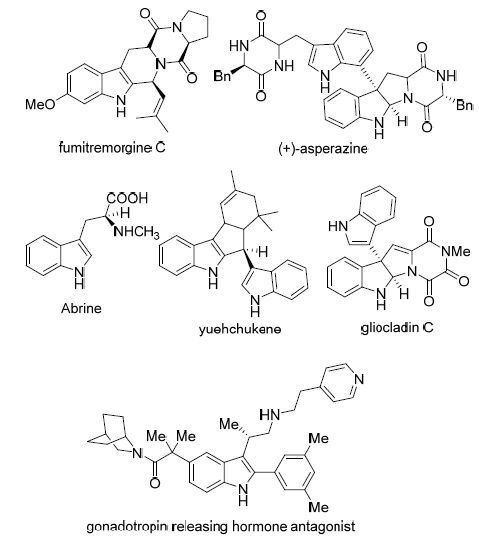
 图 1
含有吲哚的天然产物和药物分子
Figure 1.
Drugs and natural products containing indole
图 1
含有吲哚的天然产物和药物分子
Figure 1.
Drugs and natural products containing indole
引用本文:
朱帅, 徐鲁斌, 王亮, 肖建. 基于吲哚-3-基甲醇不对称合成光学活性吲哚衍生物研究进展[J]. 有机化学,
2016, 36(6): 1229-1240.
doi:
10.6023/cjoc201510024

Citation: Zhu Shuai, Xu Lubin, Wang Liang, Xiao Jian. Recent Advances in Asymmetric Synthesis of Optically Active Indole Derivatives from 3-Indolylmethanols[J]. Chinese Journal of Organic Chemistry, 2016, 36(6): 1229-1240. doi: 10.6023/cjoc201510024
Citation: Zhu Shuai, Xu Lubin, Wang Liang, Xiao Jian. Recent Advances in Asymmetric Synthesis of Optically Active Indole Derivatives from 3-Indolylmethanols[J]. Chinese Journal of Organic Chemistry, 2016, 36(6): 1229-1240. doi: 10.6023/cjoc201510024
基于吲哚-3-基甲醇不对称合成光学活性吲哚衍生物研究进展
English
Recent Advances in Asymmetric Synthesis of Optically Active Indole Derivatives from 3-Indolylmethanols
Abstract:
The electrophilic intermediate, vinylogous imine or vinylogous iminium, can be in situ generated from 3-indolyl- methanols under acidic conditions. With the aid of chiral catalysts, miscellaneous nucleophiles can attack these electrophilic intermediates to afford enantioenriched and biologically important 3-substituted indole derivatives. The recent advances of preparation of optically active indole derivatives from 3-indolylmethanols via asymmetric alkylation, asymmetric reduction and asymmetric rearrangement are summarized.
-
Key words:
- 3-indolylmethanol
- / optically active
- / indole derivatives
- / asymmetric synthesis
-
在过去的一个半世纪里,吲哚及其衍生物因在材料科学[1]、农用化学品[2]以及药物化学[3]中的广泛应用得到了科学家们的持续关注和深入研究. 尤其在药物化学领域,许多含有吲哚骨架的天然产物和药物分子具有重要的生理活性,在人类的生产生活中扮演着重要角色(图 1)[4]. 例如,Asperazine对人类预防白血病有重要作用[5]; Fumitremorgin C (FTC)是一种特殊的化学增敏剂[6]; 从九里香(Murruya paniculuru)中分离出来的yuehchukenel有很强的免疫抑制活性[7]; 相思子碱(Abrine)能够降低葡萄球菌毒素所致的肩部炎症反应[8]; gliocladin C对小鼠P388淋巴细胞性白血病细胞有细胞毒性[9]; GnRH及其类似物通过影响下丘脑-垂体-性腺轴,对子宫内膜异位症、子宫肌瘤、前列腺癌和两性性早熟等病症有很好的治疗效果[10]等. 因此,高效、立体选择性地合成光学活性吲哚类化合物已成为化学家们追求的目标[11]. 其中,因其结构的多样性以及所具有的独特生物活性和药理活性使光学活性3-取代吲哚衍生物的合成成为该领域的研究热点[12~16].
图 1
含有吲哚的天然产物和药物分子
Figure 1.
Drugs and natural products containing indole
吲哚是一种富电子芳香杂环化合物,其C(3)位置具有很强的亲核性,通常化学家们用吲哚与亲电试剂的傅克烷基化反应合成 3取代吲哚类化合物[17]. 对于亲核试剂,则可以通过与类乙烯基亚胺或类乙烯基亚胺离子(vinylogous imine或vinylogous iminium)中间体反应合成3-取代吲哚衍生物. 这些中间体可以通过消除3-取代吲哚苄位的离去基团原位生成[18],然后被亲核试剂捕获得到3-取代吲哚衍生物(Scheme 1). 这类反应不但与傅克烷基化反应互为补充,而且适用范围更加广泛. 目前已经报道的离去基团有砜基、磺酰基、卤素、羟基 等[19]. 与其他离去基团相比,吲哚-3-基甲醇有显著的优点: (1)底物容易制备: 吲哚-3-基甲醇可以通过简单的傅克烷基化反应来制备,而其它底物则需要以吲哚-3-基甲醇为原料进一步衍生才能得到; (2)反应环境友好: 反应唯一的副产物为水,而用其它底物时会生成磺酰胺、亚磺酸盐等对环境有污染的副产物; (3)羟基离去条件温和: 在酸性条件下羟基很容易离去生成亲电性离子中间体; (4)不对称反应容易控制: 手性酸碱都可以作为催化剂控制反应的立体选择性. 基于上述优点,吲哚-3-基甲醇在不对称合成光学活性3-取代吲哚衍生物得到了广泛的应用.
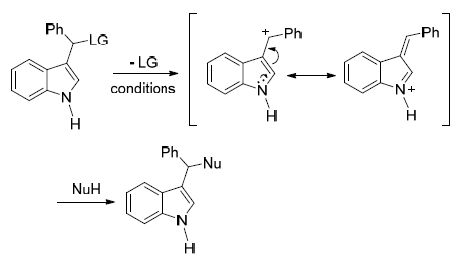 图 图式1
通过捕捉类乙烯基亚胺离子合成3-取代吲哚衍生物
Figure 图式1.
Synthesis of 3-substituted indole derivatives via capture of vinylogous iminium intermediates
图 图式1
通过捕捉类乙烯基亚胺离子合成3-取代吲哚衍生物
Figure 图式1.
Synthesis of 3-substituted indole derivatives via capture of vinylogous iminium intermediates
在吲哚-3-基甲醇参与的反应中,由吲哚与醛生成的吲哚-3-基甲醇A和与靛红生成的醇B应用最为广泛(Scheme 2). 在酸性条件下,吲哚-3-基甲醇A或B质子化后脱去水得到各自对应的类乙烯基亚胺离子,然后与亲核试剂反应得到3-取代吲哚衍生物C或D. 吲哚-3-基甲醇B生成的类乙烯基亚胺离子中间体由于受邻位羰基拉电子效应的影响活性更高,具有更强的亲电性.
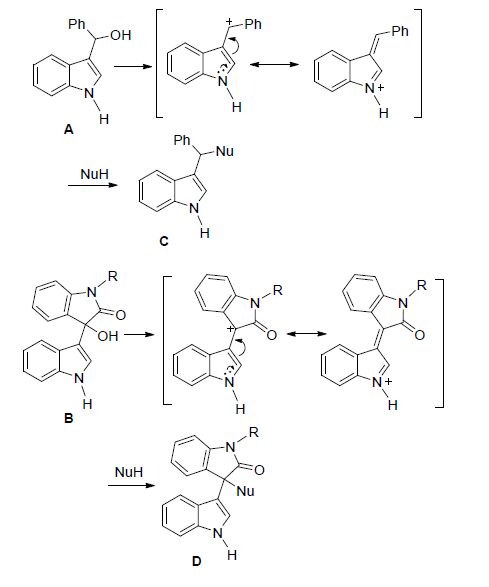 图 图式2
两种常用吲哚-3-基甲醇的比较
Figure 图式2.
Comparison of two types of 3-indolylmethanols which are commonly used
图 图式2
两种常用吲哚-3-基甲醇的比较
Figure 图式2.
Comparison of two types of 3-indolylmethanols which are commonly used
吲哚-3-基甲醇与亲核试剂的不对称反应可以通过两种策略来实现: 一种是通过手性抗衡阴离子导向的不对称催化(ACDC)策略,这种策略目前主要通过手性磷酸来实现. 手性磷酸催化吲哚-3-基甲醇质子化后脱水得到类乙烯基亚胺离子中间体,继而与其形成紧密手性离子对控制反应的立体选择性(图 2,1~5). 另一种策略是利用手性催化剂与亲核试剂反应原位生成手性亲核中间体实现反应的立体选择性控制(图 2,6~12). 本文全面总结了近几年发展起来的由这两种催化策略实现的吲哚-3-基甲醇的不对称烷基化反应、还原反应以及重排反应.
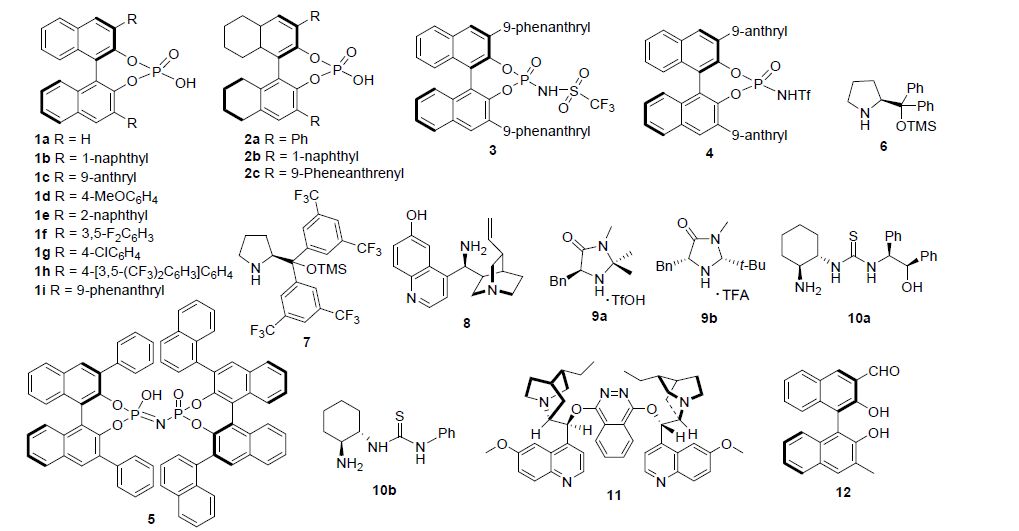 图 2
用于本文相关反应的手性催化剂
Figure 2.
Chiral catalysts used in this paper
图 2
用于本文相关反应的手性催化剂
Figure 2.
Chiral catalysts used in this paper
1 不对称烷基化反应
1.1 羰基化合物的不对称烷基化反应
1.2 不对称傅克烷基化反应
1.1.2 酮或氧化吲哚作为亲核试剂
2012年,郭其祥课题组[35]使用手性磷酸1d作为催化剂实现了吲哚-3-基甲醇31与环己酮39的不对称α烷基化反应,以高产率、优异的立体选择性得到目标产物40 (Scheme 11). 由于吲哚-3-基甲醇31中羰基的存在,类乙烯基亚胺(离子)中间体I的亲电性大大增强,因此反应活性相对较低的酮也可以很好地发生烷基化. 在这个反应中,手性磷酸作为双功能催化剂与两个底物之间形成双重氢键是手性控制的关键因素. 当吲哚中N原子被苄基保护时,反应的效率和立体选择性大大降低. 其原因可能是由于苄基存在阻止了手性磷酸与吲哚形成氢键.
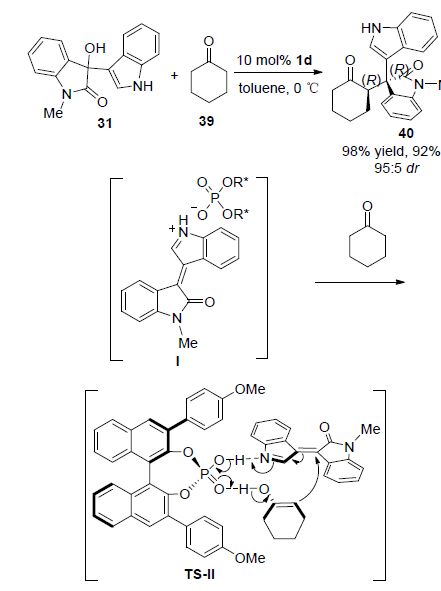 图 图式11
手性磷酸催化的酮的不对称α烷基化反应
Figure 图式11.
Chiral phosphoric acid catalyzed asymmetric α- alkylation of ketones
图 图式11
手性磷酸催化的酮的不对称α烷基化反应
Figure 图式11.
Chiral phosphoric acid catalyzed asymmetric α- alkylation of ketones
刘利课题组[36]利用手性双金鸡纳碱11和三氟甲磺酸铜的协同催化体系实现了吲哚-3-基甲醇18和N-Boc-3-苄基-2-氧化吲哚41的不对称烷基化反应(Eq. 2),以高收率、优异的立体选择性得到了手性3,3’-二取代的氧化吲哚衍生物42.
1.1.1 烯胺作为亲核试剂
2009年,Cozzi课题组[20]报道了2-甲基吲哚-3-基甲醇14与醛13的不对称α烷基化反应. 在室温条件下,使用MacMillan催化剂9b,以理想的产率和较好的立体选择性得到目标产物15 (Scheme 3). 该反应在低温时可得到更高的ee值,但产率相对较低. 其反应机理为: 在酸催化下,手性二级胺与醛缩合形成手性烯胺I后进攻碳正离子中间体Ⅱ得到最终产物. 2013年纪 顺俊、汪顺义课题组[21]也报道了类似的反应,他们使用吲哚-3-基甲醇16为底物,在MacMillan催化剂9a催化下实现了醛的不对称α烷基化反应,得到目标产物17 (Scheme 3),产率和ee值均很高. 值得一提的是,该反应使用了乙腈和水的混合溶剂.
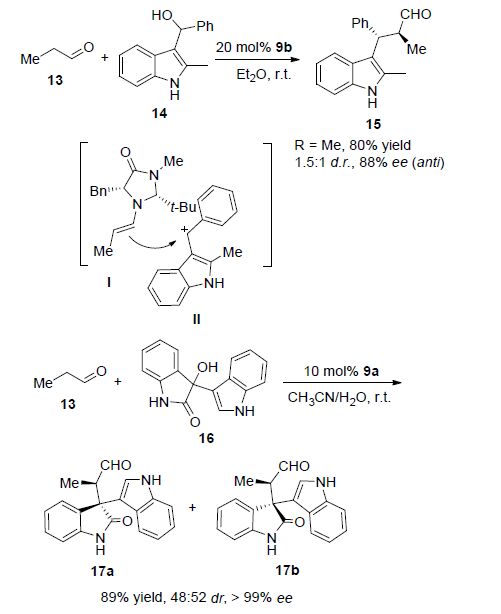 图 图式3
MacMillan催化剂催化的醛的α位不对称烷基化反应
Figure 图式3.
Asymmetric alkylation of aldehydes catalyzed by MacMillan catalyst
图 图式3
MacMillan催化剂催化的醛的α位不对称烷基化反应
Figure 图式3.
Asymmetric alkylation of aldehydes catalyzed by MacMillan catalyst
Loh课题组[22]使用Jørgensen催化剂7和TfOH的协同催化体系,实现了吲哚-3-基甲醇18与醛13的不对称α烷基化反应,以较好的产率和极高的立体选择性(99% ee)得到目标产物19 (Eq. 1). TfOH在反应中有两个作用: 一是促进手性烯胺的生成,二是促使吲哚-3-基甲醇18脱水形成类乙烯基亚胺(离子)中间体.
鉴于吲哚-3-基甲醇的脱水也可以在路易斯酸催化下实现[23],我们课题组在Jørgensen催化剂7和InBr3的协同催化下实现了吲哚-3-基甲醇20与醛13的不对称α烷基化反应,立体选择性很高(Scheme 4)[24a]. 进一步研究发现,该反应还可以在三氟乙醇中实现无酸条件下的不对称α烷基化,在这里三氟乙醇起到了酸活化的作 用[24b].
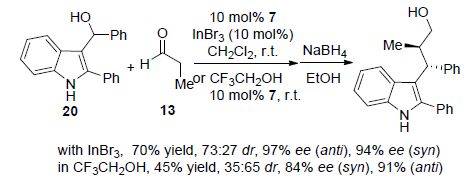 图 图式4
由协同催化体系催化的醛的不对称α烷基化反应
Figure 图式4.
Asymmetric α-alkylation of aldehydes catalyzed by cooperative systems
图 图式4
由协同催化体系催化的醛的不对称α烷基化反应
Figure 图式4.
Asymmetric α-alkylation of aldehydes catalyzed by cooperative systems
2010年,陈应春课题组[25]通过串联不对称烷基化/分子内亚胺基Ene反应制备了环戊并二氢吲哚22 (Scheme 5). 该反应的关键步骤是不饱和醛21与Jørgensen催化剂6反应得到手性β,γ-二烯中间体Ⅱ,然后与类乙烯基亚胺(离子)中间体I作用.
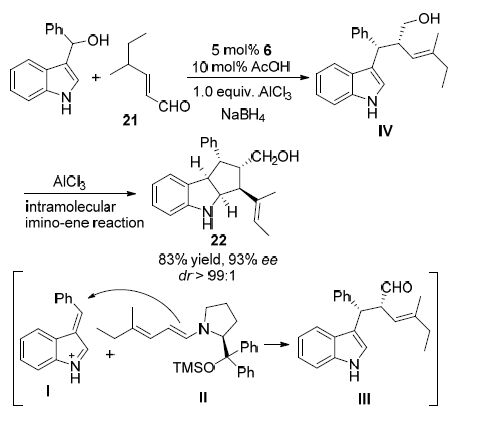 图 图式5
吲哚-3-基甲醇的串联不对称烷基化/分子内亚氨基- Ene反应
Figure 图式5.
Cascade asymmetric alkylation of aldehydes/intra- molecular imino-ene reaction of 3-indolylmethanol
图 图式5
吲哚-3-基甲醇的串联不对称烷基化/分子内亚氨基- Ene反应
Figure 图式5.
Cascade asymmetric alkylation of aldehydes/intra- molecular imino-ene reaction of 3-indolylmethanol
2012年,该课题组[26]报道了Jørgensen催化剂6催化的2-甲基吲哚-3-基甲醇14与α,β-不饱和醛23的不对称[4+2]环加成反应,高立体选择性地合成了手性四氢咔唑24 (Scheme 6). 在这个反应中,原位生成的类乙烯基亚胺(离子)中间体I的2位甲基上脱去一个质子后得到二烯体Ⅱ,然后与手性α,β-不饱和亚胺中间体Ⅲ发生[4+2]环加成反应. 值得注意的是,N-甲基保护的吲哚底物没有得到目标产物.
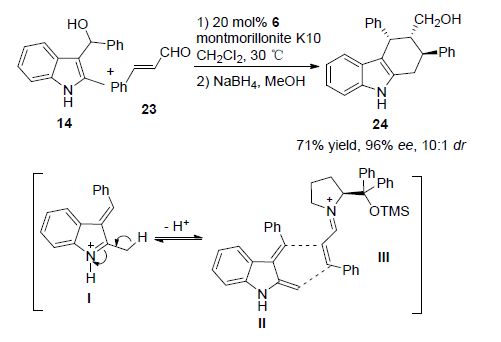 图 图式6
2-甲基吲哚-3-基甲醇和α,β-不饱和醛的不对称[4+2]环加成反应
Figure 图式6.
Asymmetric [4+2] reaction of 2-methyl-3-indolyl- methanols with α,β-unsaturated aldehydes
图 图式6
2-甲基吲哚-3-基甲醇和α,β-不饱和醛的不对称[4+2]环加成反应
Figure 图式6.
Asymmetric [4+2] reaction of 2-methyl-3-indolyl- methanols with α,β-unsaturated aldehydes
郭其祥课题组[27]通过醛的α烷基化和双傅克烷基化反应,一锅法构建了环戊烷并吲哚骨架27 (Scheme 7). 首先,手性硫脲10b和氨基酸25催化吲哚-3-基甲醇18与异丁醛26发生α烷基化得到化合物I,然后手性磷酸1a催化I与苄基保护的吲哚发生分子间傅克烷基化反应得到化合物Ⅱ,然后在1a催化剂作用下原位生成类乙烯基亚胺(离子)中间体Ⅲ,最后通过分子内傅克烷基化反应得到27. 研究证实,一锅法反应的产率比分步反应更高.
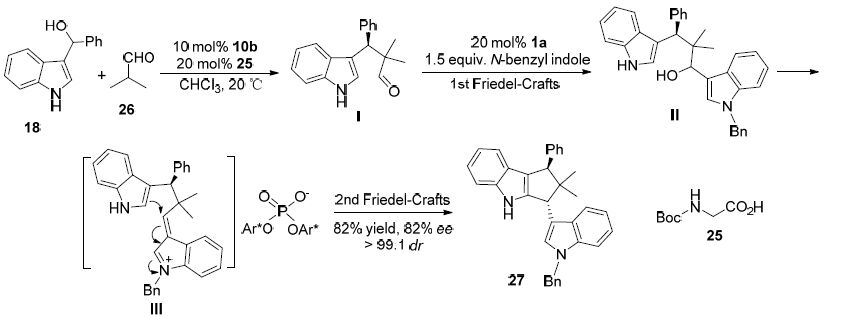 图 图式7
串联不对称醛烷基化以及双傅克烷基化反应
Figure 图式7.
Cascade asymmetric α-alkylation of aldehyde and dual Friedel-Crafts reactions
图 图式7
串联不对称醛烷基化以及双傅克烷基化反应
Figure 图式7.
Cascade asymmetric α-alkylation of aldehyde and dual Friedel-Crafts reactions
随后该课题组[28]又报道了手性磷酸催化的吲哚-3-基甲醇18与3-烯基吲哚28的[3+2]环加成反应,以较高的产率得到了目标产物,然而该反应的立体选择性却很低(Scheme 8). 在这个反应中,类乙烯基亚胺(离子)中间体I经过分子间共轭加成得到亚胺中间体Ⅱ,再经分子内共轭加成反应得到目标产物,反应立体选择性低的原因可能是在分子内傅克烷基化反应中,反应位点和手性催化剂中心距离较远的缘故. 石枫课题组在手性磷酸催化下,以较好的产率和高立体选择性实现了吲哚-3-基甲醇29分别与2位[29]或7位[30]烯基吲哚30或32的[3+2]环加成反应(Scheme 8).
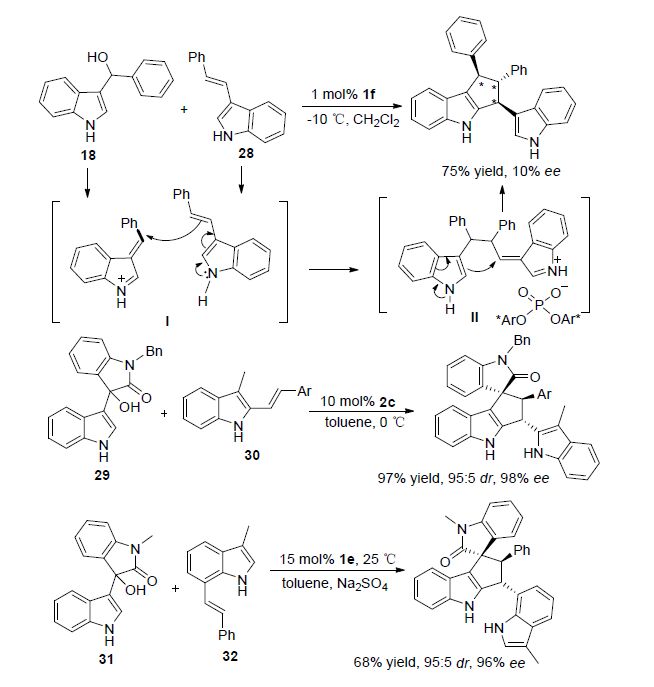 图 图式8
不对称吲哚-3-基甲醇与吲哚烯的[3+2]环加成反应
Figure 图式8.
Asymmetric [3+2] cycloaddition of vinylindoles with 3-indolylmethanols
图 图式8
不对称吲哚-3-基甲醇与吲哚烯的[3+2]环加成反应
Figure 图式8.
Asymmetric [3+2] cycloaddition of vinylindoles with 3-indolylmethanols
龚流柱课题组[31]利用H8-BINOL衍生的手性磷酸2a催化烯酰胺33和吲哚-3-基甲醇18反应,以高产率和优异的立体选择性得到了目标产物34 (Scheme 9). 随后,该课题组[32]还以吲哚-3-基甲醇16和烯酰胺35为底物,在手性磷酸1e催化下高立体选择性地得到了亲核取代产物36,并将这个反应应用到了(+)-falicanthine的全合成中(Scheme 9). 在上述反应中,手性磷酸作为一种双功能催化剂,通过与烯胺和类乙烯基亚胺(离子)中间体形成双重氢键来控制反应时的立体选择性(如过渡态TS-I、TS-Ⅱ所示). 石枫课题组[33]用吲哚-3-基甲醇31和烯胺37也实现了类似的不对称反应(Scheme 9).
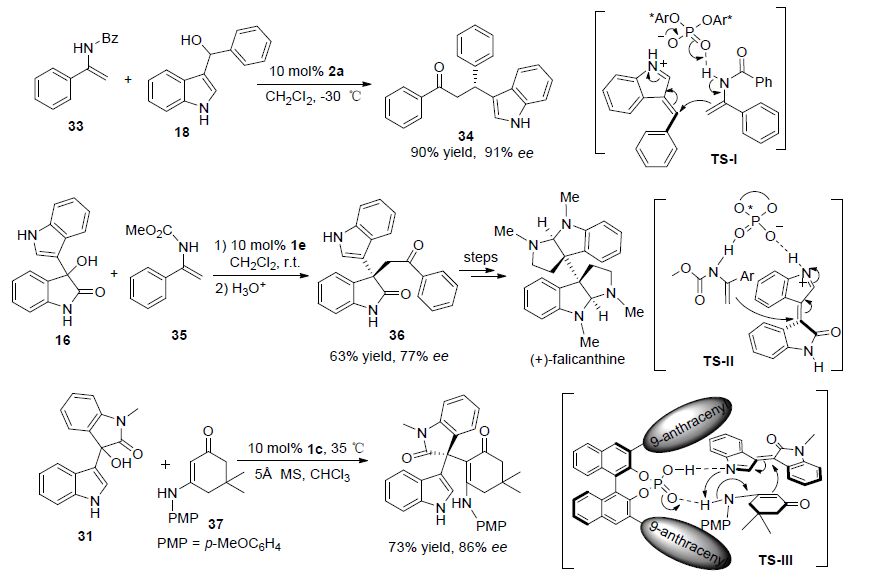 图 图式9
手性磷酸催化的吲哚-3-基甲醇与烯胺的不对称烷基化反应
Figure 图式9.
Chiral phosphoric acids catalyzed asymmetric alkylation of enamides with 3-indolylmethanols
图 图式9
手性磷酸催化的吲哚-3-基甲醇与烯胺的不对称烷基化反应
Figure 图式9.
Chiral phosphoric acids catalyzed asymmetric alkylation of enamides with 3-indolylmethanols
龚流柱课题组[34]利用手性伯胺8和手性磷酸1c协同催化实现了吲哚-3-基甲醇16与醛38的不对称α烷基化反应,高产率、高立体选择性地合成了带有多个官能团的3,3-二取代氧化吲哚(Scheme 10). 与仅用手性磷酸催化相比,手性伯胺和手性磷酸的协同催化可以明显增强反应的立体选择性,该方法已经成功地应用到(+)-gliocladin C的全合成中.
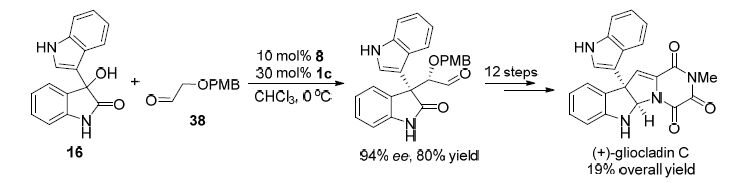 图 图式10
手性磷酸和手性胺协同催化的醛的不对称α-烷基化反应
Figure 图式10.
Chiral phosphoric acid and amine cooperatively catalyzed asymmetric α-alkylation of aldehyde
图 图式10
手性磷酸和手性胺协同催化的醛的不对称α-烷基化反应
Figure 图式10.
Chiral phosphoric acid and amine cooperatively catalyzed asymmetric α-alkylation of aldehyde
1.1.4 1,3偶极子作为亲核试剂
2014年,石枫课题组[40]报道了手性磷酸催化的类乙烯基亚胺(离子)中间体I与氮叶立德Ⅱ之间的不对称[3+3]偶极环加成反应,以较高收率和立体选择性得到手性螺环产物50 (Scheme 14,a). 在手性磷酸催化下,类乙烯基亚胺(离子)中间体I和氮叶立德中间体Ⅱ通过串联共轭加成/Pictet-Spengler反应实现了螺环产物的构建,这两步反应都是通过手性磷酸与中间体形成双重氢键实现反应立体选择性的控制. 中间体氮叶立德Ⅱ与类乙烯基亚胺离子I可分别由对硝基苯甲醛48/2-氨基丙二酸酯49和吲哚-3-基甲醇29原位生成. 当用靛红代替对硝基苯甲醛时也可以实现类似的反应(Scheme 14,b)[41].
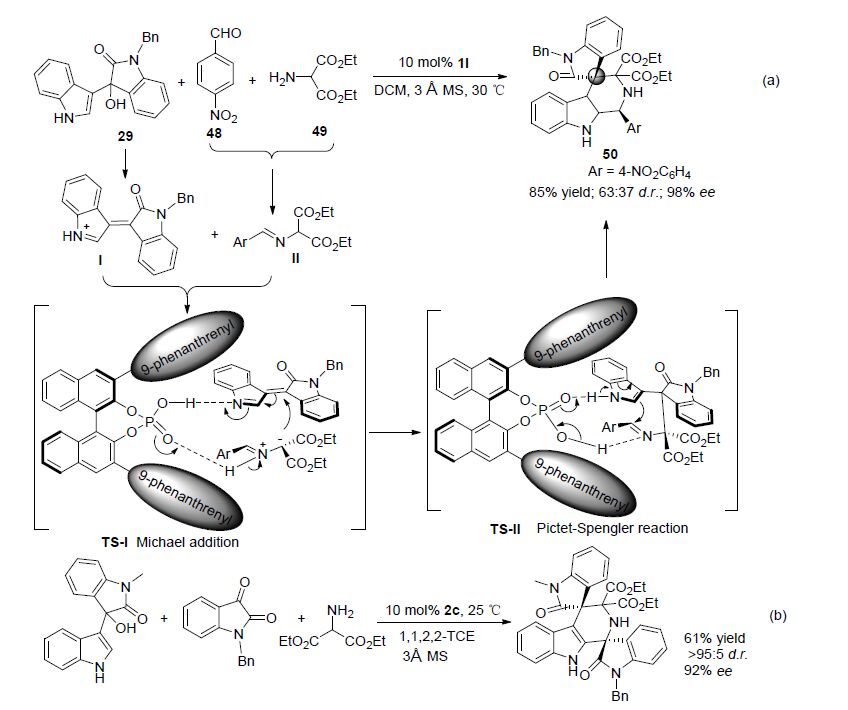 图 图式14
吲哚-3-基甲醇与α-氨基醛酮的不对称烷基化反应
Figure 图式14.
Asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols
图 图式14
吲哚-3-基甲醇与α-氨基醛酮的不对称烷基化反应
Figure 图式14.
Asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols
1.1.3 活泼亚/次甲基作为亲核试剂
α-氨基醛同样可以通过吲哚-3-基甲醇作为烷基化试剂来进行α-烷基化制备非天然的α-氨基酸衍生物. 郭其祥课题组[37]通过手性硫脲10a和对硝基苯甲酸(PNBA)协同催化实现了吲哚-3-基甲醇18与α-氨基醛43的不对称α烷基化反应,以99%收率和96% ee得到目标产物(Scheme 12). 随后,该课题组[38]还发展了手性醛10a与3,5-二硝基苯甲酸(DNBA)协同催化的吲哚-3-基甲醇18与2-氨基丙二酸酯44的不对称α烷基化反应,以高产率和优异的立体选择性得到了色氨酸衍生物45. 该反应的机理为: 在DNBA的存在下,2-氨基丙二酸酯44与手性醛12缩合形成氮叶立德I,催化剂12上的酚羟基与类乙烯基亚胺(离子)中间体Ⅱ形成分子间氢键(如过渡态Ⅲ所示),随后氮叶立德立体选择性地进攻中间体Ⅱ得到Ⅳ,最后水解得到产物45并实现催化剂再生(Scheme 12).
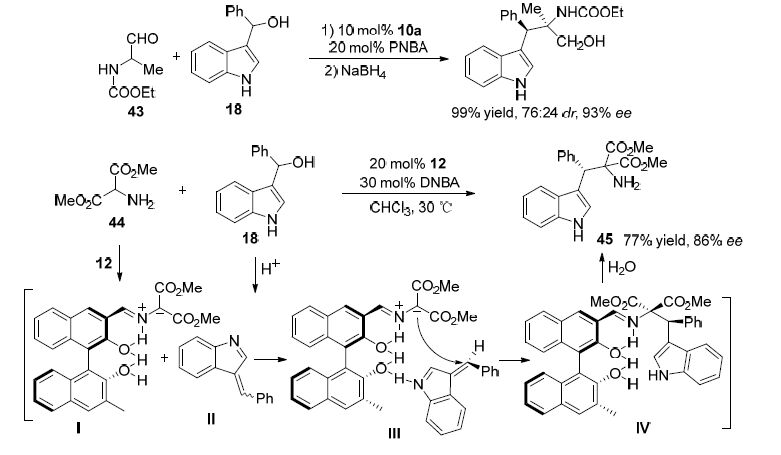 图 图式12
吲哚-3-基甲醇与α-氨基醛的不对称烷基化反应
Figure 图式12.
Asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols
图 图式12
吲哚-3-基甲醇与α-氨基醛的不对称烷基化反应
Figure 图式12.
Asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols
2015年,马军安课题组[39]报道了手性磷酸1h催化的β-酮酸46和吲哚-3-基甲醇31的不对称烷基化反应(Scheme 13),以高产率和高立体选择性得到亲核取代脱羧产物47. 其手性催化机理为: β-酮酸的羧基与手性磷酸的磷酰氧键形成分子间氢键,同时类乙烯基亚胺(离子)中间体与手性磷酸阴离子形成紧密离子对(如过渡态I所示),继而β-酮酸的烯醇式能够立体选择性地进攻类乙烯基亚胺(离子)中间体,再经过脱羧后得到烷基化产物.
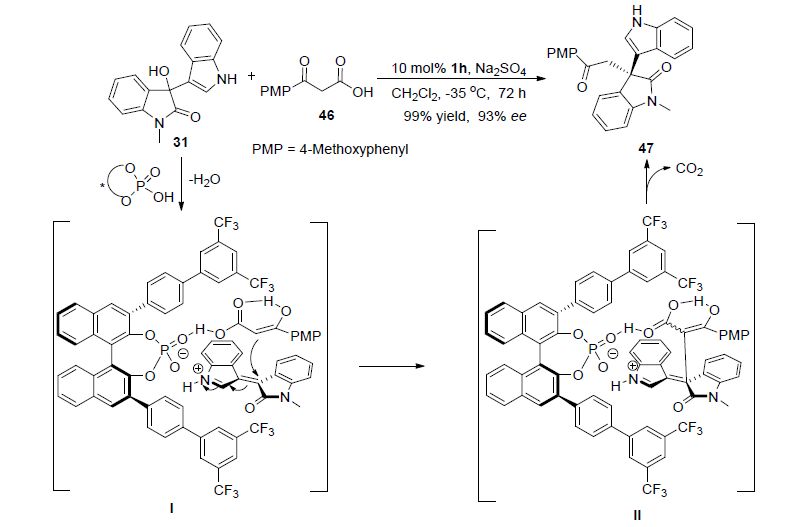 图 图式13
吲哚-3-基甲醇与β-酮酸的不对称烷基化反应
Figure 图式13.
Asymmetric alkylation of β-ketone carboxylic acid with 3-indolylmethanols
图 图式13
吲哚-3-基甲醇与β-酮酸的不对称烷基化反应
Figure 图式13.
Asymmetric alkylation of β-ketone carboxylic acid with 3-indolylmethanols
1.2.2 苯的傅克烷基化
游书力课题组[45]以吲哚58与2-甲酰基联苯59为底物,在手性磷酸1b催化下成功实现了立体选择性的双傅克烷基化串联反应,得到了光学活性的芴衍生物60 (Scheme 17). 手性磷酸在两次傅克烷基化反应中分别起着不同的作用. 在第一次傅克烷基化反应中,手性磷酸通过氢键来活化羰基; 在第二次傅克烷基化反应中,手性磷酸起双重作用,首先是质子化中间体I的羟基并脱水产生中间体Ⅱ,二是通过手性磷酸阴离子和类乙烯基亚胺(离子)中间体Ⅱ形成紧密手性离子对控制反应的立体选择性. 他们还发现[46]当用N-三氟甲磺酰基取代的手性磷酸4催化该反应时,也能以良好的收率和高达94%的e e值得到目标产物60. 值得一提的是,N-三氟甲磺酰基取代的手性磷酸4与手性磷酸1b催化该反应得到的立体选择性正好相反.
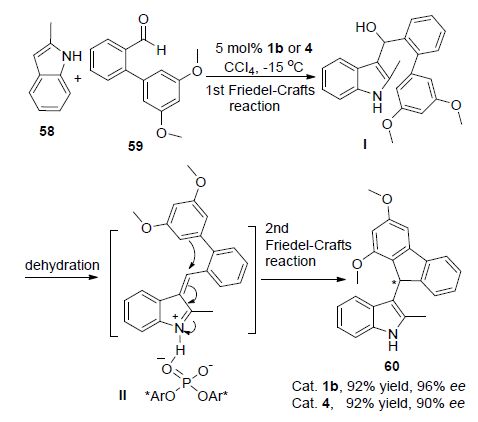 图 图式17
手性磷酸催化的串联双傅克烷基化反应
Figure 图式17.
Chiral phosphoric acid catalyzed tandem double Friedel-Crafts reaction
图 图式17
手性磷酸催化的串联双傅克烷基化反应
Figure 图式17.
Chiral phosphoric acid catalyzed tandem double Friedel-Crafts reaction
1.2.1 吲哚/吡咯的傅克烷基化
Rueping课题组[42]报道了手性磷酰胺3催化的吲哚与不饱和酮酸酯52的不对称傅克烷基化反应,以中等的立体选择性得到双吲哚产物53 (Scheme 15). 该反应机理为: 吲哚51与不饱和酮酸酯52先进行傅克烷基化反应原位生成吲哚-3-基甲醇I,然后在酸作用下生成类乙烯基亚胺(离子)中间体Ⅱ,再与另一分子吲哚发生傅克烷基化反应得到双吲哚产物53. 反应的较低的立体选择性归因于氮原子被甲基保护,阻碍了手性磷酸催化剂与吲哚NH形成氢键.
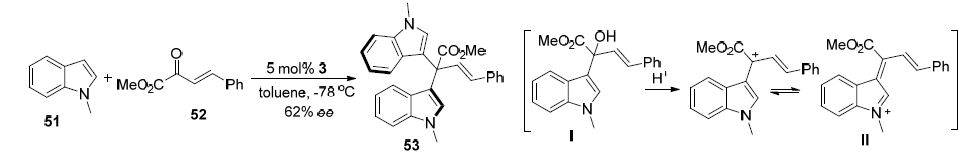 图 图式15
吲哚与不饱和酮酸酯的不对称烷基化反应
Figure 图式15.
Asymmetric Friedel-Crafts alkylation of indole with unsaturated keto-ester
图 图式15
吲哚与不饱和酮酸酯的不对称烷基化反应
Figure 图式15.
Asymmetric Friedel-Crafts alkylation of indole with unsaturated keto-ester
TMS保护的吲哚-3-基甲醇也可以转变成类乙烯基亚胺(离子)中间体,继而与富电子芳香化合物进行傅克烷基化反应. 姜毅君课题组[43]利用手性双磷酸5为催化剂,用很低的催化剂量(1 mol%)就能够高立体选择性地催化底物54与吡咯/吲哚的不对称傅克烷基化反应,得到一系列吡咯基或双吲哚取代的手性三芳基甲烷55a和55b (Scheme 16). 反应同样是通过类乙烯基亚胺(离子)中间体与手性磷酸阴离子形成紧密手性离子对来实现反应立体选择性控制.
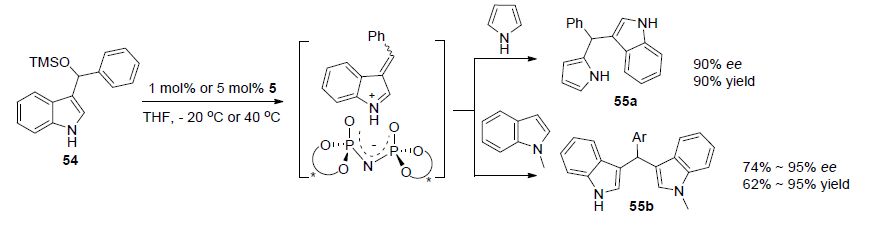 图 图式16
手性双磷酸催化的TMS保护吲哚-3-基甲醇的傅克烷基化反应
Figure 图式16.
Chiral diphosphoric acids catalyzed Friedel-Crafts of TMS-protected 3-indolylmethanols
图 图式16
手性双磷酸催化的TMS保护吲哚-3-基甲醇的傅克烷基化反应
Figure 图式16.
Chiral diphosphoric acids catalyzed Friedel-Crafts of TMS-protected 3-indolylmethanols
石枫课题组[44]以3-甲基吲哚56为亲核试剂,在手性磷酸1g催化下高产率、高立体选择地合成了具有季碳手性中心的3,3'-二吲哚基氧化吲哚57,产率可达99%,对映体比例为91:9 (Eq. 3).反应通过3-甲基吲哚56中的N—H键与手性磷酸形成分子间氢键实现反应活性和立体选择性. 当底物31中吲哚N—H键被甲基保护后,就得不到目标产物. 氧化吲哚N—H键不影响反应活性但降低反应的立体选择性,若N—H键被甲基保护后则对反应的立体选择性有利. 这是因为氧化吲哚和3-甲基吲哚N—H键都可以与手性磷酸形成氢键并且相互竞争,降低了手性磷酸与3-甲基吲哚上N—H键形成氢键的几率,因此会降低反应的立体选择性.
2 不对称还原反应
周永贵等[47]的工作表明,通过手性H8-BINAP和三氟乙酸钯催化的2-甲基吲哚-3-基甲醇14的不对称氢化反应能够以很高的立体选择性得到在药物化学上有重要应用的2,3-二取代的吲哚啉骨架61 (Scheme 18,a). 在对甲苯磺酸作用下,吲哚-3-基甲醇14脱水得到类乙烯基亚胺离子中间体I,然后经过1,4-氢加成得到2,3-二取代的吲哚中间体Ⅱ,再经酸化后得到亚胺离子中间体Ⅲ,最后经过1,2-氢加成得到2,3-二取代吲哚啉61. 随后的研究证实,上述反应亦可“一锅法”得到手性2,3-二取代吲哚61 (Scheme 18,b) [48].
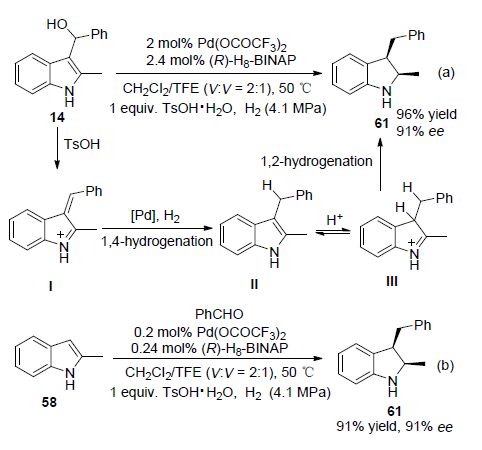 图 图式18
手性钯络合物催化的吲哚-3-基甲醇的不对称氢化反应
Figure 图式18.
Chiral palladium complex catalyzed asymmetric hydrogenation of 3-indolylmethanols
图 图式18
手性钯络合物催化的吲哚-3-基甲醇的不对称氢化反应
Figure 图式18.
Chiral palladium complex catalyzed asymmetric hydrogenation of 3-indolylmethanols
3 不对称重排反应
Antilla课题组[49]在手性磷酸2b催化下成功实现了吲哚-3-基二醇62的不对称频哪醇重排反应,以高收率和优异的立体选择性得到手性α-吲哚酮63 (Eq. 4). 值得注意的是,当氮原子被甲基、苄基或烯丙基保护时,同样可以得到很好的立体选择性和产率,这有别于上面所讨论的通过抗衡离子导向的不对称催化(ACDC)策略来控制反应过程中的立体选择性. 该反应可能的机理是手性磷酸和二醇的两个羟基形成双重氢键,脱去一分子水后得到的类乙烯基亚胺(离子)中间体上剩余的羟基与手性磷酸的磷酰氧形成分子间氢键,进而控制苯基迁移的立体选择性.
4 结论与展望
本文综述了吲哚-3-基甲醇通过不对称烷基化反应、不对称还原反应以及不对称重排反应三种不同的反应模式合成光学活性3-取代吲哚衍生物的方法. 以上所有反应都要经过碳正离子中间体或者类乙烯基亚胺(离子)中间体,反应都具有产率高、立体选择性高、副产物少等特点,预计这些反应将在含有吲哚骨架复杂分子的全合成中获得广泛应用. 尽管手性仲胺、手性磷酸或手性硫脲等催化剂已成功应用到光学活性3-取代吲哚衍生物的合成中,但依然存在着催化剂价格昂贵,催化剂用量较大以及底物普适性不强等问题. 因此发展底物容易制备、普适性强、高选择性的有机催化协同体系以及发展新的反应模式和反应策略,仍然是这个领域亟待解决的问题. 鉴于手性吲哚-3-基甲醇衍生物的重要性以及当前对环境保护的日益重视,我们也期待更高效和更绿色的合成方法出现,例如水相反应以及无酸体系下的不对称反应等[50].
-
-
[1]
Lo, K. K.-W.; Tsang, K. H.-K.; Hui, W.-K.; Zhu, N. Chem. Commun. 2003, 2704.
-
[2]
Plimmer, J. R.; Gammon, D. W.; Ragsdale, N. N. Encyclopedia of Agrochemicals, Vol. 3, John Wiley & Sons, New York, 2003.
-
[3]
Ramirez, A.; Garcia-Rubio, S. Curr. Med. Chem. 2003, 10, 1891.
-
[4]
Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489. doi: 10.1021/cr900211p
-
[5]
Goveky, S. P.; Overman, L. E. Tetrahedron 2007, 63, 8499. doi: 10.1016/j.tet.2007.05.127
-
[6]
Lancianesi, S.; Palmoeri, A.; Petrini, M. Chem. Rev. 2014, 114, 7108. (b) Shiri, M.; Zolfigol, M. A. Chem. Rev. 2010, 110, 2250. (c) Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Chem. Soc. Rev. 2010, 39, 4449.
-
[7]
Ramesh, C.; Kavala, V.; Kuo, C. W.; Rama, R. B.; Yao, C. F. Eur. J. Org. Chem. 2010, 2010, 3796. doi: 10.1002/ejoc.v2010:20
-
[8]
Furstner, A.; Radkowski, K.; Peters, H. Angew. Chem., Int. Ed. 2005, 44, 2777. doi: 10.1002/anie.200462215
-
[9]
Usami, Y.; Yamaguchi, J.; Numata, A. Heterocycles 2004, 63, 1123.
-
[10]
Zhan, Z. P.; Yang, R. F.; Lang, K. Tetrahedron Lett. 2005, 46, 3859. doi: 10.1016/j.tetlet.2005.03.174
-
[11]
Auria, M. Tetrahedron 1991, 47, 9225.
-
[12]
Rabindran, S. K.; Ross, D. D.; Doyle, L. A.; Yang, W. D.; Greenberger, L. M. Cancer Res.2000 ,60, 47.
-
[13]
Bergman, J.; Venemalm, L. Tetrahedron Lett. 1988, 29, 2993. doi: 10.1016/0040-4039(88)85068-8
-
[14]
Richou, R. M.; Lallouette, P.; Richou, H. C.R. Acad. Sci. 1967, 264, 2426.
-
[15]
Usami, Y.; Yamaguchi, J.; Numata, A. Heterocycles 2004, 63, 1123.
-
[16]
Conn, P., M.; Crowley, W. F., Jr. Annu. Rev. Med. 1994, 45, 391. doi: 10.1146/annurev.med.45.1.391
-
[17]
Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed.2009, 48, 9608. (b) Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Chem. Soc. Rev. 2010, 39, 4449. (c) Zeng, M.; You, S.-L. Synlett 2010, 1289 doi: 10.1002/anie.200901843
-
[18]
Lyttle, D. A.; Weisblat, D. I. J. Am. Chem. Soc. 1947, 69, 2118. (b) Semenov, B. B.; Granik, V. G. Pharm. Chem. J. 2004, 38, 287. (c) Palmieri, A.; Petrini, M.; Shaikh, R. R. Org. Biomol. Chem. 2010, 8, 1259. (d) Wang, L.; Chen, Y.-Y.; Xiao, J. Asian J. Org. Chem. 2014, 3, 1036. doi: 10.1021/ja01201a015
-
[19]
Kataja, A. O.; Masson, G. Tetrahedron 2014, 70, 8783. doi: 10.1016/j.tet.2014.06.101
-
[20]
Cozzi, P. G.; Benfatti, F.; Zoli, L. Angew. Chem., Int. Ed. 2009, 48, 1313. doi: 10.1002/anie.200805423
-
[21]
Zhang, Y.; Wang, S.-Y.; Xu, X.-P.; Jiang, R.; Ji, S.-J. Org. Biomol. Chem. 2013, 11, 1933.
-
[22]
Xiao, J.; Zhao, K.; Loh, T.-P. Chem. Asian J. 2011, 6, 2890.
-
[23]
Bandini, M.; Tragni, M. Org. Biomol. Chem. 2009, 7, 1501. (b) Emer, E.; Sinisi, R.; Capdevila, M. G.; Petruzziello, D.; De Vincentiis, F.; Cozzi, P. G. Eur. J. Org. Chem. 2011, 2011, 647. (c) Cozzi, P.; Gualandi, A. Synlett 2013, 24, 281. (d) Kumar, R.; Eycken, E. V. V. d. Chem. Soc. Rev. 2013, 42, 1121. doi: 10.1039/b823217b
-
[24]
Xiao, J. Org. Lett. 2012, 14, 1716. (b) Xiao, J.; Zhao, K.; Loh, T.-P. Chem. Commun. 2012, 48, 3548.
-
[25]
Han, B.; Xiao, Y.-C.; Yao, Y.; Chen, Y.-C. Angew. Chem., Int. Ed. 2010, 49, 10189.
-
[26]
Xiao, Y.-C.; Zhou, Q.-Q.; Dong, L.; Liu, T.-Y.; Chen, Y.-C. Org. Lett. 2012, 14, 5940.
-
[27]
Xu, B.; Guo, Z. L.; Jin, W. Y.; Wang, Z. P.; Peng, Y. G.; Guo, Q. X. Angew. Chem., Int. Ed. 2012, 51, 1059. doi: 10.1002/anie.v51.4
-
[28]
Zhang, C.; Zhang, L.-X.; Qiu, Y.; Xu, B.; Zong, Y.; Guo, Q.-X. RSC Adv. 2014, 4, 6916.
-
[29]
Tan, W.; Li, X.; Gong, Y.-X.; Ge, M.-D.; Shi, F. Chem. Commun. 2014, 50, 15901
-
[30]
Shi, F.; Zhang, H.-H.; Sun, X.-X.; Liang, J.; Fan, T.; Tu, S.-J. Chem. Eur. J. 2015, 21, 3465
-
[31]
Guo, Q.-X.; Peng, Y.-G.; Zhang, J.-W.; Song, L.; Feng, Z.; Gong, L.-Z. Org. Lett. 2009, 11, 4620.
-
[32]
Guo, C.; Song, J.; Huang, J.-Z.; Chen, P.-H.; Luo, S.-W.; Gong, L.-Z. Angew. Chem., Int. Ed. 2012, 51, 1046.
-
[33]
Tan, W.; Du, B.-X.; Li, X.; Zhu, X.; Shi, F.; Tu, S.-J. J. Org. Chem. 2014, 79, 4635
-
[34]
Song, J.; Guo, C.; Adele, A.; Yin, H.; Gong, L. Z. Chem. Eur. J. 2013, 19, 3319. doi: 10.1002/chem.201204522
-
[35]
Song, L.; Guo, Q.-X.; Li, X.-C.; Tian, J.; Peng, Y.-G. Angew. Chem., Int. Ed. 2012, 124, 1935.
-
[36]
Ren, C.-L.; Zhang, T.; Wang, X.-Y.; Wu, T.; Ma, J.; Xuan, Q.-Q.; Wei, F.; Huang, H.-Y.; Wang, D.; Liu, L. Org. Biomol. Chem. 2014, 12, 9881. doi: 10.1039/C4OB02035A
-
[37]
Guo, Z.-L.; Xue, J.-H.; Fu, L.-N.; Zhang, S.-E.; Guo, Q.-X. Org. Lett. 2014, 16, 6472.
-
[38]
Xu, B.; Shi, L.-L.; Zhang, Y.-Z.; Wu, Z.-J.; Fu, L.-N.; Luo, C.-Q.; Zhang, L.-X.; Peng, Y.-G.; Guo, Q.-X. Chem. Sci. 2014, 5, 1988.
-
[39]
Ma, J.-A.; Dong, X.-D.; Li, S.; Guo, R.; Nie, J. Org. Lett. 2015, 17, 1389.
-
[40]
Shi, F.; Tu, S. J.; Zhu, R.Y. Chem. Eur. J. 2014, 20, 2597. doi: 10.1002/chem.v20.9
-
[41]
Dai, W.; Lu, H.; Li, X.; Shi, F.; Tu, S.-J. Chem. Eur. J. 2014, 20, 11382.
-
[42]
Rueping, M.; Nachtsheim, B. J.; Moreth, S. A.; Bolte, M. Angew. Chem., Int. Ed. 2008, 47, 593. doi: 10.1002/(ISSN)1521-3773
-
[43]
Zhuo, M.-H.; Jiang, Y.-J.; Fan, Y.-S.; Gao, Y.; Liu, S.; Zhang, S. Org. Lett. 2014, 16, 1096.
-
[44]
Sun, X.-X.; Du, B.-X.; Zhang, H.-H.; Ji, L.; Shi, F. ChemCatChem 2015, 7, 1211.
-
[45]
Sun, F.-L., Gu. Q.; Zeng, M.; You, S.-L. Chem. Eur. J. 2009, 15, 8709.
-
[46]
Wang, S.-G.; Han, L.; Zeng, M.; Sun, F.-L.; Zhang, W.; You, S.-L. Org. Biomol. Chem. 2012, 10, 3202.
-
[47]
Wang, D.-S.; Tang, J.; Zhou, Y.-G.; Chen, M.-W.; Yu, C.-B.; Duan, Y.; Jiang, G.-F. Chem. Sci. 2011, 2, 803.
-
[48]
Duan, Y.; Chen, M.-W.; Ye, Z.-S.; Wang, D.-S.; Chen, Q.-A.; Zhou, Y.-G. Chem. Eur. J. 2011, 17, 7193.
-
[49]
Liang, T.; Zhang, Z.; Antilla, J. C. Angew. Chem., Int. Ed. 2010, 49, 9734. doi: 10.1002/anie.201004778
-
[50]
Non-asymmetric alkylation and arylation of 3-indolylmethanol were implemented in our group under catalyst-free condition with water and trifluoroethanol as reaction media: (a) Wen, H.; Wang, L.; Xu, L.; Hao, Z.; Shao, C.-L.; Wang, C.-Y.; Xiao, J. Adv. Synth. Catal. 2015, 357, 4023. (b) Xiao, J.; Wen, H.; Wang, L.; Xu, L.; Hao, Z.; Shao, C.-L.; Wang, C.-Y. Green Chem. 2016, 18, 1032. doi: 10.1002/adsc.201500500
-
[1]
-

图式1 通过捕捉类乙烯基亚胺离子合成3-取代吲哚衍生物
Scheme 1 Synthesis of 3-substituted indole derivatives via capture of vinylogous iminium intermediates

图式2 两种常用吲哚-3-基甲醇的比较
Scheme 2 Comparison of two types of 3-indolylmethanols which are commonly used

图式3 MacMillan催化剂催化的醛的α位不对称烷基化反应
Scheme 3 Asymmetric alkylation of aldehydes catalyzed by MacMillan catalyst

图式4 由协同催化体系催化的醛的不对称α烷基化反应
Scheme 4 Asymmetric α-alkylation of aldehydes catalyzed by cooperative systems

图式5 吲哚-3-基甲醇的串联不对称烷基化/分子内亚氨基- Ene反应
Scheme 5 Cascade asymmetric alkylation of aldehydes/intra- molecular imino-ene reaction of 3-indolylmethanol

图式6 2-甲基吲哚-3-基甲醇和α,β-不饱和醛的不对称[4+2]环加成反应
Scheme 6 Asymmetric [4+2] reaction of 2-methyl-3-indolyl- methanols with α,β-unsaturated aldehydes

图式7 串联不对称醛烷基化以及双傅克烷基化反应
Scheme 7 Cascade asymmetric α-alkylation of aldehyde and dual Friedel-Crafts reactions

图式8 不对称吲哚-3-基甲醇与吲哚烯的[3+2]环加成反应
Scheme 8 Asymmetric [3+2] cycloaddition of vinylindoles with 3-indolylmethanols

图式9 手性磷酸催化的吲哚-3-基甲醇与烯胺的不对称烷基化反应
Scheme 9 Chiral phosphoric acids catalyzed asymmetric alkylation of enamides with 3-indolylmethanols

图式10 手性磷酸和手性胺协同催化的醛的不对称α-烷基化反应
Scheme 10 Chiral phosphoric acid and amine cooperatively catalyzed asymmetric α-alkylation of aldehyde

图式11 手性磷酸催化的酮的不对称α烷基化反应
Scheme 11 Chiral phosphoric acid catalyzed asymmetric α- alkylation of ketones

图式12 吲哚-3-基甲醇与α-氨基醛的不对称烷基化反应
Scheme 12 Asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols

图式13 吲哚-3-基甲醇与β-酮酸的不对称烷基化反应
Scheme 13 Asymmetric alkylation of β-ketone carboxylic acid with 3-indolylmethanols

图式14 吲哚-3-基甲醇与α-氨基醛酮的不对称烷基化反应
Scheme 14 Asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols

图式15 吲哚与不饱和酮酸酯的不对称烷基化反应
Scheme 15 Asymmetric Friedel-Crafts alkylation of indole with unsaturated keto-ester

图式16 手性双磷酸催化的TMS保护吲哚-3-基甲醇的傅克烷基化反应
Scheme 16 Chiral diphosphoric acids catalyzed Friedel-Crafts of TMS-protected 3-indolylmethanols

图式17 手性磷酸催化的串联双傅克烷基化反应
Scheme 17 Chiral phosphoric acid catalyzed tandem double Friedel-Crafts reaction
-


 下载:
下载:



















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 3316
- HTML全文浏览量: 1082
 下载:
下载:
