域最重要的基元器件之一, OFETs具有制备工艺简单、成本低廉、轻质和柔性等优点, 其潜在应用于射频电子商标、智能卡、传感器、逻辑电路以及电子纸等光电设备中[1~4].由于其巨大的市场前景, 全球多家研究机构都相继开展了相关研究.经过三十多年的努力, OFETs在有机半导体材料、理论与应用方面取得了系列突破, 目前欧美日韩多国已投资OFETs相关的应用研究.
作为OFETs的核心组成部分, 有机半导体的发展很大程度上决定着OFETs的发展.有机半导体的分子结构及其薄膜的聚集态结构决定着OFET器件载流子传输的类型和性能[1!~4].根据OFETs测试时载流子传输类型区分, 有机半导体可分为三大类: (1) p-型空穴传输材料, 以并五苯、聚噻吩、吡咯并吡咯二酮(DPP)和异靛青(IDG)类衍生物的空穴传输性能最为突出; (2) n-型电子传输材料, 通常为含强缺电子官能团的萘酰亚胺(NDI)和苝酰亚胺(PDI)类共轭有机小分子和聚合物半导体材料; (3)空穴和电子同时传输的双极性半导体材料, 其典型代表为NDI和DPP类共轭小分子和聚合物材料.按分子量大小区分, 有机体半导体材料可分为以下三类:有机小分子、齐聚物和聚合物半导体材料.对于小分子材料而言, 其优点在于结构易于修饰和纯化, 可采用真空镀膜法和溶液法制备薄膜器件和单晶/微纳米器件等.通常, 小分子真空镀膜法成膜的结晶性和有序性良好, 其器件的迁移率较高.然而, 溶液法加工小分子薄膜的均一性及其器件性能的重复性相对较差.相比之下, 聚合物半导体材料具有溶液法加工成膜性好、制备工艺简单以及成本低廉等优势, 适合采用溶液旋涂、打印和丝网印刷等技术构造柔性、大面积和高性能的有机光电子薄膜器件[1~4].因此, 开发综合性能优异的聚合物半导体材料是加速OFETs的大面积集成与产业化发展的关键所在.
近年来, 设计与合成新型的聚合物半导体已成为柔性有机电子学理论和应用研究的热点, 并且取得了系列突破[1~4].目前, p-型聚合物取向纤维膜的迁移率已超过35 cm2•V-1•s-1, n-型聚合物的迁移率超过3.0 cm2•V-1• s-1, 双极性聚合物的空穴和电子迁移率也均超过5.0 cm2•V-1•s-1.基于共轭聚合物材料的给体(D)和受体(A)结构来分, 聚合物半导体材料可分为以下三类: (1) D-D型, 即为全给体型的p-型聚合物材料, 以并噻吩和稠环噻吩类共聚物最为经典, 其空穴迁移率已超过4.0 cm2•V-1•s-1; (2) A-A型, 即为全受体型的n-型共轭聚合物材料, 电子迁移率已超过1.0 cm2•V-1•s-1; (3) D-A型, 该类聚合物的D单元通常为连二噻吩、噻吩[3, 2-b]噻吩、噻吩乙烯噻吩、硒吩、苯和萘等富电子的化合物, 而A单元通常为带强缺电子酰胺/酰亚胺官能团的NDI、PDI、DPP以及IDG.研究证实:通过合理地组合与剪裁D和A单元, 可开发出载流子传输类型不同的D-A型共轭聚合物.由于D-A型聚合物分子内强的电荷转移作用促使了聚合物分子链间的π-π堆积和薄膜的结晶性, 其迁移率在三类聚合物材料中处于领先地位.
针对这一快速发展的领域, 本文综述了近年来高迁移率聚合物半导体材料及其OFETs器件性能的最新进展情况.首先, 简要介绍了有机场效应晶体管的器件结构、工作原理以及性能参数.然后, 分类总结和评述了p-型、n-型和双极性聚合物半导体材料的最新进展, 并对各类聚合物半导体材料的分子设计思路、器件制备及其性能参数进行了重点评述.
1 有机场效应晶体管的结构
有机场效应晶体管主要由衬底(substrate)、绝缘层(Dielectric layer)、有机半导体层(Organic layer)、栅电极(Gate electrode)和源-漏电极(Source/Drain electrode)组成[4].通常选用高掺杂的硅作为衬底和栅电极.绝缘层通常选用介电性能良好的二氧化硅、三氧化铝、氮化硅、氧化铪等无机绝缘材料, 也可以选用聚甲基丙烯酸甲酯、聚丙烯腈和聚甲基硅倍半氧烷等高分子绝缘材料.有机半导体层通常为有机半导体薄膜、单晶和微纳米线等.其中, 有机薄膜可采用真空镀膜、溶液旋涂、丝网印刷以及溶液打印等方法制备.而有机单晶和微纳米线则采用气相生长法或溶剂挥发生长等方法制备.通常选用高导电率的金、铂、银、铜等金属作为器件的源-漏电极, 也可选用石墨烯和聚吡咯等导电材料.
根据电极和有机半导体活性层的相对位置不同, OFETs可以分为底栅极结构和顶栅极结构, 见图 1.当栅电极位于介电层和有机半导体层的底部时, 为常用的底栅极结构OFETs, 见图 1a和图 1b; 当栅电极位于介电层和有机半导体层的顶部时, 为常用的顶栅极结构OFETs, 见图 1c和图 1d.其中, n-型双极性OFETs通常选用顶栅结构OFETs结构, 因为一定厚度的栅电极和介电层薄膜可有效地隔绝空气中H2O和O2对半导体活性层的侵害, 有利于提高OFETs器件的迁移率和稳定性等.此外, 根据有机半导体层和源-漏电极间的相对位置, 底栅结构的OFETs又可分为上电极结构和下电极结构.当源-漏电极位于有机半导体层顶部时, 为常用的上电极结构, 见图 1a和图 1d.通常, 上电极结构能保证有机半导体层与源-漏电极间形成良好接触, 有利于载流子的注入和传输.当源-漏电极位于有机半导体层的底部时, 为下电极结构, 见图 1b和图 1c.其中, 下电极OFETs可采用光刻技术制备源-漏电极, 因此下电极OFETs与光刻技术具有良好兼容性, 可采用溶液法加工技术制备大面积有机电路.
图 1
常见的OFET器件结构平面示意图
Figure 1.
OFETs structures in this perspective
2 有机场效应晶体管工作原理和性能参数
OFETs是一种电压控制型器件, 它利用栅极电压来调控绝缘层电容的耦合大小, 从而调控沟道内半导体活性层的导电能力.我们以增强型OFETs为例, 简要介绍其原理:在一定的源漏电场的作用下, 且栅极电压(VG)为零时, 沟道内活性层的电导能力很低, 从源极注入并被漏极所收集的电流几乎为零, 此时OFETs处于关态; 当施加VG后, 由于绝缘层的电容耦合作用会在绝缘层和半导体层的界面处诱导出大量的载流子, 然而并不是诱导出的所有载流子均可移动, 只有当VG大于阈值电压VT时, 且在源漏电压(VD)的作用下部分载流子发生定向漂移, 形成导电沟道, 此时器件处于开态, 从而实现了OFETs的开关功能.
评价OFETs性能好坏的关键参数为:迁移率(μ)、开关电流比(Ion/Ioff)、阈值电压(VT)和亚阈值斜率(S)等, 下面对这些参数进行简短地介绍.
迁移率(μ):在单位电场强度下, 载流子在单位时间内漂移的距离, 其单位为cm2•V-1•s-1.它反映了在一定电场强度下, 电子或空穴在半导体层中传输的快慢, 通过OFETs的转移曲线斜率换算得出, 见图 2b.迁移率的大小直接决定着OFETs的功率和工作频率, 对OFETs各方面的应用影响重大.
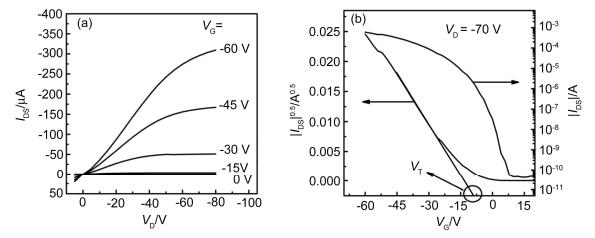 图 2
有机场效应晶体管的典型输出曲线(a)和转移曲线(b)
Figure 2.
Typical output curve (a) and transfer curve (b) of OFETs
图 2
有机场效应晶体管的典型输出曲线(a)和转移曲线(b)
Figure 2.
Typical output curve (a) and transfer curve (b) of OFETs
开关电流比(Ion/Ioff):在一定VG范围内, OFETs的开态源-漏电流和关态源-漏电流之间的比值, 它通过OFETs的转移曲线获得, 见图 2b.开关电流比的大小能够直接反映出OFETs开关性能的好坏, 该参数在有源矩阵显示和逻辑电路中非常重要.具有高的Ion/Ioff的OFETs可以获得更大的负载驱动能力、更好的稳定性和抗干扰能力.
阈值电压(VT):诱导出OFETs导电沟道的最小电压, 它反映晶体管操作电压的重要参数之一, 通过OFETs的转移曲线获得, 见图 2b.影响阈值电压大小的主要因素包括:绝缘层和半导体层界面的缺陷密度、绝缘层电容的大小以及源-漏电极与半导体层间的接触等因素.通常, 低的VT是构造低功耗电子器件的必要条件.
亚阈值斜率(S):也称为亚阈值摆幅, 其定义为亚阈值区漏端电流增加一个数量级所需要增大的栅电压, 反映了电流从“关”态切换到“开”态时电流变化的快慢程度, 小的亚阈值斜率说明晶体管的打开速度快.它和低的VT一起成为决定晶体管器件的操作电压.
有机场效应晶体管的电流-电压理论同无机场效应晶体管相似, 它的各项性能参数主要通过OFETs的输出曲线(图 2a)和转移曲线(图 2b)计算出来的.输出曲线是指IDS随VD的变化曲线, 然而晶体管的大部分性能参数都是通过转移曲线获得.按照如下所列的两个公式, 我们可求出OFETs的线性区和饱和区的迁移率.对于线性区迁移率(μ)是IDS对VG作图换算而得.而饱和区迁移率(μ)是通过
$ \sqrt {{I_{{\rm{DS}}}}} $ 对VG作图, 由切线的斜率换算出载流子迁移率的大小.
线性区电流-电压关系为:
饱和区电流-电压关系为:
其中, μ为场效应迁移率, L为沟道长度, W为沟道宽度, VD为源漏电压, VG为栅压, VT为阈值电压, Ci为绝缘层单位面积的电容率.
3 空穴传输型(p-型)聚合物半导体材料最新进展
近年来, 溶液法加工p-型聚合物半导体及其OFETs性能得到了快速发展[1~4, 8].在报道的p-型聚合物中, 聚3-烷基噻吩(P3AT)是研究最为广泛的一类聚噻吩半导体, 部分高迁移率P3AT衍生物的分子结构如图 3所示.在报道的P3AT中, 以聚3-己基噻吩(P3HT)为典型研究代表.目前, P3HT薄膜OFETs的迁移率和开关比相对较低, 器件稳定性差, 难以达到应用要求.通常, 人们可采用分子设计和器件优化来解决这些难题.常用的分子设计思路如下[1~4]: (1)在聚噻吩主链上引入等规取代的长烷基链, 确保聚噻吩衍生物的溶解性和自组装能力; (2)引入未取代的联二噻吩、联二硒吩和并噻吩等间隔单元, 可提高分子链的共平面性, 减少烷基链的取代密度, 从而降低聚噻吩衍生物的HOMO能级; (3)在聚噻吩主链中引入杂原子或杂环, 可降低聚噻吩的HOMO能级, 加强分子链间的π-π相互作用.基于以上思路, Ong等[9]报道了一类以联二噻吩为π桥的等规聚噻吩半导体PQT-12.通过在主链上引入未取代的联二噻吩, 增强了分子链的旋转自由度, 同时降低了烷基链的取代密度.因此, PQT-12获得比P3HT更好的抗氧化能力.在空气中, PQT-12退火薄膜构造的底栅OFET器件性能重复性好, 最高空穴迁移率达0.14 cm2•V-1•s-1, 电流开关比达107.然而, 随着空气湿度增加, 该器件的迁移率和开关比依然呈下降趋势[10]. 2006年, McCulloch等[11]报道了一类含噻吩[3, 2-b]噻吩的聚噻吩半导体PBTTT-14, 其HOMO能级相比P3HT降低了0.3 eV.此外, PBTTT-14退火薄膜展现出典型的液晶相行为, 其多晶薄膜晶粒尺寸达200 nm, 最高迁移率达0.72 cm2•V-1• s-1. 2009年, Bao等[12]在聚噻吩主链上引入噻唑杂环, 设计合成了一类HOMO能级低(-5.19 eV)的噻唑基共聚物PT2Tz-C12.研究表明:未取代的联二噻吩间隔单元和噻唑杂环增强了分子链间的π-π相互作用, 促使PT2Tz-C12退火薄膜形成有序的液晶薄膜.在空气中, 基于PT2Tz-C12退火薄膜构造的底栅OFETs的空穴迁移率达0.33 cm2•V-1•s-1, 同时该器件还表现出优越的空气稳定性和操作稳定性.
聚噻吩乙烯(PTV)是另一类经典的聚噻吩半导体材料, 被广泛应用于有机光电子学领域中[1-4].近年来, 双键桥连型PTV衍生物成为OFETs领域中的热门研究体系, 部分高迁移率PTV衍生物的分子结构如图 3所示. 2009年, Kim等[13]在PQT-12的主链中引入双键基团, 开发了一类结构新颖的PTV衍生物(PETV12T).双键的引入增加了聚合物骨架的旋转自由度, 减少了邻近噻吩基团之间的位阻, 拓宽了聚合物共轭骨架, 促使了PETV12T退火薄膜获得大的多晶谷粒, 其迁移率高达0.15 cm2•V-1•s-1. 2011年, Kim等[14]报道了一类烷基取代的噻吩乙烯噻吩(TVT)基共聚物PC12TV12T.该聚合物不仅具有大的共平面骨架和强的自组装能力, 而且还表现出优良的溶解性和加工性, 其溶液法加工OFETs的空穴迁移率高达1.0 cm2•V-1•s-1. 2014年, Fei等系统地研究了PTV衍生物主链上烷基链取代位置对材料光电性能的影响.研究发现:当烷基取代在乙烯双键同侧, 所开发的PTV吩衍生物PTVT-TT的主链共平面性最好, 利于高性能电荷传输.基于PTVT-TT构造的薄膜OFET器件的最高迁移率达4.6 cm2•V-1•s-1, 为目前报道的D-D型聚合物半导体迁移率的最高值.随后, Kim等[16]报道了一类含并三噻吩和双键π桥的全给体型聚合物半导体PTVT-DTT, 最高迁移率达3.91 cm2•V-1•s-1.以上研究结果充分证实:通过合理的分子剪裁和器件优化, D-D型聚合物半导体的迁移率可以同D-A型聚合物半导体相媲美.
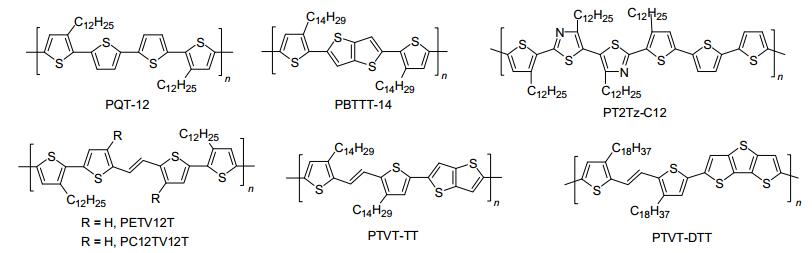 图 3
p-型聚噻吩半导体的分子结构
Figure 3.
Molecular structures of p-type polythoiphene semiconductors
图 3
p-型聚噻吩半导体的分子结构
Figure 3.
Molecular structures of p-type polythoiphene semiconductors
D-A型共轭聚合物材料, 被誉为第三代半导体聚合物材料, 近年来已成为有机光电领域研究的热点[1-4].将D-A型聚合物半导体材料应用于OFETs中, 通常能够获得理想的器件性能.在D-A型聚合物场效应晶体管材料设计过程中, 可选择的D单元较为丰富, 而A单元相对稀少, 理想A单元的设计与合成极富有挑战.近年来, 最为经典的A单元如下:苯并噻二唑(BT)、吡咯并吡咯二酮(DPP)、苯并二噻二唑(BBT)、萘二酰亚胺(NDI)和异靛青(IDG)等缺电子单元.根据A单元不同, 下面我们对近期报道的部分高迁移率p-型聚合物半导体材料进行分类介绍, 其结构和性能见图 4~6和表 1.
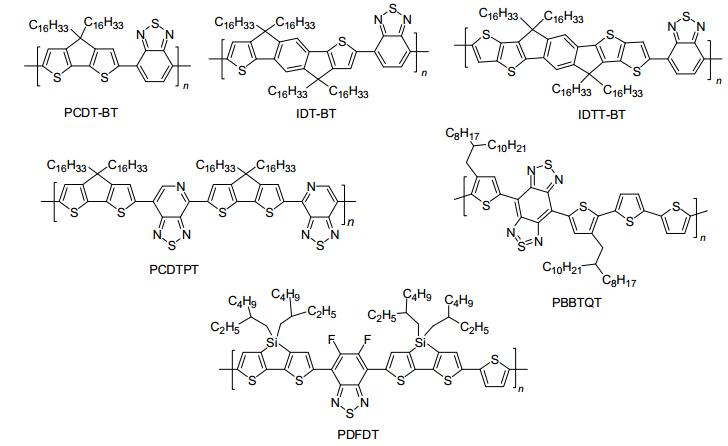 图 4
p-型聚苯并噻二唑衍生物的分子结构
Figure 4.
Molecular structures of p-type polymeric semiconductors containing benzothiadiazole derivatives
图 4
p-型聚苯并噻二唑衍生物的分子结构
Figure 4.
Molecular structures of p-type polymeric semiconductors containing benzothiadiazole derivatives
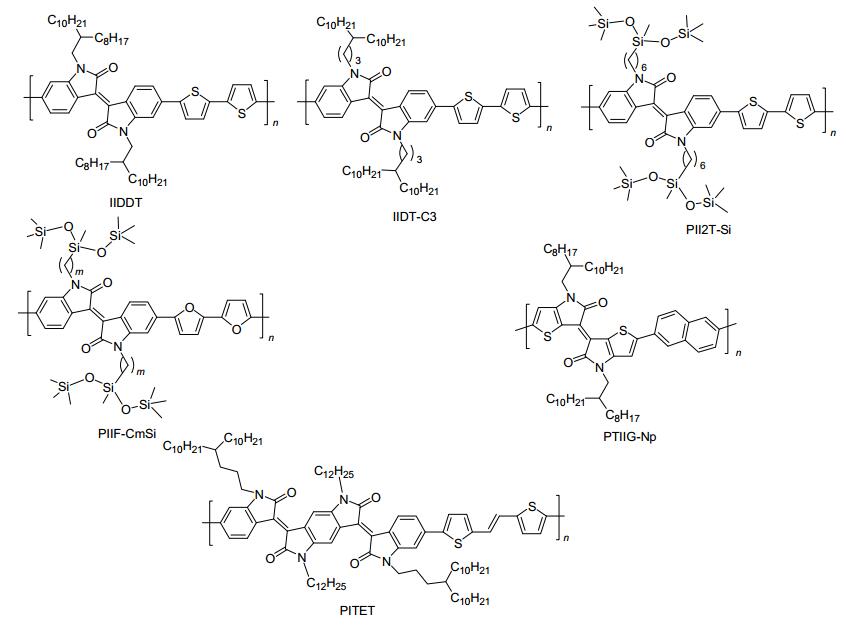 图 5
p-型聚异靛衍生物的分子结构
Figure 5.
Molecular structures of p-type polymeric semiconductors containing isoindigo derivatives
图 5
p-型聚异靛衍生物的分子结构
Figure 5.
Molecular structures of p-type polymeric semiconductors containing isoindigo derivatives
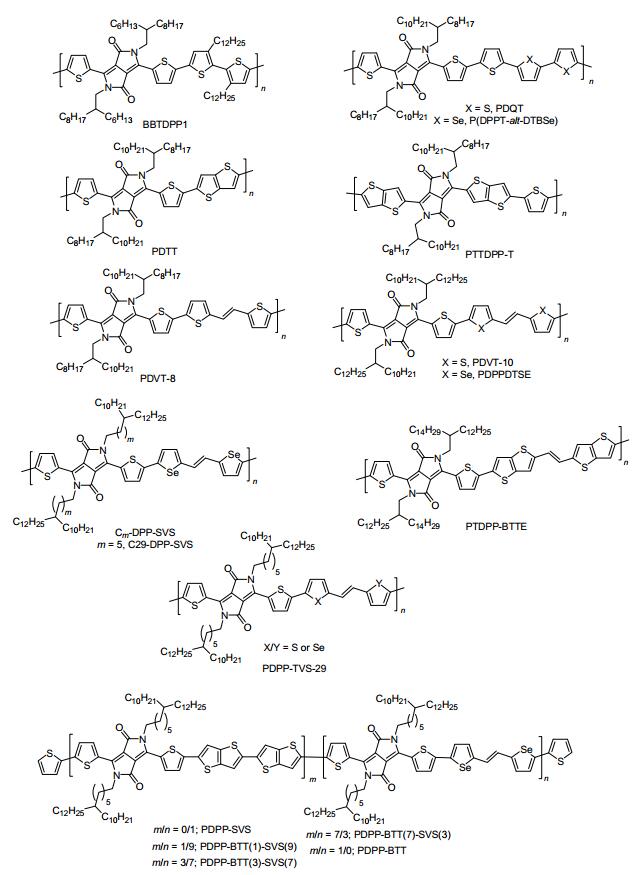 图 6
p-型DPP类聚合物半导体的分子结构
Figure 6.
Molecular structures of p-type polymeric semiconductors containing DPP derivatives
图 6
p-型DPP类聚合物半导体的分子结构
Figure 6.
Molecular structures of p-type polymeric semiconductors containing DPP derivatives
 表 1
p-型共轭聚合物半导体材料的性能参数
Table 1.
Electronic parameters for p-type polymer semiconductors
表 1
p-型共轭聚合物半导体材料的性能参数
Table 1.
Electronic parameters for p-type polymer semiconductors
Polymer Mn/Mw/(kg•mol-1) HOMO/eV LUMO/eV μhole/(cm2•V-1•s-1) Ion/Ioff Device structure Ref. PQT-12 17.3/22.9 — — 0.14 >107 BGTC [9, 10] PBTTT-14 33.0/59.6 -5.10 — 0.72 >106 BGBC [11] PT2Tz-C12 —/— -5.19 — 0.33 >106 BGTC [12] PETV12T 15.0/25.5 -5.20 -3.35 0.15 >104 TGBC [13] PC12TV12T 24.9/— -5.02 — 1.05 >104 TGBC [14] PTVT-TT 20/58 -4.97 — 4.60 104~105 TGBC [15] PTVT-DTT 23.0/52.9 -4.80 -3.20 3.91 >103 TGBC [16] PCDT-BT 35.0/— — — 3.30 105~106 BGBC [17, 18] IDT-BT 80/160 -5.40 — 3.60 >106 TGBC [19, 20] IDTT-BT 76.0/197.6 -5.40 -3.70 8.70 >104 TGBC [21] PCDTPT 140.0/— — — 36.3 >104 BGBC [22~25] PBBTQT 65.9/132.4 -4.60 -3.80 2.50 >103 BGBC [26] PDFDT 14.0/33.6 -5.24 -3.37 9.05 >103 TGBC [27] IIDDT 87.9/185.8 -5.70 -3.70 0.79 ~107 BGTC [30] IIDDT-C3 39.2/125.4 -5.52 -5.74 3.62 >106 BGTC [31] PII2T-Si 138/454 -5.20 -3.58 2.48 >106 BGTC [32] PIIF-C9Si 48.3/101.4 -5.25 — 4.80 ~106 BGTC [33] PTIIG-Np 21.0/102.2 -5.12 -3.49 14.4 >103 TGBC [35] PITET 37.6/89.5 -5.32 -4.05 1.92 >104 BGTC [36] PDQT 25.4/60.6 -5.2 -4.0 0.97 ~107 BGTC [42] P(DPPT-alt-DTSe) 19.3/62.8 — — 1.50 ~106 BGTC [43] PDTT 110/501 -5.2 -3.4 10.5 >106 BGBC [44, 45] PTTDPP-T 14/75 -5.06 -3.68 ≈2.0 ~105 TGBC [46] PDVT-8 70/180 -5.30 — 4.5 >105 BGBC [47] PDVT-10 73.5/183 -5.28 — 11.0 >104 BGBC [47, 48] PDPPDTSE 63.4/114.1 -5.37 — ≈5.0 >107 BGTC [49] C29-DPP-SVS 34.1/58.3 -5.14 — 17.8 >105 BGTC [50, 51] PTDPP-DTTE 127.85/731.3 -5.16 — 7.43 >105 TGSS-BGTC [52] PDPP-BTT(1)-SVS(9) 66.5/100.1 -5.49 -3.82 6.51 >104 BGTC [53] PDPP-TVS-C29 74.5/100.6 — — 8.2 >105 BGTC [54] 表 1 p-型共轭聚合物半导体材料的性能参数
Table 1. Electronic parameters for p-type polymer semiconductors苯并噻二唑(BT)及其衍生物具有良好的共平面性、强的杂原子效应和强缺电子特性等优势, 因而被广泛地用于设计、合成溶液法加工高迁移率聚合物OFETs材料. 图 4为部分高迁移率p-型聚苯并噻二唑衍生物的分子结构. 2007年, Müllen等[17]以十六烷基取代的环戊二烯并二噻吩(CDT)为电子给体, BT为电子受体, 合成了一个数均分子量为10.2 kDa的共聚物PCDT-BT, 其空穴迁移率达0.17 cm2•V-1•s-1.随后, Müllen等[18]通过分子量和烷基链优化, 基于高分子量的PCDT-BT退火薄膜构造的底栅OFETs的空穴迁移率进一步提高到3.3 cm2•V-1•s-1, 这一结果刷新了当时p-型聚合物OFETs迁移率的记录. 2010年, McCulloch等[19]以十六烷基取代的引达省(IDT)为电子给体, BT为电子受体, 合成了一类结构新颖的D-A型共聚物IDT-BT, 其退火薄膜采取face-on的排列模式, π-π堆积距离约为4.1 Å.在空气中,
采用顶栅器件结构制备的薄膜OFET器件迁移率达1.25 cm2•V-1•s-1.通过合成优化, McCulloch等[20]合成了分子量更高、分子量分布更窄的IDT-BT(Mn=80 kDa, PDI=2).经过器件优化, 其空穴迁移率进一步提高到3.6 cm2•V-1•s-1.作者分析, 高的分子量和窄的分布是提高空穴迁移率的主要原因. 2015年, McCulloch等[21]对IDT给体的分子骨架进一步拓宽, 设计并合成了一个骨架共轭更大的引达省衍生物(IDTT给体).通过与BT受体共聚, 开发了一类大π共轭型聚合物半导体IDTT-BT. X射线衍射证实: IDTT-BT退火薄膜采取face-on和edge-on的混合模式堆积.通过采用CuSCN对OFETs的金-源漏电极进行修饰, 有效地降低了IDTT-BT薄膜的空穴聚入势垒.在氮气中, 基于IDTT-BT退火薄膜构造的顶栅OFETs的迁移率高达8.7 cm2•V-1•s-1.
通过结构修饰, 化学家开发了系列结构新颖的苯并噻二唑类衍生物单元, 并成功应用于合成高迁移率聚合物半导体材料.其中, 以吡啶噻二唑(PT)、苯并二噻二唑(BBT)和二氟代苯并噻二唑(2FBT)的研究最为广泛. 2011年, 基于结构不对称的PT受体和CDT给体, Bazan等[22]巧妙地合成了一类结构等规的D-A型共聚物PCDTPT, 其数均分子量达34 kDa, 迁移率高达0.6 cm2•V-1•s-1.随后, Bazan等[23]通过制备凝胶渗透色谱仪分离出两种高子量的PCDTPT, 其薄膜器件迁移率从0.8 cm2•V-1•s-1 (Mn=100 kDa)进一步提高到2.5 cm2• V-1•s-1 (Mn=300 kDa).此外, Bazan等[23~25]探索了一种能够在衬底上获得单一取向薄膜的方法, 即夹层隧道体系.他们利用毛细管效应与重力效应协同诱导PCDTPT聚合物溶液沿衬底纳米槽的方向组装, 获得高度取向的聚合物纤维薄膜.通过调节源漏电极间的距离, 最后使得OFETs的迁移率从25.4 cm2•V-1•s-1提高到36.3 cm2•V-1•s-1 [25]. 2012年, 基于BBT强受体, Wudl等[26]报道了一类超低能隙的BBT基共聚物PBBTQT. X射线衍射证实: 260 ℃退火薄膜堆积紧密有序, 且采取edge-on模式堆积, 其π-π堆积距离小至3.5 Å;在氮气中, 其底栅薄膜OFETs的迁移率达2.5 cm2•V-1•s-1. 2015年, Noh等[27]采用烷基取代二噻吩并硅杂环戊二烯为电子给体, 2FBT为电子受体, 设计合成了一类结构新颖的D-A共聚物PDFDT.由X射线衍射可知: PDFDT退火薄膜采取edge-on和face-on混合模式堆积, 其π-π堆积距离小至3.42 Å.在氮气中, 其顶栅薄膜OFET器件迁移率最高达9.05 cm2•V-1•s-1, 为目前报道的氟代苯并噻二唑类聚合物半导体迁移率的最高值.以上研究结果证实:聚合物半导体材料的分子量、薄膜的有序度和取向度、堆积结构对聚合物薄膜OFETs的迁移率、开关比和空气稳定性等参数都起着至关重要的影响.
异靛为靛青工业染料的一种同分异构体, 它具有良好的共平面性、强的吸电子能力以及溶解性可调等优势, 因而被广泛地用于合成高性能的有机/聚合物光电功能材料[28]. 图 5为部分高迁移率p-型聚异靛衍生物的分子结构. 2010年, Reynolds等[29]首次利用烷基取代异靛单元, 设计并合成了系列结构新颖的D-A型共聚物, 并成功应用于有机太阳能电池中. 2011年, 裴坚课题组率先将异靛分子应用于聚合物场效应晶体管领域中, 设计合成了一类以异靛为核的D-A型聚合物IIDDT[30].该聚合物薄膜拥有低的HOMO能级(-5.70 eV)和紧密的edge-on模式堆积.在空气中, 基于IIDDT退火薄膜制备的OFET器件的空穴迁移率高达0.79 cm2•V-1•s-1.同时该器件还表现出良好的空气稳定性, 可在高湿度条件下(60%)的空气中稳定长达6个月之久. 2012年, 该课题组首次研究了烷基链分叉位置对异靛基聚合物IIDT-Cm的光物理、电化学、薄膜堆积形态以及OFETs性能的影响[31].发现随着分叉位置远离聚合物IIDT-Cm的主干, π-π堆积距离会逐渐变小, 当距离为三个亚甲基时(即聚合物IIDT-C3)达到了最大迁移率3.62 cm2•V-1•s-1. 2011年, Bao等[32]首次将硅氧烷基链引入到异靛基共聚物的侧链上, 合成了聚合物PII2T-Si.该硅氧烷基侧链展现出强的自组装能力, 使得PII2T-Si退火薄膜获得了超紧密的π-π堆积距离(3.57 Å), 并采取二维层状模式堆积.因此, PII2T-Si退火薄膜OFET器件的最高空穴迁移率达2.48 cm2•V-1•s-1.随后, 该课题组对比研究了硅氧烷链分叉位置对呋喃桥连异靛基共聚物PIIF-CmSi的热力学性能、能级结构、薄膜有序性、晶粒大小、堆积结构以及电荷传输性能的影响[33].当连接碳数m为9时, 聚合物PIIF-C9Si迁移率最高可达4.8 cm2•V-1•s-1.以上研究结果揭示了不同烷基链分叉位置对共轭聚合物材料性能的影响, 其研究经验对今后设计与合成高性能聚合物半导体材料具有重要的指导意义.
近年来, 化学家对异靛母核展开了系统地结构修饰, 开发了系列结构新颖的异靛衍生物及其共轭聚合物半导体材料.其中, 以噻吩异靛基聚合物半导体性能表现最为突出[34, 35]. 2010年, McCulloch等[34]首次报道了噻吩异靛及其共轭聚合物(IGT-BT)的合成方法, 并系统的研究了噻吩异靛基共轭聚合物的电荷传输性能. 2014年, 通过与萘给体共聚, Yang等[35]开发了一类高迁移率的噻吩异靛基共轭聚合物PTIIG-Np, 其薄膜OFET器件迁移率高达14.4 cm2•V-1•s-1, 为目前所报道的溶液法加工聚合物薄膜OFETs迁移率的最高值. 2015年, 裴坚等[36]对异靛母核进行了系列优化, 设计并合成了一类共轭更大的异靛衍生物电子受体(NBDOPV), 通过与噻吩乙烯噻吩(TVT)共聚, 合成了一类以NBDOPV为电子受体的D-A型共轭聚合物PITET. X射线衍射证实: 200 ℃退火薄膜展现出有序堆积的纤维相态结构, 并采取edge-on模式紧密堆积.在氮气中, 其底栅退火薄膜OFETs的空穴迁移率达1.92 cm2•V-1•s-1, 这一结果证实了NBDOPV缺电子单元为一类综合性能优异的聚合物半导体材料构建单元.
吡咯并吡咯二酮(DPP)衍生物是一类综合性能优异的工业染料, 它具有强的着色能力, 高的颜色饱和度, 优异的耐光、耐气候、耐热和耐酸碱性能, 因而被广泛的应用于印染行业. Ciba公司发明了以丁二酸酯和苯腈为原料, 一步法高效合成了苯基取代的DPP染料.由于苯基具有较大的空间位阻, 导致苯基取代DPP的骨架平面性较差, 因而苯基取代DPP被较少应用于有机光电子学领域.近年来, 以氰基噻吩、氰基呋喃、氰基硒吩、氰基噻吩并噻吩和氰基吡啶等化合物为起始原料, 开发了系列骨架共平面性更好的DPP衍生物单元[37~39], 并且被成功应用于开发有机光电材料.其中, 部分高迁移率的p-型DPP类聚合物半导体材料的分子结构如图 6所示, 以噻吩取代DPP及其共轭聚合物研究最为广泛.噻吩取代DPP单晶结构证实[40]: DPP核上羰基氧原子与邻位噻吩上的氢原子之间存在一种类似于氢键的弱相互作用, 使得噻吩取代DPP具有良好的共平面性, 为分子骨架堆积提供了强的驱动力.
2008年, Winnewisser等[41]首次将噻吩取代DPP基共聚物(BBTDPP1)应用到薄膜OFETs中, 获得优异的双极性性能, 其空穴和电子迁移率分别达0.1和0.09 cm2•V-1•s-1. 2011年, Li等[42]报道了一类以DPP为电子受体, 联四噻吩为电子给体的共聚物PDQT, 未经退火的PDQT薄膜就能获得很好的结晶性和有序性, 其空穴迁移率高达0.89 cm2•V-1•s-1; 在温和退火条件下, 其100 ℃退火薄膜的迁移率为0.97 cm2•V-1•s-1.结果表明:较低退火温度对今后开发柔性有机光电子器件具有非常重要的意义. 2011年, Choi等[43]同样报道了DPP基共聚物PDQT, 其数均分子量为58781, 其退火薄膜的最大空穴迁移率达1.04 cm2•V-1•s-1.同时, 该课题组还对比合成了一类以联二硒吩为π桥的DPP基共聚物P(DPPT-alt-DTBSe).尽管P(DPPT-alt-DTBSe)的数均分子量仅为PDQT的三分之一, 然而其退火薄膜OFETs的空穴迁移率高达1.5 cm2•V-1•s-1. AFM形貌测试和X射线衍射结果表明:相比PDQT, P(DPP-alt-DTBSe)退火薄膜具有更大的结晶谷粒和更好的长程有序性, 其π-π堆积更加紧密, 因而获得更高的迁移率. 2011年, Li等[44]首次将噻吩[3, 2-b]噻吩引入DPP基聚合物主链中, 合成了一类大π共轭的DPP基共聚物PDTT, 其数均分子量为89700. X射线衍射证实:退火薄膜展现出有序堆积的纤维相态结构, 且采取edge-on模式紧密堆积, π-π堆积距离仅为3.71 Å, 最高空穴迁移率达0.94 cm2•V-1• s-1. 2012年, Ong等[45]通过优化催化剂体系、反应溶剂和反应温度等聚合反应条件, 采用Stille偶联法高效合成了一类分子量更高(Mn=110 kDa)的PDTT, 其π−π堆积间距进一步缩短到3.43 Å, 空穴迁移率显著提高到10.5 cm2•V-1•s-1. 2011年, Bronstein等[46]以2-氰基取代噻吩并[3, 2-b]噻吩和丁二酸二异丙酯为原料, 首次合成了一类刚性更大、平面性更好的噻吩并[3, 2-b]噻吩取代DPP衍生物, 并采用Stille偶联法合成了交替型共聚物PTTDPP-T, 其顶栅退火薄膜OFET器件的最高空穴迁移率接近2.0 cm2•V-1•s-1.
2012年, 陈华杰等[47]首次将噻吩乙烯噻吩(TVT)单元引入到DPP基共聚物的主链中, 设计并合成了一类可溶液法加工的双键桥连DPP类共轭聚合物半导体(PDVT-8和PDVT-10).研究证实:带更长烷基取代的PDVT-10展现出更好的溶液加工性能, 获得更有序堆积的结晶薄膜, 以及更高的电荷传输能力.在空气中, PDVT-10的迁移率最高可达8.2 cm2•V-1•s-1, 这一结果刷新了当时p-型聚合物场效应器件迁移率的记录. X-射线衍射研究证实: PDVT-8和PDVT-10的分子骨架采用紧密的edge-on模式堆积, 这种堆积方式有利于高迁移率电荷传输; 其层间距d-d分别为19.44和21.11 Å, 分子主链间的π-π距离分别为3.72和3.66 Å, 这一相对较小的d-d和π-π堆积间距都是十分有利于高迁移率的电荷传输.值得一提的是, 采用简单的PMMA封装后, 基于PDVT-10退火薄膜构造的底栅OFETs展现出优异的空气稳定性和操作稳定性.放置在空气湿度为20%的空气中100 d后, 其迁移率依然保持在5.0 cm2•V-1•s-1左右. 2014年, 刘云圻等[48]制备了柔性PDVT-10退火薄膜OFET器件, 并研究了该柔性器件在低电压驱动条件下的电荷传输特性.当驱动电压低至-3 V时, 其PDVT-10薄膜OFET器件的空穴迁移率高达11.0 cm2•V-1•s-1, 这一迁移率值为目前报道的柔性低电压驱动聚合物OFETs的最高值.此外, 该低电压驱动器件对光和热展现出良好的刺激响应功能, 在传感器和电子皮肤等领域具有重大应用潜力.
随后, 全球多个课题组对PDVT-10的主链骨架结构和侧链助溶基团进行了系统的优化, 设计并合成了系列高迁移率的聚合物半导体材料. 2012年, Kwon等[49]合成了以硒吩乙烯硒吩(SVS)为π桥的DPP基共聚物PDPPDTSE, 并且对比研究了PDVT-10和PDPPDTSE的光电性质.研究表明:相比PDVT-10, PDPPDTSE展现出更强的分子内电荷转移和更强的π-π相互作用, 其退火薄膜OFETs的空穴迁移率高达5.0 cm2•V-1•s-1, 高于同等条件下制备和测试的PDVT-10退火薄膜OFETs的空穴迁移率(2.77 cm2•V-1•s-1).随后, 该课题组进一步研究了烷基链分叉位置对硒吩乙烯硒吩桥连DPP基共聚物Cm-DPP-SVS的光物理、能级结构、薄膜聚集态结构以及电荷传输性能的影响[50, 51], 发现当连接碳数m为5时, 对应的聚合物C29-DPP-SVS退火薄膜的π-π堆积距离非常紧密, 仅为3.58 Å, 最高迁移可达17.8 cm2•V-1•s-1. 2015年, Choi等[52]以噻吩并[3, 2-b]噻吩-乙烯-噻吩[3, 2-b]并噻吩(TT-V-TT)为π桥单元, 开发了一类主链平面性更好、高度共轭的DPP类共聚物PTDPP-DTTE.基于新开发的模板诱导溶液剪切成膜技术(Template-Guided Solution-Shearing Method, TGSS), 制备了高度有序的PTDPP-DTTE结晶薄膜, 其最高迁移率可达7.43 cm2•V-1•s-1.此外, 主链分子结构无规的PDVT-10衍生物也被成功开发, 并展现出优异的OFETs性能. 2014年, Kwon等[53]以硒吩-乙烯-硒吩(SVS)、噻吩并[3, 2-b]噻吩-噻吩并[3, 2-b]噻吩(BTT)以及噻吩取代的DPP为共聚物的构建单元, 通过调控SVS和BTT的比例, 设计并合成了系列结构无规的DPP类三元共聚物PDPP-BTT(m)-SVS(n).研究证实:当m/n=1/9时, 对应的三元共聚物PDPP-BTT(1)-SVS(9)的电荷传输性能最优.采用非卤溶剂(二甲苯)成膜制备的OFETs的最高迁移率可达6.51 cm2•V-1•s-1, 为目前报道的空穴迁移率最高的三元共聚物. 2015年, Kwon等[54]以结构不对的噻吩乙烯硒吩(TVS)作为共聚物的π桥单元, 设计并合成了主链结构无规的D-A型噻吩取代DPP类共聚物PDPP-TVS-C29.采用非卤溶剂(四氢呋喃、甲苯、邻二甲苯和四氢萘)溶液旋涂制备了PDPP-TVS-C29薄膜OFETs, 其中以四氢萘为溶剂构造的薄膜OFETs的空穴迁移率最高达8.2 cm2•V-1•s-1, 这一研究结果表明聚合物PDPP-TVS-C29在采用低毒、无卤溶剂体系打印有机光电子器件中具有重大潜力.
4 电子传输型(n-型)聚合物半导体材料最新进展
溶液法加工n-型聚合物半导体是构造逻辑互补电路及其有机光电子器件的重要组成部分[55~57].然而, 相比p-型聚合物半导体, 综合性能优异的n-型聚合物半导体报道相对较少, n-型聚合物半导体被公认为是有机电子学发展的重大挑战.部分高迁移率的n-型聚合物半导体的分子结构如图 7所示.通常, n-型聚合物半导体材料的理想LUMO能级应该小于-4.0 eV, 这样有利于电子从高功函数的金电极(5.1 eV)注入到半导体活性层中[55].但大部分聚合物的LUMO能级与Au电极不匹配, 导致电子注入效率低.尽管可采用低功函数的Ca、Mg和Al金属电极来改善注入势垒, 但这些金属易与H2O/O2反应, 不利于器件的长期使用.为解决器件稳定性差这一难题, 最常用的合成方法就是能级结构调控[55-57]:通过在单体上引入强缺电子的卤原子、氰基以及氟代烷基链等吸电子基团, 可有效地调节其共聚合物的LUMO能级, 然而强极性基团的引入通常会降低聚合物的溶液加工性能, 导致聚合物的成膜性变差.此外, 空气中H2O/O2极易捕获导电沟道中活性层的电子, 导致器件迁移率降低甚至失去活性.此外, 聚合物薄膜中存在大量的缺陷, 这一因素将会严重影响电子的注入和传输.根据不同受体单元分类, 我们将对部分高迁移率的n-型聚合物半导体材料进行介绍, 其结构和性能分别见图 7和表 2.
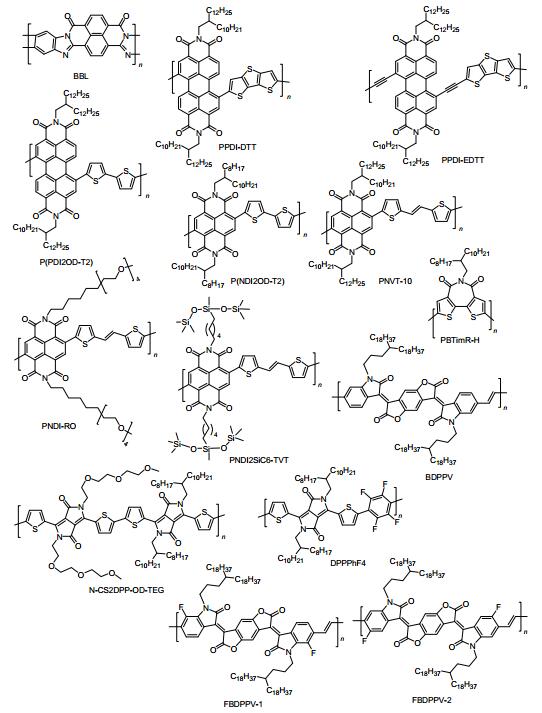 图 7
n-型聚合物半导体的分子结构
Figure 7.
Molecular structures of n-type polymeric semiconductors
表 2
n-型共轭聚合物半导体材料的性能参数
Table 2.
Electronic parameters for n-type polymer semiconductors
图 7
n-型聚合物半导体的分子结构
Figure 7.
Molecular structures of n-type polymeric semiconductors
表 2
n-型共轭聚合物半导体材料的性能参数
Table 2.
Electronic parameters for n-type polymer semiconductors
Polymer Mn/Mw (kg/mol) HOMO/eV LUMO/eV μelectron/(cm2•V-1•s-1) Ion/Ioff Device structure Ref. BBL —/— — -4.0~-4.4 0.10 105 BGBC [58, 59] PPDI-DTT 10.0/15.0 -5.90 -3.90 0.013 >104 BGTC [60] PPDI-EDTT 15.0/17.0 -5.80 -4.0 0.075 106 BGBC [61] P(PDI2OD-T2) 32.0/96.0 -5.61 -3.96 0.002 ~105 BGTC [62] P(NDI2OD-T2) 32.0/172.0 -5.36 -3.91 3.20 >104 TGBC [62, 63, 65] PNVT-10 70.0/138.6 -5.42 -4.0 1.80 106 TGBC [66, 67] PNDI-RO 24.3/70.2 -5.30 -4.01 1.64 ~105 BGTC [68] PNDI2SiC6-TVT 15.0/49.2 -5.36 -3.96 1.04 ~103 TGBC [69] PBTimR-H 7.2/14.3 -6.28 -3.47 0.19 >106 TGBC [70, 71] N-CS2DPP-OD-TEG 67.3/314.0 -5.38 -3.66 3.0 >104 TGBC [72] DPPPhF4 16.3/— -5.65 -4.18 2.36 ~104 BGTC [73] BDPPV 37.6/89.5 -6.12 -4.10 1.10 >105 TGBC [77] FBDPPV-1 66.3/128.9 -6.19 -4.26 1.70 >105 TGBC [78] FBDPPV-2 53.8/164.7 -6.22 -4.30 0.81 >105 TGBC [78] 表 2 n-型共轭聚合物半导体材料的性能参数
Table 2. Electronic parameters for n-type polymer semiconductors2003年, Jenekhe等[58, 59]报道了第一个n-型聚合物半导体BBL.研究发现:该聚合物具有很强的刚性和分子间作用力.采用甲基磺酸溶液旋涂制备的BBL退火薄膜OFET器件的电子迁移率高达0.1 cm2•V-1•s-1, 开关电流比达105.然而BBL难以溶解于普通有机溶剂中, 必须在强酸性溶剂中才能够缓慢溶解, 因而限制了BBL的应用.因此, 在设计聚合物半导体材料的过程中, 我们必须考虑在聚合物骨架上引入柔性助溶基团以改善聚合物半导体材料的溶解性和加工特性.
芳香酰亚胺是一类溶解性可调、电子亲和能力强、共平面性好的n-型聚合物半导体构建单元, 以苝酰亚胺、萘酰亚胺和噻吩酰亚胺为典型代表[4].通过改变酰亚胺N原子上的助溶取代基团, 化学家可方便地调控酰亚胺类共聚物的溶解性、加工性、薄膜形貌以及骨架堆积方式. 2007年, Zhan等[60]首次报道了以并三噻吩(DTT)为电子给体和苝酰亚胺(PDI)为电子受体的n-型共聚物PPDI-DTT.该聚合物表现出良好的溶液加工特性、优良的热稳定性和低的LUMO能级-3.9 eV.以铝作为源/漏电极, 其底栅OFET器件的电子迁移率高达0.013 cm2•V-1•s-1.随后, 该课题组采用Sonogashira偶联法合成了一类以DTT为电子给体和PDI为电子受体的三键桥连PDI基聚合物半导体PPDI-EDTT[61].对比PPDI-DTT研究表明:聚合物PPDI-EDTT主链中的三键单元有效地降低了DTT和PDI单元间的位阻, 提高了PPDI-EDTT的共平面性, 降低了LUMO能级值.在空气中, 基于PPDI-EDTT退火薄膜制备的OFETs的电子迁移率高达0.075 cm2•V-1•s-1.然而, 单键相连的PPDI-DTT退火薄膜OFETs在空气中未能测出任何性能.
2008年, Facchetti等[62]采用Stille偶联法合成了一个以PDI为电子受体和联二噻吩为电子给体的n-型共聚物P(PDI2OD-T2), 其底栅OFET器件的电子迁移率为0.002 cm2•V-1•s-1.同时, 他们采用Stille偶联法合成了一个以NDI为受体和联二噻吩为给体的n-型共聚物P(NDI2OD-T2)[62].在氮气中, 基于P(NDI2OD-T2)退火薄膜制备的底栅OFETs的电子迁移率高达0.06 cm2• V-1•s-1, 高出P(PDI2OD-T2)的电子迁移率30倍.作者分析:二溴苝酰亚胺衍生物存在两种难以分离的异构体, 所以导致所合成的P(PDI2OD-T2)的主链结构无规, 不利于聚合物主链的骨架有序排列; 然而, 二溴萘酰亚胺衍生物容易纯化, 没有异构体, 这样所合成的聚合物P(NDI2OD-T2)的主链结构规整度高, 有利于主链之间的相互堆积, 从而获得更高的电子迁移率. 2010年, Facchetti等[63]研究了不同聚合物绝缘层材料对其P(NDI2OD-T2)退火薄膜顶栅OFETs性能的影响.由于聚合物绝缘层的良好封装效果减小了H2O/O2对导电沟道中活性层电子传输的负面影响, P(NDI2OD-T2)的电子迁移率从0.06 cm2•V-1•s-1进一步提高到0.45~0.85 cm2•V-1•s-1, 同时该类顶栅OFET器件表现出良好的空气稳定性.
相比传统的钯催化Stille偶联法和Suzuki偶联法, 直接芳香化法具有合成步骤简单、节能环保等优势.近年来, 钯催化直接芳香化法被广泛地应用于合成新型有机/聚合物π-共轭材料[64]. 2015年, Sommer等[65]采用直接芳香化法和Stille偶联法对比合成了系列数均分子量相当的NDI类共聚物P(NDI2OD-T2).研究证实:采用直接芳香化法合成的P(NDI2OD-T2)主链结构的缺点密度低, 器件重复性好; 当Mn=15 kDa时, 基于直接芳香化法合成的P(NDI2OD-T2)退火薄膜构造的顶栅OFETs的电子迁移率高达1.58 cm2•V-1•s-1, 高于Stille偶联法合成的P(NDI2OD-T2)的电子迁移率(e=0.82 cm2•V-1• s-1); 当Mn=32 kDa时, 基于直接芳香化法合成的P(NDI2OD-T2)的电子迁移率进一步提高到3.0 cm2• V-1•s-1, 与Stille偶联法合成的P(NDI2OD-T2)的电子迁移率相当(e=3.2 cm2•V-1•s-1), 这一电子迁移率值为目前所报道的n-型聚合物半导体材料的最高值之一.
2013年, 陈华杰等[66]首次将噻吩乙烯噻吩(TVT)单元引入到NDI类共轭聚合物的主链中, 设计并合成了大π共轭型聚合物PNVT-8和PNVT-10.研究结果表明:相比P(NDI2OD-T2), 双键的引入不仅使PNVT-8和PNVT-10获得了有利于空穴和电子注入的HOMO/ LUMO能级, 而且还加强了聚合物分子骨架间的π-π堆积, 获得了多晶谷粒更大、表面形貌更有序的聚合物薄膜.其中, 基于长链烷基取代的PNVT-10薄膜构造的顶栅场效应晶体管器件的最高空穴和电子迁移率分别达到0.32和1.57 cm2•V-1•s-1, 同时该器件还表现出优越的空气稳定性, 放置在空气中30天后, 其空穴和电子迁移率依然分别高于0.1和1.0 cm2•V-1•s-1.基于PNVT-10薄膜, 溶液法加工倒相器电路成功制备, 其增溢值突破155, 充分展示了该类聚合物半导体材料在柔性有机光电子器件中的应用潜力.此外, Kwon等[67]同时报道了D-A型NDI类共聚物PNVT-10.在空气中, 其顶栅退火薄膜OFETs获得优异的双极性电荷传输性能, 其空穴和电子迁移分别0.15和1.4 cm2•V-1•s-1.通过采用碳酸铯修饰金源-漏电极, 基于PNVT-10退火薄膜构造的顶栅OFETs展现出空气稳定的n-型器件性能, 最高电子迁移率高达1.8 cm2•V-1•s-1.随后, Kwon和Yang等对PNVT系列共聚物都进行了侧链修饰, 分别报道了含四聚乙二醇衍生物侧链的PNDI-RO[68]和含硅烷氧基侧链的PNDI2SiC6-TVT[69].通过器件优化, PNDI-RO和PNDI2SiC6-TVT的电子迁移率分别高达1.64和1.04 cm2•V-1•s-1.
二噻吩酰亚胺(BTI)衍生物为另外一类综合性能优异的n-型聚合物半导体材料构建单元. 2008年, Marks等[70]首次报道合成了BTI及其相应的均聚物PBTimR-L.循环伏安测试证实: PBTimR-L薄膜展现出强而可逆的多重还原峰, 其LUMO能级处在-3.47 eV左右.在氮气中, 基于PBTimR-L退火薄膜构造的底栅OFETs的最高电子迁移率可达0.011 cm2•V-1•s-1.后来, 该课题组通过合成优化, 进一步开发了分子量更高的BTI基均聚物PBTimR-H, 其顶栅退火薄膜OFETs的电子迁移率进一步提高到0.19 cm2•V-1•s-1[71].这些研究结果表明, 二噻吩酰亚胺衍生物是一类经典的n-型聚合物半导体材料构建单元.
近年来, 一系列强缺电子单元桥连DPP共聚物被成功开发为高迁移率的n-型聚合物半导体. 2012年, Patil等[72]首次将齐聚乙二醇衍生物链引入到噻吩取代DPP的侧链上, 采用Suzuki偶联法合成了带两种不同烷基链的DPP-DPP基共聚物N-CS2DPP-OD-TEG.在氮气中, 基于N-CS2DPP-OD-TEG退火薄膜构造的底栅OFETs展现出较好的双极性电荷传输性能, 其空穴和电子迁移率均在0.01 cm2•V-1•s-1左右.通过器件优化, 基于N-CS2DPP-OD-TEG退火薄膜构造的顶栅OFETs显示了优异的n-型场效应性能, 其电子迁移率最高可达3.0 cm2•V-1•s-1. 2013年, Jo等[73]以噻吩取代DPP和四氟代苯为构建单元, 设计并合成了DPP基共聚物DPPPhF4.研究发现:多个氟原子的引入显著降低了DPPPhF4的LUMO能级(-4.18 eV), 提高了DPPPhF4的电子亲和能力和分子链间的π-π相互作用.在氮气中, 基于DPPPhF4退火薄膜构造的底栅OFET器件展现出了优异的n-型器件性能, 其最高电子迁移率达2.36 cm2•V-1• s-1.
聚对苯撑乙烯(PPV)及其衍生物是一类研究最为广泛的p-型聚合物半导体材料[74, 75].通过低功函金属(如Ca)和特殊的介电层材料进行器件优化, PPV也能够显示比较低的电子迁移率, 约10-5~10-4 cm2•V-1•s-1水平[76]. 2013年, 裴坚课题组首次报道了一类新型的含有苯并二呋喃二酮的PPV衍生物, 简称BDPPV[77].在空气中, 其顶栅OFET器件展现出优异的n-型电荷传输性能, 其电子迁移率高达1.1 cm2•V-1•s-1, 高出传统PPV聚合物大约4个数量级.值得一提的是, 该器件放置在空气中30天后的电子迁移率依然高达0.31 cm2•V-1•s-1, 为目前所报道的第一个空气稳定的n-型聚合物半导体材料.作者分析: (1)苯并二呋喃二酮中的碳基能够与苯环上的氢形成分子内氢键, 有利于提高聚合物的平面性和分子链间的π-π相互作用; (2)选用4-十八烷基二十二烷基链作为BDPPV的侧链, 一方面解决了BDPPV的溶解性和加工性, 另一面有利于增强分子链间π-π相互作用, 从而提高了载流子迁移率. 2014年, 该课题组引入氟原子修饰BDOPV骨架, 设计并合成了氟原子取代位置不同的两类2FBDOPV基共聚物FBDPPV-1和FBDPPV-2[78].研究表明: (1)氟原子的引入有效地降低了聚合物的LUMO能级, 使得FBDPPV-1和FBDPPV-2获得理想的LUMO能级值; (2)氟原子的引入对有效地锁定了聚合物主链的骨架构象, 增强了聚合物分子链间的π-π相互作用, 提高了薄膜的长程有序性; (3)氟原子取代位置不同, 对聚合物构象锁定效应不同, 最终导致他们的器件性能有所差异.在空气中, 基于FBDPPV-1和FBDPPV-2退火薄膜构造的顶栅OFET器件展现出空气稳定的n-型器件性能, 其最高电子迁移率分别达1.7和0.81 cm2•V-1•s-1.
5 双极性聚合物半导体材料最新进展
双极性聚合物半导体材料在有机柔性显示和逻辑互补电路等领域具有潜在的应用价值, 深受科研人员的关注.传统的双极性逻辑互补电路的构造过程复杂, 需要采用掩模板法将两种不同的p-型和n-型有机/聚合物半导体材料精确沉积在亚微米级衬底上[79].采用单一组分的双极性材料集成在亚微米级衬底上的方法将简化器件制备工艺和降低器件制作的成本[79].双极性聚合物半导体材料的设计除了考虑聚合物给体/受体单元的共平面性和对称性以外, 该类材料的性能还受材料的溶解性、化学稳定性、分子量、能级结构、结晶性以及堆积排列方式等因素的影响.因此, 设计与合成综合性能优异的双极性聚合物半导体材料是一项极富有挑战的课题.
从材料能级工程角度考虑[4]:双极性聚合物半导体材料的理想HOMO能级应处在-5.0~-5.5 eV之间, 这样能与常用金电极的功函数(5.1 eV)匹配良好, 利于空穴的注入; 最理想的LUMO能级应该处于-3.8~-4.3 eV之间, 这样有利于电子的注入.然而, 由于聚合物材料的LUMO能级一般都处在-2.5~-3.5 eV之间, 这样与金电极的功函数相差太大, 导致电子注入势垒太大, 不利于电子的注入和传输.通常会采用低功函数的Ca和Mg电极来改善这类材料的电子注入势垒, 但是这些金属电极在空气中极易氧化变质.常用的合成策略如下:在聚合物主链中引入强缺电子基团, 开发新型的D-A型聚合物, 可以有效地调节聚合物的LUMO能级到理想值附近.为了隔绝水和氧对OFETs导电沟道的影响, 通常的解决方法就是构造顶栅结构的OFETs, 或者是增加一层Al2O3的封装层.
近年来, 基于DPP受体单元, 化学家设计开发了系列高迁移率的双极性聚合物半导体材料.其中, 部分双极性聚合物半导体材料的结构和性能如图 8和表 3所示. 2009年, Janssen等[80]报道了一个含联三噻吩电子给体的双极性DPP基聚合物PDPP3T, 其空穴和电子迁移率分别达0.04和0.01 cm2•V-1•s-1. 2012年, Chen等[81]将PDPP3T中的一个共聚单元噻吩换成硒吩, 合成了PSeDPP.研究发现:硒吩的引入增强了分子间的相互作用和电子亲和力, 使得PSeDPP获得优异的双极性性能.在氮气中, 基于PSeDPP退火薄膜构造的底栅OFET器件的空穴和电子迁移率分别达1.62和0.14 cm2•V-1• s-1. 2012年, Sirringhaus等[82]通过溶剂清洗和O2等离子体处理金电极的方法, 有效地调控了金源/漏电极的能级结构和载流子的注入势垒.研究发现:相比O2等离子体处理的金电极功函数(5.0~5.5 eV), 溶剂清洗的金电极功函数降低至4.7~4.9 eV, 因而有效地降低了电子的注入势垒, 提高了噻吩并[3, 2-b]噻吩桥连DPP基共聚物PDTT的电子注入能力.在氮气中, 基于PDTT退火薄膜构造的顶栅OFETs的空穴和电子迁移率分别为1.36和1.56 cm2•V-1•s-1. 2012年, Yang等[83, 84]报道了一系列带硅氧烷侧链的DPP基共聚物PTDPPSe-SiCm, 系统地研究了硅氧烷侧链分叉位置对PTDPPSe-SiCm系列共聚物的光物理、能级结构、薄膜聚集态结构以及电荷传输性能的影响.研究表明:硅氧烷基侧链增强了聚合物薄膜的自组装能力, 使得PTDPPSe-SiCm退火薄膜采取三维有序的层状模式堆积, 因而有利于高迁移率的载流子传输; 当连接碳数为5时, 聚合物PTDPPSe-SiC5展现出最优的双极性电荷传输性能, 最高空穴和电子迁移率分别达8.84和4.34 cm2•V-1•s-1, 为目前所报道的迁移率最高的双极性聚合物半导体材料.
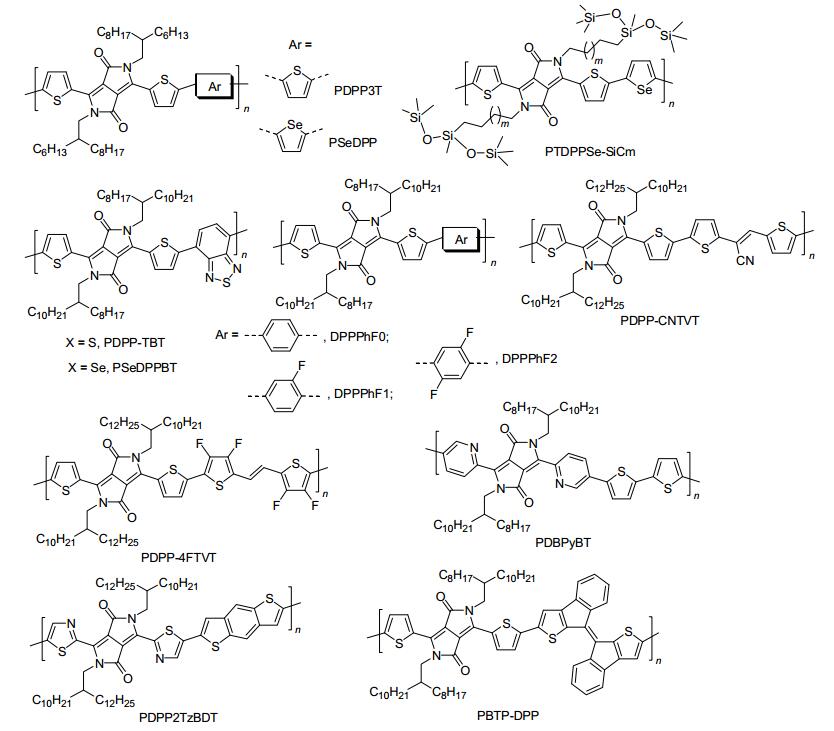 图 8
双极性DPP类聚合物半导体的分子结构
Figure 8.
Molecular structures of ambipolar DPP-based polymeric semiconductors
表 3
双极性共轭聚合物半导体材料的性能参数
Table 3.
Electronic parameters for ambipolar polymer semiconductors
图 8
双极性DPP类聚合物半导体的分子结构
Figure 8.
Molecular structures of ambipolar DPP-based polymeric semiconductors
表 3
双极性共轭聚合物半导体材料的性能参数
Table 3.
Electronic parameters for ambipolar polymer semiconductors
Polymer Mn/Mw (kg/mol) HOMO/eV LUMO/eV μhole/(cm2•V-1•s-1) μelectron/(cm2•V-1•s-1) Device structure Ref. PDPP3T 54.0/170.1 -5.17 -3.61 0.04 0.01 BGBC [80] PSeDPP 96.6/204.8 -5.09 -3.46 1.62 0.14 BGTC [81] PDTT 50.0/193.5 -5.33 -4.07 1.36 1.56 TGBC [82] PTDPPSe-SiC5 23.3/70.4 -5.10 -3.49 8.84 4.34 BGTC [83, 84] PDPP-TBT 42.4/60.2 -5.20 -4.0 0.35 0.40 BGTC [85] PSeDPPBT 30.0/69.0 -5.16 -3.84 0.46 0.84 TGBC [86] PDPP-CNTVT 125.2/189.1 -5.77 -3.92 0.749 7.0 TGBC [87] PDPP-4FTVT 59.9/294.1 -5.36 -3.50 3.40 5.86 TGBC [88] PDBPyBT 26.3/93.6 -5.69 -4.33 6.30 2.78 BGBC [89] PDPP2TzBDT —/— — -4.0 5.47 5.33 BGTC [90] PBTP-DPP 32.8/91.0 -5.48 -3.98 0.68 0.13 BGBC [91] PNIBT 113.0/376.3 -5.10 -3.70 0.04 0.003 BGTC [79] PBBT12DPP 8.8/14.1 -4.55 -3.90 1.17 1.32 BGBC [92] PBBTTT 15.1/29.3 -4.36 -3.80 1.0 0.7 BGBC [93] PFII2T 75.7/195.3 -5.46 -3.96 1.85 0.43 TGBC [94] PCII2Se 58.6/124.8 -5.57 -3.84 1.05 0.72 TGBC [95] IGT-BT 40.0/90.0 -4.80 -3.88 0.16 0.14 TGBC [34] P-BPDTT 16.0/33.3 -5.0 -3.70 1.24 0.82 BGBC [96] PBAI-TBT 41.2/101.7 -4.91 -3.63 1.50 0.41 BGBC [97] BDOPV-TT 20.9/59.7 -5.70 -3.80 1.70 1.37 TGBC [98] PINDFBT 43.1/206.9 -5.65 -3.84 0.51 0.50 BGBC [99] 表 3 双极性共轭聚合物半导体材料的性能参数
Table 3. Electronic parameters for ambipolar polymer semiconductors近年来, 化学家将强吸电子取代基(氰基、卤原子以及杂原子)引入到DPP基共轭聚合物的主链中, 提高了聚合物的电子亲和力, 设计并合成了系列高迁移率的双极性聚合物半导体材料[1~4]. 2010年, Li等[85]将一缺电子的苯并噻二唑单元(BT)引入到噻吩取代DPP基共聚物的主链中, 合成了一类噻吩取代DPP基共聚物PDPP-TBT.在氮气中, 基于PDPP-TBT退火薄膜构造的底栅OFETs的最高空穴和电子迁移率分别高达和0.35和0.40 cm2•V-1•s-1. 2012年, Kronemeijer等[86]开发一类新型的硒吩取代DPP核, 通过与苯并噻二唑受体共聚, 合成了聚合物PSeDPPBT, 其空穴和电子迁移率分别达0.46和0.84 cm2•V-1•s-1.另外, Jo等[73]开发了系列多氟代苯桥连DPP基共聚物DPPPhF0, DPPPhF1和DPPPhF2, 并系统地研究了主链中氟取代数量对电荷传输性能影响.研究发现:不含氟取代的聚合物DPPPhF0和带一个氟取代的DPPPhF1表现出平衡的双极性传输特征, 其空穴和电子迁移率大约为0.30~0.40 cm2•V-1• s-1.基于PDVT-10母体结构, Kwon等[87]在噻吩乙烯噻吩(TVT)骨架上引入强吸电子的氰基基团, 设计并合成了PDVT-10的衍生物PDPP-CNTVT.研究发现:相比PDVT-10, 氰基的引入增强了PDPP-CNTVT的电子亲和能力, 同时降低了HOMO/LUMO能级分别至-5.77和-3.92 eV, 因而有利于空穴和电子的注入和传输.在氮气中, 基于PDPP-CNTVT退火薄膜构造的顶栅OFET器件展现出优异的双极性电荷传输性能, 其空穴和电子迁移率分别高达0.749和7.0 cm2•V-1•s-1. 2015年, 耿延候课题组采用直接芳香化法直接共聚了四氟代噻吩乙烯噻吩(4FTVT)和噻吩取代DPP单体, 设计并合成了四氟代PDVT-10衍生物PDPP-4FTVT[88].相比PDVT-10, PDPP-4FTVT具有更低的HOMO/LUMO能级, 更小的π-π堆积距离(3.53 Å).在氮气中, 基于PDPP-4FTVT退火薄膜构造的底栅OFET器件的空穴和电子迁移率分别高达3.40和5.86 cm2•V-1•s-1, 为目前所报道的基于直接芳香化法合成的聚合物半导体迁移率的最高值. 2014年, Li等[89]开发了一类骨架共平面性优异的吡啶取代DPP缺电子单元, 通过与联二噻吩共聚, 合成了一类高性能的双极性聚合物半导体材料PDBPyBT, 其空穴和电子迁移率分别达2.78和6.30 cm2•V-1•s-1.作者分析:优异的双极性性能归因于理想的能级结构(HOMO/ LUMO=-5.69/-4.33 eV)、良好的骨架共平面性、以及紧密有序的π-π堆积结构. 2015年, 李伟伟等[90]以噻唑取代DPP基共聚物PDPP2TzBDT为底栅OFET器件的活性层材料, 对比研究了PDPP2TzBDT薄膜器件和微米线器件的性能差异.在氮气中, PDPP2TzBDT退火薄膜OFET器件的空穴和电子迁移率仅为0.14和0.11 cm2•V-1•s-1.然而, PDPP2TzBDT微米线OFET器件展现出更加优异的双极性传输性能, 其空穴和电子迁移率分别高达5.47和5.33 cm2•V-1•s-1, 为目前报道的双极性微米线聚合物OFET器件. 2015年, 基于骨架扭曲的二茚并[2, 1-b]噻吩(BTP)电子受体单元, 陈华杰课题组[91]报道了一类结构新颖的BTP桥连DPP基共聚物PBTP-DPP.研究证实:尽管BTP单元的分子骨架扭曲, 但所合成的BTP类聚合物PBTP-DPP依然获得紧密堆积的结晶薄膜, 其π-π堆积距离仅为3.61 Å.同时, 适当的骨架扭曲解决了大π共轭聚合物溶解性和加工性差的难题.在氮气箱中, 聚合物PBTP-DPP获得良好的双极性电荷传输性能, 其空穴和电子迁移率分别达0.68和0.13 cm2•V-1•s-1.
近年来, 结构新颖的杂环和稠环被成地应用于开发高迁移率的双极性聚合物半导体材料[1~4], 以萘酰亚胺、苯并双噻二唑(BBT)、异靛衍生物、Pechmann染料衍生物和苯并二呋喃二酮衍生物(BDOPV)的研究最为广泛, 如图 9所示. 2009年, Jenekhe等[79]报道了一个以烷氧基取代联二噻吩为电子给体, NDI为电子受体的双极性聚合物半导体材料PNIBT.通过在联二噻吩上引入烷氧基团, 成功地升高了PNIBT的HOMO能级(-5.10 eV), 同时维持其LUMO能级在-3.70 eV左右, 这样保证了空穴和电子同时从金电极注入到PNIBT活性层中, 获得双极性传输性能.在氮气中, 基于PNIBT退火薄膜制备的底栅OFET器件的空穴和电子迁移率分别达0.003和0.04 cm2•V-1•s-1.此外, Jenekhe等[79]基于单一PNIBT退火薄膜构造了第一个双极性聚合物倒相器件, 其增益值高达30. 2012年, Wudl等[92]报道了一个以强缺电子的BBT与噻吩取代DPP为共聚物单元的聚合物半导体材料PBBT12DPP.尽管PBBT12DPP的数均分子量只有8800, 但其富杂原子的主链结构和强的D-A分子内相互作用促使了PBBT12DPP分子链间的π-π相互作用和电荷传输.在氮气中, PBBT12DPP退火薄膜OFET器件展现出优良的双极性传输特征, 其空穴和电子迁移率分别达1.17和1.32 cm2•V-1•s-1.随后, Wudl等[93]选用噻吩[3, 2-b]噻吩为电子给体和BBT为电子受体, 开发了一类超低带隙的双极性聚合物PBBTTT, 其LUMO能级为-3.8 eV, 十分有利于电子的注入与传输.此外, 该聚合物退火薄膜展现出有序的层状堆积结构, 其π-π堆积距离仅3.53 Å.在氮气中, PBBTTT退火薄膜OFET器件获得平衡的双极性传输特征, 其空穴和电子迁移率分别达0.7和1.0 cm2•V-1•s-1.
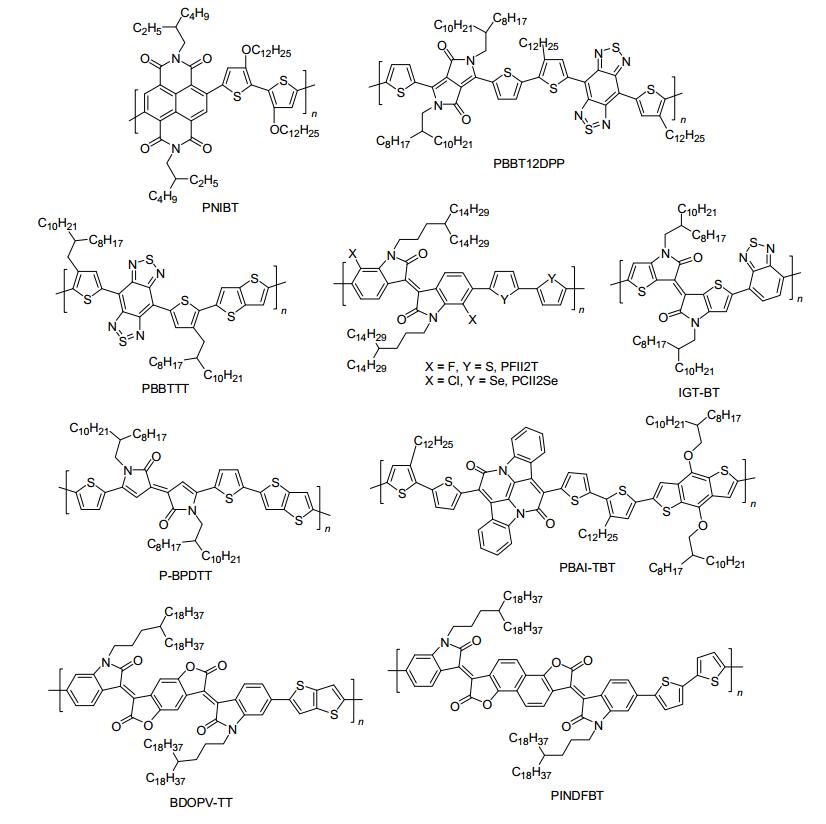 图 9
双极性聚合物半导体的分子结构
Figure 9.
Molecular structures of ambipolar polymeric semiconductors
图 9
双极性聚合物半导体的分子结构
Figure 9.
Molecular structures of ambipolar polymeric semiconductors
异靛及其衍生物为另外一类经典的双极性聚合物半导体构建单元. 2012年, 裴坚课题组开发了一类新型的二氟代异靛衍生物, 通过与联二噻吩给体共聚, 成功地合成了一类氟代异靛基共聚物PFII2T[94].研究发现:主链中氟原子的引入降低了PFII2T的LUMO能级(-3.96 eV), 同时促使PFII2T退火薄膜形成紧密的层状堆积结构, 其π-π堆积距离仅为3.55 Å, 有利于双极性电荷传输.在氮气中, PFII2T退火薄膜OFETs的最高空穴和电子迁移率分别为1.85和0.51 cm2•V-1•s-1.然而, 不带氟取代的异靛基聚合物PII2T的电子迁移率仅为0.07 cm2•V-1•s-1.随后, 该课题组又开发了一类新型的氯代异靛衍生物, 通过与联二硒给体共聚, 成功合成了氯代异靛基共聚物PCII2Se[95], 其最高空穴和电子迁移率分别为1.05和0.72 cm2•V-1•s-1.以上研究结果表明:氯代和氟代是一类简单而且有效的调控手段, 可用来调节聚合物的能级结构、薄膜聚集态结构以及电荷传输性能等.
近年来, 基于异靛单元, 化学家展开了系统的结构修饰, 设计并合成了系列综合性能优异的异靛衍生物单元, 并成功应用于开发聚合物场效应晶体管材料. 2012年, Ashraf等[34]将异靛外部的苯环用噻吩替代, 合成了噻吩取代异靛受体单元(IGT).通过与苯并噻二唑共聚, 开发了一类结构新颖的共轭聚合物IGT-BT.研究发现:主链上噻吩与噻吩相连, 提高了IGT-BT的共平面性, 增加分子链间的相互作用, 其π-π堆积距离仅为3.6 Å.此外, IGT-BT分子内强的D-A效应降低了聚合物的LUMO能级, 获得有利于双极性电荷传输的能级结构.在氮气中, 基于IGT-BT退火薄膜构造的顶栅OFET器件的最高空穴和电子迁移率分别为0.16和0.14 cm2• V-1•s-1. 2013年, 张德清课题组[96]首次将Pechmann染料应用OFETs领域中, 设计并合成一类高迁移率的聚合物半导体材料P-BPDTT, 其HOMO/LUMO能级分别为-5.0和-3.7 eV, 利于空穴和电子的同时注入.在氮气中, 基于P-BPDTT退火薄膜构造的底栅OFET器件获得平衡的双极性性能, 其空穴和电子迁移率分别达1.24和0.82 cm2•V-1•s-1, 这一结果证实: Pechmann染料及其衍生物单元是一类综合性能优异的双极性聚合物半导体构建单元. 2014年, Liu等[97]开发了一类大π共轭的稠合靛蓝衍生物(BAI), 同时报道了一类以BAI为电子受体的D-A型共轭聚合物PBAI-TBT.在氮气中, 基于PBAI-TBT退火薄膜构造的底栅OFET器件的最高空穴和电子迁移率分别为1.5和0.41 cm2•V-1•s-1, 为目前报道的双极性性能最好的靛蓝基聚合物半导体材料. 2015年, 裴坚课题组报道了以苯并二呋喃二酮衍生物(BDOPV)为电子受体, 噻吩并[3, 2-b]噻吩为电子给体的D-A型共聚物BDOPV-TT.该聚合物结晶薄膜采取edge-on的方式紧密堆积排列, 其π-π堆积距离仅为3.45 Å, 利于高迁移率电荷传输.在空气中, 基于BDOPV-TT退火薄膜构造的顶栅OFETs展现出平衡的双极性器件性能, 其空穴和电子迁移率分别高达1.70和1.37 cm2•V-1•s-1[98].随后, 通过拓宽BDOPV的分子骨架结构, Li等[99]设计并合成了一类大π共轭的萘并二呋喃二酮电子受体单元(INDF).相比强缺电子的BDOPV单元, INDF具有相对弱的缺电子能力和更大的π共轭骨架, 利于开发高迁移率的双极性聚合物半导体材料.通过与联二噻吩共聚, Li等开发了一类空穴和电子平衡传输的双极性聚合物半导体材料PINDFBT.在氮气中, 基于PINDFBT退火薄膜构造的底栅OFETs的空穴和电子迁移率分别达0.51和0.50 cm2•V-1•s-1.
6 总结与展望
聚合物半导体材料及其薄膜OFETs作为有机光电子学的一个重要分支, 近年来取得了快速发展.目前, 聚合物场效应晶体管在迁移率和开关比等方面都取得了系列突破, 在射频电子商标、智能卡、传感器、逻辑电路以及电子纸等领域的应用展现出美好的前景.然而, 大部分高性能的聚合物OFET器件都需要在氮气箱中或真空状态下进行测试, 其器件的操作稳定性和空气稳定性通常都较差.要真正走向应用, 聚合物半导体材料及其OFETs必须同时具备以下条件: (1)合成路线简单, 原料广泛易得, 材料易于纯化, 这样才能降低制作成本; (2)具有良好的光、热稳定性, 以及优良的抗氧化能力; (3)具有很好的溶解性和成膜性, 容易采用溶液法加工制备出大面积、高质量的聚合物薄膜; (4)不但具有高的迁移率和开关比, 而且要同时具有良好的空气稳定性和操作稳定性.
综合性能优异的聚合物半导体材料应具备良好的共平面性和对称性、优异的溶解性和化学稳定性、高的分子量和窄的分子量分布、合适的能级结构、高的结晶性以及紧密有序的堆积排列方式等等.因此, 我们在设计聚合物半导体材料时, 可以从几个方面着手: (1)构造共平面性能好的聚合物骨架结构, 以加强分子链间的轨道重叠和π-π堆积, 利于自组装成长程有序性好的聚合物多晶薄膜; (2)开发分子量高和分子量分布窄的聚合物半导体材料, 以提高聚合物薄膜的自组装能力以及器件性能; (3)调控助溶烷基链的种类、长短以及支化点的位置等, 以调控聚合物的溶解性、分子量、聚集态结构以及电荷传输性能; (4)在主链中引入杂原子或杂环, 以开发能级结构理想、结晶度高、长程有序性好、π-π堆积紧密的聚合物半导体材料; (5)构造D-A型交替共轭聚合物骨架, 通过分子链内强的极化作用和分子内电荷转移有效地驱动分子链间的π-π堆积, 提升其薄膜OFET器件的电荷传输能力; (6)采用直接芳香化法合成结构规整度高、主链缺陷密度少的高迁移率聚合物半导体材料.尽管该领域依然存在众多问题, 但聚合物半导体材料的可灵活修饰性赋予我们广阔的设计空间, 挑战与机遇并存.
-
-
[1]
夏昕, 雷霆, 裴坚, 刘晨江, 有机化学, 2014, 34, 1905. doi: 10.6023/cjoc201403052Xia, X.; Lei, T.; Pei, J.; Liu, C. J. Chin. J. Org. Chem. 2014, 34, 1905 (in Chinese). doi: 10.6023/cjoc201403052
-
[2]
万刚, 付宇昂, 郭佳宁, 向中华, 化学学报, 2015, 73, 557. doi: 10.6023/A15020106Wan, G.; Fu, Y. A.; Guo, J. N.; Xiang, Z. H. Acta Chim. Sinica 2015, 73, 557 (in Chinese). doi: 10.6023/A15020106
-
[3]
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208. doi: 10.1021/cr100380z
-
[4]
Guo, X.; Facchetti, A.; Marks, T. J. Chem. Rev. 2014, 114, 8943. doi: 10.1021/cr500225d
-
[5]
Tsumura, A.; Koezuka, H.; Ando, T. Appl. Phys. Lett. 1986, 49, 1210. doi: 10.1063/1.97417
-
[6]
Tang, C. W. Appl. Phys. Lett. 1986, 48, 183. doi: 10.1063/1.96937
-
[7]
Tang, C. W.; Vanslyke, S. A. Appl. Phys. Lett. 1987, 51, 913. doi: 10.1063/1.98799
-
[8]
桑明, 曹四振, 赖文勇, 黄维, 化学学报, 2015, 73, 770.Sang, M.; Cao, S. Z.; Lai, W. Y.; Huang, W. Acta Chim. Sinica 2015, 73, 770 (in Chinese).
-
[9]
Ong, B. S.; Wu, Y.; Liu, P.; Gardner, S. J. Am. Chem. Soc. 2004, 126, 3378. doi: 10.1021/ja039772w
-
[10]
Chabinyc, M. L.; Endicott, F.; Vogt, B. D.; DeLongchamp, D. M.; Lin, E. K.; Wu, Y.; Liu, P.; Ong, B. S. Appl. Phys. Lett. 2006, 88, 113514/1. doi: 10.1063/1.2181206
-
[11]
McCulloch, I.; Heeney, M.; Bailey, C.; Genevicius, K.; MacDonald, I.; Shkunov, M.; Sparrowe, D.; Tierney, S.; Wagner, R.; Zhang, W.; Chabinyc, M. L.; Kline, R. J.; McGehee, M. D.; Toney, M. F. Nat. Mater. 2006, 5, 328. doi: 10.1038/nmat1612
-
[12]
Kim, D. H.; Lee, B. L.; Moon, H.; Kang, H. M.; Jeong, E. J.; Park, J.; Han, K. M.; Lee, S.; Yoo, B. W.; Koo, B. W.; Kim, J. Y.; Lee, W. H.; Cho, K.; Becerril, H. A.; Bao, Z. N. J. Am. Chem. Soc. 2009, 131, 6124. doi: 10.1021/ja8095569
-
[13]
Lim, B.; Baeg, K. J.; Jeong, H. G.; Jo, J.; Kim, H.; Park, J. W.; Noh, Y. Y.; Vak, D.; Park, J. H.; Park, J. W.; Kim, D. Y. Adv. Mater. 2009, 21, 2808. doi: 10.1002/adma.200803700
-
[14]
Kim, J.; Lim, B.; Baeg, K. J.; Noh, Y. Y.; Khim, D.; Jeong, H. G.; Yun, J. M.; Kim, D. Y. Chem. Mater. 2011, 23, 4663. doi: 10.1021/cm2021802
-
[15]
Fei, Z.; Pattanasattayavong, P.; Han, Y.; Schroeder, B.; Yan, F.; Kline, R.; Anthopoulos, T.; Heeney, M. J. Am. Chem. Soc. 2014, 136, 15154. doi: 10.1021/ja508798s
-
[16]
Jang, S.; Kim, I.; Kim, J.; Khim, D.; Jung, E.; Kang, B.; Lim, B.; Kim, Y.; Jang, Y.; Cho, K.; Kim, D. Chem. Mater. 2014, 26, 6907. doi: 10.1021/cm502486n
-
[17]
Zhang, M.; Tsao, H. T.; Pisula, W.; Yang, C.; Mishra, A. K.; Müllen, K. J. Am. Chem. Soc. 2007, 129, 3472. doi: 10.1021/ja0683537
-
[18]
Tsao, H. N.; Cho, D. M.; Park, I.; Hansen, M. R.; Mavrinskiy, A.; Yoon, D. Y.; Graf, R.; Pisula, W.; Spiess H. W.; Müllen, K. J. Am. Chem. Soc. 2011, 133, 2605. doi: 10.1021/ja108861q
-
[19]
Zhang, W.; Smith, J.; Watkins, S. E.; Gysel, R.; McGehee, M.; Salleo, A.; Kirkpatrick, J.; Ashraf, S.; Anthopoulos, T.; Heeney, M.; McCulloch, I. J. Am. Chem. Soc. 2010, 132, 11437. doi: 10.1021/ja1049324
-
[20]
Zhang, X.; Bronstein, H.; Kronemeijer, A. J.; Smith, J.; Kim, Y.; Kline, R. J.; Richter, L. J.; Anthopoulos, T. D.; Sirringhaus, H.; Song, K.; Heeney, M.; Zhang, W.; McCulloch, I.; Delongchamp, D. M. Nat. Commun. 2013, 4, 2238.
-
[21]
Zhang, W.; Han, Y.; Zhu, X.; Fei, Z.; Feng, Y.; Treat, N.; Faber, H.; Stingelin, N.; McCulloch, I.; Anthopoulos, T.; Heeney, M. Adv. Mater. 2015, DOI: 10.1002/adma.201504092.
-
[22]
Ying, L.; Hsu, B. B. Y.; Zhan, H.; Welch, G. C.; Zalar, P.; Perez, L. A.; Kramer, E. J.; Nguyen, T. Q.; Heeger, A. J.; Wong, W. Y.; Bazan, G. C. J. Am. Chem. Soc. 2011, 133, 18538. doi: 10.1021/ja207543g
-
[23]
Tseng, H. R.; Ying, L.; Hsu, B. B.; Perez, L. A.; Takacs, C. J.; Bazan, G. C.; Heeger, A. J. Nano Lett. 2012, 12, 6353. doi: 10.1021/nl303612z
-
[24]
Tseng, H.-R.; Phan, H.; Luo, C.; Wang, M.; Perez, L. A.; Patel, S. N.; Ying, L.; Kramer, E. J.; Nguyen, T.-Q.; Bazan, G. C.; Heeger, A. J. Adv. Mater. 2014, 26, 2993. doi: 10.1002/adma.201305084
-
[25]
Luo, C.; Kyaw, A. K. K.; Perez, L. A.; Patel, S.; Wang, M.; Grimm, B.; Bazan, G. C.; Kramer, E. J.; Heeger, A. J. Nano Lett. 2014, 14, 2764. doi: 10.1021/nl500758w
-
[26]
Fan, J.; Yuen, J. D.; Cui, W. B.; Seifter, J.; Mohebbi, A. R.; Wang, M.; Zhou, H.; Heeger, A. J.; Wudl, F. Adv. Mater. 2012, 24, 6164. doi: 10.1002/adma.v24.46
-
[27]
Nketia-Yawson, B.; Lee, H. S.; Seo, D.; Yoon, Y.; Park, W. T.; Kwak, K.; Son, H. J.; Kim, B. S.; Noh, Y. Y. Adv. Mater. 2015, 27, 3045. doi: 10.1002/adma.201500233
-
[28]
Wang, E.; Mammo, W.; Andersson, M. R. Adv. Mater. 2014, 26, 1801. doi: 10.1002/adma.v26.12
-
[29]
Stalder, R.; Mei, J.; Reynolds, J. R. Macromolecules 2010, 43, 8348. doi: 10.1021/ma1018445
-
[30]
Lei, T.; Cao, Y.; Fan, Y.; Liu, C.; Yuan, S.; Pei, J. J. Am. Chem. Soc. 2011, 133, 6099. doi: 10.1021/ja111066r
-
[31]
Lei, T.; Dou, J.; Pei, J. Adv. Mater. 2012, 24, 6457. doi: 10.1002/adma.v24.48
-
[32]
Mei, J.; Kim, D. H.; Ayzner, A. L.; Toney, M. F.; Bao, Z. J. Am. Chem. Soc. 2011, 133, 20130. doi: 10.1021/ja209328m
-
[33]
Mei, J.; Wu, H.; Diao, Y.; Appleton, A.; Wang, H.; Zhou, Y.; Lee, W. Y.; Kurosawa, T.; Chen, W.; Bao, Z. Adv. Funct. Mater. 2015, 25, 3455. doi: 10.1002/adfm.v25.23
-
[34]
Ashraf, R. S.; Kronemeijer, A. J.; James, D. I.; Sirringhaus, H.; McCulloch, I. Chem. Commun. 2012, 48, 3939. doi: 10.1039/c2cc30169e
-
[35]
Kim, G.; Kang, S. J.; Dutta, G. K.; Han, Y. K.; Shin, T. J.; Noh, Y. Y.; Yang, C. D. J. Am. Chem. Soc. 2014, 136, 9477. doi: 10.1021/ja504537v
-
[36]
Cao, Y.; Yuan, J.; Zhou, X.; Wang, X.; Zhuang, F.; Wang, J.; Pei, J. Chem. Commun. 2015, 51, 10514. doi: 10.1039/C5CC02026C
-
[37]
Shi, S.; Xie, X.; Gao, C.; Shi, K.; Chen, S.; Yu, G.; Guo, L.; Li, X.; Wang, H. Macromolecules 2014, 47, 616. doi: 10.1021/ma402107n
-
[38]
Shi, S.; Shi, K.; Yu, G.; Li, X.; Wang, H. RSC Adv. 2015, 5, 70319. doi: 10.1039/C5RA14721B
-
[39]
Nielsen, C. B.; Turbiez, M.; McCulloch, I. Adv. Mater. 2013, 25, 1859. doi: 10.1002/adma.v25.13
-
[40]
Lee, O. P.; Yiu, A. T.; Beaujuge, P. M.; Woo, C. H.; Holcombe, T. W.; Millstone, J. E.; Douglas, J. D.; Chen, M. S.; Fréchet, J. M. J. Adv. Mater. 2011, 23, 5359. doi: 10.1002/adma.201103177
-
[41]
Bürgi, L.; Turbiez, M.; Pfeiffer, R.; Bienewald, F.; Kirner, H. J.; Winnewisser, C. Adv. Mater. 2008, 20, 2217. doi: 10.1002/(ISSN)1521-4095
-
[42]
Li, Y.; Sonar, P.; Singh, S. P.; Soh, M. S.; Meurs, M.; Tan, J. J. Am. Chem. Soc. 2011, 133, 2198. doi: 10.1021/ja1085996
-
[43]
Ha, J. S.; Kim, K. H.; Choi, D. H. J. Am. Chem. Soc. 2011, 133, 10364. doi: 10.1021/ja203189h
-
[44]
Li, Y.; Singh, S. P.; Sonar, P. Adv. Mater. 2010, 22, 4862. doi: 10.1002/adma.201002313
-
[45]
Li, J.; Zhao, Y.; Tan, H.; Guo, Y.; Di, C.; Yu, G.; Liu, Y.; Lin, M.; Lim, S. H.; Zhou, Y.; Su, H.; Ong, B. S. Sci. Rep. 2012, 2, 754.
-
[46]
Bronstein, H.; Chen, Z.; Ashraf, R. S.; Zhang, W.; Du, J.; Durrant, J. R.; Tuladhar, P. S.; Song, K.; Watkins, S. E.; Geerts, Y.; Wienk, M. M.; Janssen, R. A. J.; Anthopoulos, T.; Sirringhaus, H.; Heeney, M.; McCulloch, I. J. Am. Chem. Soc. 2011, 133, 3272. doi: 10.1021/ja110619k
-
[47]
Chen, H.; Guo, Y.; Yu, G.; Zhao, Y.; Zhang, J.; Gao, D.; Liu, H.; Liu, Y. Adv. Mater. 2012, 24, 4618. doi: 10.1002/adma.v24.34
-
[48]
Liu, X.; Guo, Y.; Ma, Y.; Chen, H.; Mao, Z.; Wang, H.; Yu, G.; Liu, Y. Adv. Mater. 2014, 26, 3631. doi: 10.1002/adma.v26.22
-
[49]
Kang, I.; An, T. K.; Hong, J.; Yun, H. J.; Kim, R.; Chung, D. S.; Park, C. E.; Kim, Y. H.; Kwon, S. K. Adv. Mater. 2013, 25, 524. doi: 10.1002/adma.201202867
-
[50]
Kang, I.; Yun, H. J.; Chung, D. S.; Kwon, S. K; Kim, Y. H. J. Am. Chem. Soc. 2013, 135, 14896. doi: 10.1021/ja405112s
-
[51]
Back, J. Y.; Yu, H.; Song, I.; Kang, I.; Ahn, H.; Shin, T. J.; Kwon, S. K.; Oh, J. H.; Kim, Y. H. Chem. Mater. 2015, 27, 1732. doi: 10.1021/cm504545e
-
[52]
Shin, J.; Hong, T. R.; Lee, T. W.; Kim, A.; Kim, Y. H.; Cho, M. J.; Choi, D. H. Adv. Mater. 2014, 26, 6031. doi: 10.1002/adma.201401179
-
[53]
Yun, H. J.; Lee, G.; Chung, D. S.; Kim, Y. H.; Kwon, S. K. Adv. Mater. 2014, 26, 6612. doi: 10.1002/adma.v26.38
-
[54]
Choi, H. H.; Baek J. Y.; Song, E.; Kang, B.; Cho, K.; Kwon, S. K.; Kim, Y. H. Adv. Mater. 2015, 27, 3626. doi: 10.1002/adma.v27.24
-
[55]
Zaumseil, J.; Sirringhaus, H. Chem. Rev. 2007, 107, 1296. doi: 10.1021/cr0501543
-
[56]
Usta., H.; Facchetti, A.; Marks, T. J. Acc. Chem. Res. 2011, 44, 501. doi: 10.1021/ar200006r
-
[57]
Tang, M.; Bao, Z. Chem. Mater. 2011, 23, 446. doi: 10.1021/cm102182x
-
[58]
Babel, A.; Jenekhe, S. A. J. Am. Chem. Soc. 2003, 125, 13656. doi: 10.1021/ja0371810
-
[59]
Briseno, A. L.; Mannsfeld, S. C. B.; Shamberger, P. J.; Ohuchi, F. S.; Bao, Z.; Jenekhe, S. A.; Xia, Y. Chem. Mater. 2008, 20, 4712. doi: 10.1021/cm8010265
-
[60]
Zhan, X.; Tan, Z.; Domercq, B.; An, Z.; Zhang, X.; Barlow, S.; Li, Y.; Zhu, D.; Kippelen, B.; Marder, S. R. J. Am. Chem. Soc. 2007, 129, 7246. doi: 10.1021/ja071760d
-
[61]
Zhao, X.; Ma, L.; Zhang, L.; Wen, Y.; Chen, J.; Shuai, Z.; Liu, Y.; Zhan, X. Macromolecules 2013, 46, 2152. doi: 10.1021/ma302428x
-
[62]
Chen, Z.; Zheng, Y.; Yan, H.; Facchetti, A. J. Am. Chem. Soc. 2008, 131, 8.
-
[63]
Yan, H.; Chen, Z.; Zheng, Y.; Newman, C.; Quinn, J. R.; Dötz, F.; Kastler, M.; Facchetti, A. Nature 2009, 457, 679. doi: 10.1038/nature07727
-
[64]
Mercier, L. G.; Leclerc. M. Acc. Chem. Res. 2013, 46, 1597. doi: 10.1021/ar3003305
-
[65]
Matsidik, R.; Komber, H.; Luzio, A.; Caironi, M.; Sommer, M. J. Am. Chem. Soc. 2015, 137, 6705. doi: 10.1021/jacs.5b03355
-
[66]
Chen, H.; Guo, Y.; Mao, Z.; Yu, G.; Huang, J.; Zhao, Y.; Liu, Y. Chem. Mater. 2013, 25, 3589. doi: 10.1021/cm401130n
-
[67]
Kim, R.; Amegadze, P.; Kang, I.; Yun, H.; Noh, Y.; Kwon, S.; Kim, Y. Adv. Funct. Mater. 2013, 23, 5719. doi: 10.1002/adfm.v23.46
-
[68]
Kim, R.; Kang, B.; Sin, D.; Choi, H.; Kwon, S.; Kim, Y.; Cho, K. Chem. Commun. 2015, 51, 1524. doi: 10.1039/C4CC08381D
-
[69]
Kim, Y.; Long, D.; Lee, J.; Kim, G.; Shin, T.; Nam, K.; Noh, Y.; Yang, C. Macromolecules 2015, 48, 5179. doi: 10.1021/acs.macromol.5b01012
-
[70]
Letizia, J. A.; Salata, M. R.; Tribout, C. M.; Facchetti, A.; Ratner, M. A.; Marks, T. J. J. Am. Chem. Soc. 2008, 130, 9679. doi: 10.1021/ja710815a
-
[71]
Guo, X.; Ortiz, R. P.; Zheng, Y.; Hu, Y.; Noh, Y. Y.; Baeg, K. J.; Facchetti, A.; Marks, T. J. J. Am. Chem. Soc. 2011, 133, 1405. doi: 10.1021/ja107678m
-
[72]
Kanimozhi, C.; Yaacobi-Gross, N.; Chou, K.; Amassian, A.; Anthopoulos, T. D.; Pathil, S. J. Am. Chem. Soc. 2012, 134, 16532. doi: 10.1021/ja308211n
-
[73]
Park, J. H.; Jung, E. H.; Jung, W. J.; Jo, W. H. Adv. Mater. 2013, 25, 2583. doi: 10.1002/adma.201205320
-
[74]
Blom, P. W. M.; Jong, M. J. M.; Munster, M. G. Phys. Rev. B 1997, 55, 655.
-
[75]
Meijer, E. J.; De, L. D. M.; Setayesh, S.; Veenendaal, E. V.; Huisman, B. H.; Blom, P. W. M.; Hummelen, J. C.; Scherf, U.; Klapwijk, T. M. Nat. Mater. 2003, 2, 678. doi: 10.1038/nmat978
-
[76]
Chua, L.-L.; Zaumseil, J.; Chang, J.-F.; Ou, E. C. W.; Ho, P. K. H.; Sirringhaus, H.; Friend, R. H. Nature 2005, 434, 194. doi: 10.1038/nature03376
-
[77]
Lei, T.; Dou, J.; Cao, X.; Wang, J.; Pei, J. J. Am. Chem. Soc. 2013, 135, 12168. doi: 10.1021/ja403624a
-
[78]
Lei, T.; Xia, X.; Wang, J.; Liu, C.; Pei, J. J. Am. Chem. Soc. 2014, 136, 2135. doi: 10.1021/ja412533d
-
[79]
Kim, F. S.; Guo, X.; Watson, M. D.; Jenekhe, S. A. Adv. Mater. 2010, 22, 478. doi: 10.1002/adma.v22:4
-
[80]
Bijleveld, J. C.; Zoombelt, A. P.; Mathijssen, S. G. J.; Wienk, M. M.; Turbiez, M.; de Leeuw, D. M.; Janssen, R. A. J. J. Am. Chem. Soc. 2009, 131, 16616. doi: 10.1021/ja907506r
-
[81]
Lin, H. W.; Lee, W. Y.; Chen, W. J. Mater. Chem. 2012, 22, 2120. doi: 10.1039/C1JM14640H
-
[82]
Chen, Z.; Lee, M. J.; Ashraf, R. S.; Gu, Y.; Albert-Seifried, S.; Nielsen, M. M.; Schroeder, B.; Anthopoulos, T. D.; Heeney, M.; McCulloch, I.; Sirringhaus, H. Adv. Mater. 2012, 24, 647. doi: 10.1002/adma.201102786
-
[83]
Lee, J.; Han, A. R.; Kim, J.; Kim, Y.; Oh, J. H.; Yang, C. J. Am. Chem. Soc. 2012, 134, 20713. doi: 10.1021/ja308927g
-
[84]
Lee, J.; Han, A.; Yu, H.; Shin, T.; Yang, C.; Oh, J. J. Am. Chem. Soc. 2013, 135, 9540. doi: 10.1021/ja403949g
-
[85]
Sonar, P.; Singh, S. P.; Li, Y. N.; Soh, M. S.; Dodabalapur, A. Adv. Mater. 2010, 22, 5409. doi: 10.1002/adma.201002973
-
[86]
Kronemeijer, A. J.; Gili, E.; Shahid, M.; Rivnay, J.; Salleo, A.; Heeney, M.; Sirringhaus, H. Adv. Mater. 2012, 24, 1558. doi: 10.1002/adma.201104522
-
[87]
Yun, H.; Kang, S.; Xu, Y.; Kim, S.; Kim, Y.; Noh, Y.; Kwon, S. Adv. Mater. 2014, 26, 7300. doi: 10.1002/adma.v26.43
-
[88]
Gao, Y.; Zhang, X.; Tian H.; Zhang, J.; Yan, D.; Geng, Y.; Wang, F. Adv. Mater. 2015, 27, 6753. doi: 10.1002/adma.201502896
-
[89]
Sun, B.; Hong, W.; Yan, Z.; Aziz, H.; Li, Y. Adv. Mater. 2014, 26, 2636. doi: 10.1002/adma.v26.17
-
[90]
Xiao, C.; Zhao, C.; Zhang, A.; Jiang, W.; Janssen, R.; Li, W.; Hu, W.; Wang, Z. Adv. Mater. 2015, 27, 4963. doi: 10.1002/adma.201502617
-
[91]
Li, C.; Mao, Z.; Chen, H.; Zheng, L.; Huang, J.; Zhao, B.; Tan, S.; Yu, G. Macromolecules 2015, 48, 2444. doi: 10.1021/acs.macromol.5b00067
-
[92]
Yuen, J. D.; Fan, J.; Seifter, J.; Lim, B.; Hufschmid, R.; Heeger, A. J.; Wudl, F. J. Am. Chem. Soc. 2011, 133, 20799. doi: 10.1021/ja205566w
-
[93]
Fan, J.; Yuen, J. D.; Wang, M. F.; Seifter, J.; Seo, J. H.; Mohebbi, A. R.; Zakhidov, D.; Heeger, A. J.; Wudl, F. Adv. Mater. 2012, 24, 2186. doi: 10.1002/adma.201103836
-
[94]
Lei, T.; Dou, J.; Ma, Z.; Yao, C.; Liu, C.; Wang, J.; Pei, J. J. Am. Chem. Soc. 2012, 134, 20025. doi: 10.1021/ja310283f
-
[95]
Lei, T.; Dou, J.; Ma, Z.; Liu, C.; Wang, J.; Pei, J. Chem. Sci. 2013, 4, 2447. doi: 10.1039/c3sc50245g
-
[96]
Cai, Z.; Luo, H.; Qi, P.; Wang, J.; Zhang, G.; Liu, Z.; Zhang, D. Macromolecules 2014, 47, 2899. doi: 10.1021/ma5003694
-
[97]
He, B.; Pun, A.; Zherebetskyy, D.; Liu, Y.; Liu, F.; Klivansky, L.; McGough, A.; Zhang, B.; Lo, K.; Russell, T.; Wang, L.; Liu, Y. J. Am. Chem. Soc. 2014, 136, 15093. doi: 10.1021/ja508807m
-
[98]
Zhou, X.; Ai, N.; Guo, Z.; Zhuang, F.; Jiang, Y.; Wang, J.; Pei, J. Chem. Mater. 2015, 27, 1815. doi: 10.1021/acs.chemmater.5b00018
-
[99]
Deng, Y.; Sun, B.; He, Z.; Quinn, J.; Guo, C.; Li, Y. Chem. Commun. 2015, 51, 13515. doi: 10.1039/C5CC03917G
-
[1]
-

图 2 有机场效应晶体管的典型输出曲线(a)和转移曲线(b)
Figure 2 Typical output curve (a) and transfer curve (b) of OFETs

图 3 p-型聚噻吩半导体的分子结构
Figure 3 Molecular structures of p-type polythoiphene semiconductors

图 4 p-型聚苯并噻二唑衍生物的分子结构
Figure 4 Molecular structures of p-type polymeric semiconductors containing benzothiadiazole derivatives

图 5 p-型聚异靛衍生物的分子结构
Figure 5 Molecular structures of p-type polymeric semiconductors containing isoindigo derivatives

图 6 p-型DPP类聚合物半导体的分子结构
Figure 6 Molecular structures of p-type polymeric semiconductors containing DPP derivatives

图 8 双极性DPP类聚合物半导体的分子结构
Figure 8 Molecular structures of ambipolar DPP-based polymeric semiconductors

图 9 双极性聚合物半导体的分子结构
Figure 9 Molecular structures of ambipolar polymeric semiconductors
表 1 p-型共轭聚合物半导体材料的性能参数
Table 1. Electronic parameters for p-type polymer semiconductors
Polymer Mn/Mw/(kg•mol-1) HOMO/eV LUMO/eV μhole/(cm2•V-1•s-1) Ion/Ioff Device structure Ref. PQT-12 17.3/22.9 — — 0.14 >107 BGTC [9, 10] PBTTT-14 33.0/59.6 -5.10 — 0.72 >106 BGBC [11] PT2Tz-C12 —/— -5.19 — 0.33 >106 BGTC [12] PETV12T 15.0/25.5 -5.20 -3.35 0.15 >104 TGBC [13] PC12TV12T 24.9/— -5.02 — 1.05 >104 TGBC [14] PTVT-TT 20/58 -4.97 — 4.60 104~105 TGBC [15] PTVT-DTT 23.0/52.9 -4.80 -3.20 3.91 >103 TGBC [16] PCDT-BT 35.0/— — — 3.30 105~106 BGBC [17, 18] IDT-BT 80/160 -5.40 — 3.60 >106 TGBC [19, 20] IDTT-BT 76.0/197.6 -5.40 -3.70 8.70 >104 TGBC [21] PCDTPT 140.0/— — — 36.3 >104 BGBC [22~25] PBBTQT 65.9/132.4 -4.60 -3.80 2.50 >103 BGBC [26] PDFDT 14.0/33.6 -5.24 -3.37 9.05 >103 TGBC [27] IIDDT 87.9/185.8 -5.70 -3.70 0.79 ~107 BGTC [30] IIDDT-C3 39.2/125.4 -5.52 -5.74 3.62 >106 BGTC [31] PII2T-Si 138/454 -5.20 -3.58 2.48 >106 BGTC [32] PIIF-C9Si 48.3/101.4 -5.25 — 4.80 ~106 BGTC [33] PTIIG-Np 21.0/102.2 -5.12 -3.49 14.4 >103 TGBC [35] PITET 37.6/89.5 -5.32 -4.05 1.92 >104 BGTC [36] PDQT 25.4/60.6 -5.2 -4.0 0.97 ~107 BGTC [42] P(DPPT-alt-DTSe) 19.3/62.8 — — 1.50 ~106 BGTC [43] PDTT 110/501 -5.2 -3.4 10.5 >106 BGBC [44, 45] PTTDPP-T 14/75 -5.06 -3.68 ≈2.0 ~105 TGBC [46] PDVT-8 70/180 -5.30 — 4.5 >105 BGBC [47] PDVT-10 73.5/183 -5.28 — 11.0 >104 BGBC [47, 48] PDPPDTSE 63.4/114.1 -5.37 — ≈5.0 >107 BGTC [49] C29-DPP-SVS 34.1/58.3 -5.14 — 17.8 >105 BGTC [50, 51] PTDPP-DTTE 127.85/731.3 -5.16 — 7.43 >105 TGSS-BGTC [52] PDPP-BTT(1)-SVS(9) 66.5/100.1 -5.49 -3.82 6.51 >104 BGTC [53] PDPP-TVS-C29 74.5/100.6 — — 8.2 >105 BGTC [54]  下载: 导出CSV
下载: 导出CSV
表 2 n-型共轭聚合物半导体材料的性能参数
Table 2. Electronic parameters for n-type polymer semiconductors
Polymer Mn/Mw (kg/mol) HOMO/eV LUMO/eV μelectron/(cm2•V-1•s-1) Ion/Ioff Device structure Ref. BBL —/— — -4.0~-4.4 0.10 105 BGBC [58, 59] PPDI-DTT 10.0/15.0 -5.90 -3.90 0.013 >104 BGTC [60] PPDI-EDTT 15.0/17.0 -5.80 -4.0 0.075 106 BGBC [61] P(PDI2OD-T2) 32.0/96.0 -5.61 -3.96 0.002 ~105 BGTC [62] P(NDI2OD-T2) 32.0/172.0 -5.36 -3.91 3.20 >104 TGBC [62, 63, 65] PNVT-10 70.0/138.6 -5.42 -4.0 1.80 106 TGBC [66, 67] PNDI-RO 24.3/70.2 -5.30 -4.01 1.64 ~105 BGTC [68] PNDI2SiC6-TVT 15.0/49.2 -5.36 -3.96 1.04 ~103 TGBC [69] PBTimR-H 7.2/14.3 -6.28 -3.47 0.19 >106 TGBC [70, 71] N-CS2DPP-OD-TEG 67.3/314.0 -5.38 -3.66 3.0 >104 TGBC [72] DPPPhF4 16.3/— -5.65 -4.18 2.36 ~104 BGTC [73] BDPPV 37.6/89.5 -6.12 -4.10 1.10 >105 TGBC [77] FBDPPV-1 66.3/128.9 -6.19 -4.26 1.70 >105 TGBC [78] FBDPPV-2 53.8/164.7 -6.22 -4.30 0.81 >105 TGBC [78]
下载: 导出CSV
表 3 双极性共轭聚合物半导体材料的性能参数
Table 3. Electronic parameters for ambipolar polymer semiconductors
Polymer Mn/Mw (kg/mol) HOMO/eV LUMO/eV μhole/(cm2•V-1•s-1) μelectron/(cm2•V-1•s-1) Device structure Ref. PDPP3T 54.0/170.1 -5.17 -3.61 0.04 0.01 BGBC [80] PSeDPP 96.6/204.8 -5.09 -3.46 1.62 0.14 BGTC [81] PDTT 50.0/193.5 -5.33 -4.07 1.36 1.56 TGBC [82] PTDPPSe-SiC5 23.3/70.4 -5.10 -3.49 8.84 4.34 BGTC [83, 84] PDPP-TBT 42.4/60.2 -5.20 -4.0 0.35 0.40 BGTC [85] PSeDPPBT 30.0/69.0 -5.16 -3.84 0.46 0.84 TGBC [86] PDPP-CNTVT 125.2/189.1 -5.77 -3.92 0.749 7.0 TGBC [87] PDPP-4FTVT 59.9/294.1 -5.36 -3.50 3.40 5.86 TGBC [88] PDBPyBT 26.3/93.6 -5.69 -4.33 6.30 2.78 BGBC [89] PDPP2TzBDT —/— — -4.0 5.47 5.33 BGTC [90] PBTP-DPP 32.8/91.0 -5.48 -3.98 0.68 0.13 BGBC [91] PNIBT 113.0/376.3 -5.10 -3.70 0.04 0.003 BGTC [79] PBBT12DPP 8.8/14.1 -4.55 -3.90 1.17 1.32 BGBC [92] PBBTTT 15.1/29.3 -4.36 -3.80 1.0 0.7 BGBC [93] PFII2T 75.7/195.3 -5.46 -3.96 1.85 0.43 TGBC [94] PCII2Se 58.6/124.8 -5.57 -3.84 1.05 0.72 TGBC [95] IGT-BT 40.0/90.0 -4.80 -3.88 0.16 0.14 TGBC [34] P-BPDTT 16.0/33.3 -5.0 -3.70 1.24 0.82 BGBC [96] PBAI-TBT 41.2/101.7 -4.91 -3.63 1.50 0.41 BGBC [97] BDOPV-TT 20.9/59.7 -5.70 -3.80 1.70 1.37 TGBC [98] PINDFBT 43.1/206.9 -5.65 -3.84 0.51 0.50 BGBC [99]
下载: 导出CSV
-










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 7842
- HTML全文浏览量: 1885
 下载:
下载:
