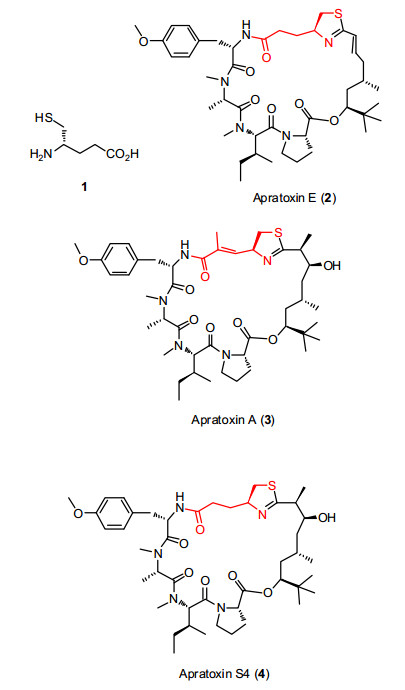
 图 1
(S)-4-氨基-5-巯基戊酸及含有此单元的活性化合物结构
Figure 1.
Structures of (S)-4-amino-5-mercaptopentanoic acid and some bioactive compounds bearing this unit
图 1
(S)-4-氨基-5-巯基戊酸及含有此单元的活性化合物结构
Figure 1.
Structures of (S)-4-amino-5-mercaptopentanoic acid and some bioactive compounds bearing this unit
引用本文:
吴平, 马春华, 胥森瀚, 李英霞, 翟延君, 张伟. (S)-4-氨基-5-巯基戊酸的简便合成方法[J]. 有机化学,
2015, 36(1): 191-195.
doi:
10.6023/cjoc201509005

Citation: Wu Ping, Ma Chunhua, Xu Senhan, Li Yingxia, Zhai Yanjun, Zhang Wei. Facile Synthesis of (S)-4-Amino-5-mercaptopentanoic Acid[J]. Chinese Journal of Organic Chemistry, 2015, 36(1): 191-195. doi: 10.6023/cjoc201509005
Citation: Wu Ping, Ma Chunhua, Xu Senhan, Li Yingxia, Zhai Yanjun, Zhang Wei. Facile Synthesis of (S)-4-Amino-5-mercaptopentanoic Acid[J]. Chinese Journal of Organic Chemistry, 2015, 36(1): 191-195. doi: 10.6023/cjoc201509005
(S)-4-氨基-5-巯基戊酸的简便合成方法
摘要:
(S)-4-氨基-5-巯基戊酸(Glutamate thiol, GluSH)是谷氨酸α-羧基被亚甲基巯基取代的衍生物, 也可被视为半胱氨酸的衍生物.该氨基酸是海洋抗肿瘤环酯肽Apratoxin E及其结构优化物的重要组成片段.报道(S)-4-氨基-5-巯基戊酸两种简便合成方法.第一种方法是以巯基和氨基均被保护的D-半胱氨酸为原料, 先与米氏酸缩合生成β-酮酯, 然后还原消除酮羰基、脱羧形成内酰胺, 最后去除巯基和氨基上的保护基以及将内酰胺环打开, 以4步76.0%的总收率得到所需产物.第二种方法以氨基和γ-羧基被保护的谷氨酸为原料, 先将α-羧基还原为伯醇, 然后借助Mitsunobu反应引入巯基, 最后一锅脱除氨基、巯基和羧基上的保护基得到(S)-4-氨基-5-巯基戊酸.
-
关键词:
- (S)-4-氨基-5-巯基戊酸
- / γ-氨基酸
- / Mitsunobu反应
- / 合成
English
Facile Synthesis of (S)-4-Amino-5-mercaptopentanoic Acid
Abstract:
(S)-4-Amino-5-mercaptopentanoic acid (glutamate thiol, GluSH) is a derivative of glutamic acid with α-carboxyl group substituted by methanethiol group. Meanwhile, it could be considered as a derivative of cysteine. This amino acid is an important fragment of marine cytotoxic cyclic depsipeptide apratoxin E. Two facile synthetic methods of (S)-4-amino-5-mercaptopentanoic acid are described. For the first strategy, protected D-cysteine derivate was used as starting material. After coupled with Meldrum's acid, the resulting β-keto ester underwent reductive elimination and thermal decarboxylative ring closure to afford 5-substituted pyrrolidinone (γ-lactam). After removal of protecting groups and ring-open, the target compound was obtained in 76.0% overall yield. For the second method, commercial available Boc-Glu(OBzl)-OH was used as starting material. The thiol group was introduced by Mitsunobu reaction after converting the carboxylic acid to its corresponding alcohol. Then the protecting groups of amino, thiol, and carboxylic acid were removed smoothly in one pot to yield (S)-4-amino-5-mercaptopentanoic acid.
-
Key words:
- (S)-4-amino-5-mercaptopentanoic acid
- / γ-amino acids
- / Mitsunobu reaction
- / synthesis
-
与天然氨基酸相比, 非天然氨基酸大多具有独特的理化性质, 在化学、药物及材料等领域的研究中获得了广泛的应用.(S)-4-氨基-5-巯基戊酸(Glutamate thiol, 简写作GluSH, 1)是谷氨酸α-羧基被亚甲基巯基取代的产物, 也可被视为半胱氨酸的衍生物(图 1).文献[1, 2]报道, GluSH是谷氨酰氨肽酶(glutamyl aminopeptidase)的抑制剂, 在降血压药物的研发中有潜在的应用价值.(S)-4-氨基-5-巯基戊酸也是具有显著生物活性的海洋天然产物Apratoxin E(2)的组成片段[3].Luesch研究小组[4]利用该片段替换Apratoxin A(3)原(S, E)-2-甲基-4-氨基-5-巯基-2-戊烯酸后, 得到其衍生物4.该衍生物保持了Apratoxin A抗肿瘤活性的同时, 降低了其毒副作用.此外, (S)-4-氨基-5-巯基戊酸还是多个潜在药物的重要中间体[5~7].已有文献报道了4-氨基-5-巯基戊酸或其类似物的合成方法[4, 8, 9].若按照现有方法制备(S)-4-氨基-5-巯基戊酸, 则路线偏长, 或者关键步骤收率偏低.因此, 发展该化合物的高效合成方法具有重要的意义.
图 1
(S)-4-氨基-5-巯基戊酸及含有此单元的活性化合物结构
Figure 1.
Structures of (S)-4-amino-5-mercaptopentanoic acid and some bioactive compounds bearing this unit
1 结果与讨论
以天然氨基酸为原料制备非天然氨基酸策略主要有碳链延长法和官能团转换法两种, 我们分别将这两种策略应用于(S)-4-氨基-5-巯基戊酸的合成.
我们首先尝试了碳链延长的策略, 即以D-半胱氨酸衍生物5为起始原料的合成路线(Scheme 1).化合物5与米氏酸缩合, 反应结束后, 经过简单的洗涤、浓缩即可得到β-酮酯6.在酸性条件下用硼氢化钠将6中的酮羰基还原消除后, 所得产物7在甲苯中回流即可生成内酰胺8.该步反应结束后将反应液减压浓缩至干, 然后加入浓盐酸再回流1 h, 浓缩后重结晶, 即可得到(S)-4-氨基-5-巯基戊酸盐酸盐(1).
 图 图式1
目标化合物的合成路线一
Figure 图式1.
The first synthetic route for the target compound
图 图式1
目标化合物的合成路线一
Figure 图式1.
The first synthetic route for the target compound
由于化合物5价格较高, 因此我们又尝试了官能团转化的策略, 即以廉价易得的试剂9为起始原料的路线(Scheme 2).将谷氨酸衍生物9游离的α-羧基活化为混合酸酐后还原为伯醇11, 然后由Mitsunobu反应引入巯基得到1a/1b后, 再在酸性条件下一锅脱除氨基、巯基和羧基上的保护基, 得到(S)-4-氨基-5-巯基戊酸的盐酸盐1.
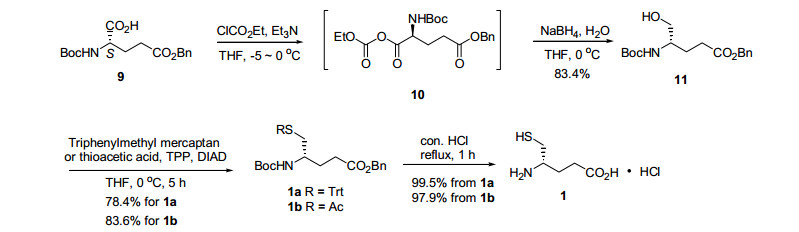 图 图式2
目标化合物的合成路线二
Figure 图式2.
The other synthetic route for the target compound
图 图式2
目标化合物的合成路线二
Figure 图式2.
The other synthetic route for the target compound
以上两种方法各有特色.方法一最大的优点是每一步反应的后处理都极其简单, 反应时间短、收率高, 且中间体和目标产物均为白色固体, 有利于产物的纯化, 整条路线仅需在最后一步进行重结晶即可.其不足之处在于, 所用起始原料为价格昂贵的D-半胱氨酸衍生物, 从而使这条路线的制备成本偏高.
方法二以商品化且廉价的谷氨酸衍生物9为原料, 仅需要三步(两锅)反应即可转化为半胱氨酸衍生物1a/1b, 而且可以根据需要选择性脱除单一官能团上的保护基.此外, Mitsunobu反应除了可以将羟基转变成巯基外, 还能转变成其他多种基团, 如氨基等.所以, 该路线在衍生物的多样性上优于第一种方法.其不足之处在于, Mitsunobu反应之后产物为粘稠液体, 只能通过柱层析进行纯化, 在大量制备时较为繁琐.如果以粗产品直接进行水解反应, 则目标产物的纯化难度有所增加, 即使多次重结晶依然难以获得较高纯度的目标产物.
以上两种方法均能顺利制备(S)-4-氨基-5-巯基戊酸, 经HPLC检测, 其纯度均高于98%.为了检测目标产物的光学纯度, 我们使用手性柱对不同方法制备的(S)-4-氨基-5-巯基戊酸进行了ee值的测定, 结果如表 1所示.不同方法制备的目标产物ee值均高于99%, 说明两种方法均不会引起明显的消旋化.
 表 1
不同方法制备的目标产物1的数值
Table 1.
The data of the target product 1 prepared by different methods
表 1
不同方法制备的目标产物1的数值
Table 1.
The data of the target product 1 prepared by different methods
方法 中间体 1收率/% 比旋光度a 纯度/% ee/% 1 8 76.0 +26.6 >98 >99 2 1a 65.1 +28.1 >98 >99 2 1b 68.3 +27.2 >98 >99 a测定条件为: 25 ℃, 0.5 g/100 mL. 表 1 不同方法制备的目标产物1的数值
Table 1. The data of the target product 1 prepared by different methods2 结论
本文研究了两种用于制备(S)-4-氨基-5-巯基戊酸的方法.第一种方法是以巯基和氨基均被保护的D-半胱氨酸为原料, 先与米氏酸缩合生成β-酮酯, 然后还原消除酮羰基、脱羧形成内酰胺, 最后去除巯基和氨基上的保护基以及将内酰胺环打开, 以4步76.0%的总收率得到所需产物.该路线各步反应时间短、收率较高, 中间体易于纯化, 但所用原料价格较高, 经济性略差.第二种方法是以氨基和γ-羧基保护的谷氨酸为原料, 先将α-羧基还原为伯醇, 然后借助Mitsunobu反应引入巯基, 最后在酸性条件下一锅脱除氨基、巯基和羧基上的保护基, 以4步65.1%~68.3%的总收率得到(S)-4-氨基-5-巯基戊酸.该方法所用原料及试剂均廉价易得, 且借助Mitsunobu反应有利于实现产物的多样化衍生, 但中间体需以柱层析进行纯化, 大量制备时操作略显繁琐.
3 实验部分
3.1 仪器与试剂
Bruker AM 400型核磁共振仪(瑞士Bruker公司), 以CDCl3或D2O为溶剂, TMS为内标; Agilent LCMSD质谱仪(美国Agilent公司); AB 5600+Q高分辨质谱仪(美国AB SCIEX公司); Rudolph Autopol IV-T型自动旋光仪(美国鲁道夫).
薄层层析硅胶、柱层析硅胶购自烟台江友硅胶开发有限公司, N-Boc-S(Trt)-D-半胱氨酸(5)、N-Boc-γ-苄酯谷氨酸(9)购自吉尔生化(上海)有限公司, 其余试剂购自上海泰坦科技股份有限公司旗下探索平台.
3.2 化合物的合成
3.2.1 中间体6的合成
依次将Boc-S-三苯甲基-D-半胱氨酸(5.00g, 10.79 mmol)、米氏酸(1.71 g, 11.86 mmol)溶于100 mL干燥二氯甲烷中, 冰浴下加入4-二甲氨基吡啶(1.98 g, 16.18 mmol).搅拌10 min后, 加入二环己基碳二亚胺(2.45 g, 11.86 mmol), 并继续在此温度下搅拌4 h, 直至LC-MS和TLC显示反应完全.加入饱和氯化铵淬灭反应, 并依次用1 mol/L盐酸、饱和碳酸氢钠及饱和氯化钠洗涤, 无水硫酸钠干燥.过滤, 滤液减压浓缩后得淡棕黄色粉末状固体, 乙醚洗涤两次后, 得5.67 g白色粉末状固体6, 产率89.2%.该中间体可直接用于下步反应.Rf=0.49 [V(石油醚):V(乙酸乙酯)=1:2];m.p.92~95 ℃; [α]D24-47.3(c 1.0, CHCl3); 1H NMR(400 MHz, CDCl3)δ: 1.40(s, 9H), 1.71(s, 6H), 2.59(br, 1H), 2.59(br, 1H), 2.75(br, 1H), 5.02(br, 1H), 5.55(br, 1H), 7.16~7.39(m, 15H); 13C NMR(100 MHz, CDCl3)δ: 202.5, 193.7, 170.8, 167.6, 159.5, 154.9, 148.6, 144.3, 144.0, 129.6, 129.5, 128.9, 127.1, 105.7, 91.4, 84.5, 80.4, 67.5, 66.7, 66.1, 66.0, 52.2, 43.5, 33.9, 32.5, 31.1, 28.4, 28.0, 27.1, 26.9, 15.4;HRMS(ESI-TOF)calcd for C33H35NNaO7S [M+Na]+ 612.2026, found 612.2005.
3.2.5 中间体1a的合成
将三苯基膦(3.93 g, 15.00 mmol)溶于50 mL无水四氢呋喃中, 冰浴条件下滴加偶氮二甲酸二异丙酯(2.98 mL, 15.00 mmol).30 min后将上步产物11(3.23 g, 10.00 mmol)和三苯基甲硫醇(4.15 g, 15.00 mmol)溶于20 mL无水四氢呋喃, 并滴加至此反应体系中.室温下继续搅拌5 h直至LC-MS显示反应完全.饱和氯化铵淬灭反应, 减压浓缩除去大部分溶剂后, 用100 mL乙酸乙酯稀释, 有机相用5%硫酸氢钾水溶液洗至水相近无色.硅胶柱层析[V(石油醚):V(乙酸乙酯)=10:1]得4.56 g无色透明粘稠液体, 产率78.4%.Rf=0.5 [V(石油醚):V(乙酸乙酯)=4:1];[α]D25-6.8(c 0.72, CHCl3); 1H NMR(400 MHz, CDCl3)δ: 1.41(s, 9H), 1.57~1.69(m, 1H), 1.71~1.81(m, 1H), 2.25~2.32(m, 4H), 3.64(s, 1H), 4.46(d, J=8.4 Hz, 1H), 5.08(s, 2H), 7.18~7.40(m, 20H); 13C NMR(100 MHz, CDCl3)δ: 172.9, 155.2, 144.6, 135.8, 129.6, 128.5, 128.2, 127.9, 126.7, 79.3, 66.6, 66.3, 60.4, 49.4, 37.1, 31.0, 29.6, 28.4, 21.1, 14.2;HR
MS(ESI-TOF)calcd for C36H39NO4S [M+Na]+604.2492, found 604.2492.
3.2.2 中间体7的合成
将中间体6 (5.67 g, 9.60 mmol)溶于100 mL二氯甲烷, 并加入冰醋酸(6.79 mL, 118.69 mmol).冰浴条件下分批加入硼氢化钠(1.02 g, 26.98 mmol), 继续搅拌1 h直至TLC显示反应完全.反应液依次用1 mol/L盐酸、饱和碳酸氢钠及饱和氯化钠洗涤, 无水硫酸钠干燥.过滤, 滤液减压浓缩后得4.74 g白色粉末状固体7, 产率85.6%.该中间体可直接用于下步反应.Rf=0.24 [V(石油醚):V(乙酸乙酯)=2:1];m.p.98~99 ℃; [α]D24+7.0(c 0.5, CHCl3); 1H NMR(400 MHz, CDCl3)δ: 1.40(s, 9H), 1.73(s, 3H), 1.77(s, 3H), 2.07~2.19(m, 2H), 3.92(d, J=5.0 Hz, 1H), 4.01(td, J=8.9, 3.3 Hz, 1H), 4.56(d, J=8.2 Hz, 1H), 7.20~7.24(m, 3H), 7.27~7.31(m, 6H), 7.40~7.42(m, 6H); 13C NMR(100 MHz, CDCl3)δ: 171.3, 165.5, 166.5, 155.7, 144.6, 129.7, 128.2, 127.0, 105.1, 79.9, 67.1, 60.6, 49.4, 48.5, 44.6, 37.5, 33.1, 31.7, 29.9, 28.8, 28.4, 26.0, 25.8, 25.1, 22.8, 21.2, 14.4, 13.5;HRMS(ESI-TOF)calcd for C33H37NNaO6S [M+Na]+ 598.2234, found 598.2200.
3.2.3 中间体8的合成
将上步反应所得中间体7溶于100 mL甲苯, 回流3 h直至TLC显示反应完全.减压浓缩得3.90 g白色固体8, 产率100 %, 该中间体可直接用于下步反应.Rf=0.51 [V(石油醚):V(乙酸乙酯)=2:1];m.p.181~183 ℃; [α]D24-65.2(c 1.1, CHCl3); 1H NMR(400 MHz, CDCl3)δ: 1.46(s, 9H), 1.76~1.82(m, 1H), 1.93~2.04(m, 1H), 2.26~2.36(m, 3H), 2.54(dd, J=11.4, 3.1 Hz, 1H), 4.11~4.17(m, 1H), 7.19~7.24(m, 3H), 7.25~7.32(m, 6H), 7.41~7.43(m, 5H); 13C NMR(100 MHz, CDCl3)δ: 173.5, 148.8, 143.8, 128.8, 127.4, 126.2, 82.4, 66.1, 55.9, 34.5, 30.6, 27.4, 21.5;HRMS(ESI-TOF)calcd for C29H31NNaO3S [M+Na]+ 496.1917, found 496.1910.
3.2.4 中间体11的合成
将Boc-Glu(OBzl)-OH(5.50 g, 16.30 mml)溶于80 mL无水THF中, 加入三乙胺(2.80 mL, 32.60 mmol), 并在氩气保护下降温至-5 ℃.向其滴加氯甲酸乙酯(1.75 mL, 29.30 mmol), 立即有大量盐析出.滴毕, 在-5 ℃下继续搅拌30 min, 分批加入硼氢化钠(1.85 g, 48.9 mmol), 再滴加500 μL水.至反应液无气泡生成后, 撤去冷媒使其自然升温, 1 h后LC-MS显示反应完全.缓慢滴加1 mol/L盐酸淬灭反应, 减压浓缩除去大部分有机溶剂.浓缩液用乙酸乙酯稀释, 依次用饱和碳酸氢钠和饱和氯化钠溶液洗涤, 无水硫酸钠干燥.过滤, 滤液减压浓缩得粘稠物.加入乙醚溶解, 在冰浴条件下边搅拌边缓慢滴加正己烷至大量白色固体析出.减压抽滤, 滤饼用V(石油醚):V(乙酸乙酯)=10:1洗涤后真空干燥, 得4.40 g白色粉末状固体, 产率83.4%.Rf=0.58 [V(石油醚):V(乙酸乙酯)=1:1];[α]D25-10.5(c 1.0, MeOH){文献值[10][α]D25-10(c=1.0, MeOH)}; 熔点及化合物表征数据与文献[10]一致.
3.2.6 中间体1b的合成
按照中间体1a的合成方法, 用硫代乙酸代替三苯基甲硫醇, 其余均不变.得3.19 g无色透明粘稠液体, 产率83.6%.Rf=0.4 [V(石油醚):V(乙酸乙酯)=5:1];[α]D25-14.0(c 1.0, CHCl3){文献值[8] [α]D25-8.8(c 0.8, CHCl3)}; 化合物表征数据与文献[8]一致.
3.2.7 (S)-4-氨基-5-巯基戊酸盐酸盐(1)的合成
将中间体8 (50.0 mg, 0.10 mmol)悬浮于2 mL浓盐酸并回流1 h.C-MS显示反应完全后, 乙酸乙酯洗涤(5 mL×3)除去脂溶性杂质, 水相减压浓缩所得粗产品经丙酮洗涤后得20.1 mg白色固体1, 产率100%.若以1a或1b为原料, 产率分别为99.5%和97.9%.m.p.178~180 ℃; HRMS(ESI-TOF)calcd for C5H12NO2S [M+H]+ 150.0583, found 150.0579;化合物表征数据与文献[6]一致.
辅助材料(Supporting Information) 所有新化合物的1H NMR、13C NMR和HRMS图谱, 已知化合物的1H NMR图谱以及目标化合物光学纯度测定方法.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
Wilk, S.; Thlirston, L. S. Neuropeptides 1990, 16, 163. doi: 10.1016/0143-4179(90)90129-M
-
[2]
Claperon, C.; Rozenfeld, R.; Iturrioz, X.; Inguimbert, N.; Okada, M.; Roques, B.; Maigret, B.; Llorens-Cortes, C. Biochem. J. 2008, 416, 37. doi: 10.1042/BJ20080471
-
[3]
Matthew, S.; Schupp, P. J.; Luesch, H. J. Nat. Prod. 2008, 71, 1113. doi: 10.1021/np700717s
-
[4]
Chen, Q. Y.; Liu, Y.; Cai, W.; Luesch, H. J. Med. Chem. 2014, 57, 3011. doi: 10.1021/jm4019965
-
[5]
Comegna, D.; Paola, I.; Saviano, M.; Del Gatto, A.; Zaccaro, L. Org. Lett. 2015, 17, 640. doi: 10.1021/ol503664t
-
[6]
Bergeron, R. J.; Wiegand, J.; Weimar, W. R.; Vinson, J. R.; Timothy, B.; Jörg, Y.; Guo, Wei.; McManis, J. S. J. Med. Chem. 1999, 42, 95. doi: 10.1021/jm980340j
-
[7]
Jobron, L.; Hummel, G. Org. Lett. 2000, 2, 2265. doi: 10.1021/ol006019o
-
[8]
Longobardo, L.; Cecere, N.; DellaGreca, M.; de Paola, I. Amino Acids 2013, 44, 443. doi: 10.1007/s00726-012-1352-5
-
[9]
Smrcina, M.; Majer, P.; Majerová, E.; Guerassina, T. A.; Eissenstat, M. A. Tetrahedron 1997, 53, 12867. doi: 10.1016/S0040-4020(97)00840-5
-
[10]
Truchot, C.; Wang, Q.; Sasaki, N. A. Eur. J. Org. Chem. 2005, 9, 1765.
-
[1]
-

图 1 (S)-4-氨基-5-巯基戊酸及含有此单元的活性化合物结构
Figure 1 Structures of (S)-4-amino-5-mercaptopentanoic acid and some bioactive compounds bearing this unit
表 1 不同方法制备的目标产物1的数值
Table 1. The data of the target product 1 prepared by different methods
方法 中间体 1收率/% 比旋光度a 纯度/% ee/% 1 8 76.0 +26.6 >98 >99 2 1a 65.1 +28.1 >98 >99 2 1b 68.3 +27.2 >98 >99 a测定条件为: 25 ℃, 0.5 g/100 mL.  下载: 导出CSV
下载: 导出CSV
-




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1898
- HTML全文浏览量: 423
 下载:
下载:
